

Abstract
arrest mutants have pleiotropic phenotypes, ranging from an early arrest of oogenesis to irregular embryonic segmentation defects. One function of arrest is in translational repression of oskar mRNA; this biochemical activity is presumed to be involved in other functions of arrest. To identify genes that could provide insight into how arrest contributes to translational repression or that may be targets for arrest-dependent translational control, we screened deficiency mutants for dominant modification of the arrest phenotype. Only four of the many deficiencies tested, which cover ∼30% of the genome, modified the starting phenotype. One enhancer, identified fortuitously, is the Star gene. Star interaction with arrest results in excess Gurken protein, supporting the model that gurken is a target of repression. Two modifiers were mapped to individual genes. One is Lk6, which encodes a protein kinase predicted to regulate the rate-limiting initiation factor eIF4E. The second is Delta. The interaction between arrest and Delta mimics the phenotype of homozygous Delta mutants, suggesting that arrest could positively control Delta activity. Indeed, arrest mutants have significantly reduced levels of Delta protein at the interface of germline and follicle cells.
TRANSLATIONAL control has long been recognized as an important regulatory process in the early stages of animal development. Classical studies focused on the dramatic change in translation activity that occurred after fertilization in amphibians (Davidson 1986). More recent discoveries revealed transcript-specific forms of regulation, such as the coordinated activation of large classes of mRNAs by interaction of cytoplasmic polyadenylation element (CPE)-binding protein with the CPE and subsequent extension of the poly(A) tail (Mendez and Richter 2001). Control events that may be specific for small numbers of mRNAs have emerged from the analysis of body patterning in Drosophila, where the transcripts encoding determinants that define the dorsoventral and anteroposterior axes of the egg are subject to elaborate programs of localization and translational regulation (Lipshitz and Smibert 2000). For the bicoid mRNA, which encodes the anterior determinant, translation appears to be activated as a consequence of poly(A) tail extension shortly after fertilization (Sallés et al. 1994). The oskar (osk) and gurken (grk) mRNAs, which encode proteins whose positions specify posterior and dorsal fates, respectively, are under more complex forms of control. Transcription of osk mRNA begins very early in oogenesis, but Osk protein does not appear at high levels until mid-oogenesis when the osk mRNA becomes localized to the posterior pole of the oocyte (Kim-Ha et al. 1995; Markussen et al. 1995; Rongo et al. 1995). The translational repression of osk mRNA prior to its localization is achieved in part through the action of Bruno (Bru), a protein that binds to regulatory sequences, Bruno response elements, in the osk 3′-UTR (Kim-Ha et al. 1995; Webster et al. 1997). Bru also appears to regulate the grk mRNA, as Bru can bind to grk mRNA and overexpression of a bru cDNA leads to dorsoventral patterning defects consistent with a reduction in Grk activity (Kim-Ha et al. 1995; Norvell et al. 1999; Filardo and Ephrussi 2003). At present the mechanism by which Bru represses translation is only poorly understood.
Bru is encoded by the arrest (aret) gene (Schüpbach and Wieschaus 1991; Webster et al. 1997). Bru protein was characterized and named before the discovery that it corresponds to aret and is widely known by the name Bru. In this article we refer to the gene and mutants as aret and the gene products as Bru. The phenotype of aret mutants suggests that Bru is likely to regulate other mRNAs in addition to the known targets, since some aspects of the phenotype are not readily attributed to misregulation of either osk or grk. During wild-type oogenesis cysts of 16 germline cells, which remain interconnected because of incomplete cytokinesis, are formed in the germarium. Individual cysts bud off and become enveloped by a layer of somatic follicle cells to create an egg chamber. One germline cell in each cyst becomes specified as the oocyte, while the remaining 15 become nurse cells (Spradling 1993). In strong aret mutants, such as aretQB, the cysts contain variable numbers of germline cells that complete cytokinesis and fail to differentiate and specify an oocyte (Schüpbach and Wieschaus 1991; Parisi et al. 2001). Overexpression of osk or grk does not cause such a phenotype (Smith et al. 1992; Neuman-Silberberg and Schüpbach 1994; Ghiglione et al. 2002), suggesting that aret regulates expression of one or more other mRNAs.
Some alleles of aret have weaker phenotypes that also reveal the pleiotropic character of the gene (Schüpbach and Wieschaus 1991; Webster et al. 1997). In intermediate allelic combinations, such as aretQB/aretPD or aretQB/aretPA, morphologically normal egg chambers form and an oocyte is specified, but after stage 6 or 7 the egg chambers become necrotic. Again, overexpression of either osk or grk does not lead to this phenotype. Females of the weakest allelic combinations, such as aretPD/aretPA, produce eggs that can be fertilized and develop to a late stage of embryogenesis. Many of the embryos have irregular anteroposterior patterning defects revealed by alterations of the ventral denticle belts: some individual segments are missing or adjacent segments are partially fused. Although overexpression of osk or grk does cause embryonic patterning defects, these defects are very specific and consistent and are unlike those of the aret mutants.
One genetic approach that could be useful in learning more about the function of Bru is an interaction screen, in which mutants are tested for their ability to modify aret− phenotypes. This approach has been used with considerable success to identify loss-of-function mutants that dominantly modify an existing phenotype (e.g., Simon et al. 1991). An interaction screen offers the possibility of providing insights into two of the main questions about the function of Bru. How does Bru repress translation? And what additional mRNAs are regulated by Bru? A reduction in the dosage of a gene that acts in the same process as Bru could enhance or suppress the aret phenotype. Similarly, a reduction in the level of an mRNA normally repressed by Bru could suppress the aret phenotype. Here we describe the results of such a screen.
MATERIALS AND METHODS
Fly stocks:
aret mutants (Schüpbach and Wieschaus 1991) were from Trudi Schüpbach. Deficiency mutants, P-element insertion mutants and the Star1 (S1) allele were obtained from the Bloomington Stock Center. P[eIF4E S251D] was from Paul Lasko. nosGAL4VP16 (Van Doren et al. 1998) was from Ruth Lehmann. matα4-GAL4-VP16#6 was obtained by jumping the P element in matα4-GAL4-VP16 V32a (Martin and St. Johnston 2003), obtained from Daniel St. Johnston. The S1 aretPD and aretPD P[eIF4E S251D] chromosomes were constructed by recombination using standard genetic methods.
Deficiency screen:
The cross scheme shown in Figure 1 was used to generate adults of the desired genotypes. After 3–4 days in well-yeasted vials, females were dissected and the ovaries fixed in 4% formaldehyde in PBS for 20–30 min. After rinsing several times in PBS plus 0.1% Triton X-100 (PBT), the ovaries were mounted in Vectastain (Vector Labs, Burlingame, CA) containing DAPI to label nuclei. Ovaries were examined using a Nikon epifluoresence microscope.
