

Abstract
Understanding how genetic variation is maintained begins with a comprehensive description of what types of genetic variation exist, the extent and magnitude of the variation, and patterns discernable in that variation. However, such studies have focused primarily on DNA sequence data and have ignored genetic variation at other hierarchical levels of genetic information. Microarray technology permits an examination of genetic variation at the level of mRNA abundance. Utilizing a round-robin design, we present a quantitative description of variation in mRNA abundance in terms of GCA (general combining ability or additive variance). We test whether genes significant for GCA are randomly distributed across chromosomes and use a nonparametric approach to demonstrate that the magnitude of the variation is not random for GCA. We find that there is a paucity of genes significant for GCA on the X relative to the autosomes. The overall magnitude of the effects for GCA on the X tends to be lower than that on the autosomes and is contributed by rare alleles of larger effect. Due to male hemizygosity, GCA for X-linked phenotypes must be due to trans-acting factors, while GCA for autosomal phenotypes may be due to cis- or trans-acting factors. The contrast in the amount of variation between the X and the autosomes suggests that both cis and trans factors contribute to variation for expression in D. simulans with the preponderance of effects being trans. This nonrandom patterning of genetic variation in gene expression data with respect to chromosomal context may be due to hemizygosity in the male.
STUDIES describing and quantifying genetic variation, and putting it in an evolutionary context, have a rich history, beginning with pioneering investigations of protein variation almost 40 years ago (Harris 1966; Hubby and Lewontin 1966; Lewontin and Hubby 1966). This work suggested that widespread overdominance was not the major force maintaining genetic variation and was criticized as incomplete, because only a fraction of the variation present in a DNA sequence can be detected on a protein gel. Subsequently, an intensive examination of genetic variation at the sequence level ensued, including development of statistical tests of neutrality (Hudson et al. 1987). While these studies have demonstrated large amounts of variation and the pervasiveness of selection, they have raised additional questions, namely how selective events shape DNA sequence variation (Andolfatto 2001). Moreover, a complete picture of genetic variation includes far more than sequence variation, but should span hierarchical levels of genetic information systems. Recently, descriptions of variation in abundance and distribution of mRNA have been pioneered (Jin et al. 2001; Arbeitman et al. 2002; Ranz et al. 2003; Rifkin et al. 2003), but due to the expense of such studies, they have necessarily been limited in scope. Here we present a systematic study of variation in levels of mRNA in males of Drosophila simulans using a round-robin breeding design, which allows us to make inferences about additivity and epistatic variation in mRNA abundance.
Gene expression studies have identified genes whose expression varies among individuals (Wolfinger et al. 2001; Oleksiak et al. 2002). However, the genetic architecture of the phenotype of gene expression remains to be described, particularly whether or not expression exhibits quantifiable additive genetic variation. To understand the genetic architecture of gene expression, a controlled crossing scheme is used, where the specific mating design allows for estimation and testing of inferences about the underlying genetic architecture of the phenotype of interest. With this in mind, we modified a classical quantitative genetic breeding design: the diallel (Griffing 1956a,b). Diallel designs can generate empirical estimates of general combining ability (GCA) and specific combining ability (SCA). SCA is not considered further here, as it cannot be independently estimated for our modified diallel crossing design. GCA is equivalent to breeding value. GCA is a description of a genotype's performance as a parent relative to the other genotypes within the set and is expressed as a deviation from the overall mean of parents in the set. In general GCA may be thought of as an approximation of additive genetic variance, provided that additive-by-additive epistasis is very small: 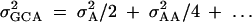 (Lynch and Walsh 1998). Accordingly, diallels may be used to obtain an upper estimate for heritability, by making the simplifying assumption that additive-by-additive epistasis is negligible relative to additive variation.
(Lynch and Walsh 1998). Accordingly, diallels may be used to obtain an upper estimate for heritability, by making the simplifying assumption that additive-by-additive epistasis is negligible relative to additive variation.
Full diallels have the disadvantage of being labor and resource intensive, because the number of crosses to be assayed equals the number of parents squared. This is why we chose a partial diallel design (Kempthorne and Curnow 1961; Lynch and Walsh 1998). In partial diallel designs, better estimates of GCA will result when the number of parental lines is maximized (Lynch and Walsh 1998). In this study, for instance, we had available resources for 30 microarray hybridizations. As three replicates per genotype is a minimum for array analysis (Black and Doerge 2002), this allows selection of 10 genotypes. We elected to perform a one-way round-robin design (10 unrelated lines with line 1 crossed to line 2, line 2 crossed line 3, etc., concluding with line 10 crossed to line 1) to maximize the number of unique parents (Figure 1).
Figure 1.—

Diagram of crossing design, showing hemizygosity and unique representation of each X in the sample, as compared to heterozygosity and multiple representation of autosomes. Different hatching patterns correspond to different parental stocks. The sex chromosomes are at the left side of the pedigree, with the X chromosome telocentric and the Y chromosome acrocentric. For example, the first cross represents the cross progeny of SIM6 (open chromosomes) and SIM11 (hatched chromosomes). SIM11 also contributes to the next cross, hence the sharing of the hatched pattern. Finally the last cross represents the completion of the round robin, with the reappearance of SIM6 (open chromosomes) in combination with the tenth line, SIM105.
