

Abstract
This article presents an analysis of a model of isolation by distance in a continuous, two-dimensional habitat. An approximate expression is derived for the distribution of coalescence times for a pair of sequences sampled from specific locations in a rectangular habitat. Results are qualitatively similar to previous analyses of isolation by distance, but account explicitly for the location of samples relative to the habitat boundaries. A separation-of-timescales approach takes advantage of the fact that the sampling locations affect only the recent coalescent behavior. When the population size is larger than the number of generations required for a lineage to cross the habitat range, the long-term genealogical process is reasonably well described by Kingman's coalescent with time rescaled by the effective population size. This long-term effective population size is affected by the local dispersal behavior as well as the geometry of the habitat. When the population size is smaller than the time required to cross the habitat, deep branches in the genealogy are longer than would be expected under the standard neutral coalescent, similar to the pattern expected for a panmictic population whose population size was larger in the past.
NATURAL populations are often geographically structured. That is, there is frequently a correlation between the geographic locations of parents and their offspring, resulting in the accumulation of genetic differences between local populations. The connection between genealogies and geography has long been a subject of interest in population genetics. In particular, many efforts have been made to construct methods for making geographic and demographic inferences about populations from patterns of genetic diversity.
The best-studied models of geographic population structure assume that the population is divided into a number of subpopulations, or demes. In the “island model” (Wright 1931; Maruyama 1970a), migration between demes occurs through a common migrant pool. Because the destination of a migrant is independent of its origin, this model lacks explicit geography. Geographically explicit models are typically some variant of the stepping-stone model (Kimura and Weiss 1964). In these models, demes are arrayed in a lattice, with migration limited to adjacent demes, or biased in favor of nearby demes. Models of continuous habitats, in which geographic structure is more commonly referred to as “isolation by distance” (Wright 1943), are in some sense a special case of a stepping-stone model. If we enforce strict density regulation and let the deme size become small, the stepping stone becomes a lattice model, with one individual occupying each position in the lattice. Models of this sort have been developed by Malécot (1968) and Weiss and Kimura (1965) and specifically in the case of a two-dimensional habitat of finite extent by Maruyama (1971) and Malécot (1975).
Most analyses of stepping-stone models have relied on one of two types of assumption to secure mathematical tractability. One class of models (the infinite stepping stone) assumes an infinite array of demes (Weiss and Kimura 1965; Nagylaki 1974a; Sawyer 1976, 1977). These models are useful for considering the short-term relationship between genetic and geographic distance, but on longer timescales they predict infinite divergence between individuals (Sawyer 1976; Griffiths 1981; Wilkinson-Herbots 1998). The other class of models invokes periodic boundary conditions (Maruyama 1970b, 1971; Nagylaki 1974b, 1977; Strobeck 1987; Slatkin 1991). In these models, the ends of the array of demes are joined together to form the “circular stepping stone” in one dimension. In two dimensions, the array of demes takes on the shape of a torus. Other analyses have undertaken the problem of a finite array of demes and the effect of range boundaries on the genetic structure of populations (Maruyama 1970c,d, 1972; Fleming and Su 1974; Malécot 1975; Nagylaki and Barcilon 1988). The general result of these analyses is that proximity to the edge of the deme array increases probabilities of identity and the covariance in gene frequency across demes.
Much of the recent work on geographic inference is based on the coalescent (Kingman 1982a,b; Hudson 1983; Tajima 1983). Coalescent models focus on the history of a particular sample, rather than on the population as a whole. In this framework, models of population structure are analyzed to make probabilistic statements about the times to common ancestry for samples drawn from specific geographic locations. Observed patterns of genetic diversity, then, can be used to infer parameter values (e.g., migration rate, population size) that best describe particular populations within a given model. In the context of geographically structured populations, this approach has been formalized as the structured coalescent (Notohara 1990; Wilkinson-Herbots 1998). Analysis of genealogical processes in a continuous habitat faces a particular challenge, pointed out by Felsenstein (1975). If not all organisms reproduce, and offspring appear in locations close to where their parents were, the population becomes clumped. However, most analyses have assumed a uniform population density that is inconsistent with this mode of reproduction. A common method for addressing this inconsistency (and the one that is followed in this analysis) is to assume strong local density regulation that forcibly maintains the uniform population density. A fully occupied lattice model represents an extreme form of local density regulation.
Traditional (precoalescent) population genetics models have focused on measures such as the probability of identity and the covariance in gene frequency. These measures are closely tied to the distribution of coalescence times (Slatkin 1991); however, the formulation of the problem within the coalescent framework provides results that make greater use of the information in DNA sequence data. The question of how best to infer migration patterns in a two-dimensional habitat has been considered by a number of authors (Slatkin and Barton 1989; Slatkin and Maddison 1990; Barton and Wilson 1996; Rousset 1997). The effects of habitat range boundaries on coalescence times have been investigated in the one-dimensional stepping-stone model by Hey (1991) and Herbots (1994)(pp. 66 and 145–146), who found shorter coalescence times nearer to the edges of the array, consistent with the effects on probability of identity and covariance in gene frequency. The analysis of Wilkins and Wakeley (2002) in the continuous one-dimensional model found a similar relationship between sampling location and coalescence time.
The goal of the work described in this article is to derive explicit expressions for the distribution of coalescence times for pairs of sequences sampled from specific locations within a finite two-dimensional habitat. Geographic structuring arises in this model as a result of limited intergenerational dispersal, or gene flow. In terms of the coalescent process, we can imagine tracing the locations of the ancestors of sequences in our sample backward in time. The locations of these ancestral lineages are modeled as a random walk in the habitat, where the size of the steps is determined by the rate of gene flow. Two lineages coalesce if they are derived from the same individual in a given generation. Under strict density regulation, this is equivalent to saying that the lineages coalesce if they approach the same location at the same time. The proximity within which two lineages must approach each other to coalesce is determined by the population density. To construct the distribution of coalescence times, we need to consider the locations of the two lineages back through time, conditional on their not yet having coalesced. Because of this conditioning, the two random walks are not independent of each other. The dependence of each random walk on the location of the other lineage is the source of the difficulty of obtaining mathematical expressions for this process.
The problem of coalescence times was treated originally under different terms by Wright (1943), but has been addressed most extensively by Barton and Wilson (1995)(1996). Their work derives recursion equations for the probability of coalescence for pairs of sequences. In the recent past, coalescence probabilities are determined only by local dynamics. However, for deeper portions of the genealogy, coalescence probabilities are influenced by the fact that populations inhabit finite ranges. Barton and Wilson present an expression for their solution for the toroidal habitat and indicate how an expression can be derived under reflecting boundary conditions, where the habitat would be rectangular. More recent work derives recursion equations in a continuous habitat for different degrees of local density regulation (Barton et al. 2002).
The challenge of using genealogical data to infer geographic structure lies both in the fact that those analytic expressions that can be derived are often unwieldy and in the fact that only a fraction of the genealogical history contains information relevant to geographic structure. As Barton and Wilson point out, the deeper branches in genealogical trees will often represent lineages that have crossed the species range multiple times. The fact that geographically relevant information is restricted to recent parts of the genealogy is the basis of the success of rare allele methods of estimating gene flow (Slatkin 1985). Other analyses of models of gene flow have also found deep branches in the genealogies to be uninformative (Slatkin and Maddison 1990). Recent simulation work on genealogical structures in continuous populations (Irwin 2002) illustrates the poor correspondence between processes occurring at the geographic level and the deeper branches of genealogies. Under higher migration rates, deep genealogical divisions show little correlation with geography. Under low migration, these deep divisions can suggest specific barriers to gene flow where none actually exist.
A number of models of population structure have recently been studied with some success using a separation-of-timescales approach. These analyses treat the coalescent process in the recent past as a function of the details of the population structure and the sampling scheme. In the more distant past, the genealogical process is assumed to be independent of the sampling scheme. Wakeley (1999) has referred to these two processes as the “scattering phase” and the “collecting phase,” respectively, and has successfully used this method to analyze island-type models with large numbers of demes (Wakeley 1998, 1999, 2000, 2001; Wakeley and Aliacar 2001; Wakeley and Lessard 2004). Similar methods have been used to describe the coalescent process in plants with selfing (Nordborg 1997, 2000; Nordborg and Donnelly 1997; Möhle 1998).
Separation of timescales is most powerful when the scattering phase is very short compared with the collecting phase, so that it may be treated as essentially instantaneous. However, even when these conditions do not hold, the technique may permit the generation of tractable, if approximate, expressions to describe genealogies. A further affordance of the separation-of-timescales approach accrues if the collecting phase can be described using the equations developed for the standard neutral coalescent model (SNCM; Kingman 1982a,b; Hudson 1983; Tajima 1983).
Coalescent simulations (presented below) indicate that a range of conditions exists over which the genealogical process in a continuous two-dimensional habitat converges approximately on the SNCM. That is, the long-term genealogical behavior can be described by some effective population size Ne that is independent of the initial sampling scheme. It is possible to construct a separation-of-timescales-based description of the genealogical process by combining this description of the long-term behavior with a location-dependent description of the short-term coalescence probabilities. This approach results in explicit expressions for the distribution of coalescent times for a pair of sequences. This distribution is a function of the sampling locations, dispersal rate, population density, and habitat geometry.
