

Abstract
Phylogenetic analysis of sequences from gene families and homologous genes from species of varying divergence can be used to identify conserved noncoding regulatory elements. In this study, phylogenetic analysis of 5′-noncoding sequences was optimized using rab17, a well-characterized ABA-responsive gene from maize, and five additional rab16/17 homologs from sorghum and rice. Conserved 5′-noncoding sequences among the maize, sorghum, and rice rab16/17 homologs were identified with the aid of the software program FootPrinter and by screening for known transcription-factor-binding sites. Searches for 7 of 8 (7/8)bp sequence matches within aligned 5′-noncoding segments of the rab genes identified many of the cis-elements previously characterized by biochemical analysis in maize rab17 plus several additional putative regulatory elements. Differences in the composition of conserved noncoding sequences among rab16/17 genes were related to variation in rab gene mRNA levels in different tissues and to response to ABA treatment using qRT-PCR. Absence of a GRA-like element in the promoter of sorghum dhn2 relative to maize rab17 was correlated with an ∼85-fold reduction of dhn2 RNA in sorghum shoots. Overall, we conclude that phylogenetic analysis of gene families among rice, sorghum, and maize will help identify regulatory sequences in the noncoding regions of genes and contribute to our understanding of grass gene regulatory networks.
THE annotation of genome coding regions, intron/exon boundaries, and noncoding regulatory sequences is a central challenge in genome research. Annotation is significantly improved when genome sequences from related species are available for comparison (Boffelli et al. 2003; Thomas et al. 2003; Weitzman 2003). Comparative analysis of the human and mouse genome sequences revealed that ∼5% of these genomes are under functional constraint (Waterston et al. 2002). Surprisingly, only ∼1.5% of the sequences under selection correspond to protein-coding sequences, underscoring the importance of noncoding regulatory sequences in genome function. Partly in response to this finding, the human genome project ENCODE was initiated to identify and elucidate the functions of the noncoding regulatory portions of the human genome sequence (Collins et al. 2003). Recent progress on sequencing plant genomes is creating a similar opportunity to identify and understand the function of noncoding regulatory sequences that regulate plant genes (Hao et al. 1998; Arabidopsis Genome Initiative 2000; Chandler and Brendel 2002; Rice Chromosome 10 Sequencing Consortium 2003).
The noncoding regulatory portion of eukaryotic genomes controls gene function through modulation of transcription initiation, RNA processing, RNA stability, translation, and chromatin structure. Promoter cis-regulatory elements that provide binding sites for transcription-factors (TFs) are of particular interest because they regulate gene transcription, guide development, and form the basis of gene regulatory networks (Davidson et al. 2003). Like animal promoters, plant promoters contain regulatory modules composed of combinations of cis-elements that mediate changes in transcription in response to internal and external input. For example, an ∼350-bp region of the promoter of maize rab17 contains a minimum of nine TF-binding sites that mediate responses to ABA and dehydration and regulate gene expression during seed and vegetative development (Busk et al. 1997). Cis-elements are also important to define because phenotypic variation can be caused by mutations in these sequences. For example, sequence differences in the teosinte branched-1 promoter are correlated with changes in gene expression, morphology, and development associated with the evolution of cultivated maize from teosinte (Wang et al. 1999; Clark et al. 2004). Similarly, sequence differences in a putative cis-element of the AP1 promoter have been proposed to be responsible for variation in vernalization requirements in wheat (Yan et al. 2003).
The Arabidopsis thaliana genome encodes ∼1500 transcription factors of which ∼45% are unique to plants (Riechmann et al. 2000). Information about the binding sites for plant transcription factors is increasing rapidly (see the TRANSFAC database at http://www.gene-regulation.com/; PLACE at http://www.dna.affrc.go.jp/htdocs/PLACE/; PlantCARE at http://intra.psb.ugent.be:8080/PlantCARE/; and AGRIS at http://arabidopsis.med.ohio-state.edu). The discovery and characterization of TF-binding sites often involve electrophoretic mobility shift assays, DNAseI footprinting analysis, and site-directed mutation studies. Scaling these biochemical approaches for genome-wide analysis of cis-elements is challenging. Noncoding regulatory elements can also be identified through computational analysis of promoters of coregulated genes (Tavazoie and Church 1999; Hughes et al. 2001). An increasing number of microarray-based gene expression studies in plants are helping to identify regulons and the underlying cis-element modules that mediate gene expression patterns in plants (Harmer et al. 2000; Sung et al. 2001).
A complementary way to identify noncoding regulatory sequences involves phylogenetic analysis of promoter sequences of homologous genes from species of varying divergence (Ansari-Lari et al. 1998; Thacker et al. 1999; Hardison 2000). The rationale for this approach is based on the finding that expression of homologous genes in different species is often similar, which suggests the retention of common regulatory elements. The regulatory sequences associated with homologous genes from diverged species can be identified because they are more conserved than the surrounding nonfunctional sequences. Computational approaches have been developed to facilitate phylogenetic searches for regulatory sequences (Fickett and Wasserman 2000; Tompa 2001; Halfon et al. 2002; Rebeiz et al. 2002; Lenhard et al. 2003; Rombauts et al. 2003; Frith et al. 2004). Successful implementation of these search programs requires an understanding of species phylogeny and an initial assessment of useful search parameters suitable for comparison of genes from diverged species to reduce the incidence of random sequence matching among nonfunctionally conserved sequences (Tautz 2000). This depends on a number of factors, but species separated for 15–430 MY have been successfully analyzed using phylogenetic analysis (Colinas et al. 2002; Mueller et al. 2002). Comparison of highly diverged species reduces the problem of random sequence matching; however, studies of more closely related species often provide the most information since extended evolution of regulatory regions and biological functions reduces the ability to detect regulatory sequences (Colinas et al. 2002).
Phylogenetic analysis has been used to identify conserved noncoding sequences (CNS) in plant genes in a number of studies. A study of 22 cruciferous species spanning ∼45 MY of divergence allowed the identification of CNS corresponding to known cis-elements associated with Chs and Apetala (Koch et al. 2001). Phylogenetic shadowing of AGAMOUS genes in 29 Brassicaceae species identified several known and putative cis-elements in introns (Hong et al. 2003). A study of orthologous gene sequences from A. thaliana and cauliflower, species separated for 14.5–20.4 MY (Colinas et al. 2002), identified approximately one highly significant 25-bp CNS (75% conserved) per gene.
Similar results have also been obtained through phylogenetic analysis of genes from grass species. Comparative analysis of phytochrome A gene promoters from sorghum, maize, and rice revealed CNS that spanned known cis-regulatory sequences (Morishige et al. 2002). Kaplinsky et al. (2002) compared the noncoding sequences of seven orthologous genes from rice, maize, and other grasses representing ∼50 MY of divergence and concluded that plant CNS are generally shorter than mammalian CNS from species of similar divergence. A follow-up study by this group on 52 homologous maize/rice gene pairs found that CNS spanning >14 bp are often located in introns and associated with regulatory genes (Inada et al. 2003). Similarly, a study involving >300 grass gene comparisons concluded that 20 bp (with 70% sequence matching or greater) was the minimal length needed to identify significant CNS among grass orthologs (Guo and Moose 2003). Unfortunately, the CNS identified in the studies above often did not span TF-binding sites known to regulate the target genes. The known TF-binding sites were missed because the size and conservation of these sites (6–10 bp) was below the sequence lengths used to eliminate random matching among sequences.
The goal of this study was to determine how to use phylogenetic analysis to identify cis-elements including 6- to 10-bp TF-binding sites that control gene expression in grass species. To do this, we developed a modified phylogenetic approach that facilitates the discovery of regulatory elements using a multi-stage process that includes analysis of several members of a gene family. The study focused on a family of ABA-responsive rab16/17 genes from sorghum, maize, and rice, species separated for ∼16–20 MY (sorghum, maize) and ∼50 MY (sorghum, maize vs. rice; Doebley et al. 1990). The rab16/17 genes encode a group of related ∼16- to 17-kD dehydrins that help protect plants from injury during dehydration (Close 1997). Maize rab17, a well-characterized ABA-responsive gene (Busk et al. 1997; Busk and Pages 1998; Kizis and Pages 2002), was used as a reference to determine if phylogenetic analysis was producing useful results. The identification of previously discovered and several new putative regulatory elements in the current phylogenetic study of rab16/17 genes indicates that this approach will be useful for annotation of sorghum, maize, and rice gene regulatory sequences.
MATERIALS AND METHODS
Plant growth and treatment:
Sorghum bicolor cultivar BTx623, Zea mays cultivar B73, and Oryza sativa cultivar LeMont plants were grown hydroponically under constant aeration in 0.5× Hoagland's nutrient solution in a 12-hr-day growth chamber at 31° day/22° night temperature with 50% constant humidity. At 8 days (sorghum and maize) or 11 days (rice) seedlings were treated with (±)-cis, trans-abscisic acid (Sigma, St. Louis) by spiking ABA into the hydroponic solution to a final concentration of 125 μm. Control plants were mock treated with identical solutions lacking ABA. Tissue was harvested at 3 and 27 hr post-treatment, flash frozen in liquid N2, and stored at −80°.