When two genotypes share an allele affecting the quantitative trait, the trait will be more similar between these genotypes than in the rest of population. We consider gene transcript level to be a quantitative trait, which thus will be coexpressed between related genotypes. Two F1 genotypes from a round-robin crossing scheme among independently derived lines will share their autosomal alleles when, and only when, one of their parents is identical by descent. Because each parental line was used as a male in one mating and as a female in the second mating (see Figure 1), F1 male genotypes will never share an X chromosome. Note that the term allele sharing is used here to indicate identity by descent in relation to the reference population (Lynch and Walsh 1998). Therefore, if we see coexpression of an X-linked gene between genotypes sharing autosomes, it must be due to trans effects of autosomal genes on the transcript level of the focal X chromosome gene. Similarly, the coexpression of an autosomal gene might be due to trans effects of other autosomal genes. However, the focal gene transcript level might also be affected by its own structure, i.e., by alterations in gene promoter, introns, or variations on RNA stability (cis effects). As those are shared between genotypes sharing autosomes, such cis variation will also contribute to coexpression of an autosomal focal gene.
Given the different biological interpretations of GCA between the X chromosomes and autosomes, we separated genes depending on their chromosomal context. Also, we compared the proportions of significant genes found on the X relative to the autosomes for GCAs; and we compared the relative magnitude of the GCA effects detected on the X relative to the autosomes. We find that there is a significant reduction in GCA on X, as predicted by the biological interpretations of GCA. We also find that the magnitude of GCA is different depending on chromosomal context, by using a nonparametric approach that is analogous to the tests utilizing frequency distributions of sequence polymorphisms (Fu and Li 1993; Fu 1996; Tajima et al. 1998). We consider this a first step in trying to develop hypotheses and tests around inheritance of expression variation.
MATERIALS AND METHODS
Drosophila lines:
The parental stocks of D. simulans were obtained from flies caught in Wolfskill orchard (Winters, CA) and were subjected to full-sib mating for 25–29 generations to create nearly homozygous stocks. The 10 parental stocks (randomly sampled and independently derived from a large natural reference population) were crossed together in 10 combinations to create heterozygous lines such that each parent was present twice, once as a dam and once as a sire. Crosses included (dam × sire): SIM6 × SIM11, SIM11 × SIM36, SIM36 × SIM 37, SIM37 × SIM43, SIM43 × SIM61, SIM61 × SIM77, SIM77 × SIM85, SIM85 × SIM99, SIM99 × SIM105, and SIM105 × SIM6. Crossing design is illustrated in Figure 1. Flies were kept in vials of standard cornmeal medium with yeast, five pairs per vial, at 25° on a continuous light cycle for 1 generation. Parents were cleared after 5 days. Five 1- to 2-day-old males of the first parental stock were mated with five 1- to 2-day-old females of the second parental stock, etc., as described above. Virgin females and males of the resulting heterozygous line were collected and then frozen after 3 days. Lines and their rearing are described in more detail in Nuzhdin et al. (2004).
Two complete blocks of flies were reared (i.e., two vials per F1 genotype). Flies grown in one of these vials were split into two groups, with all subsequent steps performed independently (RNA extraction, labeling, and hybridization), to maximize replication of the steps where most of the error is contributed (Long et al. 2001). Thus a total of three independently extracted, labeled, and hybridized samples were analyzed. Correlation among replicates from the same vial was not different from correlation among replicates from different vials, confirming that differences among individual flies and technical error of the array experiment, rather than differences among vials, accounted for most of the residual variation.
Microarrays and image analysis:
Total RNA was extracted from whole carcasses of adult males, aged 4–7 days posteclosion, and labeled according to Affymetrix protocols. Affymetrix Drosophila Genechips were used for the hybridizations. Data were quantified using Affymetrix MAS5 software. Hybridizations and data quantification were conducted by the University of California, Davis, Genomics Core Facility. Average difference values for each chip were normalized to the chip median and then log transformed. There were 6080 genes considered “absent” for all hybridizations; these were excluded from all analyses. Chip primers were designed on the basis of the genome sequence of D. melanogaster, while hybridizations were performed with RNA extracted from the sibling species, D. simulans. This introduces a potential problem in that poor hybridization could be confused with low expression values. However, the sequence divergence should not bias the estimates of variance in transcript level for the genes found expressed (n = 7886). It has previously been shown for Affymetrix arrays that the sequence divergence between D. melanogaster and D. simulans has not resulted in consistent reduction in expression level (Nuzhdin et al. 2004). We cannot absolutely argue that sequence variation contributes to estimated variation in expression level. However, it is unlikely that sequence divergence between D. melanogaster and D. simulans is contributing to genetic variation in D. simulans. Furthermore, cross-species hybridizations have been shown to be reproducible in more distantly related species (Moody et al. 2002).
