

Abstract
The three mammalian D-type cyclins are thought to promote progression through the G1 phase of the cell cycle as regulatory subunits of cyclin-dependent kinase 4 and 6. In addition, they have been proposed to control the activity of various transcription factors without a partner kinase. Here we describe phenotypic consequences of null mutations in Cyclin D, the single D-type cyclin gene in Drosophila. As previously observed with null mutations in the single Drosophila Cdk4 gene, these mutations do not primarily affect progression through the G1 phase. Moreover, the apparently indistinguishable phenotypes of double (CycD and Cdk4) and single mutants (CycD or Cdk4) argue against major independent functions of Cyclin D and Cdk4. The reduced cellular and organismal growth rates observed in both mutants indicate that Cyclin D-Cdk4 acts as a growth driver.
D-TYPE cyclin genes are present in plant and animal genomes. Their products function as regulatory subunits of specific cyclin-dependent kinases (cdks). Major physiological substrates of the D-type cyclin-cdk complexes are the retinoblastoma tumor suppressor protein family members (Rbfs), which are known to act as negative regulators of cell proliferation in plant and animal cells (Adams 2001; Ortega et al. 2002). Rbfs can bind to E2F transcription factors and recruit complexes with nucleosome remodeling, histone deacetylase, and methylase activities (Stevaux and Dyson 2002). Many E2F target genes are required for progression through the cell cycle and their expression is inhibited by Rbfs (Dimova et al. 2003). Phosphorylation by D-type cyclin-cdk complexes suppresses this inhibitory activity of Rbfs. In addition, mammalian D-type cyclin-cdk complexes can also suppress Rbfs by titrating cdk inhibitors of the CIP/KIP family away from cdk2 complexes, which phosphorylate and thereby contribute to inhibition of Rbfs (Adams 2001; Ortega et al. 2002). Intriguing observations have argued for D-type cyclin-cdk targets other than Rbfs (Datar et al. 2000; Boxem and van den Heuvel 2001; Xin et al. 2002; Frei and Edgar 2004), although clear cases have yet to be demonstrated.
As suggested by a growing number of reports, D-type cyclins are also thought to function independently of either a cdk partner or its kinase activity (Coqueret 2002). Such functions have been described primarily in the context of transcriptional regulation of various differentiation programs. Mammalian cyclin D1 and D2 were shown to inhibit myogenic differentiation through inactivation of MyoD transcriptional activity (Skapek et al. 1995, 1996). Cyclin D1 seems to relocalize cdk4 to the nucleus, leading to an inhibition of the myogenic transcription factor apparently by direct interaction with cdk4 independent of kinase activity (Zhang et al. 1999). More recently cyclin D1 has been shown to bind to Myb, DMP1, STAT3, and Beta2/NeuroD transcription factors and to inhibit their transcriptional activity independent of cdk4 (Ganter et al. 1998; Inoue and Sherr 1998; Horstmann et al. 2000; Bienvenu et al. 2001; Ratineau et al. 2002). In addition, cyclin D1 interacts with nuclear receptors such as the androgen and thyroid hormone receptors and represses their transcriptional activity (Knudsen et al. 1999; Reutens et al. 2001; Lin et al. 2002). In contrast, cyclin D1, but not D2 or D3, have been reported to activate estrogen receptor (ER)-mediated transcription by recruitment of coactivators after direct binding to ER (Zwijsen et al. 1997, 1998).
While it is clear, therefore, that at least some D-type cyclins can effectively modulate transcriptional activation in a cdk-independent manner, the physiological role of these effects remains to be clarified. For instance, the results obtained with knock-in mice in which the cyclin D1 coding regions were replaced by those of cyclin E have questioned the physiological significance of the interaction between cyclin D1 and ER (Geng et al. 1999). In these knock-in mice, cyclin-E expression under cyclin D1 control was found to rescue the mammary gland proliferation defect known to result from loss of cyclin D1 function. As cyclin E lacks the ability to stimulate ER transcriptional activity, proliferation of the mammary epithelium is unlikely to depend on the interaction between ER and cyclin D1 observed after overexpression in transfection experiments.
