

Abstract
The existing statistical methods for mapping quantitative trait loci (QTL) assume that the phenotype follows a normal distribution and is fully observed. These assumptions may not be satisfied when the phenotype pertains to the survival time or failure time, which has a skewed distribution and is usually subject to censoring due to random loss of follow-up or limited duration of the experiment. In this article, we propose an interval-mapping approach for censored failure time phenotypes. We formulate the effects of QTL on the failure time through parametric proportional hazards models and develop efficient likelihood-based inference procedures. In addition, we show how to assess genome-wide statistical significance. The performance of the proposed methods is evaluated through extensive simulation studies. An application to a mouse cross is provided.
QUANTITATIVE trait analysis plays an important role in the understanding of genetic variations in plants and animals. Mapping quantitative trait loci (QTL) can lead to improvements in economic traits, such as yield and quality in crop plants and milk production in cows. QTL mapping in animals can also provide valuable insights into the genetic etiologies of complex human diseases (Hilbert et al. 1991; Jacob et al. 1991; Shepel et al. 1998).
Much of the modern statistical methodology for QTL mapping in experimental crosses originates from the seminal work of Lander and Botstein (1989). The Lander-Botstein interval-mapping method postulates that QTL occur at a series of positions within a set of adjacent marker intervals and that the trait value depends on the QTL genotype through a linear regression model. The distance between each pair of genetic markers is assumed known. The method steps through the genome in specified increments, say every 1 or 2 cM, and calculates the likelihood-ratio statistic for testing no QTL present at each position. The position with the largest value of the likelihood-ratio statistic is declared to be the candidate QTL location provided that the value exceeds a certain threshold level. It is widely recognized that the interval-mapping method has higher power and requires fewer progenies than the single-marker analysis (Lander and Botstein 1989; Haley and Knott 1992; Zeng 1994). Doerge et al. (1997) described in greater detail this method and various extensions.
Most of the existing QTL-mapping methods require that the phenotype be normally distributed and fully observed. These assumptions are likely to be false when the phenotype pertains to the survival time or failure time. The Weibull distribution and other skewed distributions with long right tails are more appropriate than the normal distribution. Furthermore, the failure time is often subject to censoring so that the trait value is known only to be beyond the censoring time. An example of failure time in plant experiments is the flowering time, which may be censored due to limited duration of the experiment; see Ferreira et al. (1995). In animal studies, the failure times of interest include time to tumor and time to death (i.e., survival time), which may be subject to censoring because of limited study duration or death due to unrelated causes. One particular example is a mice cross presented by Broman (2003), in which the trait of interest is time to death after a bacterial infection and in which 30% of the mice are still alive at the end of the study period. Symons et al. (2002) presented another interesting study, in which the phenotype is the time until terminal illness due to tumor for Eμ-v-abl transgenic mice.
The incompleteness of the trait values presents major challenges in the application of the interval-mapping approach. Broman (2003) considered a cure model in which the mice that are alive at the end of the study are regarded as cured and in which the survival times among the deaths follow a log-normal distribution. This is a specialized model, which can deal only with the situations in which the potential censoring times are equal among all study subjects. Symons et al. (2002) utilized a variant of the expectation-maximization (EM) algorithm (Lipsitz and Ibrahim 1998) to map QTL with censored observations. This method is computationally intensive and its properties have not been investigated.
In this article, we provide a simple and rigorous extension of the interval-mapping approach of Lander and Botstein (1989) to censored quantitative traits. Specifically, we formulate the effects of QTL on the failure time through proportional hazards models (Kalbfleish and Prentice 2002, Sect. 2.3). We then develop efficient likelihood-based methods for locating QTL and estimating the effects of QTL. In addition, we show how to assess genome-wide statistical significance by extending the analytical results of Lander and Botstein (1989) and Dupuis and Siegmund (1999) and by developing an accurate and efficient Monte Carlo procedure. We conduct extensive simulation studies to evaluate the performance of the proposed methods. Finally, we provide an application to the mice data of Broman (2003).
METHODS
Interval mapping:
In this section, we develop an interval-mapping method for potentially censored failure-time traits in an F2 intercross population by modeling a single QTL. Expanding the results to other crosses is straightforward. Consider n progenies from an intercross between two inbred strains. Let Ti denote the quantitative trait for the ith subject, which pertains to a failure time that can potentially be censored and thus incompletely observed. Let Ci be the censoring time for the ith subject. The observation on the trait value of the ith subject consists of two components: Yi = min(Ti, Ci) and Δi = I(Ti ≤ Ci), where I(𝒜) is the indicator function for event 𝒜. The failure time Ti is fully observed only when it is uncensored, i.e., Δi = 1.
Suppose that we have data on a set of genetic markers with a known genetic map. Let Mi denote the multipoint marker genotype data for the ith subject. We consider a putative QTL locus d in the genome with two possible alleles q and Q from the two inbred parents and define Gi = −1, 0, or 1 according to whether the ith subject has genotype qq, Qq, or QQ, respectively, at the QTL. We specify a proportional hazards model for the effects of the QTL genotype on the failure time such that, conditional on the QTL genotype Gi, the hazard function of Ti takes the form
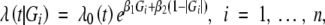 |
1 |
where β1 and β2 pertain to the additive and dominant effects of the QTL, and λ0(·) is an unknown baseline hazard function (Kalbfleish and Prentice 2002, Sect. 2.3). In this article, we assume a parametric model for λ0. In particular, we consider a Weibull hazard function 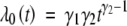 , γ1 > 0, γ2 > 0 (Kalbfleish and Prentice 2002, p. 33).