THE MODEL
The model analyzed here is a two-dimensional analog of the one-dimensional model considered by Wilkins and Wakeley (2002). A population of N haploid individuals is uniformly distributed over a two-dimensional habitat of area A. Most of the analysis focuses on a rectangle whose length and width are given by L1 and L2, where L1 ≥ L2, although some consideration is also given to a torus that is the direct product of two circles of lengths L1 and L2, where once again L1 ≥ L2. These two geometries can also be thought of as two different treatments of the boundary conditions on the same L1 × L2 rectangle. In both cases, the purpose of the boundary conditions is to retain migrating lineages within the habitat. In the “rectangular” case, reflecting boundary conditions are assumed. That is, if migration would have carried a lineage a certain distance beyond the habitat boundary, that lineage moves to a location an equally far distance from the boundary, but inside the habitat, as if the lineage had bounced off the boundary. In the toroidal case, periodic boundary conditions are assumed. A lineage exiting the habitat in this case would reenter through the boundary at the opposite end of the rectangle. These two formulations are illustrated in Figure 1.
Figure 1.—

Habitat geometry and boundary conditions. The two-dimensional habitat considered in this article is a rectangle of dimensions L1 × L2, where L1 ≥ L2. Two different treatments of the boundaries are considered. In the “rectangular” model, reflecting boundary conditions are assumed, and a lineage exiting the habitat reenters on the same side. In the “toroidal” model, periodic boundary conditions are assumed, and a lineage reaching the boundary reenters the habitat from the opposite side.
The population density ρ is equal to N/A. Population density regulation is absolute, so the structure is that of a lattice model, with exactly one individual occupying each point on the lattice every generation. This is equivalent to the lattice model investigated by Slatkin and Maddison (1990) and by Barton and Wilson (1995)(1996) for absolute local density regulation. Generations are nonoverlapping, with each individual producing a large number of propagules, which are dispersed according to a bivariate normal distribution with variance σ2 in each direction. A large number of propagules thus arrive at each point in the lattice in every generation, and one of those propagules is chosen at random to produce the adult at that location.
In the coalescent process, samples are taken from particular geographic locations. The locations of lineages corresponding to the ancestors of those samples are modeled as a random walk, where each step is drawn from a two-dimensional normal distribution with variance σ2 in each direction. The probability that the two lineages coalesce in the previous generation is a function of the distance between them. Barton et al. (2002) show that the effective density is inversely proportional to the integral of this probability over distance. Under the assumptions of Gaussian dispersal and perfect density regulation, the effective density is equal to the actual density ρ, and coalescence can be assumed to occur when the two lineages simultaneously fall within an area occupied by only one individual (1/ρ). Under these conditions, the neighborhood size, Nb, is one over the probability that two lineages starting from the same location both fall within an area of 1/ρ after each has taken one step in its random walk. As Nb becomes large, its value approaches 4πρσ2 (Wright 1943).
In the recent past (the scattering phase), the distribution of coalescence times for a pair of sequences depends on their relative sampling locations, the neighborhood size, and the location of nearby habitat boundaries. In the more distant past (the collecting phase), the coalescent process becomes independent of the original sampling scheme and depends only on properties of the population as a whole. For certain parameter values, the collecting phase is reasonably approximated by the standard equations for the coalescent process under the SNCM. In these cases, the long-term effective population size depends on the local dispersal behavior and the habitat geometry. For clarity, only the major results are presented in the text, along with comparisons of the analytic results to simulations. Derivations and more technical discussion have been relegated to the appendix.
THE SCATTERING PHASE
I begin by considering two sequences sampled from the same location in a habitat without boundaries. The coalescent behavior for two samples from the same location under strict density regulation is derived by Barton and Wilson (1995)(1996). In terms of the parameters as defined above, the probability that the two lineages share a common ancestor t generations in the past is given by
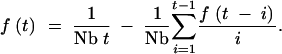 |
1 |
Equation 1 is in the form of a recursion relationship. That is, to calculate the probability of coalescence in generation t, it is first necessary to calculate the probabilities of coalescence in generations 1 through t − 1, which becomes unwieldy for large values of t. An approximate expression for this probability, which does not require recursive calculation, is derived in the appendix. The corresponding equation for the cumulative probability distribution (the probability that two sequences sampled from the same location share a common ancestor no more than T generations in the past) is given by Equation A14 and has the following form:
 |
2 |
In Equation 2, γ1 is Euler's gamma (∼0.5772). Approximate values for the subsequent γi terms are given by expression (A12) in the appendix. This expression becomes less accurate as the neighborhood size decreases. In particular, it is not valid if Nb < Log(T). The complexity of these equations arises from the nonindependence of the two random walks. Specifically, if the two lineages have not yet coalesced by a particular time T, the distance between them will, on average, be greater than would be expected from two independent random walks. This interference between lineages is, ultimately, the source of the elevation in effective population size seen in geographically structured populations.
To evaluate the range of parameter values over which the approximations invoked in this analysis are valid, the results of Equation 2 were compared with simulations. Figure 2 presents the cumulative density function (CDF) for coalescence of a pair of sequences sampled from adjacent lattice points in an unbounded habitat. Data are presented for several different values of the neighborhood size. In each case, simulation results are presented along with the distribution described by the equations presented in this article. These results show that the method fails as the neighborhood size becomes small (<20), even at smaller values of T. The probability of identity for a pair of samples from the same location was also calculated for a number of different parameter combinations by taking the sum of (F(T) − F(T − 1))e−2μT over all T based on Equation 2. This comparison assumes a per-generation mutation rate of μ under an infinite-sites model of mutation. These values were compared against Malécot's approximation under the same conditions (Equation 13 of Barton et al. 2002). For all values tested, the two estimates differed by no >0.22%.
Figure 2.—

Comparison of analytic results to simulation for pairs of sequences sampled from adjacent lattice points. This graph compares simulation results (dashed lines) with results derived from Equation 2 (solid lines) for different values of Nb. Initial spacing between the two samples is 1/ , corresponding to the distance separating two neighboring points on the two-dimensional lattice. Agreement is good for values of Nb >10. No boundary effects are included in either the simulations or the analytic results.
, corresponding to the distance separating two neighboring points on the two-dimensional lattice. Agreement is good for values of Nb >10. No boundary effects are included in either the simulations or the analytic results.
Description of the coalescence of two samples drawn from different locations is both an easier and a harder problem. The addition of a nonzero distance between the samples complicates the equations, but the degree of lineage interference is less, so fewer terms need be considered to generate an acceptable approximation. For sufficiently large values of Nb, the probability density function for coalescence of a pair of sequences separated by a distance of 2ξ (where ξ is in units of σ) is given by Equation 3 (A17 in the appendix),
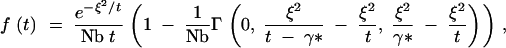 |
3 |
where γ* = 0.562, and Γ indicates the incomplete gamma function, which is given by Equation A18.
Equation 3 was compared to Malécot's approximation for probabilities of identity (Equation 15 of Barton et al. 2002) for a number of parameter combinations just as Equation 2 was. Equation 3 represents a cruder approximation than Equation 2, and the deviation from Malécot's approximation is correspondingly larger. For a neighborhood size of 50 and a separation between samples of 10σ, the deviation ranged from 12.4% (μ = 10−3) to 21.4% (μ = 10−5). For Nb = 500, the deviations were much smaller, varying between 1.2% (μ = 10−3) and 2.2% (μ = 10−5). The deviations were nearly identical for a separation of 20σ. The increasing error at smaller neighborhood sizes results from the fact that Equation 3 incompletely accounts for the interference between lineages, and this interference is greatest when the neighborhood size is small. The larger errors associated with lower mutation rates result from the fact that Equation 3 becomes more inaccurate farther in the past, and older coalescence times are more relevant to the probability of identity when the mutation rate is small. When the mutation rate was set to 10−5, coalescence times >200,000 generations in the past contributed measurably to the probability of identity based on Equation 3. With μ = 10−3, the probability of identity was not significantly influenced by coalescent events >10,000 generations in the past. The separation-of-timescales approach employed here means that we will generally require accuracy from Equation 3 only at the smaller values of t. The accuracy of Equation 3 over the duration of the scattering phase is illustrated in the context of the overall method for particular cases below (Figures 8–10).
Figure 8.—

Effect of image number on the coalescence time probability distribution. The distribution of coalescence times was determined using three different choices for the number of images (m = 4, 17, and 50). The habitat is a 10 × 10 square with ρ = 100 and σ = 0.3 (N = 10,000; Nb = 113). The samples are from adjacent locations at (1.95, 2.0) and (2.05, 2.0). The effective population size from Equation 6 is 10,669. The transition times were determined from Equation 7: τ = 308 for m = 4; τ = 454 for m = 17; τ = 759 for m = 50. Crosses indicate the probability distribution determined from 100,000 simulated coalescence times. Older coalescence rates represent the average rate over a range centered on that marker. For example, the mark at t = 30 represents the average rate from t = 25–35; the mark at t = 3000 is the average from t = 2500–3500. The predicted coalescence probability distribution is very similar for the different values of m except in the vicinity of the transition between the scattering and collecting phases. The dip in the analytic values at t = 2 results from the fact that this probability distribution was derived by subtracting the CDF at t − 1 from the CDF at t. The CDF determined by Equation 2 significantly underestimates the probability of coalescence for very small values of T, and the value of the CDF at T = 1 was taken simply to be 1/Nb.
