

Abstract
Estimating the genetic and environmental variances for multivariate and function-valued phenotypes poses problems for estimation and interpretation. Even when the phenotype of interest has a large number of dimensions, most variation is typically associated with a small number of principal components (eigen-vectors or eigenfunctions). We propose an approach that directly estimates these leading principal components; these then give estimates for the covariance matrices (or functions). Direct estimation of the principal components reduces the number of parameters to be estimated, uses the data efficiently, and provides the basis for new estimation algorithms. We develop these concepts for both multivariate and function-valued phenotypes and illustrate their application in the restricted maximum-likelihood framework.
QUANTIFYING variation in multivariate phenotypes presents four basic difficulties. First, standard methods require estimation of a large number of parameters. With k traits, there are k(k + 1)/2 genetic variances and covariances and typically an equal or larger number of parameters that describe environmental sources of variation, measurement error, etc. Limitations on the sizes of data sets and correlations among the variables cause the estimates to lose precision rapidly as the number of traits measured increases. This issue is a particular concern in evolutionary genetics, where the numbers of individuals measured are usually modest. Second, computational constraints can be limiting with large data sets. In dairy cattle, for example, it is not unusual to have several measurements taken on each of hundreds of thousands of individuals. A third issue involves numerical difficulties caused by sampling error. These can produce estimates of covariance matrices that are not within the parameter space and unstable estimates of individual variances and covariances (Hill and Thompson 1978; Hayes and Hill 1981). A fourth problem is interpretation. Patterns of covariation involving three or more variables are not readily obvious from inspecting a covariance matrix. Both the estimation and the visualization problems are particularly acute in the case of “function-valued” traits in which individuals are represented by curves, such as growth trajectories and reaction norms. Here genetic variation is naturally represented by a covariance function, rather than covariance matrix, that has an infinite number of values (Kirkpatrick and Heckman 1989).
These considerations have motivated the widespread use of data-reduction methods. The most common of these is principal components analysis (Morrison 1976). In the multivariate setting, principal components (PCs) are the eigenvectors of the covariance matrix, linear combinations of the original variables that reflect patterns of covariation in the data. In the function-valued context, the PCs are the eigenfunctions of the covariance function (Ramsay and Silverman 1997, 2002). Each eigenfunction represents a family of deformations in the shape of the average curve for the population (Kirkpatrick and Lofsvold 1992). In both the multivariate and function-valued contexts, PCs are appealing because they are statistically independent (orthogonal), describe the maximum amount of variation with the minimum number of parameters, and are easy to visualize.
Quantitative geneticists have used principal components in three ways. The first is as a tool to visualize patterns of genetic variation. In this mode, the genetic principal components are calculated from an estimate of the full genetic covariance structure (e.g., Atchley and Rutledge 1980; Kirkpatrick and Lofsvold 1992). This approach suffers from the usual problems that come with estimating a large number of parameters. The second use of principal components is to define genetic parameters to be estimated. Under special conditions, parameterizations based on principal components reduce a multivariate problem to a series of univariate ones (e.g., Hayes and Hill 1981; Meyer 1985). This approach has had limited use, however, because of the restrictive conditions it requires. A third use is to distill the original number of measurements down to a smaller number of phenotypic principal components and then estimate the genetic parameters of these PCs (e.g., Chase et al. 2002). Weaknesses with this approach are that fixed effects and selection can introduce bias into the estimates and that there is no guarantee that this is an efficient way to estimate genetic variation. For example, much information is lost when the phenotypic PCs poorly reflect patterns of genetic variation.
This article proposes putting the cart before the horse: we can estimate the leading principal components of genetic and environmental variation directly from the data, without going through the intermediate step of estimating the corresponding covariance matrix or covariance function. Several advantages follow from this direct estimation strategy. Because most genetic variation is often associated with just two or three PCs, the population can be well described with a relatively small number of parameters. With 10 traits, for example, estimating the full covariance matrix involves 55 parameters, while estimating the first two PCs involves only 19. Once the PCs have been estimated, corresponding estimates for the covariance matrix (or function) can be easily calculated. Second, the data are used efficiently. The leading PCs account for the maximum amount of variation possible with a linear combination of the trait values (Ramsay and Silverman 1997, 2002). Third, the orthogonality of PCs can be exploited in estimation algorithms (Juga and Thompson 1992). In this article we propose a stepwise algorithm in which searching for the mth PC is restricted using the results from the first m − 1 PCs, with the result that estimation becomes faster with succeeding PCs. Fourth, adding measurements of additional traits to the analysis increases the accuracy of the estimates, rather than destabilizing them by increasing the degrees of freedom. Fifth, the covariance structure estimated by the direct method is guaranteed to be positive semidefinite, which is not true of some other approaches (Hayes and Hill 1981; Kirkpatrick et al. 1990).
The direct-estimation strategy has several additional benefits when the phenotypes are function valued. By estimating a reduced number of PCs, the corresponding estimate of the covariance function is smoothed, and smoothing filters out measurement error. Different individuals can have different numbers of measurements taken at different ages. Because of the decreased computational load, it may become possible to use more desirable but more complex basis functions (such as splines) to model the covariance function. Last, the principal components for function-valued traits can be easily visualized, giving insight into patterns of variation on which selection can act (Kirkpatrick and Lofsvold 1992).
This article begins by showing how multivariate phenotypes can be represented in terms of genetic and environmental PCs, how these PCs relate to the corresponding covariance matrices, and how simplified estimates of the covariance matrices can be found using a reduced number of PCs. This idea can be applied to a wide range of estimation frameworks, including likelihood and Bayesian approaches. Next, we introduce an algorithm for fitting PCs that makes use of their orthogonality. The algorithm is again independent of the choice of statistical framework. To make the concepts of direct estimation concrete, we next show how it can be implemented using restricted maximum likelihood (REML). We then show how the direct-estimation approach extends naturally to function-valued traits. Last, we use a numerical example to highlight some of the advantages of direct estimation. Further details about the calculations underlying the direct estimation approach are given by Meyer and Kirkpatrick (2005).
REPRESENTING PHENOTYPES WITH PCs
To show how phenotypes can be represented in terms of genetic and environmental principal components, we start with the standard multivariate (MV) case with k traits. Our main goal is to estimate the additive genetic covariance matrix G, which determines the response to selection (Falconer and Mackay 1996). A second interest is to estimate the environmental covariance matrix E.
The vector phenotypic measurements for individual i can be written as the sum
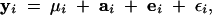 |
1 |
where μi is a mean vector (which includes the population mean and can also include effects of gender, locale, etc.), and ai is the additive genetic component (the breeding value). The vector ei represents the environmental and nonadditive genetic effects (also referred to as “permanent environmental effects” in the breeding literature). Finally, the vector εi represents the residual errors (or “temporary environmental effects”), caused, for example, by measurement error. The residual error for trait j is distributed with variance σ2εj, and we assume the residual errors for the different traits are independent. The last three terms on the right of (1) are defined to be mutually independent and have expectation 0, and we follow classical quantitative genetics by assuming they are multivariate-normally distributed. If some measurements for individual i are missing, then the corresponding elements of each vector in Equation 1 are deleted. This statistical model could be modified, for example, to include a dominance component or a different error structure.