Statistical analysis:
The mRNA expression of each gene was analyzed using an ANOVA approach tailored to the round-robin partial diallel design. Griffing (1956a)(b) proposed the following analysis of variance (ANOVA) model to partition the total mean square from a diallel design into GCA mean squares (gi, gj), SCA mean squares (sij), and reciprocal mean squares (rij): yijk = μ + gi + gj + sij + rij + εijk. Without reciprocal crosses, or specific combining ability, this model reduces to yijk = μ + gi + gj + εijk (Griffing 1956a,b; Cockerham and Weir 1977).
Accordingly we used the ANOVA model yijk = μ + GCAij + εijk, where yijk is the estimated gene expression for the kth replicate for the cross between parents i and j, μ is the overall mean, GCAij is a matrix of indicator variables for the parents, and εijk is the error term. We tested for significant GCA effects with the F ratio MSGCA/MSε. For each test we used a nominal significance level of 0.01. Variance components were estimated using a restricted maximum-likelihood approach, for models where the null hypothesis of GCA equals zero was rejected.
We tested whether the number of rejections we observed was due to chance alone using a permutation test (Edgington 1995; Good 2000). We permuted the cross designation (line numbers) relative to the arrays 1000 times to generate a distribution of values for expression that were random with respect to the line. For each permuted data set, we reanalyzed the data using the ANOVA methods described above and then counted the number of tests for GCA that exceeded the nominal 0.01 type I error rate. We then estimated the probability of having significant tests that were equal to or more than what we observed by dividing the number of permuted data sets obtained with at least that number of significant outcomes by 1000. We also computed the false discovery rate for this nominal P-value (Benjamini and Hochberg 1995). We compared the proportion of genes significant for GCA between the X chromosome and the autosomes, using a null hypothesis of equal proportions, and tested this hypothesis using a χ2 test with 1 d.f.
Assuming no additive-by-additive interactions and other higher terms, GCA may be used to estimate an upper bound on heritability for gene expression. Total phenotypic variance is 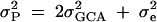 (Lynch and Walsh 1998). SCA is included in the error in our model as it is not independently estimable. For genes with significant GCA effects, heritability was calculated by the formula
(Lynch and Walsh 1998). SCA is included in the error in our model as it is not independently estimable. For genes with significant GCA effects, heritability was calculated by the formula 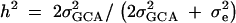 . Heritability estimates were compared between the X and the autosomes using a Wilcoxon rank sums test.
. Heritability estimates were compared between the X and the autosomes using a Wilcoxon rank sums test.
For genes found to have significant GCA overall, parental stock-specific estimates of GCA were calculated. The parental stock-specific estimates were calculated as n − 1/n − 2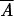 −
− 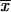 , where n is the number of parents,
, where n is the number of parents, 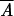 is the mean of the crosses for that parent divided by n − 1, and
is the mean of the crosses for that parent divided by n − 1, and 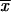 is the mean of all crosses (Falconer and Mackay 1996). To test whether the size of the parental stock-specific effects was the same across the X and the autosome, we used a nonparametric approach. We put the effect estimates in rank order, without regard to the chromosomal context. We then divided the ordered estimates into three equal groups, or tertiles. We looked at association between chromosomal context and effect size by examining the distribution of the effects in each tertile across the two classes of chromosomes, X and autosome. The null hypothesis that effect size is independent from the chromosomal context would result in an even distribution across the tertiles for both the X and the autosomes. A chi-square test was used to evaluate this null hypothesis.
is the mean of all crosses (Falconer and Mackay 1996). To test whether the size of the parental stock-specific effects was the same across the X and the autosome, we used a nonparametric approach. We put the effect estimates in rank order, without regard to the chromosomal context. We then divided the ordered estimates into three equal groups, or tertiles. We looked at association between chromosomal context and effect size by examining the distribution of the effects in each tertile across the two classes of chromosomes, X and autosome. The null hypothesis that effect size is independent from the chromosomal context would result in an even distribution across the tertiles for both the X and the autosomes. A chi-square test was used to evaluate this null hypothesis.
To examine whether the size of the effect (parental stock-specific estimate) was associated with the parental lines, we used a nonparametric approach for the X and the autosomes separately. We took all the estimates for the X chromosome, and ranked them from smallest to largest. We then divided the estimates into three equal groups (tertiles). We then examined whether the distribution of estimates across the tertiles was associated with the parental line using a chi-square test. We repeated this procedure for the autosomes. All analyses were conducted in SAS v 8.2; SAS code for these analyses is available upon request to L.M.M.
RESULTS
We describe whole-genome genetic architecture of expression variation in D. simulans by considering gene expression levels as phenotypes. Transcript levels were evaluated in RNA samples extracted from F1 adult males of 10 heterozygous lines from a round-robin mating design (see Figure 1). Three replicate hybridizations of each genotype were performed (see materials and methods), enabling estimation of genetic component of expression variation. We detected expression for 7886 genes out of 13,609 represented on Affymetrix GeneChips.