Acquisition of gene sequences related to rab16/17:
The sequence of the maize rab17 gene used in this study was previously reported (Zmrab17; X15994). The sorghum and rice ESTs most related to maize rab17 were identified using The Institute for Genome Research's eukaryotic gene ortholog database (ortholog cluster 476665; http://www.tigr.org/tbd/tgi/ego/). The sorghum EST sequence (AW747029; e−52) was used to identify sorghum BAC 21O3 by hybridization to a BAC library derived from IS3620C. Sorghum BAC 2103 was sheared (Gene Machines, San Carlos, CA) into ∼2-kb fragments and subcloned into pBluescriptII (Stratagene, La Jolla, CA). Clones that hybridized to sorghum EST AW747029 were sequenced from both ends using T3 and T7 primers. Sequences were assembled into ∼5× deep contigs containing ∼1000 bp of flanking 5′ and 3′ DNA using Sequencher software (Gene Codes, Ann Arbor, MI). The resulting genomic sequence matched a sequence of this gene previously named Sbdhn2 (GenBank U63831). Therefore the BAC-derived gene sequence obtained in this study was also named Sbdhn2 and the genomic sequence was deposited in GenBank (AY177889), where 5′-noncoding sequences correspond to nucleotides 1–1049 bp. The rice EST with the highest sequence similarity to maize rab17 (AU091664; e−55) identified five related rice genomic sequences: rab16A–D and a genomic sequence from the whole genome shotgun (WGS) database. The WGS rab sequence (AAAA01012244) was very similar to Osrab16A (97% nucleotide identity) so it was designated Osrab16A2. The 5′-noncoding sequence of the Osrab16A2 gene was included in this study (5080–6140 bp). 5′-noncoding sequences of four other members of the rice rab16 family used in this study had been previously reported (Osrab16A: Y00842, 1–1599 bp; Osrab16B: X52422, 1–1395 bp; Osrab16C: X52423, 1–1476 bp; Osrab16D: X52424, 1–685 bp).
Analysis of mRNA abundance:
RNA was isolated from root and shoot tissue separately using Trizol reagent with the suggested modification for plants (Molecular Research Center, Cincinnati). Seed RNA was extracted from dry seeds using Concert Reagent (Invitrogen, Carlsbad, CA). First-strand cDNA was made by reverse transcribing 1 μg of total RNA with random hexamers using the TAQMAN reverse transcription reagents (Applied Biosystems, Branchburg, NJ). Quantitative Real Time PCR was performed on an Applied Biosystems 7900HT machine using SYBR chemistry for Zmrab17, Osrab16A2, and Osrab16C. The generation of specific PCR products was confirmed both by melting curve and by gel analysis. FAM/TAM probes were required for specific detection of Sbdhn2, Osrab16A, Osrab16B, and Osrab16D (Synthegen, Houston). Primers and probes were designed using Primer Express software (Applied Biosystems) to allow amplification of ∼100-bp products of similar GC and Tm characteristics.
Thermal-cycling conditions were 2 min at 50° and 10 min at 95° followed by 47 cycles at 95° for 15 sec and 60° for 1 min. Assays were performed in triplicate and data were analyzed using the ABI PRISM 7900HT SDS software (Applied Biosystems). Quantification was achieved using the comparative cycle threshold (CT) method (Bieche et al. 1999), which normalizes the number of target gene copies to an endogenous reference gene (i.e., 18S rRNA, detected using the ribosomal TAQMAN kit supplied by Applied Biosystems). Fold inductions were calculated as Î(dCTcontrol-dCTABA).
Primer and probe sequences are as follows:
Sbdhn2 forward: TGGCTGCGTTGGCTCTCT
Sbdhn2 reverse: ACACTTATTCATGGACTCATCATCCTAT
Sbdhn2-FAM/TAM probe: TGGCGTGTGAAAGCCGTACTTAATCACTG
Zmrab17 forward: CCGGAGGCCACAAGGA
Zmrab17 reverse: ATCTTGTCCATAATGCCTTTCTTCTC
Osrab16A2 forward: CGAGCGCAATAAAAGGAAAAA
Osrab16A2 reverse: AGACACGGTCCGTACTGGAGAA
Osrab16A forward: CTCGGTCTGAGGATGATGGAATG
Osrab16A reverse: CCGCCCATGGCATGCT
Osrab16A FAM/TAM probe: CGGCGGCAACAAGGGCGA
Osrab16B forward: CGGCGGCCAGTTCCA
Osrab16B reverse: TGCTGGTTGTTGCCCTTGTT
Osrab16B-FAM/TAM probe: AGGGAGGACCGCAAGACCGGC
Osrab16C forward: CGTCCAGCTCGTCGTCTGA
Osrab16C reverse: CCGGTGTTCCCCATCATC
Osrab16D forward: CGGCAACCCTGCAGTGA
Osrab16D reverse: GCCGGCTCCTGGATGTG
Osrab16D-FAM/TAM probe: CACCGGAAACGCACCCACCG.
To determine the relative abundance of 16A, 16B, and 16D mRNA, RT-PCR was performed on known amounts of templates. Rice BAC OSJNBb34E03, which encodes the rab16A, rab16B, rab16C, and rab16D genes, was serially diluted and used as template for rab16A, rab16B, and rab16D primer/probe sets. These standard curves were then used to calculate primer efficiency and adjust dCT values to relative expression values.
Phylogenetic analysis:
The FootPrinter program (http://bio.cs.washington.edu/software.html) was used to identify conserved sequences among the rab genes analyzed. During optimization a wide range of search parameters were tested. Most comparisons used the following parameters: motif size, 8; maximum number of mutations, 1; maximum number of mutations per branch, 0; subregion size, 50 bp; subregion change cost, 1; allow for regulatory losses, no, except for sorghum and maize comparisons, which utilized a motif search size of 10 with no allowable mutations.
RESULTS
Alignment of related sorghum, rice, and maize rab sequences:
The maize rab17 gene promoter was selected as a reference for initial optimization of phylogenetic analysis because this promoter is well characterized (Busk et al. 1997; Kizis and Pages 2002). The nine cis-elements defined through biochemical analysis of maize rab17 (Busk et al. 1997) and the predicted TATA sequence are boxed and labeled above the rab17 sequence in Figure 1 (i.e., DRE1, ABRE1, DRE2, ABRE2, ABRE3a/3b, GRA, SPH, ABRE4, and TATA). Prior studies showed that sequences from −173 to −315 of the maize rab17 promoter contained cis-regulatory elements and TF-binding sites that are sufficient to modulate basal and ABA-induced expression of this gene in both seeds and vegetative tissues (Busk et al. 1997). Therefore, our comparison of noncoding sequences focused on ∼500 bp upstream of the coding region.
Figure 1.—

Alignment of promoter and 5′-UTR regions in sorghum dhn2, maize rab17, and rice rab16A2 genes (labeled Sb, Zm, Os). FootPrinter (http://abstract.cs.washington.edu/blanchem/FootPrinterWeb/FootPrinterInput.pl) was used to identify CNS containing 7 of 8 conserved nucleotides (7/8) between Sbdhn2/Zmrab17 and Osrab16A2 comparisons and these are highlighted in yellow and identified by numbers 1–17. CNS containing at least 10 of 10 conserved nucleotides identified by Sbdhn2 and Zmrab17 comparisons are indicated with bars above the Zmrab17 sequence and labeled with numbers 18–28. Sequence matches outside of CNS are colored blue. Biochemically defined TF-binding sites in maize (Busk et al. 1997) are boxed with thick lines and labeled above the Zmrab17 sequence, while putative regulatory elements identified through public database searches are boxed with thin lines and labeled below the Osrab16A2 sequence. Dashes indicate a gap in the alignment, while asterisks (*) represent a sequence that can align on either side of an INDEL. The transcription start site for the maize rab17 is labeled TX start and indicated with a “#.”
Phylogenetic analysis relies on the identification of conserved sequences among two or more genes that evolved from a common progenitor:
Divergence of homologous genes can occur via speciation or following gene duplication. In either case, extended regions of sequence alignment within 5′-noncoding regions are often retained between homologous genes. In this study, the 5′-noncoding sequences of three homologous genes, maize rab17, sorghum dhn2, and rice rab16A2, were initially aligned using FootPrinter, a motif discovery program designed to identify DNA elements that have evolved more slowly compared to surrounding sequences in sets of homologous genes (Blanchette et al. 2002; Blanchette and Tompa 2002; http://abstract.cs.washington.edu/blanchem/FootPrinterWeb/FootPrinterInput.pl). The alignment process was started from the initiation codon and continued incrementally to ∼500 bp upstream. The initial alignments seeded by sequence matches identified by FootPrinter were then manually edited to maximize overall alignment. The results of the alignment process are shown in Figure 1 where all matching sequences were initially colored blue. Regions of extended homology with at least a 7/8 bp match between sorghum or maize and rice were defined as CNS and highlighted in yellow (rationale provided below).
Overall, this process allowed ∼70% of the sorghum/maize 5′-noncoding sequences analyzed to be aligned, whereas only ∼30–50% of the sorghum dhn2 or maize rab17 5′-noncoding sequences could be aligned to the rice rab16A2 sequence. Two large INDELS spanning 14 and 50–54 bp in the Osrab16A2 5′-noncoding region relative to Sbdhn2/Zmrab17 were the primary cause for loss of overall alignment. The extent of sequence alignment in the Osrab16A2 promoter vs. Zmrab17 or Sbdhn2 promoters declined to ∼30% in the region 300–400 bp upstream from the translation start site. Sequences >400 bp upstream of the translation start sites became difficult to align, in part due to an increase in AT-rich sequences (data not shown). A number of INDELs ranging in size from 1 to 54 bp were used to create the alignments between maize, sorghum, and rice 5′-non-coding sequences (Figure 1). While many of these INDELS were probably introduced as an arbitrary consequence of the alignment process, overall the analysis revealed islands of conserved sequence surrounded by stretches of less-conserved sequence that have been modified extensively by insertions/deletions over the past ∼50 MY.