Figure 1.—

Cross scheme to test deficiency mutants for modification of the aret mutant phenotype. The inclusion of S1, which causes a dominant visible rough eye phenotype, on the aretPD chromosome allows us to identify the S1 aretPD/Df(2L)esc-P2-0 individuals without requiring that the third chromosome mutants be balanced or marked on their second chromosomes.
Antibody staining:
Ovaries were fixed as described above, blocked in PBT plus 5% goat serum, and incubated overnight in PBT plus 1% goat serum and the primary antibody. After several washes in PBT plus 1% goat serum, secondary antibodies were added for 2 hr, followed by several washes in PBT. In some cases DNA was counterstained with ToPro-3 (Molecular Probes, Eugene, OR) diluted 1/1000. Samples were mounted in Vectastain and imaged by confocal microscopy with a Leica TCS-SP. Primary antibodies, all from Developmental Studies Hybridoma Bank unless otherwise noted, were used at the following dilutions: mouse anti-Grk, 1/10; rat anti-Star (Uptal Banerjee), 1/10; mouse anti-Orb, 1/20; mouse anti-Hts 1B1, 1/10; mouse anti-FasIII, 1/10; rabbit anti-Lk6 (Jordan Raff), 1/500; mouse anti-Dl EC, 1/100; rabbit anti-Vas (Paul Lasko), 1/2000.
RESULTS
To identify genes that interact with aret we tested third chromosome deficiency mutants for the ability to dominantly suppress the ovarian aret phenotype. The mutants are from the “deficiency kit” maintained by the Bloomington Stock Center, and the subset that could be used (those not requiring the presence of a duplication) covers ∼74% of the chromosome. To streamline the crossing scheme the aretPD chromosome was marked with the dominant eye marker, S1, such that flies of the desired genotype could be identified despite the absence of second chromosome markers or balancer chromosomes in the deficiency stocks (see Figure 1). Serendipitously, the S1 mutation proved to dominantly enhance the aret phenotype, thus identifying an initial modifier mutant.
For each genotype the ovaries from multiple females were dissected and fixed, stained with DAPI to highlight nuclei, and examined by fluorescence microscopy. Wild-type or aretPD heterozygous females have ovaries with all stages of oogenesis represented (Figure 2A). In contrast, oogenesis in S1 aretPD/Df(2L)esc-P2-0 females is arrested at the germarial stage (Figure 2B). The vast majority of the deficiency mutants have no consistent effect on the S1 aretPD/Df(2L)esc-P2-0 oogenesis phenotype (Figure 2C; Table 1); thus the starting phenotype is not overly sensitive to variations in genetic background. Only four deficiencies elicited a clear dominant alteration of the starting phenotype, either delaying the arrest (Figure 2E) or increasing the number of germ cells in undifferentiated germaria (Figure 2D). These, as well as S1, were examined in detail. Not surprisingly, in no case was suppression of the aret phenotype complete. Instead, the arrest of oogenesis was extended to a later stage of development, but not to the point of egg laying.
Figure 2.—

Phenotypes scored in the screen. Micrographs are shown of ovarioles stained with DAPI (D) or ToPro-3 (A–C, E) to highlight nuclei. (A) Part of a wild-type ovariole, including the germarium at left and several egg chambers of increasing maturity. (B) Oogenesis is arrested at a very early stage in S1 aretPD/Df(2L)esc-P2-0 ovaries and the two ovarioles shown fail to bud off individual egg chambers. Instead, multiple undifferentiated germ cells appear in the severely truncated ovarioles. (C) An example of a third chromosome deficiency, Df(3L)pbl-X1, that has no dominant effect on the starting phenotype. (D) Df(3R)Dl-BX12 dominantly alters the starting phenotype, such that the germarium is greatly expanded to produce a large volume of germ cells surrounded by a layer of follicle cells. (E) Partial dominant suppression of the S1 aretPD/Df(2L)esc-P2-0 phenotype by Df(3R)M-Kx1. Egg chambers bud off from the germarium, although they are abnormal. Each chamber has greater than the usual number of 16 germ cells, and no oocyte is specified.
TABLE 1.
Deficiencies used in the screen
| Genotype | Breakpoints | Modification |
|---|---|---|
| Df(3L)emc-E12 | 061A; 061D03 | |
| Df(3L)Ar14-8 | 061C05–08; 062A08 | |
| Df(3L)Aprt-32 | 062B01; 062E03 | |
| Df(3L)R-G7 | 062B08–09; 062F02–05 | |
| Df(3L)M21 | 062F; 063D | |
| Df(3L)HR119 | 063C02; 063F07 | |
| Df(3L)GN34 | 063E06–09; 064A08–09 | |
| Df(3L)GN24 | 063F06–07; 064C13–15 | |
| Df(3L)ZN47 | 064C; 065C | |
| Df(3L)XDI98 | 065A02; 065E01 | |
| Df(3L)BSC27 | 065D04–05; 065E04–06 | |
| Df(3L)pbl-X1 | 065F03; 066B10 | |
| Df(3L)BSC13 | 066B12–C01; 066D02–04 | |
| Df(3L)h-i22 | 066D10–11; 066E01–02 | |
| Df(3L)Scf-R6 | 066E01–06; 066F01–06 | |
| Df(3L)29A6 | 066F05; 067B01 | |
| Df(3L)AC1 | 067A02; 067D07–13 or 067A05; 067D09–13 |
|
| Df(3L)BSC14 | 067E03–07; 068A02–06 | |
| Df(3L)lxd6 | 067E05–07; 068B02–04 | |
| Df(3L)vin2 | 067F02–03; 068D06 | |
| Df(3L)vin5 | 068A02–03; 069A01–03 | + |
| Df(3L)iro-2 | 069B01–05; 069D01–06 | |
| Df(3L)E44 | 069D02; 069E03–05 | |
| In(3LR)C190[L] | 069F03–04; 070C03–04 | |
| Df(3L)fz-GF3b | 070C01–02; 070D04–05 | |
| Df(3L)fz-M21 | 070D02–03; 071E04–05 | |
| Df(3L)BK10 | 071C; 071F | |
| Df(3L)brm11 | 071F01–04; 072D01–10 | |
| Df(3L)st-f13 | 072C01–D01; 073A03–04 | |
| Df(3L)81k19 | 073A03; 074F | |
| Df(3L)W10 | 075A06–07; 075C01–02 | |
| Df(3L)Cat | 075B08; 075F01 | |
| Df(3L)XS2182 | 076B; 076F | |