For the identification of physiologically relevant cdk-independent D-type cyclin functions, a careful comparison of organismal phenotypes resulting from loss of either D-type cyclin or partner cdk gene functions or both is clearly of interest. While genetic redundancy complicates and has so far precluded such analyses in the mouse, which has three different D-type cyclins and two partner kinases, cdk4 and cdk6, Caenorhabditis elegans and Drosophila melanogaster have single genes for cyclin D and its partner kinase cdk4. In C. elegans inactivation of cyd-1 and cdk-4 function by mutations or RNA interference results in similar but not identical phenotypes (Park and Krause 1999; Boxem and van den Heuvel 2001). It remains to be clarified whether the subtle phenotypic differences reflect partially independent cyd-1 and cdk-4 functions or differential stabilities of maternal protein contributions.
In Drosophila, we have isolated Cdk4 null alleles and have demonstrated that some mutant progeny can develop to the adult stage in complete absence of maternal and zygotic Cdk4 function (Meyer et al. 2000). Moreover, in addition to the expected involvement in the E2F-Rbf pathway, our findings demonstrated that Cdk4 is required for normal growth (accumulation of mass) of cells and the organism (Datar et al. 2000; Meyer et al. 2000). Consistent with this, overexpressed Cyclin D (CycD)-Cdk4 complexes stimulate extra growth in many cell types in the fly (Datar et al. 2000). Here we describe the isolation and phenotypic characterization of CycD null mutations. The consequences of these mutations are extremely similar if not identical to those reported for Cdk4 mutations. Moreover, double mutants do not display a more severe phenotype than single mutants. Our results therefore indicate that Drosophila Cyclin D and Cdk4 do not provide independent functions that are vitally important for cell proliferation and differentiation. In addition, they further confirm the crucial role of Cyclin D-Cdk4 in the regulation of cellular growth rates.
MATERIALS AND METHODS
Fly stocks:
The y−, w+, P{y+mDint2, w+BR.E.BR = SUPor-P}KG04817 stock was isolated by the gene disruption project of the Berkeley Drosophila Genome Project (BDGP) and kindly provided by Hugo Bellen (Baylor College of Medicine, Houston). Polymerase chain reaction (PCR) experiments confirmed the presence of a P-element insertion in this stock at the chromosomal position determined by the BDGP. For the mobilization of the transposon, we used CyO, HoP1(w+), a balancer chromosome with a hobo element containing a Δ2–3 P-transposase gene (O'Kane 1998). y−,w+, P{SUPor-P}KG04817 males were therefore crossed with w− y−/FM7i; +/CyO, HoP1(w+) females and the resulting y−,w+, P{SUPor-P}KG04817/FM7i; +/CyO, HoP1(w+) progeny were mated with FM7i/Y males. From the next generation, 258 virgin females carrying the FM7i balancer and lacking the yellow+ marker were selected and mated individually with FM7i/Y males. The y− revertant X chromosome was found to be associated with recessive lethality in only four of the resulting stocks. While we have no hints about the cause of the lethality in two of these cases, our molecular analyses of the two other chromosomes revealed deletions removing sequences of not only CycD but also CG8909, which therefore is likely an essential gene. A total of 161 of the residual stocks with y− revertant X chromosomes that were not associated with lethality were analyzed by PCR for the presence of CycD gene regions close to the P{SUPor-P}KG04817 insertion site. Males carrying revertant chromosomes from six independent stocks failed to yield the corresponding CycD fragment. With analogous PCR assays these six revertant chromosomes were further analyzed for the absence of other CycD gene regions. Finally, by sequence analysis of PCR fragments amplified from the two revertant chromosomes with the largest intragenic CycD deletions, CycD1 and CycD2, we determined the exact breakpoints. In the case of CycD1, the proximal and distal breakpoints were found to be 1011 and 892 bp upstream and downstream of the start of the CycD coding region, respectively, with 29-bp residual P-element sequences in between. The CycD2 breakpoints were 1243 and 337 bp upstream and downstream of the translational start, respectively, with two extra bases in between.
