

Abstract
We have characterized, in vitro, interactions between hippocampal neuronal cells and silica microbeads coated with synthetic, fluid, lipid bilayer membranes containing the glycosylphosphatidyl inositol (GPI)-linked extracellular domain of the postsynaptic membrane protein neuroligin-1. These bilayer–neuroligin-1 beads activated neuronal cells to form presynaptic nerve terminals at the point of contact in a manner similar to that observed for live PC12 cells, ectopically expressing the full length neuroligin-1. The synthetic membranes exhibited biological activity at neuroligin-1 densities of ∼1 to 6 proteins/μm2. Polyolycarbonate beads with neuroligin-1 covalently attached to the surface failed to activate neurons despite the fact that neuroligin-1 binding activity is preserved. This implies that a lipid membrane environment is likely to be essential for neuroligin-1 activity. This technique allows the study of isolated proteins in an environment that has physical properties resembling those of a cell surface; proteins can diffuse freely within the membrane, retain their in vivo orientations, and are in a nondenatured state. In addition, the synthetic membrane environment affords control over both lipid and protein composition. This technology is easily implemented and can be applied to a wide variety of cellular studies.
Introduction
The lipid bilayer architecture of cell membranes creates a uniquely complex surface environment, consisting of a nanoscale fluid emulsion of proteins, lipids, and cholesterol. The biological functionality of proteins within membranes is substantially affected by factors influencing both accessibility and spatial organization. For example, interactions between cells and polymer surfaces displaying the RGD peptide motif exhibit a pronounced sensitivity to the nanoscale clustering state of the RGD peptide.1 Similar effects have been observed through the use of multivalent ligands that, in the solution state, facilitate aggregation of corresponding membrane-associated receptors.2-7 Indeed, intrinsically bivalent or multivalent cross-linked antibodies to cell surface receptors are widely used to initiate signaling, and the multivalent nature of these binding interactions can be used to explore the role of protein oligomerization in signal transduction. Of relevance to the work described here, this strategy has been employed to expose a role of receptor clustering in the neurexin–neuroligin signaling system8 and in the ephrin/Eph receptor signaling system.9
Neurexins constitute a large family of neuronal cell surface receptors, with β-neurexins believed to be receptors for the postsynaptic cell surface protein, neuroligin-1.8,10 Nonneuronal cells (e.g., HEK293 or PC12) ectopically expressing neuroligin-1 (or neuroligin-2) can induce the formation of presynaptic elements in contacting axons, in vitro. Oligomerization of neuroligin is required for this function, and β-neurexin clustering is sufficient to recruit synaptic vesicles. Thus, postsynaptic neuroligin oligomers appear to induce clustering of axonal neurexins, and rearrangement of neurexins nucleates assembly of the neuronal intracellular protein complex responsible for neurotransmitter secretion.8
Planar supported membranes have been used extensively as an in vitro method of studying molecular interactions at membrane surfaces.11,12 Supported membranes can be formed by spontaneous adsorption and fusion of lipid vesicles with a solid substrate such as silica and various polymers.13-15 The process is generally selflimiting at a single bilayer, resulting in a single, continuous membrane coating on the substrate. A thin layer of water between the membrane and substrate preserves the lateral mobility of both lipids16-18 and membrane-associated proteins.19,20 Supported membranes displaying glycosylphosphatidylinositol (GPI)-linked forms of the major histocompatibility complex (MHC) and intercellular adhesion molecule (ICAM) have been used as surrogate antigen-presenting cells (APCs) for interaction with living T cells. A dynamic immunological synapse can form between the T cell and the supported membrane involving micrometer-scale spatial rearrangement of both supported membrane and T cell surface proteins.21 This hallmark example of a functional interaction between a living cell and a supported membrane clearly illustrates the dramatic extent to which the lateral mobility of proteins can be coupled to activity.
Here we describe functional interactions between a GPI-linked form of neuroligin-1 in supported membranes and primary neuronal cells in culture. Silica beads, typically 5 μm in diameter, were used as the supported membrane substrate. One reason for this choice of substrate configuration is the ease with which beads can be added to a preexisting cell culture. Membranes were formed on the silica beads by a process analogous to that used to form planar supported membranes.16,22,23 Beads coated with neuroligin-1-containing membranes activated neurons in a manner similar to live PC12 cells expressing neuroligin-1. Neuroligin-1 was active at densities of ∼1 to 6 proteins/μm2 and appeared to require a membrane environment for functionality; naked polystyrene beads to which neuroligin-1 was covalently attached failed to activate neuronal cells. This suggests that the lipid bilayer bead is superior to established methods of attaching proteins to beads for a number of possible reasons; the orientation and configuration of the protein is preserved by the bilayer, the amount of protein present on the beads can be quantitated directly, and the membrane environment preserves a fluid environment similar to that of a cell membrane, in which proteins can diffuse freely.