Figure 10.—

The coalescent in a bounded habitat. Values derived from simulations for the cumulative density function of the coalescence time for a pair of sequences are compared to values given by Equations A23 and A28. Three sampling locations from a rectangular habitat of dimension 10 × 5 were considered: (1, 1), (8, 2), and (6, 3). For each of the six pairs of locations (a–f), 100,000 coalescence times were simulated with N = 10,000 (ρ = 200). Analytic results derived from equations in the text are indicated by solid lines, and simulation results by crosses or solid and open circles. The three dispersal rate values were used, and the corresponding long-term effective population sizes were determined using Equation 6: σ = 0.15 (Nb = 56.5, Ne = 11,454, solid circles); σ = 0.25 (Nb = 157.1, Ne = 10,483, crosses); and σ = 0.4 (Nb = 402.1, Ne = 10,235, open circles). The transition times were determined from Equation 7 (a–c and f) or Equation 8 (d and e). The numbers of images used in a–f were 27, 30, 20, 18, 23, and 18, respectively.
Given these expressions for the coalescent process, we can use the method of images to construct an expression for the distribution of coalescence times for a pair of sequences. This method can be applied to an arbitrary pair of sampling locations, but is restricted to a rectangular habitat. To account for the boundary conditions, we need to assume a number of competing coalescent processes that occur simultaneously, corresponding to reflections off various habitat boundaries. Similar reasoning applies to samples drawn from separate locations. One must consider not only the coalescent process corresponding to the distance ξ between the two locations, but also each distance ξi corresponding to the distance from one sample to each reflected image of the other sample.
The total number and arrangement of images that need to be considered will depend on the relative lengths of the axes of the habitat. A minimum of eight images will be required to account for the four habitat boundaries and four corners. Additional images corresponding to multiple reflections may also need to be considered, particularly if the habitat is much longer in one dimension than in the other. In principle, the number of such images is infinite. What is important for this method is that we include all of the images that correspond to distances shorter than that of the maximally distant included image. The process of determining image distances is discussed in more detail in the appendix and is illustrated in Figure 3 for a particular hypothetical habitat. The application of the method of images to a toroidal habitat is described in detail by Barton and Wilson (1995)(1996) and in the appendix. Unfortunately, the method of images does not lead directly to a method for rigorously treating other habitat shapes. In certain cases, it may be possible to treat the recent past for nearby samples by considering only nearby boundaries. The validity of such an approach is not considered further in the present analysis, however.
Figure 3.—

Illustration of the method of images. This represents an example of how the method of images is used to construct the various components of the coalescent process. A hypothetical rectangular habitat is outlined by a thick solid line. The two sampling locations are represented by solid stars. The rectangular habitat is repeatedly reflected across each of its boundaries to yield an infinite plane of tiled images of the original habitat. The locations of the images of one of the two original samples are indicated by the other stars. The set of images used to construct the coalescent process should include the eight images in the adjacent habitat images (shaded areas). Beyond this, the choice of exactly how many images to include is somewhat arbitrary, except that the set must include all images that are closer than the farthest included image. The circle represents the minimal radius of inclusion that incorporates the images in the eight shaded regions, determined by the distance to the farthest of these (indicated by the thick dashed line to the hatched star). Any larger radius would also represent a valid choice and would simply require consideration of additional images and correspond to a longer duration of the scattering-phase description.
THE COLLECTING PHASE
The collecting phase refers to that part of the coalescent process that is independent of the original sampling scheme. It may not be immediately obvious that such a phase must exist or account for a substantial portion of the genealogy. Nor is there any reason to suspect that this phase, if it does exist, will necessarily take on a particularly simple form. However, in some models of geographic structure it has been found that most of the genealogy can be adequately described using equations derived for the coalescent process in a panmictic Wright-Fisher population of constant size without selection, which I refer to as the SNCM. Cox and Durrett (2002) suggest that this may be a general feature of the genealogical process in two dimensions. Simulations performed for this analysis suggest that there is a certain range of parameter values under which the coalescent process in the two-dimensional continuous model converges on the SNCM. The range of parameter values and the effective population size depend on the local dispersal behavior, the population density, and the size and shape of the habitat. The long-term coalescent process is better approximated by the SNCM for larger neighborhood sizes and smaller habitat distances.
Simulations were used to explore the range of parameter values over which the collecting phase converges on the SNCM. Coalescent simulations were performed on samples of size 25. In each case, 100,000 replicates were completed. The results of these simulations were analyzed as skyline plots (Pybus et al. 2000; Strimmer and Pybus 2001), that is, the average number of generations in which there were i lineages left in the sample, where 2 ≤ i ≤ 25. This average duration was then multiplied by
| i |
| 2 |
. The rate of coalescence is thus represented in terms of the effective population for which that rate would be expected under the SNCM. For each value of i, this value was plotted against the mean time at which the period of i lineages ended. For the SNCM, in which Kingman's (1982a) coalescent has time scaled by the effective population size (Ne), the expected plot would be a horizontal line near Ne. Figure 4 illustrates the convergence of the coalescent process for different sampling schemes in a square habitat. For this set of parameter values (N = 10,000; L1 = L2 = 10; ρ = 100; σ = 0.3; Nb = 113), the coalescent process converges to an effective population size of ∼10,700, regardless of the original sampling scheme. It is worth noting that the process converges in <600 generations, compared to an average tree depth of >20,000 generations. Thus, in this case, the collecting phase is well described by the SNCM and accounts for >97% of the genealogical process.
Figure 4.—

Long-term independence of the coalescent process on the sampling scheme. This skyline plot illustrates the convergence of the continuous-habitat coalescent to a process that is equivalent to the standard neutral coalescent with a constant effective population size. For each of the three curves, 100,000 replicates of the coalescent process were simulated for a sample of 25 sequences. The parameters used for the simulations were N = 10,000, ρ = 100, σ = 0.3, in a square habitat measuring 10 × 10. The mean waiting time for each coalescent event was converted into a value corresponding to the population size that would give that expected time in a panmictic population. This inverse rate is plotted on the y-axis against the average time in the past at which this coalescent event occurred. In the sampling scheme labeled “1 Location,” the 25 samples are drawn from a 5 × 5 grid of equally spaced points at the center of the habitat [location (5, 5)]. Grid spacing is 0.1, corresponding to the minimum distance separating two individuals in a two-dimensional lattice with ρ = 100. In the “5 Locations” scheme, 5 samples are drawn from a vertical cross with spacing 0.1 between samples. There are five such crosses centered on (2, 2), (2, 8), (5, 5), (8, 2), and (8, 8). In the “20 Locations” scheme, samples are drawn from a centered 5 × 5 grid with spacing of 2 [locations (1, 1), (1, 3), etc.]. These three different sampling schemes all converge on the same process, and coalescent events > ∼600 generations in the past are independent of the original sampling locations. The fact that all three converge to a horizontal line indicates that this long-term process can be approximated by Kingman's coalescent with time rescaled by the effective population size. Variances have been omitted from this and other figures for clarity of presentation, but the variance of the waiting time for each coalescent event is close to the square of the mean, consistent with the exponential distribution of waiting times expected under the SNCM.
Figure 5 shows results for rectangular habitats of a variety of length-to-width ratios. In all cases, all other parameter values are identical to those used to generate Figure 4. The total area is held constant at 100. This graph illustrates the influence of habitat geometry on the long-term coalescent process and suggests conditions under which the SNCM is an adequate description of the collecting phase. Specifically, if the length of the major axis of the habitat is too long with respect to the dispersal rate, the coalescent process does not approach the SNCM. As the ratio L1/L2 becomes very large, the coalescent process is expected to converge on that for a one-dimensional habitat. It is difficult to compare this limit explicitly with the one-dimensional solution (Wilkins and Wakeley 2002) due to the fact that as L2 becomes small, a very large number of reflections will need to be considered.
Figure 5.—

Effect of habitat geometry on the long-term coalescent process. Six skyline plots similar to those in Figure 3 are presented. In all cases, the total habitat area is 100, N = 10,000 (ρ = 100), and σ = 0.3 (Nb = 113). All habitat areas are rectangular, and the lengths L1 and L2 are given for each curve. Samples are drawn from a centered 5 × 5 grid with spacing L1/5 in one dimension and L2/5 in the other. As L1/L2 becomes large, the long-term coalescent process is no longer well approximated by a single effective population size. For a neighborhood size of 113, the condition M* > 1 holds if the ratio of lengths is <9:1. The values of M* for the plots are, from top to bottom, 0.36, 0.5625, 1.44, 2.25, 5.76, and 9.0. The four plots for which M* > 1 are close to horizontal in the collecting phase. The plots for which M* < 1 are represented by open circles and squares. The vertical crosses (10 × 10 square habitat with M* = 9.0) correspond to the conditions used to generate the skyline plots in Figure 4.
The average coalescence time for a pair of sequences drawn from a panmictic population of size N is N generations. A sequence taking a one-dimensional random walk with step size σ will have to travel a distance L1 in ∼(L1/σ)2 generations. Let us define M* as Nσ2/L21, where L1 is the length of the major axis of the habitat. If M* ≫ 1, lineages cross the habitat quickly relative to the rate of the coalescent process. If M* ≪ 1, lineage movement will be rate limiting with respect to coalescence. Thus M* can serve as an indicator of the applicability of the effective-population-size approximation for the collecting phase. The parameter values used to generate Figure 4 correspond to a value of M* = 4, and the skyline plots in Figure 5 have M* values ranging from 0.36 (for the uppermost set of points) to 9.0 (at the bottom).