Cdk43, UAS-CycD II.1, da-GAL4 G32, Cdk22, Cdk23, CycEAR95, CycEPZ5, Cdk1E1-23, Cdk1B47, CycAC8LR1, CycB2, CycB33, DPa2, DPa4, E2F191, dap4, and dapg36 have been described previously (Stern et al. 1993; Wodarz et al. 1993; Knoblich et al. 1994; Duronio and O'Farrell 1995; Royzman et al. 1997; Jacobs et al. 1998; Lane et al. 2000; Meyer et al. 2000).
Southern blot and genotyping by PCR:
Genomic DNA from w1, CycD1, or CycD2 flies was isolated and analyzed with a Southern blot according to standard procedures. Probe e and probe i (Figure 1A) were enzymatically amplified from FM7i genomic DNA using either the primer pair SA1 (5′-ATC AAG AGT AAG TTC GTA AGA TCG-3′) and SA2 (5′-CAT TAT ATC GGC CAT ACG TTCC-3′) or HJ30 (5′-GGC CTA AAG TGG CAT CTG-3′) and SA4 (5′-TGA TAT TGG CCA ATC CTA TAG TG-3′) followed by random primer labeling.
Figure 1.—

Mutations in CycD. (A) The genomic region with CycD and the flanking genes CG8909 and shibire (shi) is shown schematically. Exons of the major CycD transcript are indicated by boxes. Solid boxes represent coding regions. The most conserved cyclin box region is shaded. The deleted regions in the alleles CycD1 and CycD2, which were isolated after mobilization of the P-element insertion KG04817, are indicated by solid bars. BspHI (B) and NruI (N) sites relevant for the Southern blot experiment (B) are illustrated. In addition, the hybridization probes derived from sequences either internal (probe i) or external (probe e) of the regions deleted in CycD1 and CycD2 are indicated by black lines. Finally, position and orientation of primers (df, dr, d1r) used for genotype determinations by PCR (see C) are indicated by arrows. (B) Southern blots with genomic DNA from CycD1 (D1), CycD2 (D2), or control w1 flies (+) digested with either NruI or BspHI were hybridized with either probe i or probe e (see A). (C) Genotype determination with a multiplex PCR assay. Genomic DNA from single flies was used as a template for enzymatic amplification with a primer mix (df, dr, d1r, k4f, k4r). The pair df-dr results in amplification of a 630-bp fragment (CycD+) but from only the CycD+ and not from the CycD1 allele (see also A). The pair df-d1r efficiently amplifies a 470-bp fragment from the CycD1 allele (CycD1) and inefficiently amplifies a much larger fragment from CycD+ (not shown). The pair k4f-k4r yields a 400-bp fragment (Cdk4+) from the Cdk4+ but not from the Cdk43allele where the k4r annealing site is eliminated by an intragenic deletion (Meyer et al. 2000). The first four lanes show the PCR products obtained from representative male progeny of a cross of Cdk43 males with CycD1/+; Cdk43/+ females (Figure 3C). The last three lanes show control reactions with genomic DNA from CycD1 or Cdk43 homozygous flies or without genomic DNA template.
For genotype determination of progeny resulting from the cross illustrated in Figure 3C, genomic DNA from single flies was prepared and used as a template in a multiplex PCR containing the primers df (5′-GTA CAG GAT CTT TAA GTG CAG C-3′), dr (5′-ATC TCT TGC TCA CTG CGA TCA G-3′), d1r (5′-CGA CTT AGC ACA TAC AGC TCC-3′), k4f (5′-GAG AAC GGT GTG CCA ATG-3′), and k4r (5′-GAG CGG ATC GAC TTG CTT CAG-3′).