Results and Discussion
The aim of the experiments described below is to determine if a neuron can be induced to form synapses when brought into contact with a GPI-linked form of the postsynaptic adhesion protein, neuroligin-1, incorporated into synthetic lipid bilayers that are coated onto silica beads (Figure 1A). The GPI-linked neuroligin-1 binds to β-neurexin present in the membrane of the axons of neurons in culture. This binding interaction induces the recruitment of synaptic vesicles to the site of neuroliginneurexin interaction and the formation of a functional presynaptic terminal at the site. Figure 1B shows a bright field image of silica microbeads, coated with lipid bilayers containing GPI-linked neuroligin-1, contacting a neuron in culture.
Figure 1.
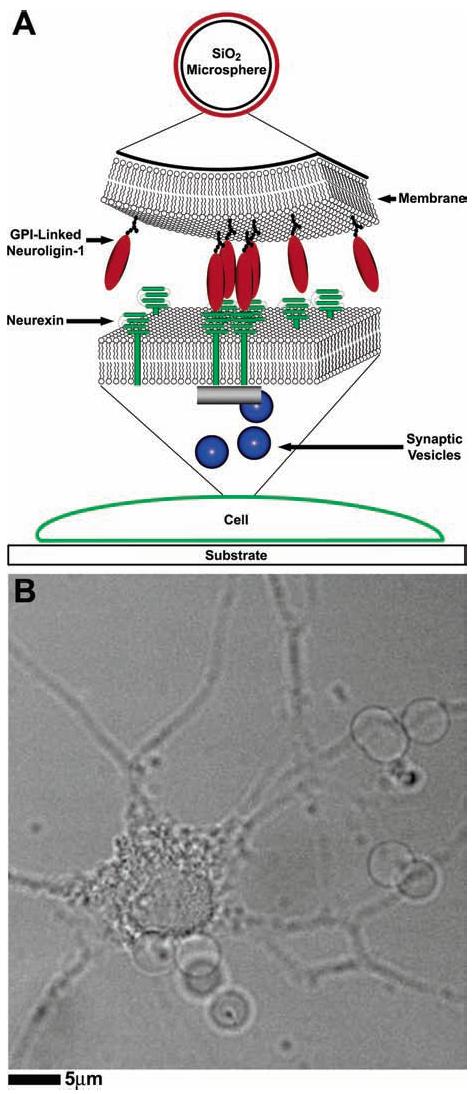
(A) Schematic diagram of the experiment: GPI-linked neuroligin-1 (red) incorporated into a synthetic lipid bilayer on a silica microbead binds to β-neurexin (green) expressed in a neuron. Formation of the neuroligin–neurexin complex results in the eventual recruitment of scaffolding proteins (grey) and exocytosis-competent synaptic vesicles (blue). (B) Membrane-coated beads were added to hippocampal cell cultures, allowed to settle by gravity, and incubated for 24 h. Transmission image of beads coated with synthetic lipid bilayers containing GPI-linked neuroligin-1, interacting with a hippocampal neuron in culture.
GPI-linked proteins can be incorporated into supported membranes, preserving both lateral mobility and biological functionality.11,19-21,24,25 Here, an engineered GPI-linked form of neuroligin-1 was incorporated into artificial bilayers on silica beads, which were then manipulated into contact with neuronal cells containing endogenous neurexin (Figure 1). Unlike more conventional methods, such as covalent binding, which can cause indiscriminate binding of proteins to surfaces, the GPI linkage favors insertion into the membrane in such a way that a proper orientation is maintained. The GPI-linked neuroligin-1 was expressed in HEK293 cells and purified by affinity chromatography. The purified protein was reconstituted into lipid vesicles via detergent dialysis, and these proteoliposomes were used to create supported membranes on 5 μm diameter silica beads.16,22,23,26 These proteomembrane-coated beads were stored in 1× PBS at 4 °C for up to 4 h. A silver-stained protein gel shows neuroligin-1 recovered from a preparation of proteoliposome-coated beads (Figure 2A). This illustrates another advantage of using membrane-coated beads; proteins incorporated onto membrane-coated beads can be easily removed to quantitate the amount of protein present and ensure that it is in a nondenatured state.