Analysis of known maize rab17 regulatory elements:
The overlap between the maize rab17 cis-elements previously defined through biochemical analysis and CNS elements was investigated as a first step toward understanding the limits of phylogenetic analysis of noncoding sequences from rice, sorghum, and maize. Aligned sequences that spanned each maize rab17 cis-element were compared in cross-species analysis (Table 1). In four of the nine elements, sequence conservation was high (7/8–8/8 bp) among rab17, dhn2, and rab16A2 (ABRE1, DRE2, ABRE2, and ABRE4). In contrast, only 4/8 bp of the DRE1 cis-element identified in maize were conserved in comparisons of rice/sorghum or rice/maize even though the core binding sequence (ACCG) for this element was present in all three species. Furthermore, Osrab16A2 apparently lacks sequences that would align to ABRE3a/3b and SPH present in the promoters of Zmrab17 and Sbdhn2; therefore, only sorghum/maize alignments were useful for detecting these regulatory elements. Similarly, the sequence corresponding to GRA was not present in sorghum in the aligned region; therefore, a sequence match was observed only between rice and maize. These results are consistent with the expectation that loss, gain, or significant change in regulatory elements among homologous genes after species separation will cause regulatory elements to be missed using phylogenetic analysis (false negatives). Information about these regulatory elements can often be obtained by carrying out phylogenetic analysis on homologous genes from more than two species spanning a range of divergence (i.e., GRA was detected in rice/maize comparisons; ABRE3a/3b and SPH were detected in sorghum/maize comparisons).
TABLE 1.
Conservation ofrab17 TF-binding sequences in rice, sorghum, and maize
| cis-elementa | Osrab16A2/Sbdhn2 | Osrab16A2/Zmrab17 | Sbdhn2/Zmrab17 |
|---|---|---|---|
| DRE1 (CACCGACG) | 4/8b | 4/8 | 8/8 |
| ABRE1 (CACGTGCC) | 7/8 | 7/8 | 8/8 |
| DRE2 (CACCGACG) | 7/8 | 7/8 | 8/8 |
| ABRE2 (ACACGTCC) | 8/8 | 8/8 | 8/8 |
| ABRE3a/3b (GTACGTGTACGTG) | — | — | 8/8, 7/8 |
| GRA (CACTGGCCGCCC) | — | 8/12 | — |
| SPH (CATGCATG) | 2/8 | 2/8 | 6/8 |
| ABRE4 (GCCACGTA) | 7/8 | 7/8 | 8/8 |
Biochemically defined from Busk et al. (1997).
The number of conserved base pairs within each element between the two species being compared.
CNS search parameters:
CNS search parameters that would minimize the loss of information (false negatives) needed to explain gene regulation were selected. Table 1 shows that at least 7/8 bp were conserved in the four cis-elements retained in rab17, dhn2, and rab16A2 (ABRE1, DRE2, ABRE2, and ABRE4). Therefore, during the optimization phase of this project, we screened the noncoding regions of rice rab16A2, sorghum dhn2, and maize rab17 genes for 7/8-bp CNS. In addition, CNS discovery was restricted initially to comparisons of sorghum/rice and maize/rice, species that diverged ∼50 MYA, because the probability of retaining a 7-bp sequence by chance in a sequence that is identical by descent in these species pairs is reasonably low (P ∼ 0.002; Kaplinsky et al. 2002). Furthermore, initial searches for 7/8-bp CNS in pairs of genes were restricted to aligned portions of the 5′-noncoding region that occur in the same relative order to increase the probability that comparisons of sequences that are identical by descent were made. TF-binding sites that were not present in the same relative order due to insertions, deletions, or rearrangements were identified in a separate search (see below).
Using this approach, 17 7/8-bp CNS were located in the sorghum/rice or maize/rice pairwise comparisons of 5′-noncoding regions (Figure 1, sequences highlighted in yellow and numbered 1–17). Eight of the 7/8-bp CNS were present in all three species (Figure 1, CNS 2, 6, 7, 9, 11, 14, 15, 16) whereas 9 7/8-bp CNSs were present only in sorghum/rice or maize/rice comparisons (Figure 1, CNS 1, 3, 4, 5, 8, 10, 12, 13, 17). Of these latter 9 CNS, 6 contained 6/8-bp matches among all three species (Figure 1, CNS 1, 3, 4, 8, 13, 17). Furthermore, the longest exact sequence match in the regions spanned by each CNS was identified to determine if CNS were part of much larger stretches of conserved sequence. The consecutive number of conserved bases per CNS ranged from 4 to 10 bp with an average identical sequence match of 6.5 bp. The rapid loss of alignment outside of CNS, including sequences flanking the nine known maize rab17 cis-elements, indicates good discrimination of 7/8-bp CNS from surrounding putative nonfunctionally constrained sequences.
The 5′-noncoding regions of sorghum dhn2 and maize rab17 genes were also subjected to phylogenetic analysis to see if useful information about regulatory elements could be obtained from this analysis. Because sorghum and maize diverged only ∼16 MYA, a scan for CNS >19 bp would be required to achieve the same discrimination as that obtained by a screen for 7-bp sequences retained in sorghum/rice or maize/rice. However, only one CNS spanning at least 20 bp was present in the sorghum/maize alignment (CNS 27). Therefore, the aligned 5′-noncoding sequences of sorghum/maize were searched for CNS that were >9 bp even though the probability of a random occurrence of a 10-bp sequence match between these species is 0.05. This search identified 11 CNS ranging in size from 10 to 20 bp with an average sequence match spanning 13 bp, much larger than most known TF-binding sites (Figure 1, CNS 18–28). The Sbdhn2/Zmrab17 CNS spanned seven of the nine known cis-elements but two cis-elements were missed using the >9-bp CNS search parameter (Figure 1, GRA, SPH). While insufficient divergence has occurred between sorghum and maize to accurately discriminate TF-binding sites, it was possible that CNS > 9 bp identified in comparisons of these species might span recently evolved cis-elements. Therefore, the Sbdhn2/Zmrab17 CNS were screened for known TF-binding sites, and these sequences were retained for further downstream analysis as described below.
Correspondence between rab17 CNS and TF-binding sites:
The relationship between 7/8-bp CNS identified through alignment of sorghum/rice and maize/rice rab 5′-noncoding sequences, known cis-elements, and putative TF-binding sites is shown in Table 2. In previous biochemical studies, nine TF-binding sites were identified in the Zmrab17 sequence region spanning −173 to −315 (Busk et al. 1997). A scan for 7/8-bp CNS among sorghum/rice or maize/rice identified five of the nine previously identified TF-binding sites (Figure 1; ABRE4, GRA, ABRE2, DRE2, ABRE1; Table 2, CNS 11, 12, 14, 15, 16). All but one of these TF-binding sites was identified in both the sorghum/rice and maize/rice comparisons, indicating a high degree of conservation. CNS 12, which matched the TF-binding site GRA, was identified only in the rice/maize alignment due to a deletion in sorghum (Figure 1). The GRA TF-binding site in maize includes the sequence (GCCGCC) that matches the binding site for AP2 factors involved in responses to jasmonate and ethylene (Brown et al. 2003). DRE1, SPH, and ABRE3a/3b were missed using the 7/8-bp criteria although the core DRE-binding sequence (ACCG) is perfectly conserved in all three species (Busk et al. 1997).
TABLE 2.
rab17 CNS sequence, position, and homology to known regulatory elements
| Maize rab17
|
Sorghum dhn2
|
Rice rab16A2
|
|||||
|---|---|---|---|---|---|---|---|
| No.a | Sequence | Position | Sequence | Position | Sequence | Position | Regulatory element |
| 1 | TTAGgagA | −38 | TTAGCTTA | −64 | TTAGCTTA | −14 | |
| 2 | gAGAA-CACg | −74 | gAGAAaCACg | −103 | aAGAA-CACc | −50 | |
| 3 | GCgAGCCAC | −86 | GCgAaCCAC | −116 | GCtAGCCAC | −66 | |
| 4 | CACCACcG | −94 | tACCACCG | −123 | CACCACaG | −88 | DRE core (ACCG) |
| 5 | CCgTCACCTGCTT | −107 | CggTC- - - -GCTT | −132 | CCtTCACCTGCTT | −104 | MYC-like (CANNTG) |
| 6 | ATgGCGGC | −120 | ATcGCGGC | −142 | ATtGCGGC | −117 | ERF (-)b(GCCGCC) |
| 7 | CcTATAAA | −127 | CcTATAAA | −150 | CtTATAAA | −124 | TATA-box |
| 8 | TCGCCgTtC | −149 | TCGCCaTCC | −175 | TCGCC-TCC | −137 | |
| 9 | TGCCGTGGC | −178 | TaCCGTGGC | −202 | TGCCGTGGC | −162 | C-ABRE (CGTG) |
| 10 | CACCCcTG | −184 | CcCCCcTa | −209 | CACCCaTG | −168 | |
| 11 | GCcCACGT | −193 | GCcCACGT | −218 | GCaCACGT | −191 | ABRE4 (CACGTA) |
| 12 | CTGGCCgCC | −218 | - - - - -cc | −236 | CTGGCCaCC | −214 | GRA (CACTGGCCGCCC); ERF |
| 13 | CCCGCCGgCG | −249 | CCCGCCcgCG | −266 | CCCGCCG-CG | −260 | |
| 14 | GCACACGTCC | −259 | GCACACGTCC | −276 | GgACACGTCC | −243 | ABRE2 (CACGTC) |
| 15 | GCCACCGaCG | −285 | GCCACCGaCG | −312 | GCCACCGcCG | −265 | DRE2 (ACCGAC) |
| 16 | gACGTGGCG | −300 | gACGTGGCG | −328 | tACGTGGCG | −273 | ABRE1 (GACGTG) |
| 17 | TTCGAGAA | −315 | TTCccGA | −343 | TTCGAGAA | −299 | |
| No.a | CNS (bp) | Sequence | Maize | Sorghum | Regulatory element | ||
| 18 | 10 | AAGCCGTGCA | −16 | −12 | C-ABRE (CGTG) | ||
| 19 | 15 | CGAGCACACAAGCAC | −63 | −88 | DBF (CACAAG) | ||
| 20 | 11 | ACCACCGGCGA | −93 | −121 | DRE core (ACCG) | ||
| 21 | 16 | GCCCAGATCCTAATCC | −167 | −193 | HD-ZIP (CCTAATCCC) | ||
| 22 | 10 | GCCCACGTAC | −193 | −218 | ABRE4 (CACGTA) | ||
| 23 | 10 | CCCAATCCAC | −211 | −236 | MYB-like (-) (CTAACCA) | ||
| 24 | 14 | GCGTACGTGTACGTG CACACGTCCCGCC |
−244 | −261 | ABRE3a/3b (TACGTGTACGTG) |
||
| 25 | 14 | GCACACGTCCCGCC | −259 | −276 | ABRE2 (CACGTC) | ||
| 26 | 13 | GCCACCGACGCAC | −285 | −311 | DRE2 (ACCGAC) | ||
| 27 | 20 | GAAGAACCGAGACTG GCGG |
−310 | −338 | DRE1/ABRE1 (ACCGAGACGTG) |
||
| 28 | 10 | GCACCGGTAA | −342 | −390 | DRE core (ACCG) | ||
Conserved sequences between maize or sorghum and rice are indicated by uppercase letters. Dashes indicate an INDEL and a gap in the alignment. Sequences of known or predicted regulatory elements are in italics.