Figure 3.—
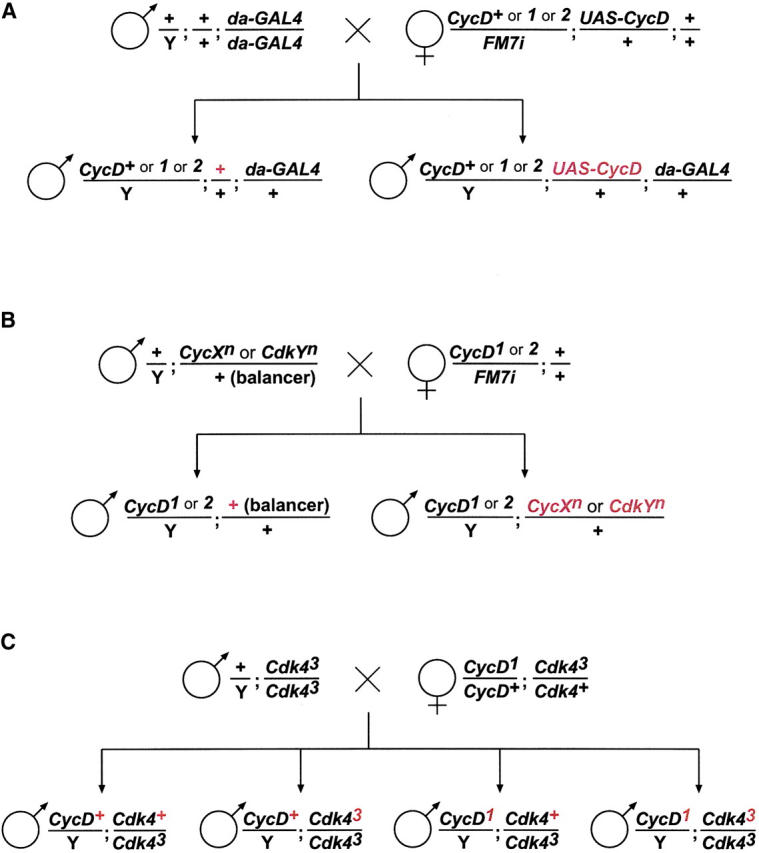
Genotype constructions: the crossing schemes realized for the phenotypic rescue experiments (A; see also Tables 1 and 2) for the analysis of genetic interactions between mutations in CycD and other cyclin/cdk genes (B; see also Table 3), as well as for the comparison of single- and double-mutant phenotypes (C; see also Table 4). Only the genotype classes of progeny analyzed phenotypically are indicated, and crucial genetic differences between them are emphasized by red print.
Clonal analyses:
Meiotic recombination was used for the construction of X chromosomes carrying P{neoFRT}18A (Xu and Rubin 1993) and either the CycD1 or the CycD2 mutation. These FLP recombinase target site (FRT) chromosomes were crossed over an FRT chromosome with P{Ubi-GFP(S65T)nls}X and P{neoFRT}18A. In addition, P{hsFLP}86E was crossed in and mitotic recombination between the X chromosomes was induced by a heat shock (25 min in a 37° water bath). Resulting clones in wing-imaginal discs were analyzed as described previously (Neufeld et al. 1998; Datar et al. 2000). Parallel control experiments were done with larvae carrying an X chromosome with P{neoFRT}18A and the wild-type CycD allele over P{Ubi-GFP(S65T)nls}X P{neoFRT}18A. Anticleaved Caspase-3 antibodies (ASP 175) were obtained from Cell Signaling Technology and used at 1:1000.
In situ hybridization and BrdU pulse labeling:
Embryos collected from stocks with CycD1 or CycD2 over the FM7c, P{ry+t7.2 = ftz/lacC}YH1 blue balancer chromosome were fixed as previously described (White 1998). For pulse labeling with 5-bromo-deoxiuridine (BrdU), embryos were permeabilized with octane and incubated for 20 min in Schneider's medium containing 1 mg/ml BrdU before fixation (Knoblich et al. 1994). Immunolabeling with mouse monoclonal antibodies against BrdU (Becton-Dickinson) in combination with secondary goat anti-mouse antibodies conjugated with Cy3 (Jackson), as well as in situ hybridizations using red fluorescent probe detection after tyramide signal amplification (TSA kit, NEN) were performed as previously described (Knirr et al. 1999; Meyer et al. 2002). For the identification of CycD mutant embryos, we applied double labeling with rabbit antibodies against β-galactosidase (ICN, Cappel) in combination with secondary goat anti-rabbit antibodies conjugated with Alexa488 (Molecular Probes, Eugene, OR).