The genetic covariance matrix G and environmental covariance matrix E are respectively equal to the variance of ai and the variance of ei across individuals sampled at random from the population. These covariance matrices can in turn be written as
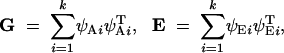 |
2 |
where ψAi is the ith eigenvector of the additive genetic covariance matrix G, and ψEi is the ith eigenvector of the environmental covariance matrix E. The eigenvectors of G are mutually orthogonal, as are those of E. Equations 2 follow immediately from the well-known spectral representation of symmetric matrices (Strang 1976).
Often eigenvectors are defined to have unit length (or norm), in which case each term in the summations of Equations 2 is modified to include an additional factor, the eigenvalues. When written in the form of Equations 2, however, the eigenvalues are absorbed into the vectors ψ. The length (norm) of each eigenvector is now equal to the square root of the corresponding eigenvalue. This parameterization is allowed because an eigenvector is determined only to within a multiplicative constant (Strang 1976). Further, doing this simplifies the calculations described below and is therefore convenient. We follow the convention that the eigenvectors are ordered in size from largest to smallest in length.
The eigenvectors ψA and ψE are the genetic and environmental PCs, respectively. We use the terms eigenvector and principal component interchangeably. These PCs are the key to our analysis. An individual's breeding values and environmental deviations for the measured traits can always be expressed as weighted sums of the genetic and environmental PCs:
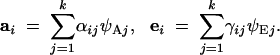 |
3 |
The vector αi = {αi1, αi2, … , αik}T is made up of the breeding values of individual i for the genetic principal components. The value of αij says how much genetic PC j contributes to the phenotype of individual i. The vector γi = {γi1, γi2, … , γik}T plays the same role for environmental PC j. Equation 3 is general: because the eigenvectors span the phenotypic space, we are guaranteed that the vectors a and e can always be written in this form (Strang 1976).
The additive genetic variance corresponding to genetic PC i is given by the square of its length (or norm),
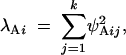 |
4 |
where ψAij is the jth element of ψAi. This quantity is an eigenvalue of the genetic covariance matrix G. The eigenvalues λEi for the environmental covariance matrix E are defined in an analogous way on the basis of the environmental PCs, the ψEi's.
By rewriting an individual's breeding value and environmental deviation in terms of the genetic and environmental PCs, we have just reparameterized Equation 1, swapping one set of variables for another. The central idea of our scheme is to simplify the estimation problem by reducing the number of terms in the sums of Equations 2 and 3 and therefore the number of parameters to be estimated. Eigenvalues typically decline rapidly in size. Consequently, the genetic and environmental covariance matrices can be well approximated by truncating the sums in Equations 2 after the first mA terms for G and the first mE terms for E, where often mA and mE may be as small as 2 or 3. We discuss how to determine appropriate values for those cutoffs below.
This then is the essence of the direct estimation approach: fitting a small number of principal components (that is, the eigenvectors that appear in Equation 2) that adequately describe the variation in the population. An important point is that our parameterization automatically ensures that the estimated covariance matrix will be positive semidefinite. That is, there can be no negative eigenvalues for a covariance function written in the form of Equation 2. This immediately eliminates a source of bias that plagues other approaches for estimating genetic parameters (Hayes and Hill 1981).
The simple idea underlying our direct estimation approach can be applied in a wide range of frameworks for statistical inference. Later we show how it can be implemented using restricted maximum likelihood. But first we outline an algorithm that can be used to fit the PCs.
AN ALGORITHM TO SEARCH FOR PCs
Here we discuss a three-step algorithm for fitting PCs that takes advantage of their orthogonality. Briefly, the algorithm is to estimate the first genetic and first environmental PCs. We then search for the estimates of the second and subsequent PCs, restricting the search to the parameter space that is orthogonal to the PCs that have already been estimated. Once an adequate number of PCs have been estimated, we finish with a final optimization in which the estimated PCs are rotated and their lengths are perturbed. We emphasize that this algorithm is not a mandatory part of the direct estimation approach: the PCs can be fitted with other search algorithms.
To estimate parameter values, we need to adopt a framework for statistical inference. In the following section we show how the direct estimation approach can be implemented with restricted maximum likelihood, but the idea could be applied with other paradigms such as Bayesian inference. In this section we use the generic phrase “optimizing the fit,” which in the likelihood framework means finding the parameter value that maximizes the likelihood.
Figure 1 sketches the algorithm in graphical form. Step 1 is to estimate the leading genetic and environmental principal components. With k traits, we search a k-dimensional space for the first genetic PC, ψA1, and for the first environmental PC, ψE1. The search continues until we converge on estimates of ψA1 and ψE1 that optimize the fit (Figure 1B).
Figure 1.—

Schematic of the search algorithm when fitting m = 2 principal components to k = 3 traits. (A) A scatterplot of the data in three dimensions, with the 95% confidence ellipses projected onto the bounding planes. (B) Step 1 fits the first PC with some error. (C) Step 2 fits a second PC in a direction that is confined to be orthogonal to the first PC. The result again has some error. (D) Step 3 rotates the two PCs and perturbs their lengths until the overall fit is optimized.
We can stop at that point or continue by estimating additional PCs. If we choose to go on, step 2 begins by searching for the second genetic PC (Figure 1C). We exploit the orthogonality property of PCs by restricting the search to the space of (k − 1) dimensions that is orthogonal to the first PC, which speeds the search. Likewise, we can choose to search for a second environmental eigenvector. The process is repeated, in each iteration decreasing by 1 the number of dimensions that must be searched. We are finally left with a set of mA genetic PCs and mE environmental PCs.
The search gets easier with each PC because the number of dimensions of the space in which we search gets smaller. Say that we want to estimate genetic PC i, ψAi, having already estimated PCs 1 to i − 1. We need fit only k − i + 1 of its k elements because the remaining i − 1 elements are determined by the constraint that this next PC must be orthogonal to the previous ones. Specifically, let elements 1 to k − i + 1 of ψAi be the elements to be estimated. A bit of algebra based on the orthogonality constraint then shows that the remaining elements k − i + 2 to k are given by
 |
5 |
where for compactness we use qi,j to denote the jth element of PC ψAi.
Step 3 is a final optimization step. When iterating step 2, the fit of each successive principal component is conditioned on the PCs that have already been estimated. That does not guarantee that the fit is optimized when all the PCs are allowed to vary simultaneously. We do, however, hope to be close to the global optimum at the end of step 2. We therefore perform small rotations on the estimated sets of genetic and environmental PCs and small perturbations on their lengths, now seeking to maximize the fit globally (Figure 1D). These “Givens rotations” are a natural perturbation because they preserve the orthogonality of the principal components (Juga and Thompson 1992; Pinheiro and Bates 1996).