, γ1 > 0, γ2 > 0 (Kalbfleish and Prentice 2002, p. 33).
Write θ = (β, γ), where β = (β1, β2) and γ = (γ1, γ2). At each locus, we may calculate πi,g = Pr(Gi = g|Mi) (g = −1, 0, 1; i = 1, … , n), which are the conditional probabilities of the QTL genotypes given the observed marker data. Under the assumptions of no crossover interference and no genotyping errors, these probabilities are determined by the genotypes of the two flanking markers and the location of the QTL; see Equation 15.2 of Lynch and Walsh (1998).
We assume that censoring is noninformative (Kalbfleish and Prentice 2002, p. 195) and that the censoring time is independent of the failure time and QTL genotype. The likelihood for the vector of parameters θ based on the complete data (Yi, Δi, Gi, Mi) (i = 1, … , n) is proportional to
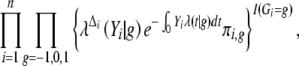 |
2 |
while the likelihood based on the observed data (Yi, Δi, Mi)(i = 1, … , n) is proportional to
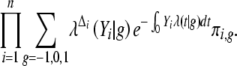 |
3 |
To obtain the maximum-likelihood estimator (MLE) of θ, we may maximize the observed-data likelihood (3) directly. An alternative approach is to apply the EM algorithm (Dempster et al. 1977) to (2). The expected value of the complete-data log-likelihood given the observed data can be shown to be, up to a constant,
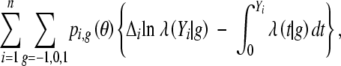 |
4 |
where
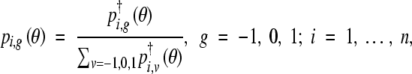 |
and
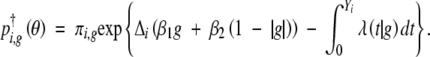 |
In the E-step, we evaluate pi,g(θ) at the current estimate of θ. The M-step can proceed in a similar manner to the case of complete data since expression (4), with θ in pi,g(θ) fixed, takes the same form as the complete-data log-likelihood. We begin the EM algorithm by assigning an initial value to θ and iterate until convergence. The initial value for β is set to 0 and that of γ to some value in the parameter space of γ. The resulting MLE is denoted by θ̂. See appendix A for further detail.
We test the null hypothesis of no QTL effects, i.e., H0: β = 0, by the likelihood-ratio statistic
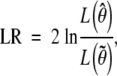 |
where L(·) is the observed-data likelihood, and θ̃ = (0, γ̃) with γ̃ being the restricted MLE of γ under H0. The LOD score is LR/(2 ln 10). Under H0, LR is asymptotically χ2-distributed with 2 d.f. (appendix B). Note that pi,g(θ), θ̂, L(θ̂), LR, and LOD all depend on the locus d through the dependence of πi,g on d. In the sequel, we include d in the expressions to emphasize their dependence on d if ambiguity arises. Note also that θ̃ and L(θ̃) do not depend on d. Thus, as in the case of standard interval mapping, the likelihood under H0 is calculated once while the likelihood under the alternative is evaluated at each location in the genome to produce a LOD curve for each chromosome. The position with the largest value of the LOD score is declared to be the QTL location provided that the value exceeds a certain threshold level. We show how to determine the threshold level in the following section.
Thresholds:
When searching the entire chromosome or whole genome for QTL, one should select a threshold level for the LOD score such that the probability (under the null hypothesis) that LOD or some other test statistic exceeds this level anywhere in the genome equals the desired false-positive rate. The pointwise significance level based on the χ2-approximation is inadequate because of the multiple tests while the Bonferroni correction is too conservative because of the dependence of the test statistics at different points in the genome. In appendix C, we show that the likelihood-ratio statistic LR(d) can be partitioned into the sum of the squares of two asymptotically independent Ornstein-Uhlenbeck processes. This result is analogous to those of Lander and Botstein (1989) and Dupuis and Siegmund (1999) and implies that the analytical approximations of thresholds for normal traits can be applied to the case of censored failure time observations. These analytical results assume that the markers are dense or equally spaced with no missing data and thus may not work well in practice. Using results of Davies (1977)(1987), Rebai et al. (1994) provided approximate thresholds in backcross (BC) and F2, which are applicable in the intermediate map density case. The calculations are formidable, even for F2, and do not accommodate missing marker data.
To overcome the limitations of the analytical approximations, we propose a novel resampling approach to determining the thresholds for genome-wide statistical significance. This approach allows arbitrary distributions of the markers as well as arbitrary test positions. It also accommodates missing marker data and dominant markers.
For evaluating the correlations of the test statistics among different locations, it is more convenient to work with the score statistic than the likelihood-ratio statistic. Let Uβ(θ̃; d) be the score function (based on the observed-data likelihood) for β at location d, which can be approximated by the sum of n independent zero-mean random vectors 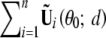 , where θ0 = (0, γ0) is the true parameter value under H0; see appendix B. Thus, n−1/2Uβθ̃; d is asymptotically zero-mean normal with covariance matrix V(d) that is the limit of
, where θ0 = (0, γ0) is the true parameter value under H0; see appendix B. Thus, n−1/2Uβθ̃; d is asymptotically zero-mean normal with covariance matrix V(d) that is the limit of 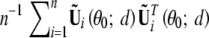 . We replace the unknown quantities in Ũi(θ0; d) by their sample estimators to yield Ûi(d) shown in appendix B. The score statistic for testing H0: β = 0 against H1: β ≠ 0 takes the form
. We replace the unknown quantities in Ũi(θ0; d) by their sample estimators to yield Ûi(d) shown in appendix B. The score statistic for testing H0: β = 0 against H1: β ≠ 0 takes the form
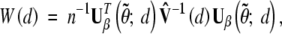 |
where 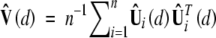 is a consistent estimator of the limiting covariance matrix V(d). It can be shown that W(d) is asymptotically equivalent to LR(d) (Cox and Hinkley 1974, Sect. 9.3).
is a consistent estimator of the limiting covariance matrix V(d). It can be shown that W(d) is asymptotically equivalent to LR(d) (Cox and Hinkley 1974, Sect. 9.3).