Analysis of fly weight and wings:
Newly eclosed progeny were collected every 24 hr, transferred into fresh yeasted vials, and cultured for an additional 3 days before freezing at −70° in tightly capped vials for a maximum of 3 days. After warming to room temperature, individual flies were weighed on a AG135 Mettler Toledo balance. Wings were dissected, deposited on a slide, and covered with a coverslip, which was fixed with nail polish after gentle flattening of the preparation with a weight. Microscopic images were captured with a CCD camera. The total wing area and the wing-hair count in a defined area of constant size was determined using IPLab software. An average cell size was estimated by dividing the analyzed area by the obtained wing-hair count. The total number of cells in the wing was estimated by multiplying the wing-hair count obtained in the defined area with the ratio between this defined area and the total wing area.
RESULTS
For the isolation of mutant CycD alleles we mobilized a P-element P{SUPor-P}KG04817 located on the X chromosome within an intergenic region ∼470 bp upstream of the major transcriptional start site of CycD (Figure 1A). Molecular characterizations by PCR, DNA sequencing, and Southern blotting (Figure 1B; see also materials and methods) revealed the presence of small deletions in two of the resulting revertant chromosomes. These deletions, CycD1 and CycD2, remove the promoter and parts of the coding region and are therefore considered to represent CycD null alleles (Figure 1). We emphasize that even the region encoding the conserved cyclin box, which mediates the association with Cdk4, is partially deleted in CycD1.
The CycD null alleles did not interfere with development of hemizygous males and homozygous females to the adult stage. The fertility of these mutant adults was severely reduced, however, precluding their propagation as a stock. Nevertheless, a few adult progeny could be obtained from CycD1 or CycD2 mutant parents, indicating that development of adults in complete absence of maternal and zygotic CycD+ function is not absolutely impossible.
CycD mutant flies were significantly smaller than CycD+ siblings. For quantification we measured the weight of hemizygous male flies. On average these males weighed 10–20% less than CycD+ control males (Tables 1 and 4). To confirm that this weight deficit resulted from a lack of CycD+ function, we ubiquitously expressed a UAS-CycD transgene in the CycD mutants as well as in CycD+ control males using da-GAL4. The resulting UAS-CycD expression was found to eliminate the weight differences between CycD+ and CycD mutant males (Table 1).
TABLE 1.
Reduced weight ofCycD mutants
| Weightb
|
|||
|---|---|---|---|
| Crossa | Genotype of male progeny | (mg) | (%) |
| 1 | CycD+; +; da-GAL4 | 0.75 ± 0.07 | 100 |
| 1 | CycD+; UAS-CycD; da-GAL4 | 0.83 ± 0.07 | 111 |
| 2 | CycD1; +; da-GAL4 | 0.64 ± 0.06 | 85 |
| 2 | CycD1; UAS-CycD; da-GAL4 | 0.83 ± 0.07 | 111 |
| 3 | CycD2; +; da-GAL4 | 0.70 ± 0.08 | 93 |
| 3 | CycD2; UAS-CycD; da-GAL4 | 0.89 ± 0.07 | 118 |
Three crosses were set up as illustrated in Figure 3A. Females with the following genotypes were used: +/FM7i; UAS-CycD/+ (cross 1), CycD1/FM7i; UAS-CycD/+ (cross 2), and CycD2/FM7i; UAS-CycD/+ (cross 3).
At least 70 flies for each genotype were weighed individually before calculation of an average weight ± standard deviation. The average weight obtained for CycD+; +; da-GAL4 was set as 100%. The average weight difference of siblings with or without UAS-CycD expression was significant in all three crosses according to t-tests (P < 10−10).