A rotation is defined by an angle and a pair of axes that determine its equatorial plane. Consider a trial set of m(m − 1)/2 rotation angles θ = {θ12, θ13, … , θ1m, θ23, …}, where θij is the rotation involving the axes defined by PCs i and j. Given a set {ψi} of m eigenvectors estimated under step 2, we calculate a new set {ψi′} that results from the rotation using
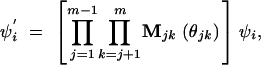 |
6 |
for i = 1, 2, … , m. The quantity in square brackets is a matrix that rotates all of the principal components. Mij(θij) is a Givens rotation matrix for principal components i and j. It is based on an m-dimensional identity matrix with the following changes: the stth element is cos(θij) if s = t = i or if s = t = j, sin(θij) if s = i < t = j or if s = j < t = i, and −sin(θij) if t = i < s = j or if t = j < s = i. For example, with m = 4 principal components, the matrix that rotates PCs 1 and 3 through an angle θ13 is
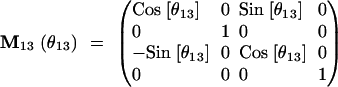 |
Step 3 also requires perturbing the lengths of the PCs. That is done by multiplying PC i by 1 + δi, where the perturbation δi that optimizes the fit will often be much smaller than 1. In total, step 3 involves searching among mA(mA − 1)/2 axes of rotation for the genetic PCs, mE(mE − 1)/2 axes for the environmental PCs, mA perturbations on the lengths of the genetic PCs, and mE perturbations on the lengths of the environmental PCs.
Figure 1 shows the result of this algorithm in an ideal case where the first two of three PCs are estimated perfectly. Even with the perfect fit there is variation in the third dimension, which is not accounted for by the two PCs. In a real application, further error is introduced because the PCs themselves will not be estimated perfectly. Below we present an example with simulated data and further discuss these two sources of error.
How many parameters have been estimated in the end? Fitting m genetic principal components for k traits is a problem that involves (mk − m(m − 1)/2) genetic parameters. If m ≪ k, the estimation problem increases approximately linearly with both m and k. This compares very favorably with estimating the unrestricted covariance matrix. That entails k(k + 1)/2 parameters, a problem that increases roughly as the square of k.
How do we decide when to stop fitting additional principal components? The residual error decreases with each new principal component that is added. But it does so at the expense of increasing the number of parameters in the statistical model: with m PCs already fitted, estimating another PC adds k − m parameters. Several methods are available to determine if the improvement is significant, including the likelihood-ratio test (Edwards 1972) and the Akaike information criterion (AIC; Akaike 1973).
ESTIMATING PCs WITH REML
To this point we have focused on an algorithm for searching parameter space, but not discussed how to evaluate the estimates. REML is a framework that offers a flexible and powerful approach (Patterson and Thompson 1971). Among its strengths are that arbitrary pedigrees can be used, bias from fixed effects (gender, age, environment, etc.) and selection is decreased, and missing data can be accommodated. Here we follow the argument of Meyer (1998) to show how REML can be applied to the direct estimation of genetic principal components. A more detailed analysis of the statistical issues is given by Meyer and Kirkpatrick (2005). Readers interested in a more general perspective on the use of likelihood and REML to estimate genetic parameters can consult Harville (1977) and Lynch and Walsh (1998)(Chaps. 26 and 27).
We want to estimate the covariance matrices G and E, which we do by estimating the genetic PCs (the ψAi) and environmental PCs (the ψEi). Fitting the model will also give us estimates of the residual error variances (the σ2εi). Our approach is based on the general linear model, or “animal model,” of quantitative genetics (Lynch and Walsh 1998, Chap. 26). Using Equations 1 and 3, the data for all of the individuals can be written as the mixed model:
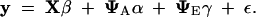 |
7 |
On the left is the vector y of kT observations, formed by concatenating the corresponding vectors for individuals. On the right, the vectors α (the breeding values for the genetic PCs), γ (the deviations for the environmental PCs), and ε (the residual errors) are formed in the same way. The first term on the right side is the vector of mean effects, which is the product of the design matrix X and the vector β of unknown fixed effects. REML is based on a transformation that removes the fixed effects from the analysis. The second and third terms on the right of (7) represent the genetic and environmental effects, respectively. The matrix ΨA is block-diagonal, where block i is a matrix whose columns are the first mA genetic PCs. (If any measurements are missing for individual i, then the corresponding rows of this submatrix are deleted.) The matrix ΨE is formed in the same way using the environmental PCs.
The likelihood L of a set of parameter values can be written in a variety of ways (reviewed by Meyer 1991). For the model of Equation 7, a useful form is
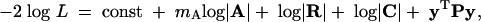 |
8 |
where |·| denotes a matrix determinant; minimizing this quantity maximizes the likelihood L. The first term on the right is a constant that does not depend on the parameters being estimated. The second term is a function of A, which is the (numerator) relationship matrix whose ijth element is twice the coefficient of coancestry between individuals i and j (e.g., 1/2 for parents and offspring, 1/4 for half-sibs, etc.). This term is constant when the number of genetic PCs being fit is fixed and so needs to be considered only when comparing models with different degrees of fit (i.e., different values of mA).
In the third term of (8), R is the kT × kT covariance matrix for the residual errors ε. It is block-diagonal, with block Ri the diagonal matrix whose jth element is σ2εj, but with rows and columns that correspond to missing measurements (if any) deleted. In the fourth term, the matrix C is
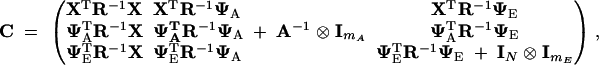 |
9 |
where ⊗ is the Kronecker (or direct) matrix product (Searle 1982), and Ii is the identity matrix with dimensions i.
The last term on the right side of Equation 8 involves the matrix
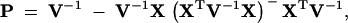 |
10 |
where − is a generalized matrix inverse (Searle 1982). V = Var[y] is a block-structured matrix whose ijth block describes the expected covariance in measurements between individuals i and j,
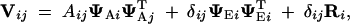 |
11 |
where δij = 1 if i = j and is 0 otherwise.
Equations 8–11 tell us how to calculate the restricted likelihood L. REML estimates for the parameters (the genetic and environmental PCs and the residual error) are those values that maximize L. In practice, the estimates are found numerically. Evaluating the equations is computationally challenging because they are nonlinear and involve the inverses and determinants of large matrices. In animal breeding applications, for example, it is not unusual for the calculations to involve solving millions of simultaneous equations (Misztal et al. 2000; Schaeffer 2004). Great effort has gone into developing efficient algorithms, however, with the result that it is now feasible to find REML estimates for even very large data sets. Meyer and Kirkpatrick (2005) discuss some of the numerical issues involved with the direct-estimation approach in more detail.