Genes for which expression was detected were analyzed as a partial diallel. We estimated GCA 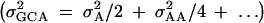 and heritability
and heritability 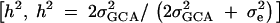 for expression of each gene. GCA is the average effect of a genotype when crossed at random to members of a population, i.e., the deviation of the set of all F1 progeny from the population mean due to their sharing one parent of a certain genotype, while their other parents are chosen from the population at random. Thus, GCA is equivalent to the breeding value and may be thought of as an approximation of additive genetic variation. If parental alleles are purely additive, then an F1 genotype will deviate from the population mean by the sum of the GCAs of its parents and due to environmental or error effects. Any additional deviation from the population mean is attributable either to dominance, i.e., intralocus interactions, of alleles or to epistasis, i.e., interlocus interactions. These deviations cannot be independently estimated with a round-robin design. Of the 7886 informative features, 663 genes had significant GCA (P < 0.01; significance testing by ANOVA is described in materials and methods). All genes with significant GCA are presented in the Supplementary Table at http://www.genetics.org/supplemental/.
for expression of each gene. GCA is the average effect of a genotype when crossed at random to members of a population, i.e., the deviation of the set of all F1 progeny from the population mean due to their sharing one parent of a certain genotype, while their other parents are chosen from the population at random. Thus, GCA is equivalent to the breeding value and may be thought of as an approximation of additive genetic variation. If parental alleles are purely additive, then an F1 genotype will deviate from the population mean by the sum of the GCAs of its parents and due to environmental or error effects. Any additional deviation from the population mean is attributable either to dominance, i.e., intralocus interactions, of alleles or to epistasis, i.e., interlocus interactions. These deviations cannot be independently estimated with a round-robin design. Of the 7886 informative features, 663 genes had significant GCA (P < 0.01; significance testing by ANOVA is described in materials and methods). All genes with significant GCA are presented in the Supplementary Table at http://www.genetics.org/supplemental/.
The false discovery rate (FDR), or percentage of genes with significant results that are expected to be false positives, was found to be 0.11 for the test of GCA with nominal significance level of 0.01. Consistent with this finding, the number of genes found with significant GCA was greater than that expected by chance alone, as determined by permutation (see materials and methods; probability of observing 660 or more rejections for GCA, P < 0.001).
There is ample evidence that the fourth chromosome has markedly reduced genetic variation relative to the autosomes due to its lack of recombination (Berry et al. 1991), and thus less expression variation is expected to be caused by cis-acting mutations on the fourth chromosome. This prediction could not be tested effectively because of the small number of genes from the fourth chromosome on the chip (62 genes, 3 of which were significant for GCA; the expected number of significant genes, given no difference from the other autosomes, was zero). Thus, these genes were excluded from further analyses. Hereafter, autosomal refers to genes from chromosomes 2 and 3 only.
We expect different numbers of genes to be significant for GCA, depending on the genes' chromosomal context. As explained in the Introduction, GCA for the X-linked genes is expected to be lower than that for the autosomes, because GCA for the autosomes includes cis and trans effects, while GCA for the X-linked genes is expected to include only trans effects (Figure 2). We compared the proportion of genes significant for GCA between the X chromosome and the autosomes (combined 2 and 3), using a null hypothesis of equal proportions, and tested this hypothesis using a χ2 test with 1 d.f. (see Table 1). The proportion of genes significant for GCA on the X is underrepresented relative to the autosomes (6.8 vs. 8.7%, respectively; P < 0.03973).
Figure 2.—

Possible sources of variation for expression for X-linked (top) and autosomal (bottom) loci. A simplified male fly with hemizygous X chromosome and a heterozygous autosome (representing 2, 3, or both) is diagrammed. The solid box indicates the gene whose expression is being measured (“target gene”). Cis-acting regulatory elements are indicated by open or hatched circles and arcs indicating the target gene; trans-acting regulatory sequences are represented by shaded or shaded-hatched boxes with dotted lines indicating the target gene.
TABLE 1.
The proportion of genes with significant GCA is underrepresented on theX relative to the autosomes (GCA: χ2 = 4.2295,P = 0.03973)
| Chromosome | Genes with significant GCA |
Total genes |
|---|---|---|
| X | 79 | 1159 |
| Autosomes | 581 | 6665 |
We estimated heritability for expression for all autosomal and X-linked genes with significant GCA (660 out of 7886; see materials and methods). Note that GCA is composed of not only additive variation, but also additive-by-additive interactions. For genes that do have significant variation for expression, the estimates for heritability are high, with a median of 0.47 and an interquartile range of 0.39–0.60. This is on the order of heritabilities for morphological traits (Houle et al. 1996). Heritability estimates are not different between genes on the X and genes on the autosome (P > 0.5), indicating that while it is less likely for genes to have significant GCA on the X, estimates of heritability are comparable between the X and the autosomes given the presence of GCA.