| Df(3L)XS543 | 076B; 077A | |
| Df(3L)kto2 | 076B01–02; 076D05 | |
| Df(3L)XS533 | 076B04; 077B | |
| Df(3L)XS572 | 076B06; 077C01 | |
| Df(3L)rdgC-co2 | 077A01; 077D01 | |
| Df(3L)ri-79c | 077B–C; 077F–78A | |
| Df(3L)ME107 | 077F03; 078C08–09 | |
| Df(3L)31A | 078A; 078E, 078D; 079B | |
| Df(3L)Pc-2q | 078C05–06; 078E03–079A01 | |
| Df(3L)Ten-m-AL29 | 079C01–03; 079E03–08 | |
| Df(3L)HD1 | 079D03–E01; 079F03–06 | |
| Df(3L)Delta1AK | 079E05–F01; 079F02–06 | |
| Df(3R)ME15 | 081F03–06; 082F05–07 | |
| Df(3R)3-4 | 082F03–04; 082F10–11 | |
| Df(3R)e1025-14 | 082F08–10; 083A01–03 | |
| Df(3R)Scr | 084A01–02; 084B01–02 | |
| Df(3R)Antp17 | 084B01–02; 084D11–12 or A06, D14 |
|
| Df(3R)p712 | 084D04–06; 085B06 | |
| Df(3R)p-XT103 | 085A02; 085C01–02 | |
| Df(3R)BSC24 | 085C04–09; 085D12–14 | |
| Df(3R)by10 | 085D08–12; 085E07–F01 | |
| Df(3R)by62 | 085D11–14; 085F06 | |
| Df(3R)M-Kx1 | 086C01; 087B01–05 | + |
| Df(3R)ry615 | 087B11–13; 087E08–11 | |
| Df(3R)ea | 088E07–13; 089A01 | |
| Df(3R)sbd105 | 088F09–89A01; 089B09–10 | |
| Df(3R)P115 | 089B07–08; 089E07–08; 020 | |
| Df(3R)DG2 | 089E01–F04; 091B01–B02 | |
| Df(3R)C4 | 089E03–04; 090A01–07 | |
| Df(3R)Cha7 | 090F01–F04; 091F05 | |
| Df(3R)Dl-BX12 | 091F01–02; 092D03–06 | + |
| Df(3R)H-B79 | 092B03; 092F13 | |
| Df(3R)e-N19 | 093B; 094 | |
| Df(3R)e-R1 | 093B06–07; 093D02 | |
| Df(3R)mbc-R1 | 095A05–07; 095D06–11 | + |
| Df(3R)crb-F89-4 | 095D07–D11; 095F15 | |
| Df(3R)crb87-5 | 095F07; 096A17–18 | |
| Df(3R)slo[8] | 096A02–07; 096D02–04 | |
| Df(3R)Espl3 | 096F01; 097B01 | |
| Df(3R)Tl-P | 097A; 098A01–02 | |
| Df(3R)D605 | 097E03; 098A05 | |
| Df(3R)3450 | 098E03; 099A06–08 | |
| Df(3R)L127 | 099B05–06; 099E04–F01 | |
| Df(3R)B81 | 099C08; 100F05 |
Star1 interacts with aret:
Ovaries of aretPD/Df(2L)esc-P2-0 females have an intermediate aret phenotype, in which oogenesis proceeds as far as stage 7 before the egg chambers degenerate (Figure 3A). When S1 is also present, in S1 aretPD/Df(2L)esc-P2-0 ovaries, the arrest of oogenesis is advanced to the germarial stage (Figure 3B). S1 has no dominant ovarian phenotype by itself (data not shown) and is thus acting as a dominant enhancer of the starting phenotype. This genetic interaction could be between S and aret, which is homozygous mutant in the affected flies, or between S and any of the genes made hemizygous by Df(2L)esc-P2-0. To distinguish between these possibilities we asked if S1 enhances the phenotype of flies in which only aret is mutant. The aretPD/aretPA allelic combination has a weak phenotype (Figure 3C), with many egg chambers producing mature oocytes that can be fertilized and progress through much of embryogenesis. When S1 is also present, the phenotype becomes more severe and oogenesis arrests at stage 6 or 7 (Figure 3D). The phenotype of aretPD/aretQB, in which an oocyte is specified but oogenesis arrests at stage 6/7 (Figure 3E), is similarly enhanced by S1. The arrest occurs earlier, at about stage 3 (Figure 3F), and no oocyte is specified (data not shown). We conclude that S1 is interacting with aret. This interaction does not appear to reflect Bru-dependent regulation of S mRNA translation, as no changes in S protein levels were detected (data not shown). The aret/S1 interaction is persistent during oogenesis, occurring at different stages. Thus either aret and S interact in an ongoing process or the severity of the defect in some initial process influences later events.
Figure 3.—

Star interacts with aret. The phenotypes of three aret allelic combinations of different strength are all dominantly enhanced by S1. In each case the aret phenotype becomes more severe, largely mimicking the effect of using a stronger aret allelic combination. (A) aretPD/Df(2L)esc-P2-0. (B) S1 aretPD/Df(2L)esc-P2-0. (C) aretPD/aretPA. (D) S1 aretPD/aretPA. (E) aretPD/aretQB. (F) S1 aretPD/aretQB.
A genetic interaction between S and aret is not surprising, since both have been implicated in expression or activity of grk. Grk is a transforming growth factor (TGF) α-like protein that is expressed as a membrane-bound form in the germline (Neuman-Silberberg and Schüpbach 1993, 1996; Serano et al. 1995). After proteolytic cleavage separates intra- and extracellular domains, the latter activates epidermal growth factor receptor (Egfr) in the overlying follicle cells (Ray and Schüpbach 1996; Ghiglione et al. 2002). S is required for the activity of Grk and appears to act in postcleavage trafficking or secretion of the protein (Ghiglione et al. 2002). Overexpression of Bru reduces the level of grk activity and the amount of localized Grk protein (Filardo and Ephrussi 2003). Thus the combined effects of reduction of both aret and S activity could well affect the level of grk activity. To determine if the amount or distribution of Grk protein is altered in S1 aretPD/aretPA ovaries, we monitored the protein in whole-mount preparations by immunofluorescence. In wild-type ovaries Grk protein accumulates in the oocyte during early stages (Figure 4A) and then becomes restricted to an anterodorsal region over the oocyte nucleus in stages 8–10. A similar Grk accumulation is observed in aret mutant ovaries (Figure 4B). When the S1 mutation is also present, in S1 aretPD/aretPA ovaries, Grk protein can now be detected at low levels in nurse cells (Figure 4C). In some preparations this nurse cell staining is concentrated at cell boundaries (Figure 4D), as if the protein is membrane associated.