Figure 2.
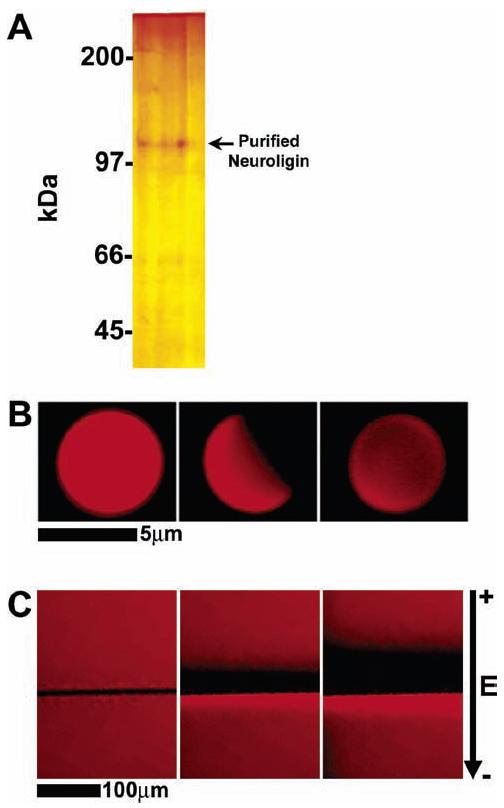
(A) Silver-stained gel of reconstituted and repurified GPI-linked neuroligin-1. Purified, recombinant GPI-linked neuroligin-1 reconstituted into proteoliposomes and subsequently repurified from them runs as a single protein band at the predicted size. (B) FRAP figure demonstrating the mobility of a neuroligin-1 containing lipid bilayer on a 5 μm diameter silica bead. Red fluorescence is due to Texas red-DPPE in the membrane: full illumination prior to bleach (left), exposure pattern during bleach (middle), full illumination 1 min after bleach (right). (C) Application of an electric field demonstrating the mobility of a lipid bilayer containing neuroligin-1 on a planar silica substrate. The strength of the electric field applied to the membrane is 18 V/cm, and the direction of the field is indicated by the arrow on the right: membrane under no electric field (left), electric field applied to membrane after 5 min (center), electric field applied to membrane after 10 min (right).
Membrane fluidity is another distinct advantage of using membrane-coated beads. Membrane-associated proteins and adhesion molecules often undergo dynamic, two-dimensional reorientation in response to binding to extracellular partners. Such molecules may require a membrane-like environment in which fluidity is preserved in order to retain in vivo functionality. Membrane fluidity of proteomembrane-coated beads was monitored in two ways: by fluorescence recovery after photobleaching (FRAP) studies of a lipid-linked fluorescent probe, Texas red-DPPE or NBD-PC, and electric field mobility observations of planar supported membranes (Figure 2). The similar diffusion characteristics of these probes in bead-supported membranes suggest that they are physically equivalent to the well-studied planar supported membranes.12,14,15
Dissociated embryonic rat hippocampal neurons were plated on polylysine-coated glass coverslips and cultured in Neurobasal medium for 10–12 days until use.27,28 For the experiment, membrane-coated beads were added to the medium and allowed to settle by gravity onto the hippocampal neurons. After incubation for an additional 24 h, both cells and beads were fixed and stained with fluorescently labeled antibodies. Interactions between the cells and beads were then analyzed using standard fluorescence microscopy techniques.
Neuronal cells can be activated through contact with cells displaying various cognate synaptic molecules on their surface, mimicking a postsynaptic neuronal partner.8,10 This is illustrated in Figure 3A, in which PC12 cells expressing the full-length neuroligin-1 are shown to trigger presynaptic differentiation in hippocampal neuronal cells. Activation consists of local clustering of neuroligin-1 on the surface of the PC12 cell at sites of contact with neuronal cells and both clustering of β-neurexin on the surface of an axonal process of the neuron and accumulation of synaptic vesicles inside the axon at the site of PC12–axon contact. PC12 cells without neuroligin-1 exhibit no discernible activating effect (not shown).
Figure 3.