We used a nonparametric rank order comparison to test whether the parental stock estimates for GCA differed in the distribution between the X and the autosomes, using a nonparametric test of trend. The estimates for GCA were rank ordered, irrespective of chromosomal context, and then divided into three equal groups (tertiles): low, containing 0–33% of the values; medium, containing 33–67% of the values, including the median value; and high, containing the top 67–100% of the values. We found that the values of GCA were distributed differently across chromosomes, such that individual alleles on the X tended to have less extreme values for GCA than did the autosomes (Table 2, P < 0.0021).
TABLE 2.
The overall distribution of the size of the parental stock-specific estimates (GCA) differs between theXand the autosomes (GCA: χ2 = 12.3404,P = 0.0021)
| GCA
|
||||
|---|---|---|---|---|
| Chromosome | First | Second | Third | Total |
| X | 241 | 307 | 242 | 790 |
| Autosomes | 1959 | 1893 | 1958 | 5810 |
| Total | 2200 | 2200 | 2200 | 6600 |
The columns under GCA indicate the tertile (first, second, or third).
Difference in the magnitude of estimates of GCA could be due to either the number of lines contributing to the estimate or the effect size of those lines. To examine whether all parental stocks had the same distribution of effect sizes with respect to chromosomal context, we calculated the GCA effect for each parental stock separately for X and autosomal loci. We then used the nonparametric trend test as described in materials and methods to test whether these effects were evenly distributed across the chromosomes. We found that more lines contribute to the estimates of GCA for autosomal than for X-linked loci, while a few lines with large effects tended to contribute to estimates of GCA for X-linked genes, suggesting that alleles were generally rare for expression variation on the X (see Table 3).
TABLE 3.
Parental stocks are different in the distribution of GCA effects found on theX(χ2 = 52.07,P < 0.0001) and the autosomes (χ2 = 230.37,P < 0.0001)
|
X chromosome
|
Autosomes
|
|||||||
|---|---|---|---|---|---|---|---|---|
| Tertiles
|
Tertiles
|
|||||||
| Parental genotypes | First | Second | Third | P-value | First | Second | Third | P-value |
| SIM6 | 22 | 24 | 33 | 0.1050 | 210 | 183 | 188 | 0.1423 |
| SIM11 | 40 | 19 | 20 | 0.0011 | 226 | 186 | 169 | 0.0029 |
| SIM36 | 34 | 27 | 18 | 0.0271 | 175 | 233 | 173 | 0.0006 |
| SIM37 | 38 | 17 | 24 | 0.0031 | 281 | 150 | 150 | <0.0001 |
| SIM43 | 30 | 29 | 20 | 0.1306 | 248 | 184 | 149 | <0.0001 |
| SIM61 | 18 | 25 | 36 | 0.0123 | 136 | 231 | 214 | <0.0001 |
| SIM77 | 17 | 33 | 29 | 0.0224 | 121 | 223 | 237 | <0.0001 |
| SIM85 | 13 | 35 | 31 | 0.0013 | 122 | 207 | 252 | <0.0001 |
| SIM99 | 25 | 30 | 24 | 0.3845 | 202 | 171 | 208 | 0.0424 |
| SIM105 | 26 | 25 | 28 | 0.6650 | 215 | 170 | 196 | 0.0211 |
The within-lines distribution of the GCA effects varies, with some lines exhibiting overall low values for GCA (i.e., Sim 11, 37, and 43) and others overall high values for GCA (i.e., Sim 61, 77, and 85).
DISCUSSION
We assayed whole-body transcript level in 10 heterozygous male cross progeny in D. simulans. We detected expression in 7886 genes out of 13,609; of the expressed genes, 663 had significant additive genetic variation for expression, while we expected 5% to be significant due to chance alone (394). Likewise, the probability of observing >660 significant tests by chance alone was determined by permutation to be P < 0.001. These results are typical for experiments in Drosophila, where 10% of genes show significant variation for expression among lines (Jin et al. 2001; Nuzhdin et al. 2004). We estimated genetic variation as GCA.
What are the sources of genetic variation for expression? Genetic variation for transcript level could be due to either cis- or trans-acting factors. A gene might contain a mutation in its cis-regulatory region, and/or the amount or activity of trans-regulating factors controlling its expression might vary. Additionally, if expression level is tissue specific, genetic variation in organ size would be detected in this experiment as genetic variation in the transcript amount. Finally, due to heterochrony between genotypes, organisms of the same age might be at different developmental stages. Thus, genetic variation for expression may have multiple underlying mechanisms. Genes should on average be affected by the organ size or heterochrony to the same degree whether they are autosomal or sex linked. However, we expect chromosomal context to be relevant to the amount of genetic variation, whether due to variation in cis- or trans-acting factors.