FUNCTION-VALUED TRAITS
As mentioned in the Introduction, traits like growth trajectories and reaction norms are function valued (FV). Because the value of the character is a function of a continuous control variable (such as age or temperature), we can view these traits as consisting of an infinite number of dimensions. The natural way to describe variation in such traits is with a covariance function whose value gives the covariance of the character between any pair of values for the control variable (Kirkpatrick and Heckman 1989). Because function-valued traits have higher dimensionality than multivariate traits, the need to find efficient descriptions of variation for FV traits is even more acute. The concepts developed above for multivariate phenotypes extend in a natural way to the FV setting with only minor changes. In what follows, we talk about age as the control variable, as when studying growth curves, but the control variable could as well be an environmental variable (e.g., temperature) or a spatial coordinate.
We begin the discussion of function-valued traits by showing how they can be represented using principal components in a way that is completely analogous with the multivariate case. We then show how the genetic parameters can be estimated using the search algorithm and via REML.
Representing FV traits with PCs:
Measurements are taken on each individual at a set of ages, and the number of measurements and the ages may differ between individuals. The additive genetic and environmental contributions to the jth measurement for individual i can be written with a minor modification of Equation 3,
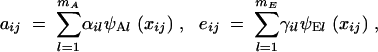 |
12 |
where xij is the age at which that measurement was taken. Comparing these expressions with (3), we see that the genetic and environmental eigenvectors, ψA and ψE, have been replaced by the genetic and environmental eigenfunctions, ψA· and ψE·. These eigenfunctions act as the principal components of the FV setting. Like their multivariate analogs, they can be used to describe variation in a population.
The genetic and covariance matrices of the multivariate setting are replaced by genetic and environmental covariance functions, G·, · and E·, ·. Their interpretation is simple: G(x1, x2) and E(x1, x2) are just the additive genetic and environmental covariances for the trait between ages x1 and x2. Like the genetic covariance matrix G for multivariate traits, the covariance function G determines how FV traits respond to selection (Kirkpatrick and Heckman 1989; Gomulkiewicz and Beder 1996). The relations between the covariance functions and the eigenfunctions are given by the analogs of Equations 2:
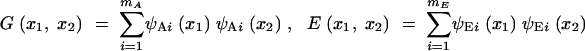 |
13 |
In general, decomposing a covariance function into a sum of eigenfunctions requires an infinite number of terms, in which case the limits of the sums in (13) and (14) are infinity. In practice, however, experience shows that most genetic and phenotypic variation is associated with the first two or three PCs (e.g., Kirkpatrick et al. 1990, 1994; Kirkpatrick and Lofsvold 1992). Thus our approach once again is to approximate the covariance structure by truncating the sums, using values of mA and mE that are as small as possible but that still give an adequate description of the population.
The new issue raised by function-valued traits is how to represent the eigenfunctions. If we do not place any constraint on their form, estimating each of them would involve searching an infinite-dimensional space. Fortunately, biological covariance functions and their eigenfunctions tend to be smooth. That means that the eigenfunctions can be approximated in a simple way. The key is to write each of them as a weighted sum of a set of basis functions, {φj(·)}:
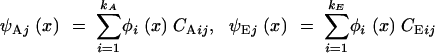 |
14 |
(Kirkpatrick and Heckman 1989). When suitable basis functions are chosen, experience shows that very good approximations to the eigenfunctions are often achieved with a small number of terms, say three or four.
A very broad range of basis functions could be used to represent the eigenfunctions. The computations are simplified, however, if we use orthogonal functions that have been scaled to have unit norm over the range of the control variable x, and we assume that the {φi} have those properties in what follows. One natural choice for the basis functions is Legendre polynomials (see Kirkpatrick et al. 1990; Meyer 1998). Estimates for the eigenfunctions are then polynomials of degree k − 1. An alternative possibility for the basis function is splines.
The additive genetic variances for the genetic principal components can be written in terms of the weights that appear in Equation 14:
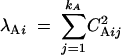 |
15 |
(Kirkpatrick and Heckman 1989). Comparing Equation 15 with Equation 4, we see that the eigenvalues for an FV trait are determined by the vector of weights CAi in the same way they are by the genetic eigenvector ψAi in the MV setting.
Our approximation for function-valued traits therefore works on two levels. First, the covariance functions are approximated by reconstructing them using only the first m principal components (eigenfunctions). Second, the principal components themselves are approximated by functions with only k degrees of freedom (for example, polynomials of degree k − 1). We might find, for example, that the data are well described by two principal components (m = 2), each of which is a cubic (k = 4).
We can now describe an individual's breeding value in terms of genetic principal components. Putting together Equations 12 and 14, we see that ai, the vector of individual i's breeding values for the trait at the ages at which it was measured, can be written in terms of αi, its vector of its breeding values for the genetic principal components,
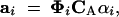 |
16 |
where [Φi]jk = φk(xij). CA is the kA × mA matrix of coefficients that appear in Equation 14. [This matrix is related to the coefficient matrix CG of Kirkpatrick et al. (1990) via CG = CA CTA.] Similarly, the environmental component of the phenotype can be written ei = ΦiCEγi. Because we have chosen to represent these components in turn as sums of orthogonal basis functions, the columns of CA are mutually orthogonal, as are the columns of CE.
We have now succeeded in representing a finite number of phenotypic observations in terms of sums of orthogonal basis functions. Estimates of the covariance functions are found by optimizing the fit of the coefficient matrices CA and CE, using the statistical framework of our choice. Those matrices then give us estimates for the PCs (Equation 14), their eigenvalues (Equation 15), and the covariance functions (Equation 13). As in the MV case, the covariance function is guaranteed to be positive-definite. In the next two subsections we show how optimizing the estimates can be accomplished using our search algorithm and the REML estimation framework.
Searching for the PCs of an FV trait:
The algorithm described earlier in the MV setting now carries over directly if we visualize the columns of the coefficient matrices CA and CE as the analogs of the eigenvectors ψA and ψE (respectively) from the multivariate case. In brief, we search sequentially for the vectors that make up the columns of CA and CE in just the same way that we searched in the multivariate setting for the eigenvectors ψA and ψE. Step 1 involves searching for the mA elements of the first column of CA and the mE elements of the first column of CE that optimize the fit. These give estimates of the first genetic and environmental PCs (eigenfunctions).
In step 2, we search sequentially for additional columns of CA and CE, which give estimates of subsequent PCs. We can exploit the fact that the columns of these matrices must be orthogonal to reduce the number of dimensions in which to search. To do that, we use the algorithm described earlier. Specifically, replace ψAi in Equation 5 with CAi, and replace ψEi with CEi, where CAi and CEi are respectively the ith columns of the matrices CA and CE. We finish with step 3, the final optimization that rotates and perturbs the lengths of the vectors that compose the columns of CA and CE, just as in the multivariate case.
Estimation with REML:
With a function-valued trait, the aim is to estimate the covariance functions G(·, ·) and E(·, ·). The approach is again based on estimating the genetic and environmental PCs, which are now the eigenfunctions ψAi(·) and ψEi(·). Fitting the model also gives estimates of the residual error variances.