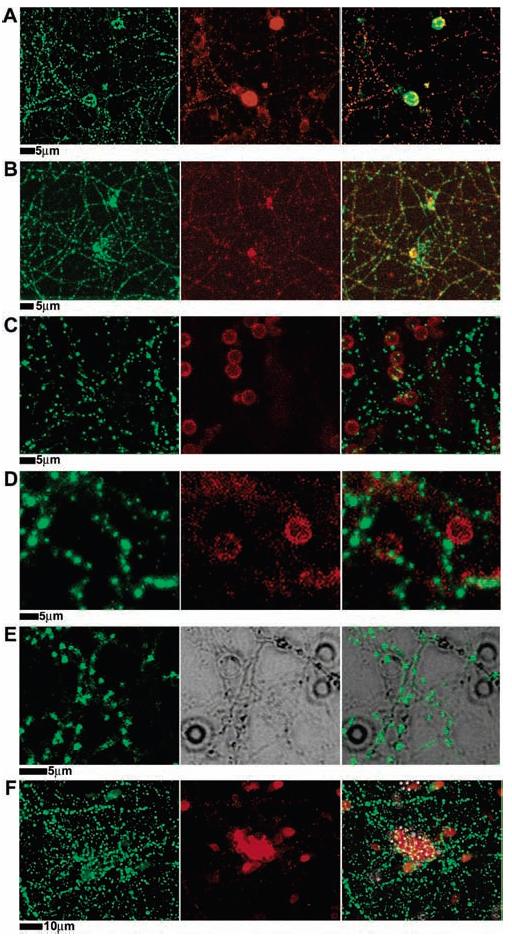
Presynaptic differentiation triggered by neuroligin-1 on the surface of cells or on lipid bilayer-coated membranes. (A) PC12 cells transfected with neuroligin-1–IRES–GFP (pseudo-colored red for consistency with other panels) were added to a 12 day old hippocampal culture, incubated overnight, then fixed and stained with antisynapsin (green in all panels) to mark presynaptic synaptic vesicles in the axons of the neuron. Synaptic vesicles accumulate, in puncta, at the site of contact with the PC12 cell. (B–F) Twelve day old hippocampal cultures interacting with beads coated with (B) synthetic lipid bilayers containing GPI-linked neuroligin-1 (neuroligin-1 stained in red), (C) synthetic lipid bilayers with no added protein (plain membranes in red), (D) synthetic lipid bilayers containing GPI-linked alkaline phosphatase (GPI-linked alkaline phosphatase in red), (E) neither synthetic lipid bilayer nor protein, bare silica beads (middle panel is DIC image of beads and neurons), and (F) with chemically coupled neuroligin-1 on polystyrene beads (neuroligin-1 in red). Cultures were fixed 24 h following introduction of beads and were stained with anti-HA antibodies for either neuroligin-1 (red), GPI-linked alkaline phosphatase (red), or synapsin (green). Microbeads coated only with synthetic lipid bilayers included 1% Texas red–DPPE in the bilayer for fluorescence imaging. Only beads with synthetic lipid bilayers containing GPI-linked neuroligin-1 induced presynaptic differentiation of the neurons, as indicated by synapsin accumulation in axons contacting the beads.
The activation of hippocampal neuronal cells by full-length neuroligin-1-expressing PC12 cells was mimicked by 5 μm diameter silica microbeads, coated with synthetic membranes containing GPI-linked neuroligin-1 external domain (Figure 3B). Neuroligin-1 on the bead coclustered with β-neurexin on the surface of the axon and synaptic vesicles inside the axon at the site of contact with the bead. Neuroligin-1 displayed on membrane-coated beads was equally effective at activating hippocampal neuronal cells as when the protein is expressed on the surface of PC12 cells. Silica glass beads coated with plain synthetic membranes without neuroligin-1 (Figure 3C), beads coated with synthetic membranes containing alkaline phosphatase, another GPI-linked protein that does not bind to proteins on neurons (Figure 3D), or bare silica glass beads (Figure 3E), all failed to activate the hippocampal neuronal cells. The effect is specific to neuroligin-1.
Polymer beads, such as polystyrene, are commonly employed to display cell surface proteins for various assays and interactions with cultured cells. This technique was tested as an alternate method to display neuroligin-1 to hippocampal neuronal cells. For this comparison, neuroligin-1 was covalently attached to 6 μm diameter polystyrene beads by means of a carbodiimide coupling reaction.29 On the basis of established data of the minimal effectiveness of this reaction, this chemical coupling strategy routinely achieved protein densities approximately 10 times greater on the polystyrene bead surface than was achieved with the membrane. Additionally, the polystyrene–carbodiimide–neuroligin-1 beads were found to adhere to HEK293 cells expressing β-neurexin (Figure 4, parts C and D) but not to cells lacking neurexin (not shown). Despite the fact that the neuroligin-1 covalently attached to the beads was capable of specific adhesion, its ability to activate the presynaptic differentiation of an axon was drastically reduced compared with that of the membrane coatings (Figure 3F). This observation illustrates the significance of a membrane environment for the natural activity of neuroligin-1.