Numbers correspond to CNS indicated in Figure 1.
(-) indicates that motif is found in the reverse orientation.
A scan of CNS for matches to other putative cis-elements/TF-binding sites contained in the TRANSFAC and PLACE databases (http://www.gene-regulation.com/; http://www.dna.affrc.go.jp/htdocs/PLACE/) showed that CNS 4 contains a DRE core-binding sequence (ACCG) in sorghum and maize but not in rice (ACAG). CNS 5 in rice/maize contains a bHLH MYC-like binding sequence (CANNT; Abe et al. 1997) whereas sorghum contains a deletion in this putative binding site. CNS 6 contains an ethylene response factor (ERF) sequence (GCCGCC) in the reverse orientation (Brown et al. 2003). CNS 7 (Cc/tTATAAA) is a putative TATA-element located in maize, sorghum, and rice, while CNS 9 (Tg/aCCGTGGC) contains a C-ABRE-binding half site (CGTGGC; Hao et al. 1998; Menke et al. 1999; Kizis and Pages 2002; Niu et al. 2002).
The 11 >9-bp CNS identified in comparisons of sorghum/maize were also searched for putative TF-binding sites (Table 2; CNS 18–28). Sbdhn2/Zmrab17 CNS 20, 22, 25, 26, and 27 overlap CNS 4, 11, 14, 15, and 16 in searches of sorghum/rice and maize/rice, respectively, and were therefore not analyzed further. Sbdhn2/Zm rab17 CNS 18 contains the C-ABRE-containing sequence (GCCGTG) similar to CNS 9, while CNS 19 spans a DBF-like binding sequence (CACAAG; Kizis and Pages 2002). CNS 21 spans the sequence CCTAATCC that has a core TAAT motif often found in HD-ZIP protein-binding sites (Wolberger 1996), while CNS 23 contains a Myb3-like motif (CTAACCA) in a reverse orientation (Abe et al. 1997). CNS 24 corresponds to ABRE3a/3b identified through biochemical analysis (Busk et al. 1997). CNS 28 spans sequences that contain the DRE core-binding site (ACCG) recognized by some AP2 transcription factors (CACCGG).
A search for TF-binding sequences was also performed by scanning the entire 5′-region of each gene for matches to TF-binding sites in the TRANSFAC and PLACE databases (http://www.gene-regulation.com/pub/databases.html#transfac; http://www.dna.affrc.go.jp/htdocs/PLACE/) to identify putative cis-elements that were missed in sorghum/rice or maize/rice analyses due to the loss or creation of regulatory elements after species separation. This search identified four additional putative TF-binding sites: a (GCCGCC) AP2-ERF binding sequence (Brown et al. 2003) immediately downstream of DRE2 in rice, a DRE-like sequence upstream of CNS 28 (ACCGAC in both maize and sorghum), and two DRE-like/AP2 binding sequences in rice (CACCGT, CACCGG) that partially overlap the GRA and SPH cis-elements in maize (Figure 1, labeled beneath Osrab16A2 sequence).
Overall, the implementation of phylogenetic analysis and TF-binding site searches described above identified 17 CNS from the Sbdhn2/Osrab16A2 or Zmrab17/Osrab16A2 searches, 6 additional unique Zmrab17/Sbdhn2 CNS that span known or putative TF-binding sites, plus 4 other putative TF-binding sites that are not supported through CNS discovery from phylogenetic analysis of either sorghum/maize by rice or sorghum by maize. Thus, a total of 27 possible CNS/TF-binding sequences were identified in the ∼400-bp 5′-noncoding region upstream of the homologous rab genes, corresponding to one putative regulatory element every ∼14 bp.
Phylogenetic analysis of additional genes related to maize rab16/17:
Gene families are created by gene duplication and therefore family members share a degree of sequence conservation and common regulation reflective of the time of divergence and forces of selection. While expression of many members of a gene family is often regulated through common regulatory pathways, specific genes of the family exhibit divergent expression under selected conditions. Therefore, phylogenetic analysis of gene families could help validate the presence of common regulatory elements and provide pairs of genes that differ by a limited subset of the regulatory elements that differentiate expression of specific members of the gene family. In these latter cases, correlation between variation in regulatory element composition and differences in gene expression could help elucidate the function of regulatory sequences. On the basis of this idea, we tested if phylogenetic analysis of four additional members of the rice rab16 gene family (rab16A-D) together with the cluster of related dhn2, rab17, and rab16A2 genes from sorghum, maize, and rice would provide useful information about CNS function.
BLASTN searches of the maize rab17 EST sequence against the nonredundant database identified several rab17 gene homologs, including rab16A (e−19), rab16B (e−19), rab16C (e−23), and rab16D (e−19). Rice rab16A–D genes are organized in close proximity to each other in a tandem array consistent with derivation by duplication (Yamaguchi-Shinozaki et al. 1989). The proteins encoded by rab16A-D are ∼65–92% similar in amino acid sequence and have domains and motifs common to the dehydrins (Close 1997). CLUSTAL analysis of protein-coding regions was used to estimate the extent of divergence among Osrab16A-D, Osrab16A2, Zmrab17, and Sbdhn2 (Figure 2). This analysis showed that three pairs of RAB proteins, encoded by Sbdhn2/Zmrab17, Osrab16A2/Osrab16A, and Osrab16B/Osrab16C, are most similar to each other and incrementally diverged from the other pairs of proteins (Figure 2). The sorghum dhn2 and maize rab17 genes diverged ∼16 MYA, providing an estimate of the time and extent of divergence between this pair of genes and other genes with similar divergence. This analysis also showed that the proteins encoded by Osrab16D and Osrab16B/C have diverged to a similar extent as Sbdhn2 and Osrab16A2 (∼50 MY). Overall, divergence among the RAB16 proteins was greater than among the initial set of RAB proteins analyzed (SbDHN2, ZmRAB17, and OsRAB16A2), suggesting that sufficient evolution had occurred to apply similar criteria for phylogenetic analysis to selected pairs of the larger set of rab16/17 gene family members.
Figure 2.—

Phylogram of rab family members in sorghum, maize, and rice. Evolutionary distance of rab family members in sorghum, maize, and rice was calculated using protein sequences with the default settings on ClustalW (http://www.ebi.ac.uk/clustalw/index.html) and is given in parentheses next to each family member.
CNS/TF-binding sites associated with the rab gene family:
The predicted promoter regions of the rab16/17 genes were subjected to phylogenetic analysis following the procedure described above. The promoters of pairs of rab genes with divergence similar to or greater than that of Osrab16A2 vs. Sbdhn2/Zmrab17 (∼50 MY) were aligned and searched for 7/8-bp CNS using FootPrinter. Following CNS discovery through pairwise analysis of genes, CNS common to more than two genes were aligned where possible. The results of this analysis are shown in Figure 3, where 7/8-bp CNS are highlighted with various colors (CNS without biochemical support, yellow; ABREs, blue; non-ABRE biochemically defined elements, brown, green, pink, and gray) and numbered above the corresponding sequence. TF-binding sites identified through biochemical analysis of Zmrab17 and Osrab16B are boxed and labeled above the Zmrab17 sequence (Figure 3, DRE1, ABRE1, C-ABRE, etc.; Ono et al. 1996; Busk et al. 1997), while sequences related to known TF-binding sites that reside outside CNS are colored red and labeled below the set of seven rab sequences [Figure 3, C-ABRE (CGTG; Ono et al. 1996), SPH (CATGC; Busk et al. 1997), CBF1 (CCGAC; Stockinger et al. 1997; Medina et al. 1999), ERF (GCCGCC; Brown et al. 2003), DRE (ACCG-core), AP2 (GCCGGT; Niu et al. 2002), bHLH MYC-like binding site (CANNTG; Abe et al. 1997), MYB (AAACAAT, CCAACC; Li and Parish 1995); PLACE/TRANSFAC databases). Some motifs were found in the reverse orientation and are indicated by the addition of (-) following the motif name. In total, four new CNS were identified through phylogenetic analysis with the additional rice rab paralogs: 16A–16D (Figure 3, CNS 11.1, 11.2, 12.1, and 13.1). CNS 11.1 identified the biochemically defined SPH element, while the remaining new CNS appear novel.
Figure 3.—

Promoter alignment of homologous rab genes from sorghum, maize, and rice. Promoter alignments were seeded by 7/8-bp CNS identified with FootPrinter before manual adjustment. CNS are highlighted and identified by number above the sequence. CNS that do not contain biochemically characterized regulatory elements are highlighted in yellow, while CNS that contain biochemically defined motifs are highlighted in blue (ABREs), brown (DRE2), green (GRA), pink (SPH), and gray (TATA). TF-binding sites are boxed and labeled above the Zmrab17, while candidate TF-binding sites are colored red and labeled below Osrab16D. Asterisks (*) represent a sequence that can align on either side of an INDEL.