The calculations described earlier for the MV setting carry over to FV traits with only trivial changes. In Equations 7–11, the matrix ΨAi is replaced by ΦiCA, and the matrix ΨEi is replaced by ΦiCE. The last thing needed is a model for the residual error. The residual error matrix for individual i is diagonal with elements 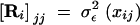 , where σ2εx is the residual (temporary environmental) error at age x. A reasonable approach is to assume that this function can be represented by a smooth function like a polynomial or spline, in which case the coefficients of that function are included among the parameters we seek to estimate (see Meyer 2001).
, where σ2εx is the residual (temporary environmental) error at age x. A reasonable approach is to assume that this function can be represented by a smooth function like a polynomial or spline, in which case the coefficients of that function are included among the parameters we seek to estimate (see Meyer 2001).
AN EXAMPLE
This section illustrates the direct estimation approach and our algorithm for fitting PCs. The numerical example uses likelihood to estimate the PCs and the covariance function of a function-valued trait. For simplicity, we use a phenotypic example in which the aim is simply to estimate the phenotypic covariance function using full maximum likelihood. Meyer and Kirkpatrick (2005) analyze a genetic example using restricted maximum likelihood.
The simulated data:
The covariance function is taken from the numerical example from Kirkpatrick et al. (1990), which in turn is based on a study of growth in mice by Riska et al. (1984). The covariance function is
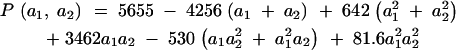 |
17 |
for 2 ≤ a1, a2 ≤ 4. The function is shown in Figure 2. Because this function is quadratic, it has only three nonzero PCs, which are shown in Figure 3. They are
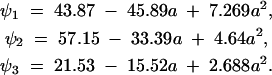 |
18 |
Figure 2.—

The covariance function used in the numerical example.
Figure 3.—

The three principal components (eigenfunctions) of the covariance function for the numerical example. The actual PCs are the thick solid curves, and typical estimates are shown as dashed curves.
(These differ from the corresponding equations in Kirkpatrick et al. 1990, p. 984, because here age is on the original scale of [2, 4], and the norms of the PCs are defined to be equal to the square root of the corresponding eigenvalues.) The eigenvalues are λ1 = 1361, λ2 = 24.54, and λ3 = 1.535.
The aim is to estimate the phenotypic covariance function P by fitting m = 1, 2, and 3 principal components. For each replicate, we simulated a population of 100 individuals, each measured at k = 5 equally spaced ages. The covariance matrix for the traits was determined by evaluating Equation 17 on a 5 × 5 lattice of points. An individual's phenotype was simulated as a vector of five measurements sampled from the corresponding multivariate normal distribution with mean zero. To each of these measurements we added an i.i.d. “temporary environmental error” (or measurement error) term with variance 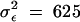 , which corresponds to between 43 and 59% of the total variance, depending on the point in the covariance function. We fit one, two, and three PCs to each sample of 100 individuals via full likelihood (because the model has no fixed effects). The likelihood was maximized using the derivative-free simplex algorithm (Nelder and Mead 1965). For each degree of fit (that is, value of m), we calculated the corresponding estimate for the covariance function using Equation 13. We also estimated the full 5 × 5 covariance matrix, which is equivalent to a multivariate analysis that ignores the ordering of the ages at which the measurements were taken. This procedure was repeated for 10,000 replicates.
, which corresponds to between 43 and 59% of the total variance, depending on the point in the covariance function. We fit one, two, and three PCs to each sample of 100 individuals via full likelihood (because the model has no fixed effects). The likelihood was maximized using the derivative-free simplex algorithm (Nelder and Mead 1965). For each degree of fit (that is, value of m), we calculated the corresponding estimate for the covariance function using Equation 13. We also estimated the full 5 × 5 covariance matrix, which is equivalent to a multivariate analysis that ignores the ordering of the ages at which the measurements were taken. This procedure was repeated for 10,000 replicates.
Measuring the accuracy of the estimates:
We evaluated our estimation approach in several ways. Our first measure is the average proportional error in the overall estimate of the covariance function reconstructed from the PCs,
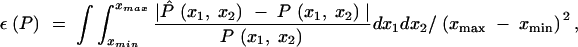 |
19 |
where the hat denotes an estimate, and the ages range from xmin = 2 to xmax = 4. We calculated the integrals numerically.
We evaluated the accuracy with which the individual principal components were estimated in two ways. A PC is a vector quantified by a direction and a length (or norm). (This holds equally for function-valued traits, where the “direction” is reflected by the shape of the PC, or eigenfunction.) A natural measure of the error in the estimated direction is the angle between the estimate and the true PC. For the multivariate case, that is
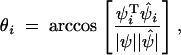 |
20 |
where |·| denotes norms of vectors. [This relation follows from the fact that the inner product of two vectors with unit norm is equal to the cosine of the angle between them (Strang 1976).] For the function-valued case, the analogous expression is
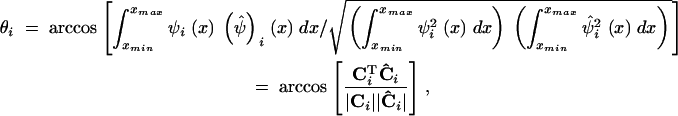 |
21 |
where Ci is the vector of coefficients for PC i that appears in Equations 14 and 15. If the direction (or shape, in the FV context) of an estimated PC is perfectly aligned with the population value, then θ is the ideal 0°, while if the estimate is a perfect failure (that is, the estimate is orthogonal to the true PC), then θ = 90°.
Our second measure for the accuracy with which the PCs were estimated is the relative error in an estimate of eigenvalue i (which is the square of the norm, or length, of PC i):
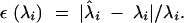 |
22 |
This measure is zero when the magnitude of the eigenvalue is estimated perfectly and otherwise is positive. We quantified the bias in estimate of the eigenvalue as
 |
23 |
This measure is zero when the eigenvalue is estimated with no bias, is negative when it is underestimated on average, and is positive when it is overestimated on average.
Finally, we quantified the relative error in the estimates of the temporary environmental variance using a statistic analogous to what we used for the eigenvalues:
 |
24 |
Results:
Table 1 shows the simulation results. The direct estimation approach does well in estimating the overall covariance function: on average, the covariances are estimated with an error of 15%. This is encouraging in view of the facts that the data sets consisted of only 100 individuals and the variance contributed by measurement error was roughly as large as that of the measurements themselves.
TABLE 1.