Figure 6 presents the results of simulations demonstrating the correspondence between M* and the uniformity of the long-term coalescent process. Each point on the graph indicates the slope of the last five points of a skyline plot like those presented in Figures 4 and 5. Slopes close to zero indicate a good correspondence to the SNCM approximation. Under the SNCM, the expected waiting time for the last coalescent event is equal to the effective population size and equal to the expected waiting time for all previous coalescent events combined. This slope thus provides a crude estimate of the fractional size of the error introduced by approximating the genealogical process in this manner. For example, if the slope is 0.25, the ratio of the expected waiting time for the last coalescent event to earlier (more recent) waiting times will be on the order of 25% greater than that expected under the SNCM. For values of M* > 1, slopes are consistently <0.05, suggesting that for these sets of parameter values, the error introduced by this approximation will be <5%. The points corresponding to the curves in Figure 5 are derived from 100,000 replicate simulations and are represented by solid circles. Crosses represent the outcome of 10,000 simulations.
Figure 6.—

M* and the long-term coalescent process. Each point is derived from one skyline plot like those presented in Figures 4 and 5. On the x-axis is plotted M*, which equals Nσ2/L21, where L1 is the length of the major axis of a rectangular habitat. On the y-axis is the slope of the last five points for each skyline plot. Solid circles correspond to the plots in Figure 5 and are derived from 100,000 replicate genealogies. Crosses correspond to plots derived from 10,000 replicates. The two horizontal lines are placed at ±0.05. For values of M* > 1, the long-term coalescent process is well described by a single effective population size. When M* < 1, the deeper branches in the genealogy are longer than what would be expected from a constant population size process. The results represent genealogies simulated under a variety of combinations of parameter values. The ratio L1/L2 was varied from 1 to 50 (with L1 ranging between 8 and 50), the density ρ was varied from 100 to 1000, and the dispersal rate σ was varied from 0.05 to 0.5, corresponding to neighborhood sizes ranging from 3 to 3000.
Analogous simulations for a toroidal habitat produced qualitatively similar results (data not shown), but with the SNCM applied to values of M* > 0.25. Intuitively, a torus whose major axis is of length L1 represents a less extended habitat than a rectangle of length L1. Specifically, a lineage need travel only a distance L1/2 to have crossed the toroidal habitat. Since M* is proportional to 1/L21, this produces the factor of four difference in the value of M* at which the long-term behavior changes from a genealogical process that is well approximated by the SNCM to one that is expected to produce longer coalescence times for the deepest branches.
The conditions under which the SNCM approximation holds bear some resemblance to the strong migration limit (Nagylaki 1980, 2000; Notohara 1993). However, the conditions considered here are less stringent. In the strong migration limit, the location of each lineage becomes independent of the locations of the other lineages. In the model considered here, where conservative migration is assumed, this would correspond to the limit in which the effective population size would approach the census size, and the duration of the scattering phase would approach zero. The regime considered here corresponds to those conditions under which the long-term coalescent process appears to be reasonably well described by the largest nonunit eigenvalue of the corresponding Markov chain transition matrix. Furthermore, a range of cases exists (where M* > 1, or, equivalently, Nb > 4πr, where r is the ratio of the lengths of the major and minor axes of the habitat) where the eigenvalues corresponding to subsequent coalescent events bear the same relation to each other as those in the SNCM do. In this range, migration is not strong enough to eliminate geographic structure completely, but the primary effect of limited gene flow is to alter the constant factor by which the coalescent process is rescaled. Figure 4 (where M* = 9) presents an example of a degree of population structure falling within this range. For more restricted gene flow (M* < 1), this rescaling is not constant throughout the coalescent process, leading to the same sort of effects on tree shape that would be expected from a population that had a larger effective population size in the past.
EFFECTIVE POPULATION SIZE
Constructing an analytic description of the collecting phase requires knowledge of the effective population size. One obvious method for determining this value is through the sort of simulations used to generate Figures 4–6. This approach possesses the advantage of explicitly verifying the assumption of convergence to the SNCM. It is also possible to derive an expression for the long-term effective population size from classical population genetics results by Maruyama (1972). For a toroidal habitat formed by the direct product of two circles of lengths L1 and L2, the effective population size is given by Equation 4 [(A36) in the appendix]:
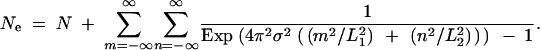 |
4 |
Computation of Equation 4 is feasible because the terms of the sum become small as either n2 or m2 becomes large. Charlesworth et al. (2003) present an excellent approximation to Equation 4 for the case where L = L1 = L2. Translated into the terms of this article, Equation 9 of Charlesworth et al. (2003) is
 |
5 |
where K = 0.24 for a Gaussian dispersal profile (Barton et al. 2002). For the case of a rectangle of lengths L1 and L2, a slightly modified version of Equation 4 yields a reasonable approximation to the long-term effective population size:
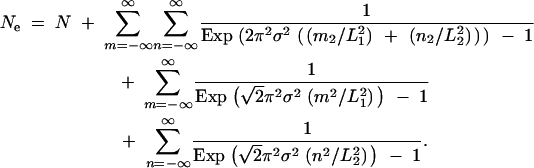 |
6 |
Equations 4 and 6 were compared with values derived from simulations (Figure 7). Each simulation-derived effective population size was taken from the last point in a skyline plot like those in Figures 4 and 5. If we plot Ne directly, it is not possible to get clear visual separation between the various curves at all values of σ simultaneously. Therefore, for clarity of presentation, simulation and analytic values are given in terms of FST (= (Ne − N)/Ne). For a fixed value of N, higher values of FST correspond to higher values of Ne. The fit is reasonable over the sets of parameter values considered. The total habitat area is the same for all curves in Figure 7.
Figure 7.—

Effective population size and habitat geometry. The long-term effective population size was determined from simulations similar to those used to generate Figure 5. For each set of parameter values, 100,000 genealogies were generated, and the effective population size was taken to be the average waiting time for the last coalescent event. These values are given as FST values [(Ne − N)/Ne] for clarity of presentation. The habitats included here are a 10 × 10 torus with N = 10,000 (solid diamonds), a 25 × 4 torus with N = 10,000 (open circles), a 10 × 10 rectangle with N = 10,000 (crosses), a 25 × 4 rectangle with N = 10,000 (solid circles), and a 10 × 10 rectangle with N = 100,000 (triangles). For each of these habitats, a number of different dispersal rates (σ) were considered and are represented along the x-axis. Expected values from Equations 4 and 6 (for toroidal and rectangular habitats, respectively) are indicated by solid lines. Agreement is good over the cases considered, including conditions for which M* < 1.
The effective population size increases with the total population size and decreases with increasing dispersal rate. In the limit of high dispersal, the population becomes effectively panmictic, and Ne is equal to N. The long-term effective population size is also a function of the habitat geometry. For fixed values of A, ρ (and therefore N), and σ, a rectangular habitat will have a larger value of Ne than a torus of the same dimensions. Intuitively, this is a consequence of the fact that two lineages can be functionally separated by a greater distance in a rectangle than in a torus. The maximum distance between any two points in an L1 × L2 rectangle is 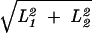 , whereas in a torus it is
, whereas in a torus it is 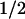
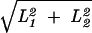 . Recalling that we have assumed that L1 ≥ L2, Ne increases with L1/L2 in both the rectangular and toroidal geometries. The intuitive explanation for this effect is similar to that for the difference between the rectangle and torus. The more protracted the rectangle, the greater the possible distance separating two lineages.
. Recalling that we have assumed that L1 ≥ L2, Ne increases with L1/L2 in both the rectangular and toroidal geometries. The intuitive explanation for this effect is similar to that for the difference between the rectangle and torus. The more protracted the rectangle, the greater the possible distance separating two lineages.
A number of the points plotted in Figure 7 correspond to conditions for which M* < 1. It is worth noting that Equations 4 and 6 appear to provide reasonable approximations for the expected waiting time for the most ancient coalescent event, even when the rest of the collecting phase is not well described by the SNCM. For example, for the 25 × 4 rectangular habitat with N = 10,000 and σ = 0.1 (M* = 0.16), the effective population size predicted by Equation 6 is 25,974. The average number of generations required to go from two lineages to the common ancestor of the entire sample in 1 million simulated genealogies was 25,894. The mean wait times for the next four most ancient coalescent events (each scaled by
| i |
| 2 |
were 22,835, 21,280, 20,290, and 19,612. This suggests that Equations 4 and 6 may provide a valid description of the collecting phase even for some cases where M* < 1, so long as our analysis is restricted to a pair of samples.
THE COALESCENCE PROBABILITY DISTRIBUTION
The only remaining problem is the choice of the time τ at which we make the transition from the scattering phase to the collecting phase. Under many models employing separation of timescales, a point exists in the genealogical process at which the collecting-phase description becomes completely accurate. In this model, by contrast, there is no finite time for which all geographic information has been lost. Rather, the genealogical process asymptotically converges to the collecting phase. Thus, there is no single correct choice for τ. I present two methods for calculating τ, each of which is dependent on the number of images considered in the analysis. Both have the feature that the results will be relatively insensitive to small changes in τ. The accuracy of the description is improved by increasing the number of images considered, but so is the computational burden. Furthermore, the approximations used become less valid as t becomes larger. The choice of the number of images, or image radius (see Figure 3), will therefore be governed by this trade-off.