Figure 4.
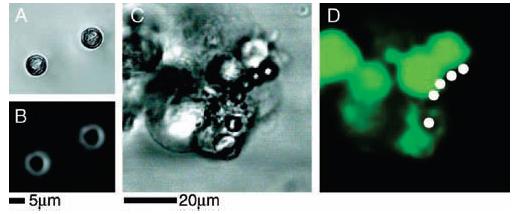
Adherence of HEK293 cells to polystyrene beads covered with chemically coupled neuroligin-1. White circles denote the positions of polystyrene beads in fluorescence images. (A, B) HEK293 cells not transfected with β-neurexin; no beads are observed to adhere to the cells and have fallen away from the coverslip after inversion of the chamber. (C) Bright field image of polystyrene beads, coupled to neuroligin-1, adhering to HEK293 cells expressing β-neurexin. In all cases, both transfected and nontransfected cells remained adhered to the glass coverslip. (D) Fluorescence image of polystyrene beads, coupled to neuroligin-1, and adhering to β-neurexin-expressing HEK293 cells.
The density of GPI-linked neuroligin-1 on the surface of the membrane-coated silica beads was determined to be between ∼1 and 6 molecules/μm2 by quantitative fluorescence. A determined number of membrane-coated beads displaying neuroligin-1, displaying a correspondingly determined total membrane surface area, were incubated with fluorescent anti-HA antibodies, which bound to an engineered HA tag on the neuroligin-1. Subsequently, the membranes containing both neuroligin-1 and the bound antibody were stripped off of the beads using a nonionic detergent, Triton X-100. The fluorescence intensity of the antibody in the resulting solution was then measured and the values compared with those from a reference set of data to determine the total amount of neuroligin-1-engaged antibody (Figures 4 and 5). From the known membrane surface area, the range of between 1 and 6 molecules/μm2 of neuroligin-1 in the membranes was determined. Given our observation that neuroligin-1 concentrates at the site of the neuronal synapse, the functional signaling area likely contains a higher protein density.
Figure 5.

Reference curve of the intensity of emitted light at a wavelength of 522 nm for different concentrations of FITC HA antibody in a 1× PBS solution with 2 vol % Triton X-100 (squares). The plain line is a linear fit to the data. Dashed lines indicate the intensity values measured for the antibody bound to neuroligin-1 incorporated in the synthetic membrane. Three samples were prepared using three different preparations of neuroligin-1, and the range of corresponding concentration of bound antibody is assumed to be directly proportional to the range of neuroligin-1 present in the synthetic membrane.
Significance
Among artificial materials, supported membranes can most closely mimic the fluid characteristic of a live cell surface. Such mimicry allows the retention of the functional aspects of natural protein behavior in vitro. This is illustrated by the functional neuroligin-1 reconstitution described here as well as previous work with immune cell surface proteins.11 In comparison to proteins displayed by direct coupling to solid surfaces, the supported membrane provides a drastically different environment. Orientation and conformation of incorporated proteins is maintained due to direct engineering of the GPI link into the protein of interest and the natural processes involved in self-assembly of the membrane bilayer. In addition, the fluidity of the membrane is likely to be an important factor in retaining in vivo functionality of membrane-associated proteins and adhesion molecules. Long-range lateral rearrangements of GPI-linked proteins in supported membranes can lead to substantial enrichments in the contact zone with a living cell. This type of reaction–diffusion process is common at cell–cell contacts. At a more subtle level, the molecular scale freedom of motion afforded within a supported membrane enables proteins to adopt differential configurations and orientations on the membrane surface. This orientational freedom may further facilitate formation of functional interactions with cognate proteins on an apposing live cell surface. The physical advantages provided by supported membranes are magnified by the general success of the GPI linkage strategy as a method of displaying cell surface proteins within an artificial membrane system. Lastly, the ease of manipulation of membrane-coated microbeads should further facilitate usage of supported membrane display strategies in a variety of live cell experiments.