The reduced size of CycD mutants was also confirmed by an analysis of wing areas (Table 2). Moreover, since every wing cell forms a microscopically detectable hair, cell numbers present in these wings can readily be estimated. These were found to be reduced in CycD mutants (Table 2). Moreover, the ratios between total wing area and cell number indicated that the size of the differentiated cells in CycD mutant wings is slightly increased (Table 2).
TABLE 2.
Wing area, cell number, and cell size inCycD mutants
| Wing areab
|
|||||
|---|---|---|---|---|---|
| Crossa | Genotype of male progeny | (mm2) | (%) | Cell numberb (%) | Cell sizeb (%) |
| 1 | CycD+; +; da-GAL4 | 1.02 ± 0.05 | 100 | 100 | 100 |
| 1 | CycD+; UAS-CycD; da-GAL4 | 1.07 ± 0.04 | 105 | 113 | 93 |
| 2 | CycD1; +; da-GAL4 | 0.90 ± 0.06 | 88 | 85 | 105 |
| 2 | CycD1; UAS-CycD; da-GAL4 | 1.05 ± 0.05 | 103 | 114 | 91 |
| 3 | CycD2; +; da-GAL4 | 0.95 ± 0.05 | 93 | 92 | 109 |
| 3 | CycD2; UAS-CycD; da-GAL4 | 1.11 ± 0.04 | 109 | 118 | 93 |
Three crosses were set up as illustrated in Figure 3A. Females with the following genotypes were used: +/FM7i; UAS-CycD/+ (cross 1), CycD1/FM7i; UAS-CycD/+ (cross 2), and CycD2/FM7i; UAS-CycD/+ (cross 3).
At least 12 wings were analyzed for each genotype (see materials and methods). The values obtained for CycD+; +; da-GAL4 were set as 100%.
To analyze the effects of loss of CycD+ function on cell growth in proliferating cells, we induced CycD−/− and CycD+/+ sister clones simultaneously by Hs-Flp-mediated mitotic recombination in a heterozygous FRT CycD−/FRT CycD+ Ub-GFP background at ∼60 hr after egg deposition. An additional 60 hr after clone induction, wing-imaginal discs were dissected and the area of Ub-GFP+/+ (CycD−/−) and Ub-GFP−/− (CycD+/+) sister clones was measured and compared (Figure 2A). In three independent experiments, CycD1 clones were found to cover only ∼50% of the area encompassed by the control sister clones. CycD2 clones covered ∼60% of the control area in two experiments. In contrast, in three control experiments with an FRT CycD+/FRT CycD+, Ub-GFP background we did not detect significant size differences between twin spots. To determine whether the decreased size of CycD mutant clones was due to increased cell death, we used a DNA stain to assess the abundance of condensed, fragmented nuclei, which are characteristic of apoptotic cells. No increase in apoptotic figures was detected. We also assayed apoptosis by immunofluorescence, using an antibody to cleaved Caspase-3. This analysis also failed to reveal increased numbers of apoptotic cells associated with the CycD1 mutant clones. To analyze the effects of loss of CycD+ function on cell size and the cell-cycle profile, we measured forward scatter and DNA content of wing-imaginal disc cells after CycD−/− clone induction and fluorescence activated cell sorting (FACS) of GFP− and GFP+ cells (Figure 2, B and C). These experiments failed to detect significant differences in cell size or cell-cycle phasing in the CycD mutant cells. In addition, the FACS analysis did not detect a significant fraction of sub-G1 cells, which would be expected if loss of CycD caused high levels of apoptosis. These findings indicate that CycD+ function is required for normal cell growth in proliferating wing-imaginal disc cells. Moreover, reduced growth in CycD1 mutant cells appears to be accompanied by a proportional delay of cell-cycle progression during G1, S, and G2 phases, since neither cell size nor cell-cycle profile was altered.