Given the different interpretations of GCA for the X and the autosomes, how do cis and trans effects contribute to these quantities? Cis modifiers of expression should contribute to GCA for the genes on the autosomes, but generally will not contribute to variation for the genes situated on the X, because each X chromosome is present only once in the experiment. Contribution of trans modifiers of expression to GCA depends on their chromosomal positions (X chromosome vs. autosomes). The positions of modifiers are unknown. However, in Saccharomyces cerevisiae the positions of modifiers and modified genes are not correlated (Brem et al. 2002; Yvert et al. 2003). If contributions of trans modifiers are similar on average for genes situated on the X chromosome and autosomes, then if genes on the X chromosome showed no GCA for expression variation, we would conclude that most, if not all, expression variation is accounted for by cis effects. If GCAs were equally frequent between chromosomes, we would conclude that most, if not all, expression variation is accounted for by trans effects.
Our data show that the proportion of genes significant for GCA on the X is 6.8%, while on the autosomes it is 8.7% (Table 1). This is consistent with cis effects having minor but significant contribution to variation in expression level in Drosophila and with trans effects being more important. A preponderance of trans effects with secondary effects of cis contributions is consistent with the findings in S. cerevisiae, where at most 30% of variation in expression level was accounted for by cis modifiers (Yvert et al. 2003). Interestingly, when we look at the effects by line and chromosome it is apparent that some lines exhibit mostly large effects (Sim 61, 77, 85) on expression, while other lines (Sim 11, 37, 43) show correspondingly mostly low effects for both X and autosomal effects. Whether or not patterns between lines are explained by common transcriptional control (trans effects) remains to be tested. Additionally, there are fewer genes with large (third tertile) parental stock-specific effects on the X compared to the autosomes (Table 2). Since cis effects are not detectable on the X, and we find that effects on the autosomes are overall larger than effects on the X, one possible interpretation is that cis effects may be larger than trans effects (Meiklejohn et al. 2003).
Before proceeding further, we must make a clear distinction between interactions among genes in the quantitative genetic sense (epistasis, Mackay 2001) and interactions among gene regulatory regions and transcription factors in molecular genetic networks (molecular interlocus interaction effects). In the quantitative genetic sense, epistasis is an interaction between the effects of alleles at two or more distinct loci, as exhibited by a different phenotype from that expected from the individual effects of these alleles. Expression variation for the genes on the X chromosome may be, but need not be, due to such interactions. Consider the following situation in which molecular interlocus interactions occur in the absence of quantitative genetic epistasis: assume that all the X chromosomes in the experiment are identical, but there is variation among the genes at the autosomal loci. One may still observe variation in X-linked gene expression levels because of the trans effects. This is due to the additive effects of the transcription factors on the autosomes, as measured by the expression phenotypes of X chromosome genes. The X chromosome genes themselves are not the genetic basis of the phenomenon in this example because there is no genetic variation at these loci and thus no epistasis in the quantitative genetic sense. In the experiment conducted here, as we cannot explicitly demonstrate differences between the X chromosomes, it is molecular interlocus interaction effects that are being observed. Hereafter, we refer to these phenomena as autosomal trans effects on the X chromosome.
Genes with a pattern of autosomal trans effects on X chromosome expression are particularly interesting to evolutionary biologists, as these are loci that would be consistent with the Dobzhansky-Muller models of speciation and would also be loci that would behave in a manner consistent with Haldane's rule. Thus, such loci are likely to be involved in speciation. Of the 79 genes with significant GCA on the X, 28 have been identified by other investigators as having anomalously fast interspecific evolution for expression (Ranz et al. 2003; Rifkin et al. 2003). Of the remaining 51 genes, 9 genes are logical candidates for speciation because of their function: three genes related to behavior [bss (Kulkarni et al. 1988), Ork1 (Nitabach et al. 2002), and CG15447 (Claridge-Chang et al. 2001)]; three genes related to immune function (CG6067, Ag5r2, and EG:BAC25B3); one gene from the fast-evolving cytochrome p450 family, cyp4d1; one gene with a paralog recently retrotransposed from the X, CanB (Betran et al. 2002); and one gene identified as a candidate for genital morphology differences between D. mauritiana and D. sechellia, scully (Macdonald and Goldstein 1999). Thus of the 79 genes with potential autosomal trans effects on X chromosome expression, 37 have independent evidence of being fast evolving and therefore are likely to be involved in speciation. Of the remaining 42 genes, we identified 11 additional genes as having exceptional evolution for expression between species in a companion article (Nuzhdin et al. 2004). All 79 of these genes, including the 48 with evidence for speciation, are presented in the Supplementary Table (column O).