Experimental Section
Materials
Lipids were obtained from Avanti Polar Lipids (Alabaster, AL). l-α egg phosphatidylcholine was received in chloroform and stored at −20 °C for up to 2 weeks. The fluorescent probe N-(Texas red sulfonyl)-1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine, triethylammonium salt (Texas red-DPPE) was purchased from Molecular Probes (Eugene, OR) in powder form and dissolved in chloroform before use. The fluorescent probe 1-palmitoyl-2-[12-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino]dodecanoyl]-sn-glycero-3-phosphocholine (NBD-PC) was purchased from Avanti Polar Lipids in chloroform and stored at −20 °C until used. Rabbit antisynapsin antibodies were purchased from Chemicon (Temecula, CA), rat anti-HA IgG fragment epitope tag 3F10 was purchased from Roche (Indianapolis, IN), and mouse antialkaline phosphatase was purchased from Sigma Chemicals (St. Louis, MO); all three were stored at 4 °C prior to use. Secondary antibodies were purchased from Jackson Immunologicals (West Grove, PA) and stored at 4 °C. Silica glass microspheres, 5 μm mean diameter, were obtained from Bangs laboratories (Fishers, IN) and stored at 4 °C under deionized H2O. Polystyrene beads, 6 μm diameter, and a carbodiimide protein-coupling kit were obtained from PolySciences Inc. (Warrington, PA) and stored at 4 °C. NTA-agarose was obtained from Qiagen (Valencia, CA).
Purification and Isolation of Protein
Recombinant HA/His-tagged GPI-anchored neuroligin-1 was expressed in HEK293 cells and isolated from cell lysates by chromatography on NTA-agarose. The protein bound to the column in phosphate-buffered saline containing NaCl (350 mM), Triton X-100 (0.5%), and imidazole (6 mM). Nonspecifically bound proteins were removed by extensive washing with PBS containing NaCl (1 M) and Triton X-100 (0.1%). The detergent was then switched to n-octyl-glucopyranoside (25 mM), and bound protein was eluted with an imidazole gradient (10–200 mM). The eluted material revealed a single protein band at the predicted size for GPI-neuroligin-1. Bands were resolved by silver staining and showed immune reactivity with specific antibodies directed against the HA tag.
Preparation of Polystyrene Beads
Beads were washed twice with carbonate buffer (0.1 M) and phosphate buffer (0.02 M), respectively, and then incubated in a carbodiimide solution (2%) for4hin order to activate surface carboxyl groups. Activated beads were washed 3 times in borate buffer (0.2 M) and mixed overnight in borate buffer (0.2 M) containing 5 μg of purified GPI-neuroligin-1. The next day, beads were incubated for 30 min in a solution of borate buffer and ethanolamine and incubated in BSA for 30 min to block unreacted sites and nonspecific binding.
Synthetic Membranes
Supported bilayers were formed by the fusion of small unilamellar vesicles (SUVs) onto clean glass microspheres. For SUV preparation, a lipid solution in chloroform was evaporated onto small round-bottomed flasks and hydrated for 60 min at 4 °C in 18.2 MΩ-cm water to a final concentration of ∼3.3 mg/mL. The lipids were probe-sonicated at 25 W for 90 s, (5 s pulses with 1 s intervals) in an ice water bath and then ultracentrifuged for 2 hat 160 000g and 4 °C. The supernatant was stored at 4 °C for up to 1 week. Proteoliposomes were formed by mixing peak fractions of purified GPI-linked neuroligin-1 in 15 mM n-octyl-glucopyranoside with preformed SUVs. The detergent was removed by dialysis against three changes of 1× PBS over the course of 18 h. The resulting proteoliposomes were stored for up to 2 days in 1× PBS at 4 °C. Bilayers were allowed to self-assemble on the microbead surface by mixing equal amounts (100 μL) of spreading solution (1:1 SUV/PBS) and microbead suspension (10% solids in deionized water) together in a 1.5 mL centrifuge tube. Excess vesicles were removed by pelleting the microbeads via pulse centrifugation and removal of the supernatant. Then, 1 mL of 18.2 MΩ-cm water was added to the pelleted microbeads and the entire mixture vortexed to allow resuspension.
Quantitation of Neuroligin-1
For neuroligin-1 quantitation, proteoliposomes were used to deposit supported membranes on 5.08 μm silica beads as described above. A typical sample contained about 7.0 ± 0.35 × 107 beads, corresponding to a total bilayer area of about 5.7 × 109 μm2. To prevent nonspecific binding of the HA antibody, the bead surface was blocked by incubation for 30 min using a 1 mg/mL solution of BSA in 1× PBS. After incubation, the beads were incubated with an excess of FITC-labeled HA antibody for another 30 min. All incubations were carried out at room temperature, and the sample tubes were placed on a rotator to prevent beads from settling and aggregating, which creates defects in the membrane. To remove all unbound antibody, the beads were washed several times by pulse centrifugation, removal of the supernatant, and resuspension in fresh 1× PBS buffer. After the last wash, the beads were resuspended in 250 μL of 1× PBS, and 2% Triton X-100 was added to remove the bilayer from the beads. After 10 min, the beads were spun down and the fluorescence corresponding to the antibody in the supernatant was measured to determine the amount of neuroligin-1 incorporated into the bilayer. Fluorescence measurements were performed on a fluorescence spectrometer (Jasco, model FP-750) with the excitation wavelength fixed at λexc = 491 nm with a 5 nm slit. The intensity of the emitted light at λem = 522 nm was detected with a slit of 10 nm for concentrations of FITC-labeled, anti-HA antibody ranging from 1 to 50 ng/mL.