The distribution of CNS/TF-binding sites among the seven rab16/17 genes analyzed is summarized in Table 3. The ABRE1 motif was present in all of the rab genes analyzed; however, most of the CNS/TF-binding sites were present in only a subset of the genes. As expected, closely related genes had more CNS/TF-binding sites in common. For example, Osrab16A2 and Osrab16A had 16/16 elements in common and Zmrab17 and Sbdhn2 shared 14 of 17 CNS/TF-binding sites. In contrast, Zmrab17 had only 8/19 CNS/TF-binding sites in common with Osrab16B and 5/18 elements in common with Osrab16D, consistent with increasing divergence among these pairs of genes (Figure 2).
TABLE 3.
Distribution of CNS/TF-binding sites amongrab genes and their role in Zmrab17 activity
| CNS-TF-binding sitesa
|
Zmrab17 activityb
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Os
|
Shoot
|
|||||||||
| TF/CNS | Zm: rab17 |
Sb: dhn2S |
16A2 | 16A | 16B | 16C | 16D | −ABA | +ABA | Embryo: +ABA |
| CNS (17) | + | + | + | + | + | 6/7 | + | |||
| DRE1 | + | + | + | + | − | − | − | o | ++ | |
| ABRE1 (16) | + | + | + | + | + | + | + | o | + | +++ |
| DRE2 (15) | + | + | + | + | + | + | − | +++ | ++ | +++ |
| ABRE2 (14) | + | + | + | + | + | + | − | o | + | ++ |
| CNS (13.1) | − | − | + | + | + | − | + | |||
| CNS (13) | + | + | + | + | − | − | − | |||
| ABRE3a/3b | + | + | − | − | − | − | − | o | + | + |
| CNS (12.1) | + | + | − | − | + | + | − | |||
| GRA (12) | + | − | + | + | − | − | − | +++ | ++ | o |
| CNS (11.2) | − | − | + | + | + | 6/7 | − | |||
| SPH (11.1) | + | + | − | − | 6/7 | + | + | − | + | + |
| ABRE4 (11) | + | + | + | + | + | + | − | + | ++ | +++ |
| CNS (10) | + | − | + | + | − | + | − | |||
| CNS (9) | + | + | + | + | − | − | + | |||
| CNS (8) | + | + | + | + | + | − | + | |||
| TATA (7) | + | + | + | + | + | + | 6/7 | |||
| CNS (6) | + | + | + | + | 6/7 | − | − | |||
| CNS (5) | + | − | + | + | − | − | − | |||
“+” indicates that the element contains a biochemically defined TF-binding site or a CNS meeting the 7 of 8 base-pair match criteria; 6/7 tracks sequences that were not identified as 7/8 CNS, yet contain a 6 of 7 base-pair match to the CNS; “−” indicates that the element is not present in that lineage.
“+++” indicates that the element is required for expression; “++” indicates that the element contributes moderately to expression; “+” indicates that the element contributes slightly to gene expression; “o” indicates that the element does not contribute to gene expression under the given condition; − indicates that the element represses gene expression (data from Busk et al. 1997).
Correlation between gene expression and CNS/TF-binding site content:
The large number of differences between the rab16/17 gene promoters, including the number, spacing, and composition of CNS and TF-binding sites, suggested that relating variation in CNS composition to differences in gene expression would be challenging. To learn how to begin making valid comparisons, rab16/17 gene mRNA accumulation in seeds or vegetative tissues of plants treated with ABA for 3 or 27 hr was quantified using real-time PCR (qRT-PCR). These tissues and treatments were selected because of the previously described impact that several rab17 TF-binding sites have on gene expression in seeds and on ABA regulation (Table 3; Busk et al. 1997).
Figure 4 shows that among the rab genes analyzed, mRNA levels increased ∼100- to 10,000-fold in roots following treatment with ABA and ∼10- to 1000-fold in shoots and that the level of rab16/17 mRNA in seeds is ∼50- to 1000-fold higher than that in roots of control vegetative plants. Induction of the rab16/17 genes by ABA is consistent with the presence of one or more ABRE sequences in all of the rab genes and “coupling elements” such as DRE2 in six of the seven genes analyzed (Shen et al. 1996). However, while all of the genes responded to ABA and all are expressed in seeds, significant variation in rab16/17 gene mRNA abundance was observed. For example, Sbdhn2 showed greater induction by ABA in shoots compared to Zmrab17, and rab16D had the smallest difference in seed vs. root mRNA level among the rab genes analyzed (Figure 4).
Figure 4.—
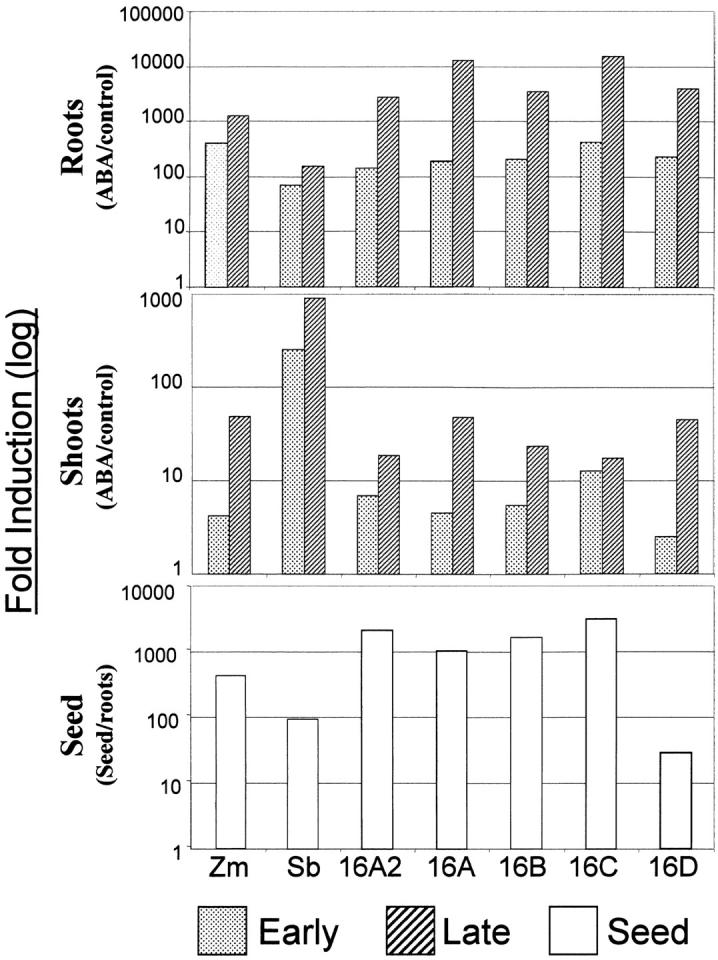
Fold RNA induction of rab gene family members in sorghum, maize, and rice in seeds and in vegetative tissue in response to ABA treatment. RNA levels of rab family members were analyzed by qRT-PCR in root and shoot tissue at 3 (early) and 27 hr (late) following ABA treatment compared to control untreated tissue. Additionally, RNA levels in seeds were determined by comparison to basal expression in control root tissue. Fold differences in mRNA levels are plotted on a log scale.
Differences in gene expression among pairs of closely related genes may be related to variation in a limited number of CNS/TF-binding sites. Variation in mRNA levels in control and ABA-induced states in different tissues and times after treatment were visualized by plotting the relative ratios (or fold differences) of mRNA abundance for pairs of genes normalized to 18S rRNA (Figure 5). As expected for the closely related Osrab16A/Osrab16A2 genes that have all of their CNS/TF-binding sites in common, the relative mRNA ratios for the genes do not vary significantly under any of the conditions examined. In contrast, the ratios of Sbdhn2/Zmrab17 mRNA abundance are similar in all tissues and treatments except control shoots where Sbdhn2 abundance is ∼85 times lower than Zmrab17 (Figure 5A). Therefore, the increased induction of Sbdhn2 mRNA by ABA in shoots compared to Zmrab17 mRNA (Figure 4) was due primarily to relatively low levels of Sbdhn2 mRNA in control shoots. Sbdhn2 and Zmrab17 have 14/17 CNS/TF-binding sites in common; however, Sbdhn2 lacks CNS 5, CNS 10, and CNS 12 (GRA; Figure 3). It has previously been demonstrated that the GRA element contributes significantly to basal Zmrab17 gene expression in unperturbed shoots (Busk et al. 1997), consistent with the results presented here.
Figure 5.—

Ratio of rab gene mRNA levels in seeds and vegetative tissues during unperturbed growth and ABA treatment. To analyze expression differences between genes, relative ratios of mRNA abundance between pairs of genes are calculated by comparing dCTs for each gene under all conditions. Relative mRNA abundance ratios for each condition are plotted for Sbdhn2/Zmrab17 (♦) and Osrab16A/Osrab16A2 (•).
A third way to visualize differences in gene expression involves the generation of standard curves so that the absolute levels of mRNA derived from different genes can be directly compared. This type of analysis was carried out for three genes, Osrab16A, Osrab16B, and Osrab16D, which are all encoded on the same rice BAC (Figure 6). This analysis showed several significant differences in gene expression. First, in shoots and seeds, Osrab16D mRNA levels are much lower than those of Osrab16A and Osrab16B. Low Osrab16D expression in these tissues is correlated with the lack of ABRE4, ABRE2, and DRE2 in the Osrab16D promoter, elements shown to contribute to basal and induced expression of Zmrab17 in shoots and seeds (Busk et al. 1997). In contrast, basal Osrab16D and Osrab16B mRNA levels in roots are similar and both genes are highly induced by ABA in this tissue. ABRE1, SPH, CNS 8, CNS 13.1, and CNS 17 are common to both genes, suggesting a role for these elements in root gene expression. Osrab16A mRNA levels were consistently higher than those of the other two rab genes analyzed, especially after 27 hr of treatment of vegetative tissues with ABA. The presence of CNS 9 and GRA in Osrab16A vs. Osrab16B, as well as several other differences in CNS/TF-binding site composition, are correlated with elevated expression of this gene.