In general, the limiting distribution of supdW(d) is not analytically tractable. We use a resampling approach similar to that of Lin et al. (1993) to approximate the null distributions of n−1/2Uβ(θ̃; d) and supdW(d). From appendix B, it is easy to see that n−1/2Uβ(θ̃; d) converges to a zero-mean Gaussian process with covariance function ψ(d1, d2) that is the limit of 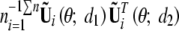 at (d1, d2) and that ψ(d1, d2) can be consistently estimated by
at (d1, d2) and that ψ(d1, d2) can be consistently estimated by 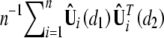 . Define
. Define 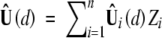 , where Zi (i = 1, … , n) are independent standard normal random variables that are independent of the observed data. Conditional on the observed data, n−1/2Û(d) is a Gaussian process with mean 0 and covariance function
, where Zi (i = 1, … , n) are independent standard normal random variables that are independent of the observed data. Conditional on the observed data, n−1/2Û(d) is a Gaussian process with mean 0 and covariance function 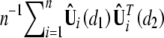 at (d1, d2), which converges to ψ(d1, d2). Thus, the conditional distribution of n−1/2Û(d) given the observed data converges to the limiting distribution of n−1/2Uβ(θ̃; d). As a result, the distribution of W(d) can be approximated by that of
at (d1, d2), which converges to ψ(d1, d2). Thus, the conditional distribution of n−1/2Û(d) given the observed data converges to the limiting distribution of n−1/2Uβ(θ̃; d). As a result, the distribution of W(d) can be approximated by that of
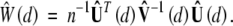 |
To approximate the distribution of supdW(d), we generate the normal random sample (Z1, … , Zn) a large number of times while holding the observed data fixed; for each sample, we calculate Ŵ(d) and supdŴ(d). The 100(1 − α)th percentile of the simulated supdŴ(d) is the threshold value for the genome-wide significance level of α. This resampling approach is computationally much more efficient than the use of permutation (Churchill and Doerge 1994) and other simulation methods because it involves only simulation of normal random variables and does not entail repeated analysis of simulated data sets.
SIMULATIONS
To investigate the operating characteristics of the proposed methods in practical situations, we performed extensive simulation studies. We generated the failure times from the Weibull distributions with baseline hazard function 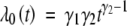 , where γ1 = 0.01 and γ2 = 2. We reparameterized γ1 and γ2 according to
, where γ1 = 0.01 and γ2 = 2. We reparameterized γ1 and γ2 according to 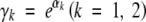 so as to ensure that the estimates of γ1 and γ2 are positive. The censoring times were generated from the uniform (0, τ) distribution, where τ was chosen to yield ∼30% censored observations. Assuming no crossover interference, we generated the marker data from the Markov chain. The interval-mapping step size was set at 1 cM.
so as to ensure that the estimates of γ1 and γ2 are positive. The censoring times were generated from the uniform (0, τ) distribution, where τ was chosen to yield ∼30% censored observations. Assuming no crossover interference, we generated the marker data from the Markov chain. The interval-mapping step size was set at 1 cM.
We considered a chromosome with a total length of 100 cM. Genetic maps with different numbers of equally spaced markers were simulated. Under H1, one QTL located at 35 cM was simulated with β1 = 0.35 and β2 = 0.30. We generated 10,000 replicates of 300 observations from an F2 population. We evaluated the finite-sample properties of the MLEs of the QTL effects at the true QTL location. The results for the estimator of the additive QTL effect are summarized in Table 1. The proposed estimator appears to be virtually unbiased. The standard error estimator reflects accurately the true variation. The confidence intervals have proper coverage probabilities. We obtained similar results for the estimator of the dominant QTL effect (data not shown). We also examined the performance of the proposed interval-mapping methods for locating the QTL and estimating the QTL effects at the location with the maximum LOD. The results are summarized in Table 2. There is little bias for the estimator of the QTL location or the estimators of the QTL effects. The mapping is more precise for denser marker maps.
TABLE 1.
Summary statistics for the estimator of the additive QTL effect at the true QTL location
| H0: β1 = 0, β2 = 0
|
H1: β1 = 0.35, β2 = 0.30
|
|||||||
|---|---|---|---|---|---|---|---|---|
| No. of markers |
Meana | SEb | SEEc | CPd | Meana | SEb | SEEc | CPd |
| 6 | 0.002 | 0.108 | 0.107 | 94.8 | 0.355 | 0.110 | 0.108 | 94.4 |
| 11 | 0.002 | 0.105 | 0.104 | 94.6 | 0.355 | 0.107 | 0.105 | 94.8 |
| 51 | −0.001 | 0.101 | 0.099 | 94.4 | 0.353 | 0.103 | 0.101 | 94.6 |
| 101 | 0.000 | 0.099 | 0.098 | 94.9 | 0.354 | 0.100 | 0.100 | 94.8 |
Mean is the mean of the parameter estimator.