Figure 2.—
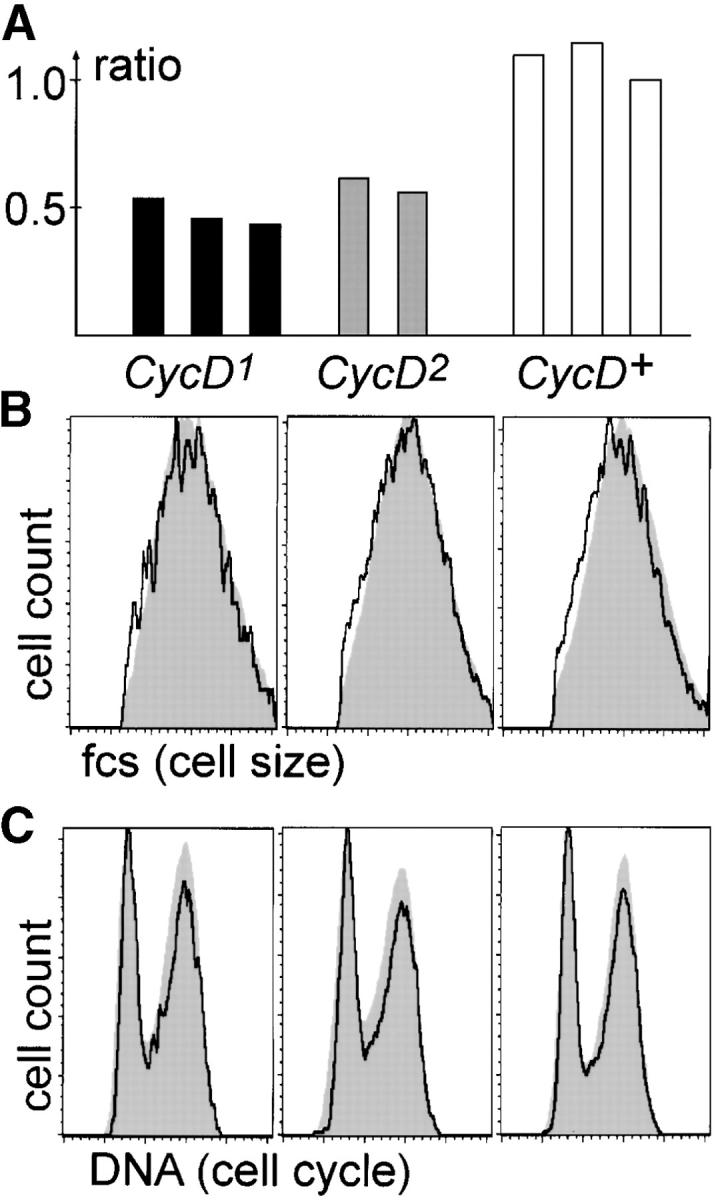
Reduced growth of CycD mutant clones. (A) Mitotic recombination was induced 60 hr after egg deposition by Hs-Flp expression in CycD1 FRT/Ub-GFP FRT (CycD1, solid bars), CycD2 FRT/Ub-GFP FRT (CycD2, shaded bars), or + FRT/Ub-GFP FRT (CycD+, open bars) larvae. Wing-imaginal discs were dissected 60 hr after clone induction and the ratio between the area covered by GFP−/− and GFP+/+ sister clones was determined. Each bar gives the ratio from an independent experiment after analysis of at least three imaginal discs with multiple twin clones. (B and C) Mitotic clones were induced as in A, followed by dissociation of imaginal discs and analysis of sorted GFP−/− (black tracings) and GFP+/+ (shaded areas) cells by flow cytometry measuring either forward scatter as an estimate for cell size (B) or DNA content (C).
To monitor cell-cycle progression during embryogenesis, we performed BrdU pulse labeling. The pattern and intensity of BrdU incorporation during embryogenesis revealed that zygotic CycD+ function is particularly important for endoreduplication. During wild-type embryogenesis, endoreduplication occurs during late stages in a spatially and temporally defined pattern in most internal organs (Smith and Orr-Weaver 1991). In CycD mutants, endoreduplication is not initiated on time in the central domain of the midgut (Figure 4, compare C and F). Moreover, the preceding endoreduplication within the anterior and posterior midgut domains is inefficient and not completed on time (Figure 4, compare C and F). In contrast, BrdU incorporation in the mitotically proliferating cells within the central nervous system appeared to be normal during these stages. In situ hybridization with probes for the E2F target genes (Duronio et al. 1995) encoding the ribonucleotide reductase subunit RnrS (Figure 4, A and D) and Cyclin E (Figure 4, B and E) indicated that transcript levels of these S-phase genes are decreased predominantly within endoreduplicating tissues.