Figure 6.—

Relative expression of rice rab16A, rab16B, and rab16D. Standard curves for qRT-PCR were generated using a dilution series of known amount of BAC DNA template to correct for differences in primer efficiency to determine absolute abundance of mRNA per gene under each condition examined. The corrected mRNA abundance for Osrab16A (♦), Osrab16B (▪), and Osrab16D (▴) is plotted for roots and shoots in control and for ABA-treated tissue at 3 and 27 hr as well as for seeds.
DISCUSSION
Rapid advances in grass genome research are providing a foundation for in-depth comparisons of gene content and organization among these species (Buell 2002; Chandler and Brendel 2002; Mullet et al. 2002). Recently, it was demonstrated that phylogenetic analysis can be used to identify conserved noncoding sequences among rice, sorghum, and maize gene orthologs (Kaplinsky et al. 2002; Morishige et al. 2002; Guo and Moose 2003; Inada et al. 2003). The ∼15–25 bp CNS discovered through these approaches were often located within introns and considered likely to regulate gene expression (Inada et al. 2003), although their location and size are inconsistent with TF-binding sites. In this study, phylogenetic analysis was carried out on a group of ABA-responsive genes related to maize rab17 that are induced in response to plant dehydration during seed development. The goal was to investigate the utility of phylogenetic methods for identifying 5′-noncoding regulatory sequences including TF-binding sites among grass genes.
Useful phylogenetic CNS search parameters based on several considerations were developed. First, the promoters of most genes contain TF-binding sites that are 6–10 bp long with only a subset of these bases under strong selection. Phylogenetic searches for CNS larger than TF-binding sites would require conservation of base pairs that are not under selection, leading to a high level of false negatives consistent with prior results (Inada et al. 2003). Second, analysis of rice, sorghum, and maize sequences spanning known TF-binding sites in rab17 indicated that 7/8-bp sequence matches in aligned regions would identify most of the binding sites that are common to the genes and the species being compared. Third, on the basis of mutation rates in the grasses (Gaut et al. 1996), the probability that species such as sorghum, maize, and rice, separated for ∼16–50 MY, have retained a 7-bp match at random in a DNA sequence that is identical by descent is relatively low (Kaplinsky et al. 2002). Moreover, comparisons of sorghum/rice and maize/rice sequences allowed good discrimination of CNS from other sequences in the promoters. On average, searches for 7/8-bp CNS identified identical sequence matches that spanned 6.5 bp and sequences surrounding CNS were usually much less conserved due to mutations, deletions, and insertions. In contrast, searches for CNS among sorghum and maize identified much longer identical sequences (13 bp) and resulted in a higher false-positive rate.
A prior phylogenetic study of grass genes concluded that it would be difficult to identify 7-bp CNS due to random sequence matches, especially among AT-rich sequences (Guo and Moose 2003). This complication was minimized in the current study in two ways. First, the search for overall sequence alignment and CNS was done incrementally, starting from the translation initiation codon and terminating when the degree of alignment and rate of CNS discovery declined significantly. Among the rab16/17 genes analyzed, overall sequence alignment and the rate of 7/8-bp CNS discovery decreased in sequences >400–450 bp upstream of the site of translation initiation. The region farther upstream contained many 7-bp AT-rich sequences similar to those reported by Guo and Moose (2003). Second, 7/8-bp CNS were required to occur in the same order relative to the translation start sites of the genes being compared, increasing the probability that the sequences were identical by descent. The final step in our approach involved searching the CNS and all other 5′-noncoding sequences for known TF-binding sites. This was done to identify additional TF-binding sites that were missed due to DNA insertions, deletions, or rearrangements since species divergence.
Application of the phylogenetic approach developed in this study for CNS discovery in sorghum dhn2, maize rab17, and rice rab16A2 genes identified 17 7/8-bp CNS in the 5′-noncoding region of these genes. In the rab17 promoter, five of the nine TF-binding sites previously defined by biochemical approaches were identified in the initial CNS alignment step, while four sites (DRE1, ABRE3a, ABRE3b, and SPH) were identified through analysis of rice rab16 paralogs or in searches for TF-binding sites (discussed below). Furthermore, 5 CNS identified in all three genes contained potential transcription-factor-binding sites identified through database searches: CNS 4 (DRE core; Niu et al. 2002), CNS 17 (putative TF-binding site in embryos; Busk et al. 1997), CNS 9 [ABRE half site (CGTGC; Izawa et al. 1993)], CNS 5 (bHLH MYC-like binding site), and a TATA-box. Two additional CNS were identified in the phylogenetic comparisons that did not span known TF-binding sequences (CNS 8 and 10). Overall, a total of 28 possible CNS/TF-binding sequences, or approximately one putative regulatory element every 14 bp, were identified in the ∼400-bp 5′-noncoding region upstream of these three rab genes. Similar results were obtained with biochemical analysis of the maize rab17 region spanning −184 to −305, where nine cis-elements, or one cis-element every 13 bp, were discovered (Busk et al. 1997).
Phylogenetic analysis of 5′-noncoding sequences among the rab17/16 gene family:
Analysis of homologous genes from widely diverged species will not detect regulatory elements that have been gained or lost by the genes being compared since divergence. This loss of information can be avoided to some extent by analyzing orthologs from more than two species or through phylogenetic “shadowing” of numerous species, including those diverged within the past 20 MY (Boffelli et al. 2003; Hong et al. 2003). In the present study, we tested an additional way to identify 5′-noncoding regulatory sequences by analyzing several members of a gene family. We assumed that members of gene families will have some regulatory elements in common and that differences in selected regulatory elements are present in specific members of the gene family. This idea is consistent with information showing that plants activate subsets of rab/dhn genes in response to different types of abiotic stress and in a range of tissues and developmental stages via specific complements of TF/ABRE interactions (Yamaguchi-Shinozaki et al. 1989; Kim et al. 1997; Choi et al. 2000; Uno et al. 2000). Moreover, we thought that differences in CNS content among gene family members could be related to variation in gene expression, providing tentative connections between CNS content and expression patterns.
To test this approach, phylogenetic analysis was carried out on five members of the rice rab16 gene family plus maize rab17 and sorghum dhn2. Phylogenetic analysis of 7/8-bp CNS among the larger group of rab/dhn genes identified many of the CNS/TF-binding sites found through analysis of three genes from rice, sorghum, and maize, providing increased evidence for the functional significance of these sequences. In addition, the analysis of the larger set of rab16/17 genes detected five CNS that were not identified in comparisons of Sbdhn2/Zmrab17 vs. Osrab16A2: a CNS located in the predicted 5′-UTR (CNTCGATC; data not shown); CNS 11.1 that spans the SPH element; and CNS 11.2, 12.1, and 13.1. On the basis of these results, we conclude that discovery of regulatory sequences by phylogenetic analysis is improved by the combined analysis of paralogs and orthologs from species spanning a range of divergence.
The alignment of CNS/TF-binding sites among the seven rab16/17 genes revealed several additional features regarding CNS composition and organization. First, the number of CNS shared by pairs of genes decreases as divergence among the genes increases. This trend probably reflects divergence in gene regulation and the accumulation of mutations that reduce our ability to detect CNS. Second, the conservation of sequences in and around CNS shared by gene family members is not perfect and could potentially contribute to differences in gene expression. For example, although ABRE1, -2, -3, and -4 all contain the same five-base ABRE core sequence (ACGTG), these binding sites differ in flanking nucleotides. Variation in sequences flanking ABRE core sequences are known to influence the interactions and binding affinities of these regulatory elements with different members of the bZIP family of transcription factors (Izawa et al. 1993; Hattori et al. 2002). Third, while the order of CNS/TF-binding sites in a region of the promoter was often conserved among the group of rab genes analyzed here, mutations, deletions, and insertions caused significant variation in the sequences and spacing between CNS.
Association of CNS content and rab gene expression:
The final part in our study assessed various methods for relating variation in CNS composition to differences in gene expression. rab genes are regulated by ABA, NaCl, cold, and other perturbations and are expressed in a wide range of cells, tissues, and developmental stages. In addition, ABA-responsive gene mRNA levels are regulated at the levels of transcription and RNA stability through regulatory elements located in the promoter as well as other regions of these genes not surveyed in this study (Finkelstein et al. 2002; Xiong et al. 2002; Himmelbach et al. 2003). Therefore, because data on rab16/17 mRNA abundance were collected only from roots, shoots, and seeds and from control and ABA-treated vegetative tissues, the associations between CNS and gene expression identified in this study will be incomplete. However, these data allowed the utility of methods for making associations between CNS content and gene expression to be explored and several associations to be tentatively identified for follow-up study.
Plots of fold changes in mRNA abundance induced by ABA or between tissues (seeds and roots) helped identify variation in rab16/17 gene expression. For example, ABA-induced expression of Sbdhn2 mRNA in shoots was greater than that of the other rab genes analyzed (Figure 4). Furthermore, analysis of the ratio of Sbdhn2 to Zmrab17 mRNA levels in basal and ABA-induced states showed that Sbdhn2 mRNA levels were low relative to Zmrab17 specifically in control shoots (Figure 5). This difference in expression was correlated with the lack of GRA and CNS 5 in Sbdhn2 relative to Zmrab17. This supports previous work in maize where mutations in the GCCGCC motif in the GRA element resulted in reduced basal expression of Zmrab17 in leaves (Busk et al. 1997). The transcription factors that bind to this element in maize or sorghum have not yet been identified. However, the (GCCGCC) ERF motif that is part of the Zmrab17 GRA binds AP2/ERBP factors that are involved in jasmonic acid/ethylene regulation in other plants (Hao et al. 1998; Brown et al. 2003).