Figure 4.—

Grk protein accumulates in nurse cells of S1 aretPD/aretPA ovaries. Stage 7 egg chambers are shown for each genotype, with nuclei in red (ToPro-3) and Grk in green. In wild-type egg chambers Grk protein is restricted to the oocyte (A). aret mutants [aretPD/Df(2L)esc-P2-0] also have detectable Grk only in the oocyte (B). When S1 is also present, in S1 aretPD/aretPA, ectopic Grk can be detected in the nurse cell cytoplasm (C) as well as the oocyte (C′, a different focal plane). Grk protein is also concentrated at nurse cell boundaries (arrows in D, a higher-magnification view of a part of C). The aret mutant combination is stronger in B [aretPD/Df(2L)esc-P2-0] than in C (aretPD/aretPA) to allow similarly staged egg chambers to be compared.
Lk6, which encodes a protein kinase predicted to phosphorylate eIF4E, interacts with aret:
Df(3R)M-Kx1, which removes parts of cytological intervals 86 and 87, dominantly suppresses the S1 aretPD/Df(2L)esc-P2-0 phenotype (Figure 2E). The interaction is with aret, rather than with S or Df(2L)esc-P2-0, since Df(3R)M-Kx1 also suppresses aretPD/aretQB (Figure 5, A and B) and aretPA/aretQB (data not shown). The interacting gene was identified by testing smaller deficiencies and P-element insertion mutants for the interaction (Figure 5C and data not shown). The P-element mutant EP(3)0886, which is inserted 5′ to the Lk6 gene (Kidd and Raff 1997; Huang and Rubin 2000), suppresses the phenotype of aretPD/aretQB (Figure 5C) and of aretPD/Df(2L)esc-P2-0 (data not shown). EP P elements, such as EP(3)0886, contain a GAL4-inducible promoter that, in conjunction with a source of GAL4, directs expression of the endogenous gene located proximal to the P element (Rorth et al. 1998), in this case Lk6. The suppression of the aret phenotype occurs in the absence of GAL4, consistent with the notion that the EP insertion by itself reduces Lk6 expression. When a germline source of GAL4 is provided, EP(3)0886 enhances the aretPD/aretQB phenotype (Figure 5D). The complementary suppression and enhancement from the same P element demonstrates that Lk6 interacts with aret. This interaction could reflect Bru-dependent control of Lk6 mRNA translation. This interpretation is unlikely, as Lk6 protein levels appear similar in wild-type and aret mutants (data not shown).
Figure 5.—

Mutation and overexpression of Lk6 leads to opposite effects on the aret phenotype. Ovaries from aretPD/aretQB females arrest oogenesis at stage 6 or 7 (A), but the arrest is delayed when the females are also heterozygous for Df(3R)M-Kx-1/+ (B). A single copy of the EP(3)0886 P-element insertion in Lk6 has a similar effect (C). Conversely, overexpression of Lk6 using EP(3)0886 and a germline GAL4 driver (matα4-GAL4-VP16#6) enhances the aret phenotype (D). The presence of the GAL4 driver alone does not change the aret phenotype (E). In all cases the aret combination was aretPD/aretQB, and the ovaries were stained with ToPro-3.
The effect of the Lk6 mutation on the aret oogenesis arrest phenotype persists throughout oogenesis, as different aret allelic combinations that arrest oogenesis at different stages all show suppression. The early arrest of aretQB/aretQB at stage 2 is extended to stage 3 or 4 when EP(3)0886 is present, and the stage 6/7 arrest of aretPD/aretQB is extended to stage 9. Furthermore, the low hatch rate of embryos from aretPA/aretPD mothers (3%, n = 300) is elevated (14%, n = 300) when the mothers also carry one copy of EP(3)0886. The suppression of aret mutants by reduction of Lk6 is paralleled by enhancement of the phenotype when Lk6 is overexpressed. In the aretPD/aretQB combination the arrest was shifted from stage 6/7 to stage 3, and with the aretPA/aretPD combination, which normally allows the completion of oogenesis, oogenesis was arrested at stage 9/10.
In addition to the oogenesis arrest phenotype, strong aret mutants can have cysts with >16 germline cells (Schüpbach and Wieschaus 1991; Parisi et al. 2001). This aspect of aret function does not appear to interact with Lk6: neither the deficiency that removes Lk6, Df(3R)M-Kx1, nor EP(3)0886 suppresses the extra germline cells phenotype (data not shown).
The Lk6 protein is predicted to phosphorylate and thus enhance activity of translation initiation factor eIF4E (Lachance et al. 2002) and could act through that pathway in suppressing the aret phenotype. Specifically, a global reduction in translation efficiency from reduced eIF4E activity could offset the enhanced translation of an mRNA negatively regulated by aret. To evaluate this possibility we asked if expression of a constitutively activated eIF4E, eIF4E S251D, in which the serine predicted to be phosphorylated by Lk6 is mutated to a negatively charged amino acid (Lachance et al. 2002), would reverse the suppression caused by the Lk6 mutant. Females of the genotype aretQB/aretPD; EP(3)0886/+ show the same timing of arrest—at stage 9—as females of the genotype aretQB/aretPD P[eIF4E S251D]; EP(3)0886/+. This result suggests that expression of the constitutively activated eIF4E has no effect on the suppression of the aret phenotype by the Lk6 mutant. However, the P[eIF4E S251D] transgene can itself suppress the aretQB/aretPD phenotype and delay arrest until stage 9. Thus either enhanced activity of eIF4E, provided by the P[eIF4E S251D] transgene, or a predicted reduction of eIF4E activity, from the Lk6 mutant, has the same effect on the aret phenotype, and interpretation of the suppression observed in the aretQB/aretPD P[eIF4E S251D]; EP(3)0886/+ females is not simple.
Additional Dfs dominantly suppress the timing of oogenesis arrest:
Only two other third chromosome deficiency mutants suppressed the timing of oogenesis arrest of S1 aretPD/Df(2L)esc-P2-0 females. One of these, Df(3R)mbc-R1, also suppressed the arrest phenotype of aretQB/aretQB, showing that it is acting on aret. Just as for Lk6-mediated suppression, there was no effect on the occurrence of cysts with >16 germline cells. We have not been able to identify a point mutant from within the region uncovered by this deficiency that displays a similar interaction.
The other suppressing deficiency, Df(3R)vin5, subsequently failed to suppress either aretQB homozygotes or aretPA/aretQB females. This deficiency presumably interacts with S1, with Df(2L)esc-P2-0, or with a combination of the mutations on the input chromosomes and was not characterized further.