SE is the standard error of the parameter estimator.
SEE is the mean of the standard error estimator.
CP is the coverage probability of the 95% confidence interval.
TABLE 2.
Sampling means of the estimators for the QTL location and for the QTL effects at the estimated QTL location in the simulation studies
| H0: β1 = 0, β2 = 0
|
H1: β1 = 0.35, β2 = 0.30
|
|||||
|---|---|---|---|---|---|---|
| QTL effects
|
QTL effects
|
|||||
| No. of markers |
QTL location (cM) |
β1 | β2 | QTL location (cM) |
β1 | β2 |
| 6 | 49.6 (34.2) | 0.001 (0.141) | −0.012 (0.235) | 36.6 (11.3) | 0.359 (0.107) | 0.304 (0.173) |
| 11 | 49.9 (32.8) | 0.000 (0.143) | −0.007 (0.233) | 35.8 (10.9) | 0.359 (0.101) | 0.300 (0.162) |
| 51 | 50.2 (31.8) | −0.001 (0.148) | −0.008 (0.251) | 35.7 (9.3) | 0.362 (0.098) | 0.314 (0.154) |
| 101 | 50.3 (31.4) | 0.001 (0.150) | −0.009 (0.255) | 35.5 (8.9) | 0.365 (0.100) | 0.319 (0.152) |
Standard errors are shown in parentheses.
We conducted additional simulation studies to evaluate the performance of the analytical and resampling methods for determining genome-wide statistical significance. We generated both equally and unequally spaced markers. We also simulated data with missing marker genotypes and dominant markers, which are more comparable with real data. We considered one chromosome with a total length of 100 cM. For the cases of unevenly spaced markers, we placed m markers at the following locations,
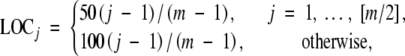 |
where LOCj is the jth marker location and [m/2] is the largest integer that is less than or equal to m/2. In these settings, the first half of the markers is denser than the second half of the markers. We generated 10,000 replicates of 300 observations from an F2 population. The dense-map and sparse-map approximations were obtained from Equation C1 in appendix C . The thresholds for the resampling method were based on 10,000 normal samples. The results are summarized in Tables 3 and 4.
TABLE 3.
Analytical and resampling-based thresholds at the targeted genome-wide significance level of α
| Empiricalb
|
Resamplingc
|
Analytical
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| H0
|
H1d
|
Dense-map
|
Sparse-map
|
||||||||
| No. of markers |
Marker patterna |
α = 5% | 1% | α = 5% | 1% | α = 5% | 1% | α = 5% | 1% | α = 5% | 1% |
| 6 | 1 | 10.05 | 13.57 | 9.80 (0.16) | 13.34 (0.25) | 9.81 (0.16) | 13.36 (0.25) | 13.37 | 17.12 | 9.15 | 12.39 |
| 11 | 1 | 10.33 | 13.90 | 10.41 (0.17) | 13.99 (0.25) | 10.42 (0.16) | 14.00 (0.25) | 13.37 | 17.12 | 10.15 | 13.53 |
| 51 | 1 | 11.79 | 15.60 | 11.47 (0.19) | 15.12 (0.27) | 11.47 (0.18) | 15.12 (0.27) | 13.37 | 17.12 | 11.80 | 15.37 |
| 6 | 2 | 9.94 | 13.65 | 9.63 (0.16) | 13.18 (0.25) | 9.64 (0.17) | 13.18 (0.25) | 13.37 | 17.12 | 9.15 | 12.39 |
| 11 | 2 | 10.65 | 14.27 | 10.23 (0.17) | 13.80 (0.25) | 10.24 (0.17) | 13.81 (0.26) | 13.37 | 17.12 | 10.15 | 13.53 |
| 51 | 2 | 11.54 | 15.08 | 11.17 (0.19) | 14.80 (0.27) | 11.17 (0.18) | 14.81 (0.26) | 13.37 | 17.12 | 11.80 | 15.37 |
Under pattern 1, markers are evenly spaced with no missing marker genotypes or dominant markers; under pattern 2, markers are unevenly spaced with 20% missing marker genotypes and 5% dominant markers.
Percentiles of the test statistic based on 10,000 simulated data sets.
Average thresholds from 10,000 simulated data sets. The values in parentheses are the standard errors of the thresholds.
QTL is located at 35 cM with β1 = 0.35 and β2 = 0.3.
TABLE 4.
Sizes/powers (%) according to the analytical and resampling-based thresholds
| Resampling
|
Analytical
|
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| H0
|
H1b
|
Dense-map
|
Sparse-map
|
||||||||||
| H0
|
H1b
|
H0
|
H1b
|
||||||||||
| No. of markers |
Marker patterna |
α = 5% | 1% | α = 5% | 1% | α = 5% | 1% | α = 5% | 1% | α = 5% | 1% | α = 5% | 1% |
| 6 | 1 | 5.6 | 1.1 | 91 | 80 | 1.1 | 0.2 | 79 | 63 | 7.5 | 1.8 | 93 | 83 |
| 11 | 1 | 4.8 | 1.0 | 93 | 82 | 1.2 | 0.2 | 84 | 69 | 5.4 | 1.2 | 93 | 83 |
| 51 | 1 | 5.5 | 1.1 | 94 | 85 | 2.6 | 0.6 | 90 | 78 | 5.0 | 1.1 | 94 | 84 |
| 6 | 2 | 5.6 | 1.2 | 77 | 57 | 1.1 | 0.2 | 56 | 36 | 7.1 | 1.8 | 80 | 62 |
| 11 | 2 | 5.9 | 1.2 | 82 | 65 | 1.5 | 0.3 | 67 | 47 | 6.3 | 1.4 | 83 | 66 |
| 51 | 2 | 5.8 | 1.2 | 85 | 68 | 2.1 | 0.5 | 75 | 56 | 4.4 | 0.9 | 82 | 65 |
Under pattern 1, markers are evenly spaced with no missing marker genotypes or dominant markers; under pattern 2, markers are unevenly spaced with 20% missing marker genotypes and 5% dominant markers.