The ratio of expression of very closely related rab genes such as Osrab16A and Osrab16A2 was similar in all basal and ABA-induced states examined (Figure 6). This result is consistent with the fact that these genes had 16/16 CNS in common. The ratios of Zmrab17 and Osrab16A2 mRNA levels were also similar under all conditions studied except in seeds. However, the CNS/TF-binding site composition of this pair of genes varies in several ways. Osrab16A2 lacks ABRE3a/3b, SPH, and CNS 12.1, contains modified GRA and DRE2 sequences, and has CNS 13.1, CBF1, and ERF sequences not present in Zmrab17 (Figure 3). This suggests that there is redundancy and/or compensating changes in the regulatory elements in these two genes.
Analysis of differences in ABA-induced expression and ratios of gene expression among pairs of genes can be done without correction for primer efficiencies. However, elements contributing to consistent differences in mRNA abundance in all tissues and states will not be detected in these analyses. Therefore, the abundance of Osrab16A, -16B, and -16D mRNAs was compared after correcting for differences in primer efficiency (Figure 6). This analysis showed that Osrab16A was expressed at higher levels than Osrab16B in all states examined. In addition, Osrab16A mRNA increased more than Osrab16B in ABA-treated roots and shoots between 3 and 27 hr. These differences in expression are correlated with the presence of GRA, CNS 13, 9, and 5, as well as loss of SPH and CNS 12.1 in Osrab16A relative to Osrab16B. Continued accumulation of Osrab16A mRNA in ABA-treated plants between 3 and 27 hr might be associated with DRE-like sequences in the DRE1 and GRA regions of this gene, which are not present in Osrab16B. The quantitative analysis of rab mRNA levels also showed that Osrab16D was expressed at relatively low levels in shoots and seeds but at levels comparable to Osrab16B in roots. Both of these genes contain ABRE1 and CNS 17, which may help explain similar levels of ABA-induced expression in roots. However, Osrab16D lacks DRE1, DRE2, ABRE2, and ABRE4 and CNS 9, 12.1, and 11.2, subsets of which are important determinants of Zmrab17 gene expression in shoots and embryos (Table 3). Interestingly, the ABRE1 element in Osrab16D is flanked by several SPH-like sequences, which have been found to mediate ABA responses in a similar configuration in the napA promoter (Ezcurra et al. 1999). Osrab16D also contains putative MYC-like and MYB-binding sequences immediately upstream of CNS 10 (Figure 3). While these elements are not phylogenetically conserved among the rab genes analyzed, it is well established that some ABA-responsive genes are modulated by bHLH transcription factors (Abe et al. 1997). Further biochemical assays will be required to test the significance of these latter putative binding sequences in Osrab16D.
An even wider phylogenetic analysis of rab and dhn gene family members among grass species could elucidate stepwise changes in gene expression, CNS/TF-binding sites, and associated phenotypes that have occurred during the ∼50 MY of evolution of the grass family. A complete analysis of the rab/dhn gene family in rice, sorghum, and maize could also help determine if differences in rab/dhn gene content and expression contribute to variation in drought tolerance among these grasses. Comparisons among orthologs from highly divergent species are most useful for TF-binding site identification, whereas phylogenetic analysis of more closely related species and gene families within species will be useful for identifying sequence regions containing more recently evolved regulatory elements. The overall grass gene CNS annotation process would benefit greatly from in-depth analysis of gene expression, better definition of TF-binding sites, and global mapping of TF-promoter associations through genome-wide chromatin immunoprecipitation assays (Lee et al. 2002). Above all, the collection of a complete set of gene sequences from sorghum and maize will be required to extract the full benefit of phylogenetic analysis of these grasses.
Acknowledgments
The authors thank Daryl Morishige for many helpful suggestions during the course of this project. This research was supported by grant nos. DBI-0110140 and DBI-9872649 from the Plant Genome Program of the National Science Foundation.
Sequence data from this article have been deposited with the EMBL/GenBank Data Libraries under accession no. AY177889.
References
- Abe, H., K. Yamaguchi-Shinozaki, T. Urao, T. Iwasaki, D. Hosokawa et al., 1997. Role of Arabidopsis MYC and MYB homologs in drought- and abscisic acid-regulated gene expression. Plant Cell 9: 1859–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansari-Lari, M. A., J. C. Oeltjen, S. Schwartz, Z. Zhang, D. M. Muzny et al., 1998. Comparative sequence analysis of a gene-rich cluster at human chromosome 12p13 and its syntenic region in mouse chromosome 6. Genome Res. 8: 29–40. [PubMed] [Google Scholar]
- Arabidopsis Genome Initiative, 2000. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408: 796–815. [DOI] [PubMed] [Google Scholar]
- Bieche, I., I. Laurendeau, S. Tozlu, M. Olivi, D. Vidaud et al., 1999. Quantitation of MYC gene expression in sporadic breast tumors with a real-time reverse transcription-PCR assay. Cancer Res. 59: 2759–2765. [PubMed] [Google Scholar]
- Blanchette, M., and M. Tompa, 2002. Discovery of regulatory elements by a computational method for phylogenetic footprinting. Genome Res. 12: 739–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchette, M., B. Schwikowski and M. Tompa, 2002. Algorithms for phylogenetic footprinting. J. Comput. Biol. 9: 211–223. [DOI] [PubMed] [Google Scholar]
- Boffelli, D., J. McAuliffe, D. Ovcharenko, K. D. Lewis, I. Ovcharenko et al., 2003. Phylogenetic shadowing of primate sequences to find functional regions of the human genome. Science 299: 1391–1394. [DOI] [PubMed] [Google Scholar]
- Brown, R. L., K. Kazan, K. C. McGrath, D. J. Maclean and J. M. Manners, 2003. A role for the GCC-box in jasmonate-mediated activation of the PDF1. Plant Physiol. 132: 1020–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buell, C. R., 2002. Current status of the sequence of the rice genome and prospects for finishing the first monocot genome. Plant Physiol. 130: 1585–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busk, P. K., and M. Pages, 1998. Regulation of abscisic acid-induced transcription. Plant Mol. Biol. 37: 425–435. [DOI] [PubMed] [Google Scholar]
- Busk, P. K., A. B. Jensen and M. Pages, 1997. Regulatory elements in vivo in the promoter of the abscisic acid responsive gene rab17 from maize. Plant J. 11: 1285–1295. [DOI] [PubMed] [Google Scholar]
- Chandler, V. L., and V. Brendel, 2002. The Maize Genome Sequencing Project. Plant Physiol. 130: 1594–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, H.-I., J.-H. Hong, J.-O. Ha, J.-Y. Kang and S. Y. Kim, 2000. ABFs, a family of ABA-responsive element binding factors. J. Biol. Chem. 275: 1723–1730. [DOI] [PubMed] [Google Scholar]
- Clark, R. M., E. Linton, J. Messing and J. F. Doebley, 2004. Pattern of diversity in the genomic region near the maize domestication gene tb1. Proc. Natl. Acad. Sci. USA 101: 700–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Close, T. J., 1997. Dehydrins: a commonality in the response of plants to dehydration and low temperature. Physiol. Plant. 100: 291–296. [Google Scholar]
- Colinas, J., K. Birnbaum and P. N. Benfey, 2002. Using cauliflower to find conserved non-coding regions in Arabidopsis. Plant Physiol. 129: 451–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins, F. S., E. D. Green, A. E. Guttmacher and M. S. Guyer, 2003. A vision for the future of genomics research. Nature 422: 835–847. [DOI] [PubMed] [Google Scholar]
- Davidson, E. H., D. R. McClay and L. Hood, 2003. Regulatory gene networks and the properties of the developmental process. Proc. Natl. Acad. Sci. USA 100: 1475–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doebley, J., M. Durbin, E. M. Golenberg, M. T. Cleg and D. P. Ma, 1990. Evolutionary analysis of the large subunit of carboxylase (rbcL) nucleotide sequence among the grasses (Gramineae). Evolution 44: 1097–1108. [DOI] [PubMed] [Google Scholar]
- Ezcurra, I., M. Ellerstrom, P. Wycliffe, K. Stalberg and L. Rask, 1999. Interaction between composite elements in the napA promoter: both the B-box ABA-responsive complex and the RY/G complex are necessary for seed-specific expression. Plant Mol. Biol. 40: 699–709. [DOI] [PubMed] [Google Scholar]
- Fickett, J. W., and W. W. Wasserman, 2000. Discovery and modeling of transcriptional regulatory regions. Curr. Opin. Biotechnol. 11: 19–24. [DOI] [PubMed] [Google Scholar]
- Finkelstein, R. R., S. S. L. Gampala and C. D. Rock, 2002. Abscisic acid signaling in seeds and seedlings. Plant Cell 14: S15–S45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frith, M. C., U. Hansen, J. L. Spouge and Z. Weng, 2004. Finding functional sequence elements by multiple local alignment. Nucleic Acids Res. 32: 189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaut, B. S., B. R. Morton, B. C. McCaig and M. T. Clegg, 1996. Substitution rate comparisons between grasses and palms: synonymous rate differences at the nuclear gene Adh parallel rate differences at the plastid gene rbcL. Proc. Natl. Acad. Sci. USA 93: 10274–10279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, H., and S. P. Moose, 2003. Conserved noncoding sequences among cultivated cereal genomes identify candidate regulatory sequence elements and patterns of promoter evolution. Plant Cell 15: 1143–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halfon, M. S., Y. Grad, G. M. Church and A. M. Michelson, 2002. Computation-based discovery of related transcriptional regulatory modules and motifs using an experimentally validated combinatorial model. Genome Res. 12: 1019–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao, D., M. Ohme-Takagi and A. Sarai, 1998. Unique mode of GCC box recognition by the DNA-binding domain of ethylene-responsive element-binding factor (ERF domain) in plant. J. Biol. Chem. 273: 26857–26861. [DOI] [PubMed] [Google Scholar]