Delta and aret interact:
Females of genotype S1 aretPD/Df(2L)esc-P2-0; Df(3R)Dl-BX12/+ have ovaries in which no cysts with the usual 16 germ cells form. Instead, in each ovariole a single layer of follicle cells surrounds a large number of undifferentiated germ cells (Figure 2D). The interacting genes responsible for the phenotype are Delta (Dl) and aret; females homozygous or trans-heterozygous for aret alleles and heterozygous for Dl9P, an amorphic allele, show a similar phenotype. Females homozygous for the strong aretQB allele, in which cysts form but no oocyte is specified, and heterozygous for Dl9P, have an ovarian phenotype similar to that observed with Df(3R)Dl-BX12 in the screen (Figure 6). In aretQB/aretQB ovaries, no oocyte is specified (Figure 6A) and individual cysts sometimes have >16 germline cells (Figure 6A′). When also heterozygous for Dl9P, almost all ovarioles now consist of the germarium and a single large egg chamber with many germline cells (the egg chamber is small in rare exceptions). This egg chamber could arise from fusion of initially separate egg chambers or from overproliferation of the germline. It is often possible to distinguish between these options, since the cell divisions that produce the germline cells have incomplete cytokinesis. Consequently, each daughter cell remains connected to its parent by a ring canal, and a branched organelle, the fusome, extends through the ring canals (Figure 6C). The number of cell divisions within a cyst can be determined by counting the number of ring canals of the oocyte. However, in the strong aret mutants cytokinesis is complete, there are no ring canals, and the fusome appears as dots (Figure 6D). In the single large egg chamber of aretQB/aretQB; Dl9P/+ ovarioles, the fusome also appears as dots (Figure 6E), and so we are unable to determine how these egg chambers form. The Dl mutation could be enhancing the frequency and severity of the germline overproliferation phenotype of aretQB. Alternatively, the aret mutations could be enhancing the egg chamber fusion phenotype of Dl mutants (Lopez-Schier and St. Johnston 2001).
Figure 6.—

Genetic interaction of Dl with aret. (A) An arrested ovariole of an aretQB/aretQB female. Some of the egg chambers show an overproliferation of the germline (identified by Vas, red staining in A–E; an example is shown at higher magnification in A′). Green staining in A–E is Hts. (B) An ovariole of an aretQB/aretQB; Dl9P/+ female. Only a single large egg chamber is present, and it has many germline cells. (C–E) Higher-magnification view to show the distribution of Hts, a fusome marker, in wild type (C), aretQB/aretQB (D), and aretQB/aretQB; Dl9P/+ (E). The fusome is branched and extends between cells in wild type (arrowheads), but appears as dots in both mutants (arrows), indicating that they have undergone complete cytokinesis.
A weaker combination of aret alleles also interacts with Dl9P. In aretPD/aretQB ovaries, oogenesis proceeds as far as stage 6/7 (Figure 7D). In aretPD/aretQB; Dl9P/+ ovaries most egg chambers have >16 germline cells (Figure 7, A–C), a situation that can again occur by overproliferation within a single egg chamber or fusion of multiple egg chambers. This genotype produces three classes of abnormal egg chambers, which are present in roughly equal numbers. In one class, a large number of germline cells of roughly equal size are enveloped by a single epithelium of follicle cells (Figure 7A). This phenotype is similar to that seen when using the stronger aret alleles, including the absence of an oocyte. The remaining two classes clearly arise from fusion. They both display partial fusion, with individual cysts failing to separate from one another, although they differ in the nature of the fusions.
Figure 7.—
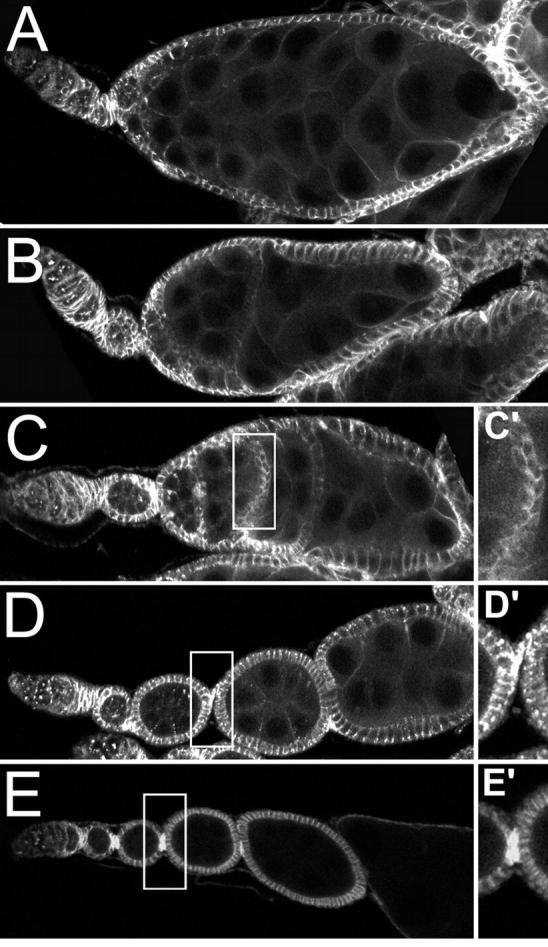
The combination of aretPD/aretQB and Dl9P/+ mimics the Dl/Dl phenotype. (A–C) The different classes of aretPD/aretQB; Dl9P/+ phenotypes. (A) A single large egg chamber that has formed by fusion or overproliferation. (B and C) Ovarioles with random or A/P incomplete egg chamber fusions, respectively. (D and E) Controls for comparison, aretPD/aretQB (D and D′) and aretQB/+; Dl9P/+ (E and E′). The absence of stalk cells between incompletely fused egg chambers is shown in C′, while stalk cells are visible in the controls (D′ and E′). Hts staining, which is strongest in somatic cells and is especially enriched in the stalk cells, is shown throughout.
In wild-type and aretPD/aretQB ovaries adjacent egg chambers are connected by several stalk cells positioned between the anterior polar cell of one egg chamber and the posterior polar cell of the next (Figure 7, D and E). For the partially fused egg chambers of aretPD/aretQB; Dl9P/+ flies, no stalk cells can be detected and the follicle cell layers of different egg chambers remain in intimate contact with one another (Figure 7C′). Some maintain a well-defined linear organization within individual ovarioles, and adjacent egg chambers are fused with each other at their anterior and posterior boundaries (Figure 7C); we refer to this class as anterior/posterior (A/P) fusions. The other class of partially fused egg chambers—called random fusions—resides in ovarioles in which the normal beads-on-a-string organization is absent. Egg chambers are positioned irregularly and can be closely apposed to multiple different egg chambers on lateral as well as anterior and posterior surfaces (Figure 7B). For both classes of partial fusion each cyst has an oocyte as determined by the presence of a single cell with a high concentration of the oocyte marker Orb (data not shown), although specific Orb staining is lost at the later stages when the cysts begin to degenerate. In the A/P fusions the oocyte is clearly at the posterior of the cyst. Defining the position of the oocyte in the random fusions is somewhat subjective, given the absence of a tandem arrangement of cysts, but in all cases the oocyte is either lateral or posterior relative to the overall polarity of the ovariole (data not shown).