The first method for calculating τ applies to samples drawn from nearby locations. For such samples, the rate of coalescence will be >1/Ne in the recent past and then decay asymptotically toward 1/Ne. The scattering-phase description using a finite number of images will provide a good approximation to the coalescent process at short timescales, but with a coalescence rate that decays to zero. It is appealing, therefore, to set τ to the point where the rate of coalescence is equivalent under the two descriptions. This point will be close to the time when the rates of coalescence are equal if we neglect interference between lineages in both processes:
 |
7 |
The term on the left-hand side of Equation 7 is summed over all image distances. Figure 8 compares the distribution of coalescence times for three different choices of the image radius for a pair of samples drawn from the adjacent locations in a rectangular habitat. The dip in the plot at t = 2 is the result of substantial inaccuracies in Equation 2 for very small values of t. If the details of the coalescence time distribution in this region are of interest, it would be better to use the recursion equation of Barton and Wilson (1995), which is given in the appendix as Equation A1. For times >10 generations in the past, Equation 2 provides a good approximation (see Figure 2).
For a pair of samples drawn from two more distant locations, there may be more than one value of τ that will satisfy Equation 7, in which case the largest value should be used as the transition time. For two samples drawn from widely separated locations, the rate of coalescence will initially be close to zero and will gradually approach 1/Ne without ever exceeding it. Thus, for some pairs of sampling locations, there will be no value of τ that will satisfy Equation 7. Considering a finite number of images, the coalescence rate given by the scattering-phase description will increase to some maximum and then decline to zero in the distant past. The coalescence rate at that maximum will be closer to 1/Ne for larger numbers of images. Under these conditions, I suggest setting τ to a value that gives this maximum, which is found by solving Equation 8 for τ:
 |
8 |
Figure 9 compares the distribution of coalescence times for a pair of samples drawn from two widely separated locations for three different choices of the number of images.
Figure 9.—

Effect of image number on the coalescence time probability distribution. This is identical to Figure 8 (N = 10,000; Nb = 113; σ = 0.3; Ne = 10,669), but with the two samples drawn from locations (2.0, 2.0) and (7.0, 9.0). Transition times were determined from Equation 8: τ = 408 for m = 10; τ = 503 for m = 16; τ = 676 for m = 39. As was the case with Figure 8, the three probability distributions are in close agreement except in the vicinity of the transition point.
Taken together, Equations 2–8 allow us to construct approximate expressions for the distribution of coalescence times for pairs of sequences drawn from a rectangular habitat. The details of constructing this distribution are provided in the appendix. To provide some validation for the approximations used, Equations A23 and A28 have been compared with Monte Carlo results for a few particular parameter combinations (Figure 10). These data illustrate the dependence of the coalescent process on location within the habitat as well as the distance between samples. As expected from previous analyses (Maruyama 1970c,d, 1972; Fleming and Su 1974; Malécot 1975; Nagylaki and Barcilon 1988; Hey 1991; Herbots 1994, pp. 66 and 145–146; Wilkins and Wakeley 2002), coalescence time increases with the distance between samples and is greater for samples drawn farther from the edge of the habitat.
DISCUSSION
There are a few different ways in which we can imagine putting the results of this analysis to use. First, the expressions derived here can provide the basis for a more sophisticated analysis of certain geographically structured populations. For example, a common method of estimating gene flow is to regress some function of observed pairwise FST values against expected values under isolation by distance (e.g., Rousset 1997). These expected values are typically derived from models that assume either an unbounded habitat or periodic boundary conditions (the circular, or toroidal, stepping stone). Results such as these could be used to incorporate additional information about sampling location into the derivation of expected pairwise values. A maximum-likelihood approach described by Tufto et al. (1996) is based on the geographical pattern of covariance of allele frequencies. Pairwise results can be used to convert demographic features (local dispersal behavior, population density, etc.) into a geographically explicit covariance matrix. Similarly, these results could be used as the basis for a more sophisticated version of any method of analysis relying on pairwise comparisons.
The fact that the separation-of-timescales approach provides a reasonably accurate approximation has implications both for computational methods of analyzing geographic structure and for the interpretation of geographic patterns of genetic diversity. Markov chain Monte Carlo (MCMC)-based approaches to the analysis of coalescent processes typically rely on having an analytic expression for the waiting time until the next event (coalescence, migration, recombination, etc.). For the simple case of n lineages in a panmictic population, there are n − 1 coalescent events. The waiting time to go from k to k − 1 lineages is exponentially distributed with mean
| k |
| 2 |
/N. Thus, one complete genealogy can be constructed simply by generating n − 1 exponential waiting times (see, e.g., Hudson 1990). The computational time required is roughly independent of the population size. One of the challenges in applying MCMC sampling in a continuous habitat is that analogous expressions for these waiting times are not readily available. Put another way, the waiting time to the next “event” is always one generation, since each lineage moves every generation. When lineages are far apart, their movement can be drawn from a Gaussian spanning several generations. However, when they are close together, every generation must be considered explicitly.
This analysis indicates a class of models for which the long-term coalescent behavior is independent of the original sampling scheme. Under the equilibrium model, there is a range of parameter values where this long-term behavior converges on the standard neutral coalescent model with some effective population size that depends on the local dispersal behavior and the geometry of the habitat. This location-independent long-term behavior typically describes the majority of the tree depth. Thus, for an arbitrary number of samples, genealogies could be generated computationally using a two-step process. The recent part of the genealogy can be generated by explicitly simulating every generation. At some point (the same point where we switch from the scattering phase to the collecting phase), this simulation process could be replaced with the conventional process based on waiting times. Such an approach might be used to improve the efficiency of computationally intensive approaches to the analysis of geographically structured data.
For many populations, the deep branches of genealogies will be shaped by nonequilibrium processes (e.g., population bottlenecks, range expansions, selection at linked loci, etc.) and by geographical heterogeneity (e.g., barriers to gene flow). A separation-of-timescales approach may lead to methods for separating isolation-by-distance effects from these sorts of nonequilibrium events. For example, MCMC integration of the scattering phase could be used to project a set of samples from particular locations onto a distribution of ancestral samples at the boundary between the scattering and collecting phases. This ancestral distribution could then be queried for patterns associated with particular demographic histories or for evidence of additional subdivision (in the form of geographic structuring of clades beyond what is explained by ongoing local dispersal). The application of multiple timescales to separate local dispersal from other factors shaping genealogical structure could provide a valuable tool in the development of statistically rigorous phylogeographic methods (Knowles and Maddison 2002; Knowles 2004).
The analysis presented here also suggests certain specific features of the genealogical process that may aid in the interpretation of empirical data. In particular, the long-term coalescent behavior observed in simulations may be relevant to efforts to use genealogical structure to infer the existence of barriers to gene flow. First, the duration of the collecting phase is longer than that of the scattering phase by a factor on the order of Nb. This means that in most populations, the scattering phase will account for only a tiny fraction of the genealogy. This is consistent with previous arguments that the deep branches in any genealogy are unlikely to carry useful information about long-term patterns of gene flow under equilibrium conditions.
I have proposed the term M* as a useful metric for characterizing the nature of the genealogical process in the collecting phase. When M* > 1, the collecting phase is well approximated by the SNCM. The wide spread of the points in the M* < 1 region of Figure 6 suggests that M* is not sufficient to fully characterize the collecting phase in those cases where it deviates significantly from the panmictic process and should therefore be used simply to characterize a particular population as falling within one of the two regimes. M* is defined as Nσ2/L21, where L1 is the length of the longer of the two habitat axes. The condition M* > 1 can be rewritten as Nb > 4πr, where r is the ratio of the lengths of the two habitat axes. Since r can be no smaller than 1, no population is expected to converge to the SNCM if Nb < 4π (∼12.5). The more extended the habitat becomes, the larger the neighborhood size must be for this convergence to occur.
The simulation work by Irwin (2002) in a one-dimensional habitat showed that under certain parameter values genealogies generated from a model of simple isolation by distance could give the appearance of a deep phylogeographic break, which could lead to the erroneous inference of a barrier to gene flow. This signature is in the form of coalescence into two geographically distinct clades, with a deep genealogical split between them. Irwin notes that the likelihood of this outcome increases as the dispersal distance (σ) or the population size (N) decreases. Inspection of Figure 4 from Irwin (2002) indicates that the region of parameter values for which deep phylogeographic breaks are likely corresponds to values of M* that are less than one.
Of course, the variance of the coalescent process is large, even in models lacking geographic structure, and deep genealogical divisions can arise by chance. In attempting to assess the likelihood of the existence of a barrier to gene flow, the best course of action is to consider multiple independently segregating loci. While individual loci may manifest deep phylogeographic breaks by chance, in the absence of linkage, multiple loci are unlikely to manifest geographically coincident deep genealogical divisions unless there is, or has been, some barrier to gene flow. However, when only a single locus is available, determination of whether M* is likely to be less than or greater than one could serve as a check on the plausibility of attributing deep phylogeographic structure to simple isolation by distance in individual cases.
The motivation behind this work has been to generate solutions for a model of geographic structure that is somewhat more realistic than many other models for which analytic solutions have been derived. At the same time, the goal has been to develop a solution that is more easily computed than one that relies on explicitly calculated recursions or Monte Carlo simulations. It is my hope that results such as these may contribute to the development of a useful middle ground between realism and tractability. The implementation of these results into an inferential framework has not been pursued here, but C programs for performing many of the basic calculations are available from the author.
Acknowledgments
I thank C. Muirhead, S. Otto, V. Savage, J. Wakeley, and an anonymous reviewer for comments on the manuscript. Special thanks go to N. Barton for detailed comments that have resulted in significant improvements to the manuscript, including the derivation of the long-term effective population size. This work was funded in part by a grant from the William F. Milton Fund.