Hippocampal Cultures
Hippocampi were removed from embryonic day 18 rats and treated with trypsin for 20 min at 37 °C, followed by washing and trituration. Dissociated cells were plated at 100 000 cells/cm2 on polylysine-coated glass coverslips and cultured in Neurobasal medium supplemented with Glutamax (2 mM) and B-27 (2%).
Polystyrene Bead Control
HEK293 cells were plated on glass coverslips and transfected with β-neurexin at an efficiency of ∼50%. Both transfected and nontransfected cells adhered strongly to the glass. Coverslips with cells were placed in a closed chamber, and polystyrene beads, to which neuroligin-1 was covalently coupled with a carboiimide linkage, were washed into the chamber and allowed to settle onto the cells. Following 20 min of bead–cell contact, the chamber was inverted, and the behavior of the beads and cells was observed.
Imaging
Images were taken with a Zeiss LSM510 confocal microscope. Laser power and photomultipliers were set such that no detectable bleed-through occurred between different channels. Eight to ten sections were taken from top to bottom of the specimen, and brightest point projections were made. Images were processed using Adobe Photoshop software (Adobe Systems, CA).
Supplementary Material
Acknowledgment
We thank Jeffrey Nye for providing the single-particle tracking measurements of neuroligin-1. This work was supported by the Chemical Sciences, Geosciences, and Biosciences Division, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC03-76SF00098.
Footnotes
Supporting Information Available: Direct measurements of neuroligin-1 mobility using single-particle tracking methods. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Maheswari G, et al. Cell adhesion and motility depend on nanoscale RGD clustering. J. Cell Science. 2000;113:1677–1686. doi: 10.1242/jcs.113.10.1677. [DOI] [PubMed] [Google Scholar]
- 2.Mammen M, Choi S-K, Whitesides GM. Polyvalent interactions in biological systems: implications for design and use of multivalent ligands and inhibitors. Angew. Chem. Int. Ed. 1998;37:2754–2794. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 3.He X-S, et al. Quantitative analysis of hepatitis C virus-specific CD8+ T cells in peripheral blood and liver using peptide-MHC tetramers. Proc. Natl. Acad. Sci. U.S.A. 1999;96:5692–5697. doi: 10.1073/pnas.96.10.5692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cochran JR, Cameron TO, Stern LJ. The relationship of MHC-peptide binding and T cell activation using chemically defined MHC class II oligomers. Immunity. 2000;12:241–250. doi: 10.1016/s1074-7613(00)80177-6. [DOI] [PubMed] [Google Scholar]
- 5.Kiessling LL, Gestwicki JE, Strong L. Synthetic multivalent ligands in the exploration of cell-surface interactions. Curr. Opin. Chem. Biol. 2000;4:696–703. doi: 10.1016/s1367-5931(00)00153-8. [DOI] [PubMed] [Google Scholar]
- 6.Gestwicki JE, Kiessling LL. Inter-receptor communication through arrays of bacterial chemoreceptors. Nature. 2002;415(3):81–84. doi: 10.1038/415081a. [DOI] [PubMed] [Google Scholar]
- 7.Bray D, Levin MD, Morton-Firth CJ. Receptor clustering as a cellular mechanism to control sensitivity. Nature. 1998;393(7):85–88. doi: 10.1038/30018. [DOI] [PubMed] [Google Scholar]
- 8.Dean C, et al. Neurexin mediates the assembly of presynaptic terminals. Nat. Neurosci. 2003;6(7):708–716. doi: 10.1038/nn1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stein E, et al. Eph receptors discriminate specific ligand oligomers to determine alternative signaling complexes, attachment and assembly receptors. Genes Dev. 1998;12:667–678. doi: 10.1101/gad.12.5.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scheiffele P, et al. Neuroligin expressed in nonneuronal cells triggers presynaptic development in contacting axons. Cell. 2000;101:657–669. doi: 10.1016/s0092-8674(00)80877-6. [DOI] [PubMed] [Google Scholar]