The parital fusion phenotypes of aretPD/aretQB; Dl9P/+ are very similar to those of Dl/Dl germline clones (see discussion). A simple interpretation of this similarity is that Dl activity is reduced in aret mutants, through a reduction in the synthesis or presentation of Dl protein. To test this prediction we examined Dl protein in wild-type and aretPD/aretQB ovaries. Dl normally accumulates at the highest levels in the membranes separating the follicular epithelium from the nurse cells and oocyte, with a lower level of cytoplasmic staining (Figure 8A). In the aret mutant the level of membrane-associated Dl is clearly reduced, although the cytoplasmic staining is not visibly different (Figure 8B). Because the role of Dl is in signaling from the germline cell to the follicle cells, the protein at the junction between these cells is expected to be the active form. Thus aret does appear to have a positive role, probably indirect, in promotion of Dl activity.
Figure 8.—

aret mutant ovaries are deficient in Dl protein. (A) Dl in wild-type (A) and aretPD/aretQB (B) egg chambers. In each case the protein was detected by immunofluorescence using a confocal microscope with the same laser power and gain settings. In the wild-type egg chambers, during stages 5–7, Dl is enriched at the surface of germline cells, especially where they appose follicle cells (A′), and dispersed throughout the cytoplasm. The overall distribution is similar in the aret mutant, but the amount of Dl protein in or closely associated with the membranes is significantly reduced (B′).
In the third class of abnormal egg chambers a large number of germline cells of roughly equal size are enveloped by a single epithelium of follicle cells (Figure 7A). This phenotype is similar to that seen when using the stronger aret alleles, including the absence of an oocyte.
DISCUSSION
We performed a screen of third chromosome deficiencies for dominant modifiers of aret mutants. About three-quarters of the third chromosome was screened, corresponding to ∼30% of the genome. Only four deficiencies dominantly modified the aret mutant phenotype, suggesting that the total number of genes in the genome with this property is small. For two of the four deficiencies we were able to identify the gene responsible for the interaction, and we fortuitously discovered a third interacting gene while preparing for the screen. We anticipated that two different types of modifiers might be detected by the screen: those in genes that act in the same process as Bru and those in genes that are themselves regulated by Bru or act in a process in which a limiting component is regulated by Bru. Characterization of the interacting genes suggests that we recovered examples of each type of modifier.
Interaction of aret and S:
Bru has been proposed to translationally regulate grk mRNA. The supporting evidence includes (i) binding of Bru to grk mRNA in vitro and indirect evidence of binding in vivo (Kim-Ha et al. 1995; Norvell et al. 1999; Filardo and Ephrussi 2003); (ii) rare dorsoventral patterning defects as a consequence of overexpression of Bru, and enhancement of this phenotype by reduction of grk gene dosage; and (iii) evidence that localized Grk is present at reduced levels when Bru is overexpressed, although unlocalized Grk appears more abundant (Filardo and Ephrussi 2003). However, there has been no evidence of excess Grk protein in aret mutants. S is required for grk activity, and it acts post-translationally in either trafficking or secretion of Grk protein (Ghiglione et al. 2002). We found that when flies were both homozygous for aret and heterozygous for S1 they accumulated Grk protein in nurse cells, while ectopic accumulation could not be detected in either aret mutants or S1 heterozygotes alone. This synthetic effect on Grk protein accumulation is simple to rationalize. In aret mutants Grk protein is excessively translated, but an S-dependent delivery step could efficiently clear the protein from the nurse cells. When S activity is reduced, a detectable level of Grk remains in the nurse cells. The distribution of the ectopic Grk, both in cytoplasm and at the nurse cell boundaries, could correspond to the sites where the protein might stall during delivery. The actual site of S action is not known, and two different sites of S concentration, in endoplasmic reticulum or on the plasma membrane, have been reported (Pickup and Banerjee 1999; Ghiglione et al. 2002). Although this explanation has some appeal, it is important to note that none of the evidence firmly establishes a role for Bru in translational repression of grk mRNA, and it remains possible that Bru could, for example, influence the site of translation rather than its efficiency.
Although the combination of S1 and aret mutations does affect Grk expression or distribution, there are no precedents that clearly demonstrate how excess or ectopic Grk would enhance the oogenesis arrest phenotype of aret mutants. Thus the explanation for the enhancement remains unknown and could involve the effects on grk or on other genes that are subject to regulation by Bru.
Is there a link between Bru and initiation of translation?
The eIF4E protein binds to the cap at the 5′ end of mRNAs. It is a rate-limiting component of translational initiation, and its activity is under tight control (Gingras et al. 1999). One form of regulation is phosphorylation, which is thought to control the mRNA cap-binding activity of eIF4E (Marcotrigiano et al. 1997; Raught et al. 2000). Several lines of correlative evidence suggest that this phosphorylation is important for cell proliferation (Bonneau and Sonenberg 1987; Raught et al. 2000), and mutation of the Drosophila eIF4E to prevent phosphorylation results in reduced viability and poor growth (Lachance et al. 2002).
A transgene expressing a mutant and constitutively activated version of eIF4E, in which the regulatory phosphorylation is mimicked by an amino acid change, can suppress the aret phenotype. This result raises the possibility that Bru has a positive role in initiation of translation. Specifically, in the aret mutant one or more target mRNAs that require Bru for activation of translation may be underexpressed, and increasing translation suppresses this defect.
However, the aret mutant phenotype is also suppressed by a mutation of Lk6 and enhanced by overexpression of Lk6. Lk6 is the Drosophila protein most closely related to mammalian mitogen-activated protein kinase-interacting protein kinase 1 (MNK1), which phosphorylates translation initiation factor eIF4E after activation by either the p44/42 or p38 MAPKs (Fukunaga and Hunter 1997; Waskiewicz et al. 1997; Lachance et al. 2002). Thus mutation of Lk6 might be expected to reduce eIF4E phosphorylation and thereby decrease translational capacity. By this view the suppression of the aret phenotype would be consistent with an interaction between eIF4E and Bru that involves the known function of Bru in translational repression. In favor of this notion Nakamura et al. (2004) have recently shown that Bru physically interacts with Cup, an eIF4E-binding protein that is required for repression of osk mRNA translation. To explore this possibility further we asked if suppression of the aret phenotype by EP(3)0886 was accompanied by a change in the levels of Osk or Grk proteins, or if homozygous EP(3)0886 females have abnormal amounts of either protein. No change was seen in either case (data not shown). Thus we do not know if the Lk6 mutation impacts the function of aret in repression of osk or grk mRNAs.