There is ample theory predicting faster fixation of advantageous alleles for the X. In addition, there are now multiple lines of empirical evidence that the X has experienced multiple selective sweeps caused by fixation of beneficial, recessive alleles. For example, though overall differences between ka and ks (the amino acid and silent substitution rates, respectively) were not observed between the X and the autosomes for the sibling species D. melanogaster and D. simulans (Bauer and Aquadro 1997), newly duplicated genes on the X have greater ka:ks ratios than recently duplicated genes on the autosomes, suggesting the fixation of beneficial recessive genes in these new duplicates (Thornton and Long 2002). Likewise, a higher ka:ks ratio has been observed on the neo-X chromosome of D. miranda than on its newly nonrecombinant homolog, the neo-Y. Rice has argued that an excess of expression variation per se is expected on the X chromosome, due to the evolution of dosage compensation (Rice 1987b,c) and/or to antagonistic fitness effects between the sexes (Rice 1987a). However, as the biological predictions underlying GCA differ for the X and the autosomes due to male hemizygosity, compelling evidence for selection on expression variation will have to wait for expression data from females.
Acknowledgments
We thank the University of California, Davis, Genomics Core Facility for their help in hybridizing the chips and Kristy Harmon for excellent technical assistance. We also thank two anonymous reviewers, Klaus Hinkelmann, and Trudy F. C. Mackay for help in clarifying analyses. This work was supported by the following: National Institutes of Health (NIH) RO1 GM59884-02 (M.L.W.), U.S. Department of Agriculture International Food and Agricultural N00149419318 (L.M.M.), NIH R01 GM61773 (S.V.N.), NIH R24 GM65513-01 (S.V.N.), Purdue University Agricultural Experiment Station (L.M.M.), and the Purdue University Academic Reinvestment Program (L.M.M.).
References
- Andolfatto, P., 2001. Adaptive hitchhiking effects on genome variability. Curr. Opin. Genet. Dev. 11: 635–641. [DOI] [PubMed] [Google Scholar]
- Arbeitman, M. N., E. E. Furlon, F. Imam, E. Johnson, B. H. Null et al., 2002. Gene expression during the life cycle of Drosophila melanogaster. Science 297: 2270–2275. [DOI] [PubMed] [Google Scholar]
- Bauer, V. L., and C. F. Aquadro, 1997. Rates of DNA sequence evolution are not sex-biased in Drosophila melanogaster and D-simulans. Mol. Biol. Evol. 14: 1252–1257. [DOI] [PubMed] [Google Scholar]
- Benjamini, Y., and Y. Hochberg, 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 57: 289–300. [Google Scholar]
- Berry, A. J., J. W. Ajioka and M. Kreitman, 1991. Lack of polymorphism on the Drosophila fourth chromosome resulting from selection. Genetics 129: 1111–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betran, E., K. Thornton and M. Long, 2002. Retroposed new genes out of the X in Drosophila. Genome Res. 12: 1854–1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black, M. A., and R. W. Doerge, 2002. Calculation of the minimum number of replicate spots required for detection of significant gene expression fold change in microarray experiments. Bioinformatics 18: 1609–1616. [DOI] [PubMed] [Google Scholar]
- Brem, R. B., G. Yvert, R. Clinton and L. Kruglyak, 2002. Genetic dissection of transcriptional regulation in budding yeast. Science 296: 752–755. [DOI] [PubMed] [Google Scholar]
- Claridge-Chang, A., H. Wijnen, F. Naef, C. Boothroyd, N. Rajewsky et al., 2001. Circadian regulation of gene expression systems in the Drosophila head. Neuron 32: 657–671. [DOI] [PubMed] [Google Scholar]
- Cockerham, C. C., and B. S. Weir, 1977. Quadratic analyses of reciprocal crosses. Biometrics 33: 187–203. [PubMed] [Google Scholar]
- Edgington, E., 1995 Randomization Tests. Marcel Dekker, New York.
- Falconer, D. S., and T. F. C. Mackay, 1996 Introduction to Quantitative Genetics. Longman, London. [DOI] [PMC free article] [PubMed]
- Fu, Y.-X., 1996. New statistical tests of neutrality for DNA samples from a population. Genetics 143: 557–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, Y.-X., and W. H. Li, 1993. Statistical tests of neutrality of mutations. Genetics 133: 693–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good, P. I., 2000 Permutation Tests: A Practical Guide to Resampling Methods for Testing Hypotheses. Springer-Verlag, New York.