QTL is located at 35 cM with β1 = 0.35 and β2 = 0.3.
The thresholds based on the resampling method are close to the empirical values, whether the data are generated under H0 or H1; consequently, the LR tests based on these thresholds have proper type I error and power. This is true of any genetic map, with or without missing marker genotypes and dominant markers. The dense-map approximations are too conservative and thus result in power loss, while the sparse-map approximations tend to be too liberal. We also assessed the approximations by Rebai et al. (1994), which turn out to be conservative when the genetic map is dense. For example, in the case of 51 markers with α = 0.05, the sizes are 2.66 and 2.26% for marker patterns 1 and 2, respectively.
APPLICATION
To illustrate our methods, we consider the mice data previously analyzed by Broman (2003). A total of 116 female mice from an intercross between the BALB/cByJ and C57BL/6ByJ strains were genotyped at 133 markers, including 2 on the X chromosome. The phenotype of interest is the time to death following infection with Listeria monocytogenes. Approximately 30% of the survival times are censored.
Broman (2003) proposed a nonparametric (NP) approach and a two-part model. The NP approach is an extension of the Kruskal-Wallis statistic (Lehmann 1975, Sect. 5.2) by assigning a prior weight (πi,g) to the rank of the ith observation for each QTL genotype group g. In this approach, the censored observations are treated as the true failure times and an average rank is assigned to those observations. In the two-part approach, Broman considered a cure model in which the mice that are alive at the end of the study are regarded as cured while the survival times among the deaths follow a log-normal distribution.
We applied the proposed methods to these data, assuming a Weibull baseline hazard, and the results are shown in Table 5 and Figures 1 and 2. The threshold for the LOD score at the 5% genome-wide significance level based on the resampling approach is 3.43, which is close to 3.27, the threshold obtained by permutation for the NP approach of Broman (2003). Our results are fairly consistent with those of Broman (2003). We detect almost the same QTL on chromosomes 5, 13, and 15 except that we detect an additional QTL on chromosome 6 rather than on chromosome 1. The QTL on chromosome 5 appears to have a strong additive effect and the hazard ratio of the survival time with genotype QQ vs. qq is ∼9.95. Genotypes qq and Qq at the QTL on chromosome 6 seem to have similar effects. The QTL on chromosome 13 appears to have both additive and dominant effects. The QTL on chromosome 15 appears to have a strong dominant effect. At most detected QTL locations, our LOD scores are larger than those of Broman's NP approach. This suggests that our approach may be more efficient in detecting QTL.
TABLE 5.
Estimates of the QTL positions and QTL effects along with the maximum LOD scores for the data on survival time following infection withListeria monocytogenes in 116 intercross mice
| Proposed method
|
Nonparametric
|
Two-part model
|
||||||
|---|---|---|---|---|---|---|---|---|
| Chromosome | Pos (cM) | LOD | β1 | β2 | Pos (cM) | LOD | Pos (cM) | LOD |
| 1 | 75 | 1.94 | −0.456 | −0.542 | 76 | 3.38 | 81 | 5.45 |
| 5 | 28 | 9.01 | 1.149 | 0.100 | 27 | 5.41 | 28 | 6.79 |
| 6 | 59 | 3.66 | 0.559 | 0.563 | 59 | 2.45 | 10 | 4.09 |
| 13 | 26 | 6.64 | −0.614 | −0.740 | 26 | 6.71 | 26 | 7.38 |
| 15 | 23 | 4.49 | 0.370 | −0.935 | 23 | 3.49 | 16 | 4.61 |
Pos, position.
Figure 1.—

The LOD scores from three QTL mapping methods for the data on survival time following infection with Listeria monocytogenes in 116 intercross mice. The threshold pertains to the 5% genome-wide significance level under the resampling method.
Figure 2.—

Plot of the −log10P-values for three QTL mapping methods for the data on survival time following infection with Listeria monocytogenes in 116 intercross mice. The P-values for the proposed method are based on 100,000 normal samples. In the region between 26 and 30 cM on chromosome 5, the P-values are <10−5 and thus are not displayed. The P-values for the NP and two-part models are based on 11,000 permutation replicates.
Figure 1 shows the LOD curves from the three methods: proposed method, nonparametric method, and two-part model. The LOD scores from the two-part model are larger than those of the other two methods at some QTL locations because there are two more free parameters in the two-part model than in the other two methods. This will decrease the power to detect QTL since a larger threshold (i.e., 4.91) is required. To evaluate different methods on a common scale, we converted the LOD curves to the estimated pointwise P-values. Figure 2 displays the values of −log10P for chromosomes 1, 5, 6, 13, and 15. Comparisons with the nonparametric method and two-part model reveal that the proposed method yields more significant results on the above chromosomes except chromosome 1. Incidentally, the resampling method is ∼100 times faster than the permutation method in this application.