- 11.Groves JT, Dustin ML. Supported planar bilayers in studies on immune cell adhesion and communication. J. Immunol. Methods. 2003;278:19–32. doi: 10.1016/s0022-1759(03)00193-5. [DOI] [PubMed] [Google Scholar]
- 12.Sackmann E. Supported membranes: Scientific and practical applications. Science. 1996;271:43–48. doi: 10.1126/science.271.5245.43. [DOI] [PubMed] [Google Scholar]
- 13.Sackmann E, Tanaka M. Supported membranes on soft polymer cushions: fabrication, characterization and applications. Trends Biotechnol. 2000;18:58–64. doi: 10.1016/s0167-7799(99)01412-2. [DOI] [PubMed] [Google Scholar]
- 14.Groves JT. Membrane array technology for drug discovery. Curr. Opin. Drug Discovery Dev. 2002;5(4):606–612. [PubMed] [Google Scholar]
- 15.Groves JT, Boxer SG. Micropattern formation in supported lipid membranes. Acc. Chem. Res. 2002;35:149–157. doi: 10.1021/ar950039m. [DOI] [PubMed] [Google Scholar]
- 16.Bayerl TM, Bloom M. Physical properties of single phospholipid bilayers adsorbed to micro glass beads. Biophys. J. 1990;58:357–362. doi: 10.1016/S0006-3495(90)82382-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koenig BW, et al. Neutron reflectivity and atomic force microscopy studies of a lipid bilayer in water adsorbed to the surface of a silicon single crystal. Langmuir. 1996;12(5):1343–1350. [Google Scholar]
- 18.Johnson SJ, et al. Structure of an adsorbed dimyristoylphosphatidylcholine bilayer measured with specular reflection of neutrons. Biophys. J. 1991;59:289–294. doi: 10.1016/S0006-3495(91)82222-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chan P-Y, et al. Influence of receptor lateral mobility on adhesion strengthening between membranes containing LFA-3 and CD2. J. Cell Biol. 1991;115(1):245–255. doi: 10.1083/jcb.115.1.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Groves JT, Wülfing C, Boxer SG. Electrical manipulation of glycanphosphatidylinisitol-tethered proteins in supported lipid bilayers. Biophys. J. 1996;71:2716–2723. doi: 10.1016/S0006-3495(96)79462-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grakoui A, et al. The immunological synapse: A molecular machine controlling T cell activation. Science. 1999;285:221–227. [PubMed] [Google Scholar]
- 22.Buranda T, et al. Biomimetic molecular assemblies on glass and mesoporous silica microbeads for biotechnology. Langmuir. 2003;19(5):1654–1663. [Google Scholar]
- 23.Baksh MM, Jaros M, Groves JT. Detection of molecular interactions at membrane surfaces through colloid phase transitions. Nature. 2004;427:139–141. doi: 10.1038/nature02209. [DOI] [PubMed] [Google Scholar]
- 24.Tözeren A, et al. Micromanipulation of adhesion of a jurkat cell to a planar bilayer membrane containing lymphocyte function-associated antigen 3 molecules. J. Cell Biol. 1992;116(4):997–1006. doi: 10.1083/jcb.116.4.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dustin ML, et al. Visualization of CD2 interaction with LFA-3 and determination of the two-dimensional dissociation constant for adhesion receptors in a contact area. J. Cell Biol. 1996;132(3):465–474. doi: 10.1083/jcb.132.3.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sapuri AR, Baksh MM, Groves JT. Electrostatically targeted intermembrane lipid exchange with micropatterned supported membranes. Langmuir. 2003;19(5):1606–1610. [Google Scholar]
- 27.Banker GA, Cowan WM. Rat hippocampal neurons in dispersed cell culture. Brain Res. 1977;126:397–342. doi: 10.1016/0006-8993(77)90594-7. [DOI] [PubMed] [Google Scholar]
- 28.Brewer GJ, et al. Optimized survival of hippocampal neurons in B-27 supplemented Neurobasal, a new serum-free medium combination. J. Neurosci. Res. 1993;35:567–576. doi: 10.1002/jnr.490350513. [DOI] [PubMed] [Google Scholar]
- 29.Gallo G, Lecfort FB, Letourneau PC. The trkA receptor mediates growth cone turning toward a localized source of nerve growth factor. J. Neurosci. 1997;17(14):5445–5454. doi: 10.1523/JNEUROSCI.17-14-05445.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.