APPENDIX
Two sequences from the same location:
I begin by deriving the distribution of coalescence times for a pair of sequences drawn from the same location. Following Barton and Wilson (1995)(1996), the probability that the two lineages coalesce t generations in the past is given by the recursion equation:
 |
A1 |
This equation can be rewritten in a nonrecursive form as
 |
A2 |
The additional terms continue to alternate sign, with each subsequent term multiplied by 1/Nb and including an additional nested sum. Because each summation is to the previous index minus 1 (e.g., the sum on j is from 1 to i − 1), the number of nonzero terms in Equation A2 is equal to t (e.g., for t = 3, only the 1/Nb, 1/Nb2, and 1/Nb3 terms are not equal to zero). The sums in Equation A2 can be eliminated using some approximate forms. Denoting the nested sum following the 1/Nbj term as ςj, Equation A2 can be written as
 |
A3 |
The ςj terms are approximated as follows:
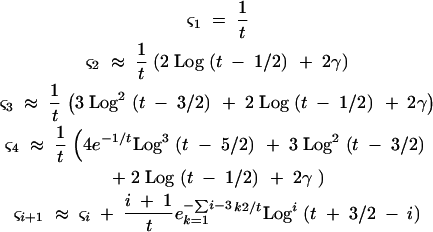 |
A4 |
In these expressions, γ is Euler's gamma (∼0.5772). The ςj terms were calculated explicitly for values of j up through seven and for values of t up through 2000. The approximations in (A4) were in reasonable agreement over this range of values, with errors of <5% over the vast majority of points. These approximations become less accurate as j increases, and there is no guarantee that the form specified by Equations A4 will hold for very large values of j. For this reason, this approach is valid only where the series of sums can be truncated. Since the lead term of ςj has the form j Logj−1(t + 3/2 − j), terms in the sum should be decreasing as long as Log(t) < Nb. Substituting these approximations into (A2) yields an expression of the form
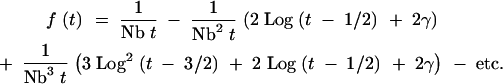 |
A5 |
Owing to the form of the approximations in A4, any term occurring in a particular ςj will appear in all subsequent terms ςj′>j. If we collect these terms together, this expression becomes
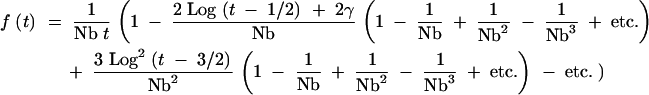 |
A6 |
We can then approximate the series of 1/Nbj terms by 1/(1 + (1/Nb)) to yield an expression of the form
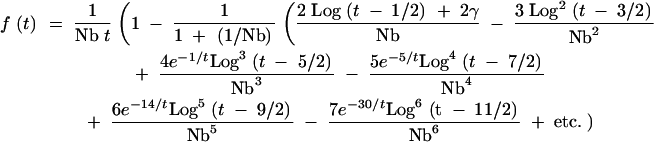 |
A7 |
In principle, this form will include terms up through
 |
A8 |
For purposes of calculation, however, it will be useful to truncate Expression A7 after a small number of terms. Once again, the magnitude of the terms in (A7) will diminish so long as Log(t) < Nb and will decrease more rapidly for smaller values of Log(t)/Nb. The corresponding CDF, or the probability that our two lineages coalesce no more than T generations in the past, is derived simply by summing Equation A7 from 1 to T:
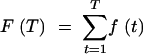 |
A9 |
The form of (A7) includes different numbers of terms for different values of t, and the inclusion of the 1/(1 + (1/Nb)) term assumes the existence of subsequent terms. However, a reasonable approximation of the CDF can be derived for larger values of T:
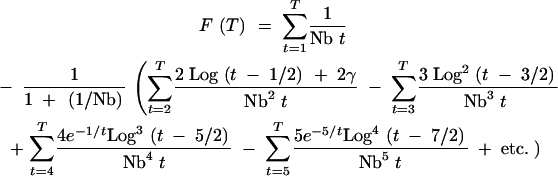 |
A10 |
The 1/Nb sum is approximately equal to (Log(T) + γ)/Nb. The “2γ” term can be similarly approximated as 2γ(Log(T) + γ − 1)/(Nb2 + Nb). The other terms asymptotically approach a similar form such that for large T Equation A10 can be written as
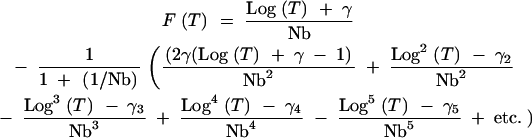 |
A11 |
Approximate values for the first few γi terms are
 |
A12 |
Equation A11 can then be rearranged to collect the Log terms:
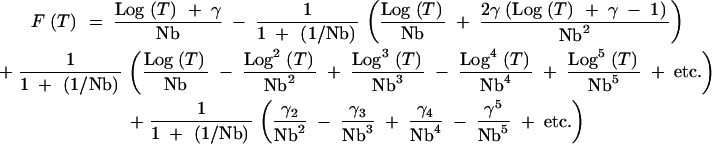 |
A13 |
This can be further simplified to
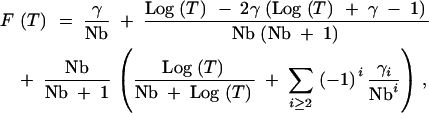 |
A14 |
where γ1 = γ. This gives a final form for the CDF that does not include any nested sums. Inspection of Equation A14 supports our prior conclusion that this approach is valid only for Log(T) < Nb, since F(T) cannot be >1. Furthermore, inspection of the series of γi terms in (A12) suggests that the terms in the sum in (A14) will decrease in magnitude so long as Nb > ∼15. A number of the approximations invoked here assume the existence of an infinite number of terms. For very small values of T, this leads to significant errors (see Figure 8), but appears to be reasonably accurate for T > 10 (Figure 2).
Two sequences from different locations:
Consider the case of two samples drawn from locations separated by a distance x. As before, I start with the recursion derived by Barton and Wilson (1995)(1996). The probability that the two lineages coalesce t generations in the past is given by
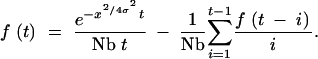 |
A15 |
If we rescale distance relative to dispersal, defining ξ = x/2σ, and assume that Nb is sufficiently large that we can neglect terms of order 1/Nb3, this can be rewritten as
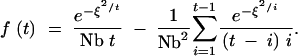 |
A16 |
Equation A16 can be approximated by
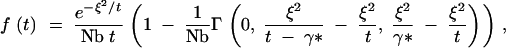 |
A17 |
where γ* is 0.562, and Γ(0, z1, z2) is the incomplete gamma function, defined as
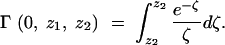 |
A18 |
Unfortunately, a general approximate expression for the CDF corresponding to Equation A17 is not readily forthcoming. The CDF can, of course, be written as
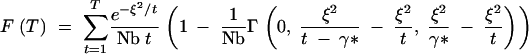 |
A19 |
For t ≫ γ*, Equation A17 can be approximated by
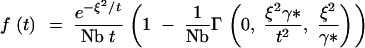 |
A20 |
Boundary effects and images:
The effect of habitat boundaries is approximated by an approach analogous to the “method of images” in electrostatics. For a pair of sequences drawn from the same location a distance z from a reflecting boundary, the coalescence probability can be modeled as the combination of two unbounded coalescent processes: one for two sequences from the same location and one for two sequences separated by a distance 2z (ξ = z/σ). This can be imagined as taking one of the two samples and creating an additional, “mirror image” sample by reflection across the boundary. The coalescent process for two sequences drawn from different locations can be constructed in an analogous manner. For two sequences that are positioned at distances x1 and x2 from the boundary and separated by a distance y parallel to the boundary, the two coalescent processes have characteristic distances  and
and  Here ξ1 corresponds to the direct distance between the two locations, and ξ2 corresponds to the distance from the location of one to the location of the mirror image of the other. Note that the distance between one sample and the mirror image of the other sample does not depend on which sample you choose to mirror across the boundary.
Here ξ1 corresponds to the direct distance between the two locations, and ξ2 corresponds to the distance from the location of one to the location of the mirror image of the other. Note that the distance between one sample and the mirror image of the other sample does not depend on which sample you choose to mirror across the boundary.
Corners of the habitat, where two boundaries meet at a right angle, are modeled similarly, with two sequential reflections, first across one boundary, and then across the other. Note that this creates three image samples, one across each of the two boundaries, and one that is the image of one of those two. Consider again the case of two sequences sampled from the same location at some distance from a rectangular corner of the habitat. If there is one boundary a distance ξ1 from the pair and a perpendicular boundary a distance ξ2 from the pair, we must consider three coalescence-at-a-distance processes using distances ξ1, ξ2, and 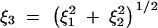 . The process of constructing image locations is illustrated by Figure 3.
. The process of constructing image locations is illustrated by Figure 3.