To get some ideas about the adequacy of the Weibull distribution in describing the Listeria data, we fitted both the semiparametric model and the Weibull model at marker D5M357, which is close to the peak of the LOD score on chromosome 5. The estimated QTL effects from the two models are very similar. It would be worthwhile to develop formal goodness-of-fit methods for assessing the adequacy of the parametric survival model at the true QTL location.
EXTENSIONS
In this section, we extend the single-QTL model to multiple QTL. The approach of interval mapping (IM) considers one putative QTL at a time. The QTL located elsewhere on the genome can have interfering effects, so that the estimators for the locations and effects of QTL may be biased and the power of detecting QTL may be compromised (Lander and Botstein 1989; Haley and Knott 1992; Zeng 1994). Boer et al. (2002) showed that the IM method fails to detect three interacting QTL with no main effects through simulation studies. A variety of approaches have been proposed for mapping multiple QTL. These methods can increase the power to detect QTL and reduce biases in the estimators of the QTL effects and locations. In this section, we consider mainly composite-interval mapping (CIM; Jansen 1993; Zeng 1993, 1994) and multiple-interval mapping (MIM; Kao et al. 1999) for censored traits.
Composite-interval mapping:
The idea of CIM is to combine IM with multiple regression analysis in mapping QTL by conditioning on markers outside a region of interest to account for the effects of other QTL. To extend the original CIM model to censored traits, we consider the following proportional hazards model,
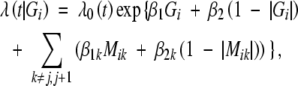 |
5 |
where j and j + 1 conform to two flanking markers bordering the putative QTL, and Mik is the indicator variable for the marker genotype, which takes values −1, 0, and 1 for genotypes aa, Aa, and AA, respectively. We may further enhance the model by considering the interaction effects between the putative QTL and controlling markers. Replacing λ(t|Gi) in (2) and (3) with (5), we obtain the complete-data and observed-data likelihood functions, respectively. As in the case of standard interval mapping, we can maximize the observed-data likelihood directly or apply the EM algorithm to obtain the MLEs. We can test H0: β = 0 at any position in the genome.
The CIM approach requires that the sample size be large relative to the number of markers included in the model. In practice, the sample size is generally not very large. Thus, Zeng (1994) suggested including in the model only those markers that are more or less evenly spaced in the genome or those preidentified markers that explain most of the genetic variation in the genome. This suggestion also applies to our setting.
Multiple-interval mapping:
The MIM approach proposed by Kao et al. (1999) uses multiple marker intervals simultaneously to fit multiple putative QTL directly in the model. Consider K QTL, Q1, … , QK, located at d1, … , dK in the genome. There are 3K possible QTL genotypes. Some of the K QTL may exhibit epistasis. We formulate the effects of the K QTL on the failure time through a proportional hazards model, such that, conditional on the joint genotype Gi = (G1i, … , GKi), the hazard function of Ti takes the form
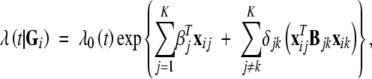 |
6 |
where xij = (Gij, 1 − |Gij|)T, δjk is an indicator variable for epistasis between Qj and Qk, and βj and Bjk pertain to the main effects and epistatic effects, respectively. The variable δjk indicates, by the values 1 vs. 0, whether or not Qj and Qk interact. Given the marker data Mi for the ith subject and assuming no crossover interference, we may calculate πi,g(g = 1, … , 3K), the conditional probabilities of the 3K possible genotypes of the K QTL. The complete-data likelihood takes the same form as Equation 2 except that the summation of g is now over (1, … , 3K). To obtain the MLEs and LOD scores, we can again apply the EM algorithm.
Since the true number and locations of the QTL are unknown, model selection is a critical issue in the MIM approach. Kao et al. (1999) suggested stepwise and chunkwise selection with the likelihood-ratio test statistic as a selection criterion to identify QTL, to separate linked QTL, and to analyze epistasis between QTL. Broman and Speed (2002) developed a modified Bayesian information criterion (Schwarz 1978) for model selection. When many QTL are included in the MIM model, the computation can be very intensive. Sen and Churchill (2001) reduced the computation burden of MIM by replacing the EM algorithm with a Monte Carlo algorithm. All these strategies can be applied to our setting.
DISCUSSION
We have described our methods in the context of an F2 population. All the proposed methods can be easily generalized to other crosses such as BC, F3, or even more complicated crosses such as combined crosses (Zou et al. 2001). We may also extend our methods to accommodate covariates, such as block factors in an agriculture field trial or cage number in a mouse cross.
Symons et al. (2002) formulated the effects of the QTL on the failure time through the semiparametric proportional hazards model. They utilized a variant of the EM algorithm developed by Lipsitz and Ibrahim (1998), in which Monte Carlo simulation is used to approximate the conditional expectation of the complete-data partial-likelihood score function given the observed data. The solution to this conditional expectation is not a maximum partial-likelihood estimator, so that the method is not statistically efficient. The Monte Carlo simulation is time-consuming. There is no formal justification for the method of Lipsitz and Ibrahim (1998) or that of Symons et al. (2002). Our method is computationally much simpler than Monte Carlo simulation. It is based on the maximum-likelihood estimator and is thus statistically efficient. Furthermore, we have established the theoretical properties of our method and assessed its empirical performance through simulation studies. The method of Symons et al. (2002) has the advantage that the baseline hazard function is unspecified.