Errors in the estimates based on fittingm = 1, 2, and 3 principal components to simulated phenotypic data sets, each consisting of 100 individuals (see text for details)
| PC1
|
PC2
|
PC3
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| m | ε(P) | ε(λ1) | Bias(λ1) | ε(θ1) | ε(λ2) | Bias(λ2) | ε(θ2) | ε(λ3) | Bias(λ3) | ε(θ3) | εσ2ε |
| 1 | 0.15 (0.095) | 0.14 (0.11) | −0.0041 (0.17) | 2.7° (1.5°) | 0.066 (0.050) | ||||||
| 2 | 0.15 (0.095) | 0.14 (0.11) | 0.010 (0.18) | 2.7° (1.4°) | 0.83 (0.70) | 0.52 (0.95) | 28° (23°) | 0.066 (0.048) | |||
| 3 | 0.15 (0.095) | 0.14 (0.11) | 0.013 (0.18) | 2.7° (1.4°) | 0.90 (0.75) | 0.60 (1.0) | 28° (23°) | 3.2 (5.2) | 1.9 (5.8) | 29° (22°) | 0.072 (0.054) |
The number of PCs fit is given by m. In each cell, the error statistic appears first, followed by its standard deviation in parentheses. Results are based on 10,000 replicate data sets.
A striking result is that the accuracy in estimating the covariance function when two or three PCs are fit is no better than when just one PC is estimated: neither ε(θ1) nor ε(λ1) changed substantially when different numbers of PCs were fit. The reason becomes clear when we look at the estimation errors for the PCs (Figure 3). The first PC is consistently well estimated. The average error in estimating its shape is trivial: ε(θ1) = 2.7°. The error in estimating the first eigenvalue, ε(λ1), is greater: on average the estimate is off by 14%. The bias for that eigenvalue, however, is extremely small. On average it is underestimated, but by <1% of its true value. This contrasts with standard multivariate methods, which can produce substantial upward biases in estimates of the leading eigenvalue (Hayes and Hill 1981).
The situation is quite different for the second and subsequent PCs. The shape (direction) of the second PC is poorly estimated, with an error of 28°, and estimates for the second eigenvalue are on average 83–90% away from their true values. The third eigenfunction fares even worse: the error in the shape is 29°, and the average error in the estimate of λ3 is 190%. But because these PCs contribute so little to the total variation, these errors have almost no impact on the accuracy of the overall estimate of the covariance function.
How do the results depend on the number of measurements taken on each individual? Not surprisingly, the accuracy of the estimate for the leading PC improves as the number of ages measured increases. As a result, the entire covariance function is better estimated. Comparing results for five ages (Table 1) with those for nine ages (not shown), the estimation error in the overall covariance function decreases by 11%. The error in estimating the leading eigenvalue declines by 10%, and the error in the estimate of the direction of PC1 by 18%. These improvements may seem modest, given that the amount of data is almost doubled. But this situation is far better than what we would see with a conventional multivariate analysis. There, the accuracy of the estimates can actually decline because the number of parameters being estimated increases so rapidly with the number of traits measured (Sales and Hill 1976a,b).
It is also interesting to compare these results with a standard multivariate analysis of the same data. In effect, the MV approach discards all information about the ages at which the measurements were taken. The average relative error in estimating the 5 × 5 covariance matrix is 20%, which is worse than the error when we estimate only a single PC but make use of the function-valued nature of the data (15%; see Table 1). The relative error in the estimate of the first eigenvalue is very similar for the standard MV approach and our new approach [ε(λ1) = 0.14 for both], but the MV approach also estimates the direction of the leading principal component with greater error than the FV approach does (5.5° vs. 2.7°). These results reinforce the impression that combining the FV and the direct estimation approaches makes efficient use of the data.
Bias in estimates of the leading eigenvalues is reduced by the new method. We used the simulated data to estimate the leading eigenvalue using the standard MV approach, that is, by calculating the eigenvalues from the estimated covariance matrix. The estimate is typically biased upward, as expected from the arguments of Hayes and Hill (1981), on average by 3.8%. But when the direct estimation method is used, bias is much reduced. Depending on the number of PCs estimated, bias under the direct estimation method is between three and nine times smaller than that under the classic MV approach.
In sum, these limited simulations suggest that the direct estimation approach is efficient at estimating the pattern of variation in a population. It is encouraging that the estimate of the first principal component is quite accurate, independent of the degree of fit, and almost free of bias. Meyer and Kirkpatrick (2005) report similarly promising results for a genetic example. We will not know how general and robust these findings are, however, until the method has been applied in a variety of settings.
DISCUSSION
The point of departure for this article is the simple observation that covariance matrices and covariance functions can be estimated directly in terms of a small number of principal components. Several benefits flow from this strategy. The data are used efficiently in the sense that the maximum amount of variation is explained with the smallest number of parameters. This reduction should speed calculations and lead to greater numerical stability of the estimates. The covariance matrix (or function) estimated by the direct method is guaranteed to be positive semidefinite. Last, estimation biases for the leading eigenvalues, which have long been recognized as a problem in classical estimation approaches, appear to be reduced substantially. These last two points obviate the need for heuristic corrections to estimates of covariance matrices, for example, the “bending” method proposed by Hayes and Hill (1981). Some of the advantages of working with genetic PCs have been recognized previously (Misztal et al. 2000; Nobre et al. 2002), but it seems they have not yet been exploited systematically.
We have seen that the orthogonality of PCs can be exploited in an algorithm to estimate the covariance structure. Pinheiro and Bates (1996) compared the efficiency of the Givens rotations (which is the third component of our algorithm) with four other parameterizations used for estimating covariance matrices from phenotypic data. They found that other parameterizations are often faster, largely because of the computational expense of the calculations involving the rotations. In large genetic analyses, however, this may not be a concern because calculating the likelihood itself will typically be a much larger part of the problem. A separate issue is whether likelihood surfaces for genetic parameters under our parameterization are conducive to numerical search. Meyer and Kirkpatrick (2005) analyzed a genetic example and found that the direct estimation approach seems to be quite efficient in that context also. Extensive simulations will be needed, however, to determine the robustness of the approach in general.
One potentially useful application of the direct PC approach is in studies of the evolution of the additive genetic variance-covariance structure, a topic of emerging interest in evolutionary genetics. A variety of statistical tools have been developed to compare covariance matrices (reviewed by Houle et al. 2002; Steppan et al. 2002). Among them are methods that focus on differences in principal components between populations. These methods are particularly powerful because PCs have straightforward interpretations and because one can test sets of nested hypotheses about how the PCs have changed (Flury 1988; Phillips and Arnold 1999). It is possible to wed the direct PC approach to these methods. One could, for example, test the correspondence between the first few PCs of two populations. These analyses would benefit from the reduced biases and increased accuracy of estimates that our direct PC approach contributes. Further work is needed on several issues, for example, to determine how the choice of the number of PCs estimated affects these analyses.
The direct PC approach may be most useful with function-valued traits. The additive genetic covariance function plays a central role in determining how FV traits respond to natural and artificial selection (Kirkpatrick and Heckman 1989; Kirkpatrick 1993; Gomulkiewicz and Beder 1996; Kirkpatrick and Bataillon 1999). Consequently, estimation and analysis of covariance functions is rapidly expanding in both evolutionary genetics and applied animal breeding (Jaffrézic and Pletcher 2000; Misztal et al. 2000; Kingsolver et al. 2001; Schaeffer 2004). Estimates of covariance functions are sensitive to error, however, and so there is substantial interest in developing methods that are fast, make efficient use of the data, and are numerically stable (Jaffrézic and Pletcher 2000; van der Werf 2002).