- Griffing, B., 1956. a Concept of general and specific combining ability in relation to diallel crossing systems. Aust. J. Biol. Sci. 9: 463–493. [Google Scholar]
- Griffing, B., 1956. b A generalised treatment of the use of diallel crosses in quantitative inheritance. Heredity 10: 31–50. [Google Scholar]
- Harris, H., 1966. Enzyme polymorphism in man. Proc. R. Soc. Lond. Ser. B 164: 298–310. [DOI] [PubMed] [Google Scholar]
- Houle, D., B. Morikawa and M. Lynch, 1996. Comparing mutational variabilities. Genetics 143: 1467–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubby, J. L., and R. C. Lewontin, 1966. A molecular approach to the study of genetic heterozygosity in natural populations. I. The number of alleles at different loci in Drosophila pseudoobscura. Genetics 54: 577–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson, R. R., M. Kreitman and M. Aguadé, 1987. A test of neutral molecular evolution based on nucleotide data. Genetics 116: 153–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, W., R. M. Riley, R. D. Wolfinger, K. P. White, G. Passador-Gurgel et al., 2001. The contributions of sex, genotype and age to transcriptional variance in Drosophila melanogaster. Nat. Genet. 29: 389–395. [DOI] [PubMed] [Google Scholar]
- Kempthorne, O., and R. N. Curnow, 1961. Partial diallel cross. Biometrics 17: 229. [Google Scholar]
- Kulkarni, S. J., A. F. Steinlauf and J. C. Hall, 1988. The dissonance mutant of courtship song in Drosophila melanogaster: isolation, behavior, and cytogenetics. Genetics 118: 267–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewontin, R. C., and J. L. Hubby, 1966. A molecular approach to the study of genetic heterozygosity in natural populations. II. Amount of variation and degree of heterozygosity in natural populations of Drosophila pseudoobscura. Genetics 54: 595–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long, A. D., H. J. Mangalam, B. Y. P. Chan, L. Tolleri, G. W. Hatfield et al., 2001. Improved statistical inference from DNA microarray data using analysis of variance and a Bayesian statistical framework—Analysis of global gene expression in Escherichia coli K12. J. Biol. Chem. 276: 19937–19944. [DOI] [PubMed] [Google Scholar]
- Lynch, M., and J. Walsh, 1998 Genetics and Analysis of Quantitative Traits. Sinauer Associates, Sunderland, MA.
- Macdonald, S. J., and D. B. Goldstein, 1999. A quantitative genetic analysis of male sexual traits distinguishing the sibling species Drosophila simulans and D. sechellia. Genetics 153: 1683–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay, T. F. C., 2001. The genetic architecture of quantitative traits. Annu. Rev. Genet. 35: 303–339. [DOI] [PubMed] [Google Scholar]
- Meiklejohn, C. D., J. Parsch, J. M. Ranz and D. L. Hartl, 2003. Rapid evolution of male-biased gene expression in Drosophila. Proc. Natl. Acad. Sci. USA 100: 9894–9899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moody, D. E., Z. Zou and L. Mcintyre, 2002. Cross species hybridization of pig RNA to human nylon microarrays. Biomed. Central Genomics 3: 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitabach, M. N., J. Blau and T. C. Holmes, 2002. Electrical silencing of Drosophila pacemaker neurons stops the free-running circadian clock. Cell 109: 485–495. [DOI] [PubMed] [Google Scholar]
- Nuzhdin, S. V., M. L. Wayne, K. L. Harmon and L. M. McIntyre, 2004. Whole genome expression profiling identifies speciation genes in Drosophila. Mol. Biol. Evol. 21: 1308–1317. [DOI] [PubMed] [Google Scholar]
- Oleksiak, M. F., G. A. Churchill and D. L. Crawford, 2002. Variation in gene expression within and among natural populations. Nat. Genet. 32: 261–266. [DOI] [PubMed] [Google Scholar]
- Ranz, J. M., C. I. Castillo-Davis, C. D. Meiklejohn and D. L. Hartl, 2003. Sex-dependent gene expression and evolution of the Drosophila transcriptome. Science 300: 1742–1745. [DOI] [PubMed] [Google Scholar]
- Rice, W. R., 1987. a The accumulation of sexually antagonistic genes as a selective agent promoting the evolution of reduced recombination between primitive sex chromosomes. Evolution 41: 911–914. [DOI] [PubMed] [Google Scholar]
- Rice, W. R., 1987. b The effect of sex chromosomes on the rate of evolution. Trends Ecol. Evol. 3: 2–3. [Google Scholar]
- Rice, W. R., 1987. c Genetic hitchhiking and the evolution of reduced genetic activity of the Y sex chromosome. Genetics 116: 161–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rifkin, S. A., J. Kim and K. P. White, 2003. Evolution of gene expression in the Drosophila melanogaster subgroup. Nat. Genet. 33: 138–144. [DOI] [PubMed] [Google Scholar]
- Tajima, F., K. Misawa and I. Hideki, 1998. The amount and pattern of DNA polymorphism under the neutral mutation hypothesis. Genetica 102/103: 103–107. [PubMed] [Google Scholar]
- Thornton, K., and M. Long, 2002. Rapid divergence of gene duplicates on the Drosophila melanogaster X chromosome. Mol. Biol. Evol. 19: 918–925. [DOI] [PubMed] [Google Scholar]
- Wolfinger, R. D., G. Gibson, E. D. Wolfinger, L. Bennett, H. Hamadeh et al., 2001. Assessing gene significance from cDNA microarray expression data via mixed models. J. Comput. Biol. 8: 625–637. [DOI] [PubMed] [Google Scholar]
- Yvert, G., R. B. Brem, J. Whittle, J. M. Akey, E. Foss et al., 2003. Trans-acting regulatory variation in Saccharomyces cerevisiae and the role of transcription factors. Nat. Genet. 35: 57–64. [DOI] [PubMed] [Google Scholar]