- Hardison, R. C., 2000. Conserved noncoding sequences are reliable guides to regulatory elements. Trends Genet. 16: 369–372. [DOI] [PubMed] [Google Scholar]
- Harmer, S. L., J. B. Hogenesch, M. Straume, H. S. Chang, B. Han et al., 2000. Orchestrated transcription of key pathways in Arabidopsis by the circadian clock. Science 290: 2110–2113. [DOI] [PubMed] [Google Scholar]
- Hattori, T., M. Totsuka, T. Hobo, Y. Kagaya and A. Yamamoto-Toyoda, 2002. Experimentally determined sequence requirement of ACGT-containing abscisic acid response element. Plant Cell Physiol. 43: 136–140. [DOI] [PubMed] [Google Scholar]
- Himmelbach, A., Y. Yang and E. Grill, 2003. Relay and control of abscisic acid signaling. Curr. Opin. Plant Biol. 6: 470–479. [DOI] [PubMed] [Google Scholar]
- Hong, R. L., L. Hamaguchi, M. A. Busch and D. Weigel, 2003. Regulatory elements of the floral homeotic gene AGAMOUS identified by phylogenetic footprinting and shadowing. Plant Cell 15: 1296–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes, J. D., P. W. Estep, S. Tavazoie and G. M. Church, 2001. Computational identification of cis-regulatory elements associated with groups of functionally related genes in Saccharomyces cerevisiae. J. Mol. Biol. 296: 1205–1214. [DOI] [PubMed] [Google Scholar]
- Inada, D. C., A. Bashir, C. Lee, B. C. Thomas, C. Ko et al., 2003. Conserved noncoding sequences in the grasses. Genome Res. 13: 2030–2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izawa, T., R. Foster and N. H. Chua, 1993. Plant bZIP protein DNA binding specificity. J. Mol. Biol. 230: 1131–1144. [DOI] [PubMed] [Google Scholar]
- Kaplinsky, N. J., D. M. Braun, J. Penterman, S. A. Goff and M. Freeling, 2002. Utility and distribution of conserved noncoding sequences in the grasses. Proc. Natl. Acad. Sci. USA 99: 6147–6151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, S. Y., H.-J. Chung and T. L. Thomas, 1997. Isolation of a novel class of bZIP transcription factors that interact with ABA-responsive and embryo-specification elements in the Dc3 promoter using a modified yeast one-hybrid system. Plant J. 11: 1237–1251. [DOI] [PubMed] [Google Scholar]
- Kizis, D., and M. Pages, 2002. Maize DRE-binding proteins DBF1 and DBF2 are involved in rab17 regulation through the drought-responsive element in an ABA-dependent pathway. Plant J. 30: 679–689. [DOI] [PubMed] [Google Scholar]
- Koch, M. A., B. Weisshaar, J. Kroymann, B. Haubold and T. Mitchell-Olds, 2001. Comparative genomics and regulatory evolution: conservation and function of the Chs and Apetala3 promoters. Mol. Biol. Evol. 18: 1882–1891. [DOI] [PubMed] [Google Scholar]
- Lee, T. I., N. J. Rinaldi, F. Robert, D. T. Odom, Z. Bar-Joseph et al., 2002. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science 298: 799–804. [DOI] [PubMed] [Google Scholar]
- Lenhard, B., A. Sandelin, L. Mendoza, P. Engstrom, N. Jareborg et al., 2003. Identification of conserved regulatory elements by comparative genome analysis. J. Biol. 2: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, S. F., and R. W. Parish, 1995. Isolation of two novel myb-like genes from Arabidopsis and studies on the DNA-binding properties of their products. Plant J. 8: 963–972. [DOI] [PubMed] [Google Scholar]
- Medina, J., M. Bargues, J. Terol, M. Perez-Alonso and J. Salinas, 1999. The Arabidopsis CBF gene family is composed of three genes encoding AP2 domain-containing proteins whose expression is regulated by low temperature but not by abscisic acid or dehydration. Plant Physiol. 119: 463–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menke, F. L., A. Champion, J. W. Kijne and J. Memelink, 1999. A novel jasmonate- and elicitor-responsive element in the periwinkle secondary metabolite biosynthetic gene Str interacts with a jasmonate- and elicitor-inducible AP2-domain transcription factor, ORCA2. EMBO 18: 4455–4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishige, D. T., K. L. Childs, L. D. Moore and J. Mullet, 2002. Targeted analysis of orthologous phytochrome A regions of the sorghum, maize, and rice genomes using comparative gene-island sequencing. Plant Physiol. 130: 1614–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller, F., P. Blader and U. Strahle, 2002. Search for enhancers: Teleost models in comparative genomic and transgenic analysis of cis regulatory elements. BioEssays 24: 564–572. [DOI] [PubMed] [Google Scholar]
- Mullet, J. E., R. R. Klein and P. E. Klein, 2002. Sorghum bicolor: an important species for comparative grass genomics and a source of beneficial genes for agriculture. Curr. Opin. Plant Biol. 5: 118–121. [DOI] [PubMed] [Google Scholar]
- Niu, X., T. Helentjaris and N. J. Bate, 2002. Maize ABI4 binds coupling element1 in abscisic acid and sugar response genes. Plant Cell 14: 2565–2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono, A., T. Izawa, N. H. Chua and K. Shimamoto, 1996. The rab16B promoter of rice contains two distinct abscisic acid-responsive elements. Plant Physiol. 112: 483–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebeiz, M., N. L. Reeves and J. W. Posakony, 2002. SCORE: a computational approach to the identification of cis-regulatory modules and target genes in whole-genome sequence data. Proc. Natl. Acad. Sci. USA 99: 9888–9893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice Chromosome 10 Sequencing Consortium, 2003 In-depth view of structure, activity, and evolution of rice chromosome 10. Science 300: 1566–1569. [DOI] [PubMed]
- Riechmann, J. L., J. Heard, G. Martin, L. Reuber, C.-Z. Jiang et al., 2000. Arabidopsis transcription factors: genome-wide comparative analysis among eukaryotes. Science 290: 2105–2110. [DOI] [PubMed] [Google Scholar]
- Rombauts, S., K. Florquin, M. Lescot, K. Marchal, P. Rouze et al., 2003. Computational approaches to identify promoters and cis-regulatory elements in plant genomes. Plant Physiol. 132: 1162–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, Q., P. Zhang and T.-H. D. Ho, 1996. ABA response complexes: composite promoter units which are necessary and sufficient for ABA induction of gene expression in barley, Hordeum vulgare. Plant Physiol. 111: 130S. [DOI] [PubMed] [Google Scholar]
- Stockinger, E. J., S. J. Gilmour and M. F. Thomashow, 1997. Arabidopsis thaliana CBF1 encodes an AP2 domain-containing transcriptional activator that binds to the C-repeat/DRE, a cis-acting DNA regulatory element that stimulates transcription in response to low temperature and water deficit. Proc. Natl. Acad. Sci. USA 94: 1035–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung, D. Y., E. Vierling and C. L. Guy, 2001. Comprehensive expression profile analysis of the Arabidopsis Hsp70 gene family. Plant Physiol. 126: 789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tautz, D., 2000. Evolution of transcriptional regulation. Curr. Opin. Genet. Dev. 10: 575–579. [DOI] [PubMed] [Google Scholar]
- Tavazoie, S., and G. M. Church, 1999. Quantitative whole-genome analysis of DNA-protein interactions by in vivo methylase protection in E. coli. Nat. Biotechnol. 16: 566–571. [DOI] [PubMed] [Google Scholar]
- Thacker, C., M. A. Marra, A. Jones, D. L. Baillie and A. M. Rose, 1999. Functional genomics in Caenorhabditis elegans: an approach involving comparisons of sequences from related nematodes. Genome Res. 9: 348–359. [PMC free article] [PubMed] [Google Scholar]
- Thomas, J. W., J. W. Touchman, R. W. Blakesley, G. G. Bouffard, S. M. Beckstrom-Sternberg et al., 2003. Comparative analyses of multi-species sequences from targeted genomic regions. Nature 424: 788–793. [DOI] [PubMed] [Google Scholar]
- Tompa, M., 2001. Identifying functional elements by comparative DNA sequence analysis. Genome Res. 11: 1143–1144. [DOI] [PubMed] [Google Scholar]
- Uno, Y., T. Furihata, H. Abe, R. Yoshida, K. Shinozaki et al., 2000. Arabidopsis basic leucine zipper transcription factors involved in an abscisic acid-dependent signal transduction pathway under drought and high-salinity conditions. Proc. Natl. Acad. Sci. USA 97: 11632–11637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, R. L., A. Stec, J. Hey, L. Lukens and J. Doebley, 1999. The limits of selection during maize domestication. Nature 398: 236–239. [DOI] [PubMed] [Google Scholar]
- Waterston, R. H., K. Lindblad-Toh, E. Birney, J. Rogers, J. F. Abril et al., 2002. Initial sequencing and comparative analysis of the mouse genome. Nature 420: 520–562. [DOI] [PubMed] [Google Scholar]
- Weitzman, J. B., 2003. Tracking evolution's footprints in the genome. J. Biol. 2: 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolberger, C., 1996. Homeodomain interactions. Curr. Opin. Struct. Biol. 6: 62–68. [DOI] [PubMed] [Google Scholar]
- Xiong, L., K. S. Schumaker and J.-K. Zhu, 2002. Cell signaling during cold, drought, and salt stress. Plant Cell 14: S165–S183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi-Shinozaki, K., J. Mundy and N.-H. Chua, 1989. Four tightly linked rab genes are differentially expressed in rice. Plant Mol. Biol. 14: 29–39. [DOI] [PubMed] [Google Scholar]
- Yan, L., A. Loukoianov, G. Tranquilli, M. Helguera, T. Fahima et al., 2003. Positional cloning of the wheat vernalization gene VRN1. Proc. Natl. Acad. Sci. USA 100: 6263–6268. [DOI] [PMC free article] [PubMed] [Google Scholar]