Two major families of methods to estimate covariance functions are currently in use. The first, which is nonparametric in spirit, represents the covariance function in terms of flexible basis functions such as polynomials. The earliest approach fit polynomials to a covariance matrix that had been previously estimated for a fixed set of ages (Kirkpatrick et al. 1990, 1994). An important advance was the introduction of the method of random regression, which escapes the need for the covariance matrix by fitting a set of basis functions to the observations for each individual (Schaeffer and Dekkers 1994; Meyer and Hill 1997; Meyer 1998). Random regression has been widely implemented using polynomials as the basis functions (Schaeffer 2004), but other basis functions such as splines have also been used (White et al. 1999; Torres 2001). Splines are numerically better behaved than polynomials, but have the drawback that they require fitting a larger number of parameters and so can become unwieldy with very large data sets.
The second family of methods begins with the assumption that the covariance function takes a simple parametric form (e.g., Pletcher and Geyer 1999; Jaffrézic et al. 2003). This constraint reduces the number of parameters and so makes the results less sensitive to estimation error (Jaffrézic and Pletcher 2000). A drawback of this approach is that there is often no strong biological justification for any particular functional form. If an inappropriate choice is made, then estimates of genetic parameters will be biased.
The direct estimation method proposed in this article may have some of the advantages of both families of methods. It makes no prior assumption about the form of the covariance function, but involves many fewer parameters than the standard random regression methods. The reduction may make it feasible to use more complex models, for example, using splines rather than polynomials to estimate the eigenfunctions.
There is, however, a drawback of our approach for some applications. Evolutionary biologists would like to know the degree to which patterns of genetic variation may constrain the potential for adaptation (Maynard Smith et al. 1985). Function-valued traits are a particularly interesting context in which to study this problem because there are in principle an infinite number of dimensions to which organisms must adapt (Kirkpatrick and Lofsvold 1992). Our approach of extracting only the major PCs is poorly suited to this kind of problem because it will often discard information about phenotypic dimensions for which there is a small but nonzero amount of genetic variation. By neglecting these dimensions, we might be falsely led to believe there is no heritable variation available for adaptation when in fact there is.
Some kinds of traits do not fall into either the multivariate or function-valued cases we discussed above. We might be interested in a set of several traits that change with age, for example, or in a trait that varies as a function of more than one continuous control variable (for example, age and environment). The direct estimation approach can be extended to these more complex kinds of phenotypes. This is an attractive idea because the number of parameters to be estimated is otherwise very large (Schaeffer 2004).
There has recently been much interest in function-valued traits among statisticians working in areas outside of quantitative genetics. Rice and Silverman (1991) introduced a nonparametric approach in which observations on individuals were fit with splines, and the PCs were derived from them. Their approach has been expanded and generalized in several respects (Ramsay and Silverman 1997, 2002). Some of the developments parallel those made independently in quantitative genetics, for example, the use of random regressions (James et al. 2000). There are, however, basic differences between these phenotypic analyses and those in quantitative genetics. Major goals of quantitative genetics are to partition variation into heritable and nonheritable components and to estimate the breeding values of individuals. Those goals motivate the standard assumption of quantitative genetics that variance components are normally distributed. In contrast, many phenotypic applications can afford to take more general nonparametric approaches (Ramsay and Silverman 1997, 2002). Nevertheless, the direct PC approach developed here may also find uses in the analysis of phenotypic data.
Acknowledgments
This work was supported by grant BFGEN.100 of Meat and Livestock Australia (to K.M.) and grants DEB-9973221 and EF-0328594 from the National Science Foundation and NER/A/S/2002/00857 from the Natural Environment Research Council (to M.K.).
References
- Akaike, H., 1973 Information theory and an extension of the maximum likelihood principle, pp. 267–281 in The Second International Symposium on Information Theory, edited by B. N. Petrov and F. Csaki. Akad. Kiadò, Budapest.
- Atchley, W. R., and J. J. Rutledge, 1980. Genetic components of size and shape. I. Dynamics of components of phenotypic variability and covariability during ontogeny in the laboratory rat. Evolution 34: 1161–1173. [DOI] [PubMed] [Google Scholar]
- Chase, K., D. R. Carrier, F. R. Adler, T. Jarvik, E. A. Ostrander et al., 2002. Genetic basis for systems of skeletal quantitative traits: principal component analysis of the canid skeleton. Proc. Natl. Acad. Sci. USA 99: 9930–9935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards, A. W. F., 1972 Likelihood: An Account of the Statistical Concept of Likelihood and Its Application to Scientific Inference. Cambridge University Press, Cambridge, UK.
- Falconer, D. S., and T. F. C. Mackay, 1996 Introduction to Quantitative Genetics, Ed. 4. Longman, London. [DOI] [PMC free article] [PubMed]
- Flury, B. D., 1988 Common Principal Components and Related Multivariate Models. Wiley, New York.
- Gomulkiewicz, R., and J. H. Beder, 1996. The selection gradient of an infinite-dimensional trait. Soc. Ind. Appl. Math. J. Appl. Math. 56: 509–523. [Google Scholar]
- Harville, D. A., 1977. Maximum likelihood approaches to variance component estimation and to related problems. J. Am. Stat. Assoc. 72: 320–338. [Google Scholar]
- Hayes, J. F., and W. G. Hill, 1981. Modification of estimates of parameters in the construction of genetic selection indices (‘bending’). Biometrics 37: 483–493. [Google Scholar]
- Hill, W. G., and R. Thompson, 1978. Probabilities of non-positive definite between-group or genetic covariance matrices. Biometrics 34: 429–439. [Google Scholar]
- Houle, D., J. Mezey and P. Galpern, 2002. Interpretation of results of common principal components analysis. Evolution 56: 433–440. [DOI] [PubMed] [Google Scholar]
- Jaffrézic, F., and S. D. Pletcher, 2000. Statistical models for estimating the genetic basis of repeated measures and other function-valued traits. Genetics 156: 913–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffrézic, F., R. Thompson and W. G. Hill, 2003. Structured antedependence models for genetic analysis of repeated measures on multiple quantitative traits. Genet. Res. 82: 55–65. [DOI] [PubMed] [Google Scholar]
- James, G. M., T. J. Hastie and C. A. Sugar, 2000. Principal component models for sparse functional data. Biometrika 87: 587–602. [Google Scholar]
- Juga, J., and R. Thompson, 1992. A derivative-free algorithm to estimate bivariate (co)-variance components using canonical transformations and estimated rotations. Acta Agric. Scand. Sect. A Anim. Sci. 42: 191–197. [Google Scholar]
- Kingsolver, J. G., R. Gomulkiewicz and P. A. Carter, 2001. Variation, selection and evolution of function-valued traits. Genetica 112–113: 87–104. [PubMed] [Google Scholar]
- Kirkpatrick, M., 1993 Evolution of size and growth in fisheries and other harvested natural populations, pp. 145–154 in The Exploitation of Evolving Resources (Lecture Notes in Biomathematics 99), edited by K. Stokes, J. M. McGlade and R. Law. Springer-Verlag, Berlin.