The proposed resampling approach to determining genome-wide significance is applicable to arbitrary genetic maps and accommodates missing marker data and dominant markers. For the CIM and MIM models, no analytical thresholds are available and the permutation method is extremely time-consuming. The proposed resampling approach can be applied to the CIM and MIM models since all the relevant formulas have been presented for an arbitrary likelihood. For the MIM method, the resampling approach produces appropriate thresholds for testing each putative QTL given the others, with or without adjustment for the fact that multiple QTL are tested simultaneously.
We have written a computer program in C to implement the proposed method. This program is available from the authors upon request.
APPENDIX A:
COMPUTATIONS OF MLE AND COVARIANCE MATRIX
In this section, we present the formulas for the M-step of the EM algorithm under the Weibull model. In addition, we provide the observed information matrix as well as a consistent estimator of the covariance matrix of MLE θ̂. In the M-step of the (k + 1)th iteration, the first and second derivatives of the expected value of the log-likelihood given the observed data and current estimate θ̂(k) are given by
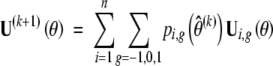 |
A1 |
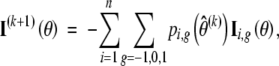 |
A2 |
where
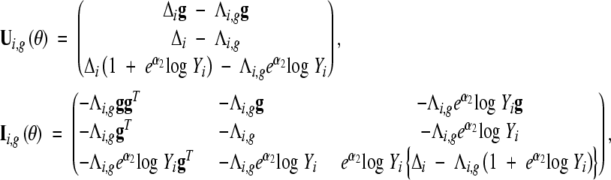 |
g = (g, 1 − |g|)T, and 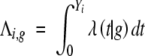 , which is the cumulative hazard function conditional on the QTL genotype g. Then, we can apply the Newton-Raphson algorithm to update the current estimate with the new maximizer θ̂(k+1).
, which is the cumulative hazard function conditional on the QTL genotype g. Then, we can apply the Newton-Raphson algorithm to update the current estimate with the new maximizer θ̂(k+1).
The observed information matrix is given by
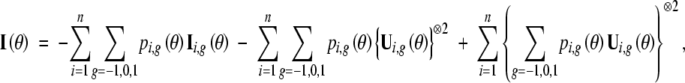 |
where for a column vector a, a⊗2 denotes the matrix aaT. Thus, a consistent estimator of the covariance matrix of θ̂ is given by the inverse of the observed information matrix evaluated at θ̂, i.e., 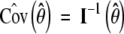 .
.
APPENDIX B:
ASYMPTOTIC PROPERTIES OF SCORE AND LIKELIHOOD-RATIO STATISTICS
In this section, we show that LR(d) is asymptotically χ22-distributed under H0 and provide the necessary ingredients for deriving the thresholds. Let U(θ; d) and I(θ; d) be the observed-data score function and information matrix at location d with the following partitions to conform with the partition (β, γ) of θ,
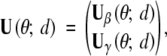 |
and
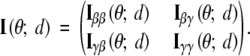 |
Under H0, n−1I(θ̃) converges to ∑(d). Denote
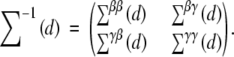 |
It can be shown that 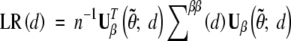 asymptotically (Cox and Hinkley 1974, Sect. 9.3). Through Taylor series expansions,
asymptotically (Cox and Hinkley 1974, Sect. 9.3). Through Taylor series expansions, 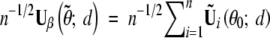 asymptotically, where
asymptotically, where 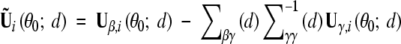 and θ0 = (0, γ0). The replacements of the unknown quantities in Ũi(θ0; d) with their sample estimators yield
and θ0 = (0, γ0). The replacements of the unknown quantities in Ũi(θ0; d) with their sample estimators yield 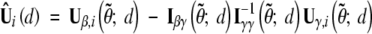 .
.
Let 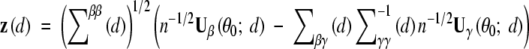 . Then z(d) converges to a normal distribution with mean 0 and an identity 2 × 2 covariance matrix. Thus, LR(d) is asymptotically distributed as χ22 under H0.
. Then z(d) converges to a normal distribution with mean 0 and an identity 2 × 2 covariance matrix. Thus, LR(d) is asymptotically distributed as χ22 under H0.
APPENDIX C:
ANALYTICAL APPROXIMATIONS OF THRESHOLDS
We show in this appendix that, for infinitely dense markers, the null distribution of LR(d) can be approximated by an Ornstein-Uhlenbeck process. Under H0, ∑(d) does not depend on d. Let d1 and d2 denote two points on the chromosome, and r be the recombination fraction corresponding to the genetic distance |d1 − d2|. Under the assumption of no crossover interference, it is easy to show that the correlation between g(d1) and g(d2) is given by
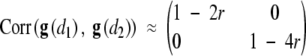 |
provided that r is small. Since Uβ,i(θ; d) = (Δi − Λ0(Yi))gi(d) under H0, we have
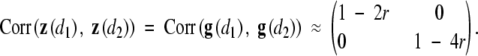 |
The above result implies that z1(d) and z2(d), the first and second components of z(d), are approximately independent Ornstein-Uhlenbeck processes with means zero and variances 1 − 2r and 1 − 4r, respectively. By the arguments of Dupuis and Siegmund (1999), the tail distribution of supdLR(d) under H0 satisfies
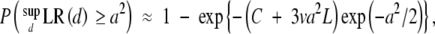 |
C1 |
where Δ is the average marker distance (in morgans), C is the number of chromosomes, L is the total length of the genome (in morgans), and v = v(a(6Δ)1/2), the definition of which can be found in Siegmund (1985). When Δ = 0, the above formula reduces to that of Lander and Botstein (1989). These results imply that all the analytical thresholds for the normal trait can be applied to our case.