For samples drawn from a location between two parallel boundaries, we must consider not only the images across each of those boundaries, but also images corresponding to reflections across both boundaries. In fact, we must consider reflections of the habitat across each of its boundaries, reflections of each of those image habitats across each of its boundaries, and so on. We can imagine the entire infinite plane tiled with habitat images, each of which has a mirror-image orientation to each of its neighbors. Specifically, if we assume a rectangular habitat ranging from 0 to L1 in one dimension and 0 to L2 in the other, the images of a point at (x1, y1) can be written as
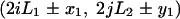 |
A21 |
for all combinations of integers i and j. If we were to assume periodic boundary conditions (a torus formed by the direct product of two circles of length L1 and L2), we could apply a similar method of images. However, rather than reflecting the rectangular habitat across each of its boundaries, the set of image habitats would be constructed by a series of translations. The infinite plane would again be tiled with habitat images, but in this case, each image would have the same orientation as the original. The locations of the images of (x1, y1) analogous to expression A21 would be
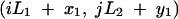 |
A22 |
Although the number of such images is infinite, in practice, a particular image will not contribute significantly to the coalescent process so long as t ≪ ξ2, as can be seen from Equation A17. Thus, the number of images that need to be considered depends on the number of generations over which geography needs to be considered explicitly. This question is addressed below. For the moment, simply note that in most circumstances, the number of images is unlikely to be large. Images corresponding to a large number of reflections represent coalescent events that occur only after a lineage has traversed the habitat multiple times, at which point the original sampling locations are likely to have become unimportant.
Given these expressions for the coalescent process, it is possible to construct an expression for the distribution of coalescence times for a pair of sequences sampled from arbitrary locations within a rectangular habitat. Accounting for the boundary conditions requires assuming multiple competing coalescent processes that occur simultaneously and correspond to reflections off various habitat boundaries. For example, consider two sequences sampled from the same location a distance ξ from a habitat boundary. The probability that coalescence does not occur is equal to the probability that coalescence described by Equation A14 does not occur and that coalescence described by Equation A19 does not occur. Each boundary must be accounted for in this way, as well as corners. For habitats that are much longer in one dimension than in the other, multiple reflections in the shorter dimension should be included, to account for the time required for diffusion to occur across the habitat in the longer dimension. For two samples drawn from different locations, the competing coalescent processes will all be described by Equation A18, with a different value of ξ corresponding to the distance between one sample location and each of the images of the other sample location.
The method of images provides a rigorous method of dealing with boundary conditions only for a few specific habitat geometries. Here I have discussed the treatment of linear boundaries that meet at a right angle, which is sufficient to characterize the coalescent process in a rectangular habitat. Similar methods could be used to derive image locations for certain other special habitats, such as an equilateral triangle. For an arbitrary two-dimensional habitat, however, multiple reflections will not tile the plane cleanly. In certain cases it might be possible, however, to describe the coalescent behavior on a very short timescale by considering only reflections off of nearby boundaries.
As indicated above, the coalescent processes characterized by the ξi terms are competitors with one another. That is, the total probability of coalescence is equal to the probability that coalescence occurs by any one of the processes. If the coalescent process is broken down into m subprocesses, each of which is characterized by a distance ξi, then the CDF is given by
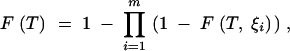 |
A23 |
where F(T, ξi) refers to Equation A14 if ξi = 0 or Equation A19 if ξi > 0. Assuming the probability of coalescence in any given generation to be small, the PDF can be written as
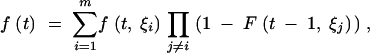 |
A24 |
where f(t, ξi) refers to Equation A7 if ξi = 0 or Equation A17 if ξi > 0.
Transition to the collecting phase:
For a pair of lineages, the probability of coalescence in a particular generation, conditional on not yet having coalesced, approaches some constant rate as t becomes large. This rate represents the largest nonunit eigenvalue of the coalescent process and is equal to 1/Ne. Unlike other models invoking a separation of timescales, in this model there is no discrete point at which the collecting-phase description becomes completely accurate. Put another way, the coalescent process does not become completely independent of the original sampling locations at any finite time. The challenge is thus to choose a transition time τ that is large enough that the process is largely independent of sampling location, but small enough to provide substantial computational savings, and to limit the scattering phase to the regime for which the approximations invoked above hold. A candidate for τ is the point where these two conditional coalescence rates are equal, where the rate of coalescence described by the scattering-phase equations is equal to (1 − F(τ))/Ne. However, since the separation-of-time scales description is not very sensitive to the exact choice of τ, we can use a much simpler formulation to derive our transition time. If we consider only terms of order 1/Nb, the conditional rate for the process described by the scattering phase becomes approximately
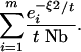 |
A25 |
The rate given by Equation A25 neglects all interaction between the two lineages. The best comparison for this rate is therefore given by the approximation to the collecting phase description that also ignores these interactions. Ignoring those interactions is equivalent to considering the unconditional distribution of each lineage location, where each of the two lineages would be equally likely to be anywhere in the population, and the conditional coalescence probability would equal 1/N. The point at which the scattering and collecting phase coalescence rates are equal is thus approximately at the value of τ that satisfies
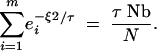 |
A26 |
This formulation means that we are concatenating the two CDF curves near the point where they are tangential to each other.
Equation A26 works well for pairs of sampling locations that are close together (ξ1 ≈ 0). For example, for a pair of points sampled from adjacent locations, the approximate coalescence rate given by expression (A25) will start at its maximum and decrease as t increases, and there will be a single value of τ for which Equation A26 will hold. For two samples at a distance, expression (A25) will first increase and then decrease with increasing t. Thus, there may be two or more values of τ for which Equation A26 will hold. In this case, the largest value of τ that satisfies (A26) should be used. Finally, if the separation between the two sampling locations is large enough, there may be no value of τ for which Equation (A26) holds. That is, the rate of coalescence may approach 1/Ne asymptotically. In this case, an alternate candidate for τ is the point where expression (A25) reaches its maximum. Setting the first derivative of (A25) equal to zero yields the following condition:
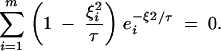 |
A27 |
The form of Equations A25–A27 makes it apparent that the transition time τ will depend on the number of image locations considered: the more image locations, the longer the duration of the scattering phase. For any given selection of images, there will be a certain amount of error introduced near the transition between the phases. Images just beyond the range of those included in the analysis will begin to contribute significantly to the coalescence probability near the end of the scattering phase. Thus, for values of T approaching τ, the CDF provided by these equations will tend to underestimate the true coalescence probability. However, the magnitude of the error is small and does not depend strongly on the exact number of images considered (see Figures 8–10). The most important considerations in constructing the set of image locations are to include at least the first reflection off of each boundary and to include all images whose corresponding distances ξi are less than the maximum distance ξmax associated with any of the included images. Depending on the particular sampling locations considered, there may be certain values of ξmax that will work better than others. The error near the transition will be smallest when the difference between ξmax and the distance ξ associated with the closest excluded image is maximized.
The complete coalescent process is then described in the following way. For T < τ, the process is described by Equations A23 and A24. For T > τ, the CDF is given by
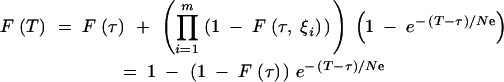 |
A28 |
and the PDF by
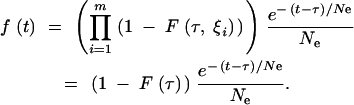 |
A29 |
Effective population size:
Application of the method, and in particular, use of Equations A28 and A29, requires knowledge of the effective population size Ne that characterizes the collecting phase. It is possible to use classical results on the rate of decrease of heterozygosity in a population to derive an explicit expression for the long-term effective population size. Maruyama (1972) provides an expression for the probability that two alleles sampled from random locations in a two-dimensional habitat are identical. Maruyama's result applies to a toroidal habitat formed by the direct product of two circles of lengths L1 and L2. This probability is equal to
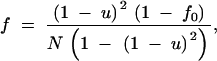 |
A30 |
where u is the mutation rate, and f0 is the probability of identity of two sequences sampled from the same location, which can be written as
 |
A31 |
Equations A30 and A31 correspond to Equations 3–8 and 3–4 in Maruyama (1972). They differ from the original equations by the use of N rather than 2N, which makes them applicable to the haploid model considered here. The S of Equation A31 represents the sum
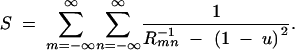 |
A32 |
For the isotropic Gaussian dispersal assumed in this analysis, the Rmn terms are
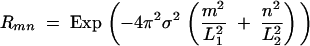 |
A33 |
Maruyama's identity coefficients are the Laplace transform of the distribution of coalescence times. That is,
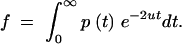 |
A34 |
If we differentiate Equation A34 with respect to u and then take the limit as u approaches zero, we get
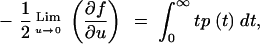 |
A35 |
where the right-hand side of Equation A35 is simply the expected coalescence time. Performing this operation on Equation A30 yields the expected coalescence time for a pair of sequences once equilibrium has been reached, which is the long-term effective population size,
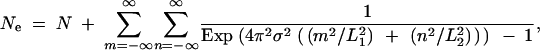 |
A36 |
where the sum in Equation A36 excludes the term where m = n = 0, which contributes the N term when the limit u → 0 is taken. Computation of A36 is practical because the terms of the sum become small as the absolute value of either m or n becomes large.
References
- Barton, N. H., and I. Wilson, 1995. Genealogies and geography. Philos. Trans. R. Soc. Lond. B 349: 49–59. [DOI] [PubMed] [Google Scholar]
- Barton, N. H., and I. Wilson, 1996 Genealogies and geography, pp. 23–56 in New Uses for New Phylogenies, edited by P. H. Harvey, A. J. Leigh Brown and J. Maynard Smith. Oxford University Press, Oxford.