- Kirkpatrick, M., and T. Bataillon, 1999. Artificial selection on phenotypically plastic traits. Genet. Res. 74: 265–270. [DOI] [PubMed] [Google Scholar]
- Kirkpatrick, M., and N. Heckman, 1989. A quantitative genetic model for growth, shape, reaction norms, and other infinite-dimensional characters. J. Math. Biol. 27: 429–450. [DOI] [PubMed] [Google Scholar]
- Kirkpatrick, M., and D. Lofsvold, 1992. Measuring selection and constraint in the evolution of growth. Evolution 46: 954–971. [DOI] [PubMed] [Google Scholar]
- Kirkpatrick, M., D. Lofsvold and M. Bulmer, 1990. Analysis of the inheritance, selection, and evolution of growth trajectories. Genetics 124: 979–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkpatrick, M., W. G. Hill and R. Thompson, 1994. Estimating the covariance structure of traits during growth and aging, illustrated with lactations in dairy cattle. Genet. Res. 64: 57–69. [DOI] [PubMed] [Google Scholar]
- Lynch, M., and J. B. Walsh, 1998 Genetics and Analysis of Quantitative Traits. Sinauer Associates, Sunderland, MA.
- Maynard Smith, J., R. Burian, S. Kauffman, P. Alberch, J. Campbell et al., 1985. Developmental constraints and evolution. Q. Rev. Biol. 60: 265–287. [Google Scholar]
- Meyer, K., 1985. Maximum likelihood estimation of variance components for a multivariate mixed model with equal design matrices. Biometrics 41: 153–165. [PubMed] [Google Scholar]
- Meyer, K., 1991. Estimating variances and covariances for multivariate animal models by restricted maximum likelihood. Genet. Sel. Evol. 23: 67–83. [Google Scholar]
- Meyer, K., 1998. Estimating covariance functions for longitudinal data using a random regression model. Genet. Sel. Evol. 30: 221–240. [Google Scholar]
- Meyer, K., 2001. Estimating genetic covariance functions assuming a parametric correlation structure for environmental effects. Genet. Sel. Evol. 33: 557–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer, K., and W. G. Hill, 1997. Estimation of genetic and phenotypic covariance functions for longitudinal data by restricted maximum likelihood. Livest. Prod. Sci. 47: 185–200. [Google Scholar]
- Meyer, K., and M. Kirkpatrick, 2005. Restricted maximum likelihood estimation of genetic principal components and smoothed covariance matrices. Genet. Sel. Evol. 36 (in press). [DOI] [PMC free article] [PubMed]
- Misztal, I., T. Strabel, J. Jamrozik, E. A. Mäntysaari and T. H. E. Meuwissen, 2000. Strategies for estimating the parameters needed for different test-day models. J. Dairy Sci. 83: 1125–1134. [DOI] [PubMed] [Google Scholar]
- Morrison, D. F., 1976 Multivariate Statistical Methods. McGraw-Hill, New York.
- Nelder, J. A., and R. Mead, 1965. A simplex method for function minimization. Comput. J. 7: 147–151. [Google Scholar]
- Nobre, P. R. C., I. Misztal, S. Tsuruta, J. K. Bertrand, L. O. C. Silva et al., 2002 Genetic evaluation of growth in beef cattle with a random regression model. Seventh World Congress on Genetics Applied to Livestock Production, INRA, Castanet-Tolosan, France, Communication 20.10.
- Patterson, H. D., and R. Thompson, 1971. Recovery of inter-block information when block sizes are unequal. Biometrika 58: 545–554. [Google Scholar]
- Phillips, P. C., and S. J. Arnold, 1999. Hierarchical comparison of genetic variance-covariance matrices. I. Using the Flury hierarchy. Evolution 53: 1506–1515. [DOI] [PubMed] [Google Scholar]
- Pinheiro, J. C., and D. M. Bates, 1996. Unconstrained parameterizations for variance-covariance matrices. Stat. Comput. 6: 289–296. [Google Scholar]
- Pletcher, S. D., and C. J. Geyer, 1999. The genetic analysis of age-dependent traits: modeling the character process. Genetics 153: 825–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsay, J. O., and B. W. Silverman, 1997 Functional Data Analysis. Springer, New York.
- Ramsay, J. O., and B. W. Silverman, 2002 Applied Functional Data Analysis. Springer, New York.
- Rice, J. A., and B. W. Silverman, 1991. Estimating the mean and covariance structure nonparametrically when the data are curves. J. R. Stat. Soc. B 53: 233–243. [Google Scholar]
- Riska, B., W. R. Atchley and J. J. Rutledge, 1984. A genetic analysis of targeted growth in mice. Genetics 107: 79–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sales, J., and W. G. Hill, 1976. a Effect of sampling errors on efficiency of selection indices. 1. Use of information from relatives for single trait improvement. Anim. Prod. 22: 1–17. [Google Scholar]
- Sales, J., and W. G. Hill, 1976. b Effect of sampling errors on efficiency of selection indices. 2. Use of information on associated traits for improvement of a single important trait. Anim. Prod. 23: 1–14. [Google Scholar]
- Schaeffer, L. R., 2004. Application of random regression models in animal breeding. Livest. Prod. Sci. 86: 35–45. [Google Scholar]
- Schaeffer, L. R., and J. C. M. Dekkers, 1994 Random regression in animal models for test-day production in dairy cattle. Proceedings of the 5th World Congress on Genetics Applied to Livestock Production, Guelph, Ontario, Canada, Vol. 18, pp. 443–446.
- Searle, S. R., 1982 Matrix Algebra Useful for Statistics. Wiley, New York.
- Steppan, S. J., P. C. Phillips and D. Houle, 2002. Comparative quantitative genetics: evolution of the G matrix. Trends Ecol. Evol. 17: 320–327. [Google Scholar]
- Strang, G., 1976 Linear Algebra and Its Applications. Academic Press, New York.
- Torres, R. A. A., 2001 Markov chain Monte Carlo methods for estimating the covariance structure of longitudinal data: an application to dairy cattle. Ph.D. Thesis, Cornell University, Ithaca, NY.
- van der Werf, J. H. J., 2002 Optimizing selection for traits along trajectories. Seventh World Congress on Genetics Applied to Livestock Production, INRA, Castanet-Tolosan, France, Communication 16.07.
- White, I. M. S., R. Thompson and S. Brotherstone, 1999. Genetic and environmental smoothing of lactation curves with cubic splines. J. Dairy Sci. 82: 632–638. [DOI] [PubMed] [Google Scholar]