Given the similar consequences on the aret phenotype of the constitutively active eIF4E and the mutant predicted to reduce eIF4E activity, the simplest explanation is that Lk6 may affect aret function by a means other than phosphorylation of eIF4E. Suppression of the aret phenotype by the mutant eIF4E clearly suggests a link between Bru and the initiation of translation, although this need not be direct.
Interaction of aret and Dl:
The combination of aretPD/aretQB with Dl9P/+ produces a variety of ovarian defects, complicating interpretation of the phenotype. Nevertheless, one striking feature is the similarity of many of the defects to those seen when Dl activity is largely or completely eliminated, suggesting that the aret mutations are enhancing the Dl phenotype. Dl is a component of the Notch/Dl signaling pathway, which acts in many signaling events in a wide range of cell types (Artavanis-Tsakonas et al. 1999). In the ovary Dl is required in the germline cells for control of differentiation and proliferation of the somatic follicle cells and for setting up anteroposterior polarity (Lopez-Schier and St. Johnston 2001; Torres et al. 2003). The earliest and, at least initially, most dramatic consequence of loss of Dl activity is the fusion of cysts—the phenotype most apparent in the aretPD/aretQB; Dl9P/+ ovaries.
Large germline clones of strong Dl mutant alleles cause a complete fusion of egg chambers into a single egg chamber with multiple cysts, reminiscent of the complete fusions described here. Smaller clones retain a more regular ovariole organization. Individual egg chambers with Dl germline clones often fuse with the adjacent anterior wild-type egg chamber. Fusion can be incomplete, resulting in a double layer of follicle cells that separate the egg chambers, much as observed for the A/P partial fusions we report. However, the similarities are not perfect. For example, Dl mutant clones upregulate FasIII in the follicular epithelium (Lopez-Schier and St. Johnston 2001), but aretPD/aretQB; Dl9P/+ egg chambers do not (data not shown). Other features of the Dl mutant phenotype, such as the defects in anteroposterior polarity (Torres et al. 2003), are difficult to detect in the aretPD/aretQB; Dl9P/+ ovaries, because of their arrest of oogenesis. The lack of perfect correspondence between the Dl germline clones and the aretPD/aretQB; Dl9P/+ ovaries is not surprising for several reasons. First, there is substantial phenotypic variation even among the Dl germline clones, if both large and small clones are considered. Second, the clones are homozygous for Dl−, while in the aret mutant background one wild-type copy of Dl remains. Third, the Dl-like defects in aretPD/aretQB; Dl9P/+ ovaries are superimposed on the aret mutant phenotype.
The simplest interpretation of our results is that the aret mutations are reducing the activity of the N/Dl signaling pathway, which in combination with mutation of one copy of Dl leads to phenotypes similar to those resulting from loss of Dl. This model is fully supported by the finding that in aret mutants the amount of Dl protein concentrated at the border between germline cells and follicle cells is reduced. What remains unclear is how this reduction occurs. Assuming that Bru is acting as a translational repressor, in the aret mutant the target protein should be present at elevated levels. By this model the target should be a gene that normally has a negative effect on Dl expression or delivery to the membrane. Alternatively, Bru could also have a role in translational activation, in which case Dl could be a direct target. This seems quite unlikely, as the Dl 3′-UTR lacks any recognizable BREs, the sequences to which Bru is known to bind. Nevertheless, a role for Bru in translational activation is possible, and the target could normally have a positive effect on provision of Dl activity.
Acknowledgments
We thank Paul Lasko, Jordan Raff, Uptal Banerjee, Erin Overstreet, and the Developmental Studies Hybridoma Bank (developed under the auspices of the National Institute for Child Health and Human Development and maintained by The University of Iowa, Department of Biological Sciences) for antibodies; Trudi Schüpbach and the Bloomington Stock Center for fly stocks; members of the Macdonald lab for discussions; and Courtney Cater and Mark Snee for comments on the manuscript. This work was supported by National Institutes of Health grant GM54409.
References
- Artavanis-Tsakonas, S., M. D. Rand and R. J. Lake, 1999. Notch signaling: cell fate control and signal integration in development. Science 284: 770–776. [DOI] [PubMed] [Google Scholar]
- Bonneau, A. M., and N. Sonenberg, 1987. Involvement of the 24-kDa cap-binding protein in regulation of protein synthesis in mitosis. J. Biol. Chem. 262: 11134–11139. [PubMed] [Google Scholar]
- Davidson, E. H., 1986 Gene Activity in Early Development. Academic Press, Orlando, FL.