- Barton, N. H., F. Depaulis and A. M. Etheridge, 2002. Neutral evolution in spatially continuous populations. Theor. Popul. Biol. 61: 31–48. [DOI] [PubMed] [Google Scholar]
- Charlesworth, B., D. Charlesworth and N. H. Barton, 2003. The effects of genetic and geographic structure on neutral variation. Annu. Rev. Ecol. Evol. Syst. 34: 99–125. [Google Scholar]
- Cox, J. T., and R. Durrett, 2002. The stepping stone model: new formulas expose old myths. Ann. Appl. Probab. 12: 1348–1377. [Google Scholar]
- Felsenstein, J., 1975. A pain in the torus: some difficulties with models of isolation by distance. Am. Nat. 109: 359–368. [Google Scholar]
- Fleming, W. H., and C.-H. Su, 1974. Some one-dimensional migration models in population genetics theory. Theor. Popul. Biol. 5: 431–449. [DOI] [PubMed] [Google Scholar]
- Griffiths, R. C., 1981. The number of heterozygous loci between two randomly chosen completely linked sequences of loci in two subdivided population models. J. Math. Biol. 12: 251–261. [Google Scholar]
- Herbots, H. M., 1994 Stochastic models in population genetics: genealogy and genetic differentiation in structured populations. Ph.D. Thesis, University of London, London.
- Hey, J., 1991. A multidimensional coalescent process applied to multi-allelic selection models and migration models. Theor. Popul. Biol. 39: 30–48. [DOI] [PubMed] [Google Scholar]
- Hudson, R. R., 1983. Properties of a neutral allele model with intragenic recombination. Theor. Popul. Biol. 23: 183–201. [DOI] [PubMed] [Google Scholar]
- Hudson, R. R., 1990 Gene genealogies and the coalescent process, pp. 1–44 in Oxford Surveys in Evolutionary Biology, Vol. 7, edited by D. J. Futuyma and J. Antonivics. Oxford University Press, Oxford.
- Irwin, D. E., 2002. Phylogeographic breaks without geographic barriers to gene flow. Evolution 56: 2383–2394. [DOI] [PubMed] [Google Scholar]
- Kimura, M., and G. H. Weiss, 1964. The stepping stone model of population structure and the decrease of genetic correlation with distance. Genetics 49: 561–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingman, J. F. C., 1982. a The coalescent. Stoch. Proc. Appl. 13: 235–248. [Google Scholar]
- Kingman, J. F. C., 1982. b On the genealogy of large populations. J. Appl. Prob. 19A: 27–43. [Google Scholar]
- Knowles, L. L., 2004. The burgeoning field of statistical phylogeography. J. Evol. Biol. 17: 1–10. [DOI] [PubMed] [Google Scholar]
- Knowles, L. L., and W. P. Maddison, 2002. Statistical phylogeography. Mol. Ecol. 11: 2623–2635. [DOI] [PubMed] [Google Scholar]
- Malécot, G., 1968 The Mathematics of Heredity. W. H. Freeman, San Francisco.
- Malécot, G., 1975. Heterozygosity and relationship in regularly subdivided populations. Theor. Popul. Biol. 8: 212–241. [DOI] [PubMed] [Google Scholar]
- Maruyama, T., 1970. a Effective number of alleles in a subdivided population. Theor. Popul. Biol. 1: 273–306. [DOI] [PubMed] [Google Scholar]
- Maruyama, T., 1970. b On the rate of decrease of heterozygosity in circular stepping-stone models of populations. Theor. Popul. Biol. 1: 101–119. [DOI] [PubMed] [Google Scholar]
- Maruyama, T., 1970. c Analysis of population structure. I. One-dimensional stepping-stone models of finite length. Ann. Hum. Genet. 34: 201–219. [DOI] [PubMed] [Google Scholar]
- Maruyama, T., 1970. d The rate of decrease of heterozygosity in a population occupying a circular or a linear habitat. Genetics 67: 437–454. [Google Scholar]
- Maruyama, T., 1971. Analysis of population structure. II. Two-dimensional stepping stone models of finite length and other geographically structured populations. Ann. Hum. Genet. 35: 179–196. [DOI] [PubMed] [Google Scholar]
- Maruyama, T., 1972. Rate of decrease of genetic variability in a two-dimensional continuous population of finite size. Genetics 70: 639–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Möhle, M., 1998. A convergence theorem for Markov chains arising in population genetics and the coalescent with selfing. Adv. Appl. Prob. 30: 493–512. [Google Scholar]
- Nagylaki, T., 1974. a The decay of genetic variability in geographically structured populations. Proc. Natl. Acad. Sci. USA 71: 2932–2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagylaki, T., 1974. b Genetic structure of a population occupying a circular habitat. Genetics 78: 777–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagylaki, T., 1977. Genetic structure of a population occupying a circular habitat. Genetics 78: 777–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagylaki, T., 1980. The strong-migration limit in geographically structured populations. J. Math. Biol. 9: 101–114. [DOI] [PubMed] [Google Scholar]
- Nagylaki, T., 2000. Geographical invariance and the strong-migration limit in subdivided populations. J. Math. Biol. 41: 123–142. [DOI] [PubMed] [Google Scholar]
- Nagylaki, T., and V. Barcilon, 1988. The influence of spatial inhomogeneities of neutral models of geographical variation. II. The semi-infinite linear habitat. Theor. Popul. Biol. 33: 311–343. [DOI] [PubMed] [Google Scholar]
- Nordborg, M., 1997. Structured coalescent processes on different time scales. Genetics 146: 1501–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordborg, M., 2000. Linkage disequilibrium, gene trees and selfing: an ancestral recombination graph with partial self-fertilization. Genetics 154: 923–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordborg, M., and P. Donnelly, 1997. The coalescent process with selfing. Genetics 146: 1185–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notohara, M., 1990. The coalescent and the genealogical process in geographically structured populations. J. Math. Biol. 29: 59–75. [DOI] [PubMed] [Google Scholar]
- Notohara, M., 1993. The strong-migration limit for the genealogical process in geographically structured populations. J. Math. Biol. 31: 115–122. [Google Scholar]
- Pybus, O. G., A. Rambaut and P. H. Harvey, 2000. An integrated framework for the inference of viral population history from reconstructed genealogies. Genetics 155: 1429–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousset, F., 1997. Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics 145: 1219–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyer, S., 1976. Results for the stepping-stone model for migration in population genetics. Ann. Prob. 4: 699–728. [Google Scholar]
- Sawyer, S., 1977. Asymptotic properties of the equilibrium probability of identity in a geographically structured population. Adv. Appl. Prob. 9: 268–282. [Google Scholar]
- Slatkin, M., 1985. Rare alleles as indicators of gene flow. Evolution 39: 53–65. [DOI] [PubMed] [Google Scholar]
- Slatkin, M., 1991. Inbreeding coefficients and coalescence times. Genet. Res. 58: 167–175. [DOI] [PubMed] [Google Scholar]
- Slatkin, M., and N. H. Barton, 1989. A comparison of three indirect methods for estimating average levels of gene flow. Evolution 43: 1349–1368. [DOI] [PubMed] [Google Scholar]
- Slatkin, M., and W. P. Maddison, 1990. Detecting isolation by distance using phylogenies of genes. Genetics 126: 249–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strimmer, K., and O. G. Pybus, 2001. Exploring the demographic history of DNA sequences using the generalized skyline plot. Mol. Biol. Evol. 18: 2298–2305. [DOI] [PubMed] [Google Scholar]
- Strobeck, C., 1987. Average number of nucleotide differences in a sample from a single subpopulation: a test for population subdivision. Genetics 117: 149–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima, F., 1983. Evolutionary relationship of DNA sequences in finite populations. Genetics 105: 437–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tufto, J., S. Engen and K. Hindar, 1996. Inferring patterns of migration from gene frequencies under equilibrium conditions. Genetics 144: 1911–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakeley, J., 1998. Segregating sites in Wright's island model. Theor. Popul. Biol. 53: 166–175. [DOI] [PubMed] [Google Scholar]
- Wakeley, J., 1999. Non-equilibrium migration in human history. Genetics 153: 1863–1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakeley, J., 2000. The effect of population subdivision on the genetic divergence of populations and species. Evolution 54: 1092–1101. [DOI] [PubMed] [Google Scholar]
- Wakeley, J., 2001. The coalescent in an island model of population subdivision with variation among demes. Theor. Popul. Biol. 59: 133–144. [DOI] [PubMed] [Google Scholar]
- Wakeley, J., and N. Aliacar, 2001. Gene genealogies in a metapopulation. Genetics 159: 893–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakeley, J., and S. Lessard, 2004. The two-locus ancestral graph in a subdivided population: convergence as the number of demes grows in the island model. J. Math. Biol. 48: 275–292. [DOI] [PubMed] [Google Scholar]
- Weiss, G. H., and M. Kimura, 1965. A mathematical analysis of the stepping stone model of genetic correlation. J. Appl. Probab. 2: 129–149. [Google Scholar]
- Wilkins, J. F., and J. Wakeley, 2002. The coalescent in a continuous, finite, linear population. Genetics 161: 873–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson-Herbots, H. M., 1998. Genealogy and subpopulation differentiation under various models of population structure. J. Math. Biol. 37: 535–585. [Google Scholar]
- Wright, S., 1931. Evolution in Mendelian populations. Genetics 16: 97–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, S., 1943. Isolation by distance. Genetics 28: 114–138. [DOI] [PMC free article] [PubMed] [Google Scholar]