References
- Boer, M. P., C. J. Braak and R. C. Jansen, 2002. A penalized likelihood method for mapping epistatic quantitative trait loci with one-dimensional genome searches. Genetics 162 951–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broman, K. W., 2003. Mapping quantitative trait loci in the case of a spike in the phenotype distribution. Genetics 163 1169–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broman, K. W., and T. P. Speed, 2002. A model selection approach for the identification of quantitative trait loci in experimental crosses (with discussion). J. R. Stat. Soc. B 64 731–775. [Google Scholar]
- Churchill, G. A., and R. W. Doerge, 1994. Empirical threshold values for quantitative trait mapping. Genetics 138 963–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox, D. R., and D. V. Hinkley, 1974 Theoretical Statistics. Chapman & Hall, London.
- Davies, R. B., 1977. Hypothesis testing when a nuisance parameter is present only under the alternative. Biometrika 64 247–254. [DOI] [PubMed] [Google Scholar]
- Davies, R. B., 1987. Hypothesis testing when a nuisance parameter is present only under the alternative. Biometrika 74 33–43. [Google Scholar]
- Dempster, A. P., N. M. Laird and D. B. Rubin, 1977. Maximum likelihood from incomplete data via the EM algorithm. J. R. Stat. Soc. B 39 1–38. [Google Scholar]
- Doerge, R. W., Z-B. Zeng and B. S. Weir, 1997. Statistical issues in the search for genes affecting quantitative traits in experimental populations. Stat. Sci. 12 195–219. [Google Scholar]
- Dupuis, J., and D. Siegmund, 1999. Statistical methods for mapping quantitative trait loci from a dense set of markers. Genetics 151 373–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira, M. E., J. Satagopan, B. S. Yandell, P. H. Williams and T. C. Osborn, 1995. Mapping loci controlling vernalization requirement and flower time in Brassica napus. Theor. Appl. Genet. 90 727–732. [DOI] [PubMed] [Google Scholar]
- Haley, C. S., and S. A. Knott, 1992. A simple regression method for mapping quantitative trait loci in line crosses using flanking markers. Heredity 69 315–324. [DOI] [PubMed] [Google Scholar]
- Hilbert, P., K. Lindpaintner, J. S. Beckmann, T. Serikawa, F. Soubrier et al., 1991. Chromosomal mapping of two genetic loci associated with blood-pressure regulation in hereditary hypertensive rats. Nature 353 521–529. [DOI] [PubMed] [Google Scholar]
- Jacob, H. J., K. Lindpaintner, S. E. Lincoln, K. Kusumi, R. K. Bunker et al., 1991. Genetic mapping of a gene causing hypertension in the stroke-prone spontaneously hypertensive rat. Cell 67 213–224. [DOI] [PubMed] [Google Scholar]
- Jansen, R. C., 1993. Interval mapping of multiple quantitative trait loci. Genetics 135 205–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalbfleish, J. D., and R. L. Prentice, 2002 The Statistical Analysis of Failure Time Data, Ed. 2. Wiley, Hoboken, NJ.
- Kao, C. H., Z-B. Zeng and R. D. Teasdale, 1999. Multiple interval mapping for quantitative trait loci. Genetics 152 1203–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander, E. S., and D. Botstein, 1989. Mapping Mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics 121 185–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann, E. L., 1975 Nonparametrics: Statistical Methods Based on Ranks. Holden-Day, San Francisco.
- Lin, D. Y., L. J. Wei and Z. Ying, 1993. Checking the Cox model with cumulative sums of martingale-based residuals. Biometrika 80 557–572. [Google Scholar]
- Lipsitz, S. R., and J. G. Ibrahim, 1998. Estimating equations with incomplete categorical covariates in the Cox model. Biometrics 54 1002–1013. [PubMed] [Google Scholar]
- Lynch, M., and B. Walsh, 1998 Genetics and Analysis of Quantitative Traits. Sinauer, Sunderland, MA.
- Rebai, A., B. Gofinet and B. Mangin, 1994. Approximate thresholds of interval mapping test for QTL detection. Genetics 138 235–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz, G., 1978. Estimating the dimension of a model. Ann. Stat. 6 461–464. [Google Scholar]
- Sen, S., and G. A. Churchill, 2001. A statistical framework of quantitative trait mapping. Genetics 159 371–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepel, L. A., H. Lan, J. D. Haag, G. M. Brasic, M. E. Gheen et al., 1998. Genetic identification of multiple loci that control breast cancer susceptibility. Genetics 149 289–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegmund, D., 1985 Sequential Analysis: Tests and Confidence Intervals. Springer-Verlag, New York.
- Symons, R. C., M. J. Daly, J. Fridlyand, T. P. Speed, W. D. Cook et al., 2002. Multiple genetic loci modify susceptibility to plasmacytoma-related morbidity in Eμ-v-abl transgenic mice. Proc. Natl. Acad. Sci. USA 99 11299–11304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng, Z-B., 1993. Theoretical basis of precision mapping of quantitative trait loci. Proc. Natl. Acad. Sci. USA 90 10972–10976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng, Z-B., 1994. Precision mapping of quantitative traits loci. Genetics 136 1457–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou, F., B. S. Yandell and J. P. Fine, 2001. Statistical issues in the analysis of quantitative traits in combined crosses. Genetics 158 1339–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]