- Filardo, P., and A. Ephrussi, 2003. Bruno regulates gurken during Drosophila oogenesis. Mech. Dev. 120: 289–297. [DOI] [PubMed] [Google Scholar]
- Fukunaga, R., and T. Hunter, 1997. MNK1, a new MAP kinase-activated protein kinase, isolated by a novel expression screening method for identifying protein kinase substrates. EMBO J. 16: 1921–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghiglione, C., E. A. Bach, Y. Paraiso, K. L. Carraway, III, S. Noselli et al., 2002. Mechanism of activation of the Drosophila EGF Receptor by the TGFalpha ligand Gurken during oogenesis. Development 129: 175–186. [DOI] [PubMed] [Google Scholar]
- Gingras, A. C., B. Raught and N. Sonenberg, 1999. eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation. Annu. Rev. Biochem. 68: 913–963. [DOI] [PubMed] [Google Scholar]
- Huang, A. M., and G. M. Rubin, 2000. A misexpression screen identifies genes that can modulate RAS1 pathway signaling in Drosophila melanogaster. Genetics 156: 1219–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd, D., and J. W. Raff, 1997. LK6, a short lived protein kinase in Drosophila that can associate with microtubules and centrosomes. J. Cell Sci. 110(2): 209–219. [DOI] [PubMed] [Google Scholar]
- Kim-Ha, J., K. Kerr and P. M. Macdonald, 1995. Translational regulation of oskar mRNA by bruno, an ovarian RNA-binding protein, is essential. Cell 81: 403–412. [DOI] [PubMed] [Google Scholar]
- Lachance, P. E., M. Miron, B. Raught, N. Sonenberg and P. Lasko, 2002. Phosphorylation of eukaryotic translation initiation factor 4E is critical for growth. Mol. Cell. Biol. 22: 1656–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipshitz, H. D., and C. A. Smibert, 2000. Mechanisms of RNA localization and translational regulation. Curr. Opin. Genet. Dev. 10: 476–488. [DOI] [PubMed] [Google Scholar]
- Lopez-Schier, H., and D. St. Johnston, 2001. Delta signaling from the germ line controls the proliferation and differentiation of the somatic follicle cells during Drosophila oogenesis. Genes Dev. 15: 1393–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotrigiano, J., A. C. Gingras, N. Sonenberg and S. K. Burley, 1997. Cocrystal structure of the messenger RNA 5′ cap-binding protein (eIF4E) bound to 7-methyl-GDP. Cell 89: 951–961. [DOI] [PubMed] [Google Scholar]
- Markussen, F.-H., A.-M. Michon, W. Breitwieser and A. Ephrussi, 1995. Translational control of oskar generates Short OSK, the isoform that induces pole plasm assembly. Development 121: 3723–3732. [DOI] [PubMed] [Google Scholar]
- Martin, S. G., and D. St. Johnston, 2003. A role for Drosophila LKB1 in anterior-posterior axis formation and epithelial polarity. Nature 421: 379–384. [DOI] [PubMed] [Google Scholar]
- Mendez, R., and J. D. Richter, 2001. Translational control by CPEB: a means to the end. Nat. Rev. Mol. Cell. Biol. 2: 521–529. [DOI] [PubMed] [Google Scholar]
- Nakamura, A., K. Sato and K. Hanyu-Nakamura, 2004. Drosophila Cup is an eIF4E binding protein that associates with Bruno and regulates oskar mRNA translation in oogenesis. Dev. Cell 6: 69–78. [DOI] [PubMed] [Google Scholar]
- Neuman-Silberberg, F. S., and T. Schüpbach, 1993. The Drosophila dorsoventral patterning gene gurken produces a dorsally localized RNA and encodes a TGFa-like protein. Cell 75: 165–174. [PubMed] [Google Scholar]
- Neuman-Silberberg, F. S., and T. Schüpbach, 1994. Dorsoventral axis formation in Drosophila depends on the correct dosage of the gene gurken. Development 120: 2457–2463. [DOI] [PubMed] [Google Scholar]
- Neuman-Silberberg, F. S., and T. Schüpbach, 1996. The Drosophila TGF-a-like protein Gurken: expression and cellular localization during Drosophila oogenesis. Mech. Dev. 59: 105–113. [DOI] [PubMed] [Google Scholar]
- Norvell, A., R. L. Kelley, K. Wehr and T. Schüpbach, 1999. Specific isoforms of squid, a Drosophila hnRNP, perform distinct roles in Gurken localization during oogenesis. Genes Dev. 13: 864–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisi, M. J., W. Deng, Z. Wang and H. Lin, 2001. The arrest gene is required for germline cyst formation during Drosophila oogenesis. Genesis 29: 196–209. [DOI] [PubMed] [Google Scholar]
- Pickup, A. T., and U. Banerjee, 1999. The role of star in the production of an activated ligand for the EGF receptor signaling pathway. Dev. Biol. 205: 254–259. [DOI] [PubMed] [Google Scholar]
- Raught, B., A. C. Gingras, S. P. Gygi, H. Imataka, S. Morino et al., 2000. Serum-stimulated, rapamycin-sensitive phosphorylation sites in the eukaryotic translation initiation factor 4GI. EMBO J. 19: 434–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray, R. P., and T. Schüpbach, 1996. Intercellular signaling and the polarization of body axes during Drosophila oogenesis. Genes Dev. 10: 1711–1723. [DOI] [PubMed] [Google Scholar]
- Rongo, C., E. R. Gavis and R. Lehmann, 1995. Localization of oskar RNA regulates oskar translation and requires Oskar protein. Development 121: 2737–2746. [DOI] [PubMed] [Google Scholar]
- Rorth, P., K. Szabo, A. Bailey, T. Laverty, J. Rehm et al., 1998. Systematic gain-of-function genetics in Drosophila. Development 125: 1049–1057. [DOI] [PubMed] [Google Scholar]
- Sallés, F. J., M. E. Lieberfarb, C. Wreden, J. P. Gergen and S. Strickland, 1994. Coordinate initiation of Drosophila development by regulated polyadenylation of maternal messenger RNAs. Science 266: 1996–1999. [DOI] [PubMed] [Google Scholar]
- Schüpbach, T., and E. Wieschaus, 1991. Female sterile mutations on the second chromosome of Drosophila melanogaster. II. Mutations blocking oogenesis or altering egg morphology. Genetics 129: 1119–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serano, T. L., M. Karlin-McGinness and R. S. Cohen, 1995. The role of fs(1) K10 in the localization of the mRNA of the TGFa homolog gurken within the Drosophila oocyte. Mech. Dev. 51: 183–192. [DOI] [PubMed] [Google Scholar]
- Simon, M. A., D. D. Bowtell, G. S. Dodson, T. R. Laverty and G. M. Rubin, 1991. Ras1 and a putative guanine nucleotide exchange factor perform crucial steps in signaling by the sevenless protein tyrosine kinase. Cell 67: 701–716. [DOI] [PubMed] [Google Scholar]
- Smith, J. L., J. E. Wilson and P. M. Macdonald, 1992. Overexpression of oskar directs ectopic activaton of nanos and presumptive pole cell formation in Drosophila embryos. Cell 70: 849–859. [DOI] [PubMed] [Google Scholar]
- Spradling, A. C., 1993 Developmental genetics of oogenesis, pp. 1–70 in The Development of Drosophila melanogaster, edited by M. Bate and A. M. Arias. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Torres, I. L., H. Lopez-Schier and D. St. Johnston, 2003. A Notch/Delta-dependent relay mechanism establishes anterior-posterior polarity in Drosophila. Dev. Cell 5: 547–558. [DOI] [PubMed] [Google Scholar]
- Van Doren, M., A. L. Williamson and R. Lehmann, 1998. Regulation of zygotic gene expression in Drosophila primordial germ cells. Curr. Biol. 8: 243–246. [DOI] [PubMed] [Google Scholar]
- Waskiewicz, A. J., A. Flynn, C. G. Proud and J. A. Cooper, 1997. Mitogen-activated protein kinases activate the serine/threonine kinases Mnk1 and Mnk2. EMBO J. 16: 1909–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster, P. J., L. Liang, C. A. Berg, P. Lasko and P. M. Macdonald, 1997. Translational repressor bruno plays multiple roles in development and is widely conserved. Genes Dev. 11: 2510–2521. [DOI] [PMC free article] [PubMed] [Google Scholar]