Figure 4.—

S-phase gene expression and progression in CycD mutants. CycD+ (A–C) and CycD1 (D–F) sibling embryos were analyzed either by in situ hybridization for expression of RnrS (A and D) or CycE (B and E) or by pulse labeling with BrdU (C and F). In CycD mutants, expression of S-phase genes is particularly weak within the endoreduplicating midgut at stage 13 (arrowheads in A and D) and stage 14 (arrow in B and E). In addition, BrdU pulse labeling indicates that endoreduplication is not initiated in the central midgut domain (arrows in C and F) and not completed in the anterior and posterior midgut domains (arrowheads) on schedule in CycD mutants.
Functional redundancies might explain the relatively minor consequences observed to result from a loss of CycD+ function. In particular, Cyclin E-Cdk2 is thought to provide functions similar to those of Cyclin D-Cdk4. To evaluate this possibility, we analyzed the effects of heterozygosity for loss-of-function mutations in various cell-cycle genes on the viability of CycD mutants (Table 3). We observed a clear effect with mutations in Cdk2 and CycE. In particular, heterozygosity for Cdk2 mutations eliminated the viability of CycD mutant males almost completely. Heterozygosity for CycE mutations reduced CycD mutant male viability as well, but less severely. Moreover, surviving CycD mutant males displayed a rough eye phenotype when heterozygous for a CycE mutation (data not shown). In contrast to the strong synthetic effects of CycE and Cdk2 mutations, we did not observe clear effects on CycD mutant viability with mutations in genes encoding Cdk1 and its cyclin subunits (CycA, CycB, CycB3; Table 3). Similarly, we did not observe clear genetic interactions between mutations in CycD and dacapo or E2F1 (Table 3). Finally, mutations in DP resulted in a slight reduction of viability (Table 3).
TABLE 3.
Genetic interactions ofCycD mutations
| Frequency of heterozygous CycD mutant male progeny (%)a |
|||
|---|---|---|---|
| Gene | Allele | CycD1 | CycD2 |
| Cdk2 | 2 | 6 | 2 |
| 3 | 0 | NDb | |
| CycE | AR95 | 35 | 18 |
| PZ5 | 61 | 46 | |
| Cdk1 | E1-23 | 103 | ND |
| B47 | 103 | ND | |
| CycA | C8LR1 | 89 | 105 |
| CycB | 2 | 110 | 90 |
| CycB3 | 3 | 114 | ND |
| dap | 4 | 128 | 98 |
| g36 | 100 | 97 | |
| E2F1 | 91 | 112 | 126 |
| DP | a2 | 64 | 75 |
| a4 | 67 | 61 | |
For an analysis of genetic interactions between mutations in CycD and genes encoding other cyclins (CycA, CycB, CycB3, CycE) or cdks (Cdk1, Cdk2), crosses were set up as illustrated in Figure 3B. In addition, alleles of E2F1, DP, and dacapo (dap) were tested analogously. After eclosion of progeny, the ratio of CycD mutant males, which were heterozygous either for another cell-cycle gene mutation or for a balancer chromosome, was determined and given after multiplication with 100. The resulting values are expected to be 100, if the mutation in CycD and the cell-cycle gene mutation analyzed do not interact. Lower values indicate that the viability of the CycD mutant males is reduced by heterozygosity for the additional cell-cycle gene mutation. At least 200 CycD mutant males were analyzed from each cross.
ND, not done.
Our characterization of CycD mutant phenotypes, described above, yielded results very similar to those found for Cdk4 mutants (Datar et al. 2000; Meyer et al. 2000, 2002). These extensive phenotypic similarities suggest that Cyclin D and Cdk4 provide their major functions exclusively together in a complex. Association of Drosophila Cyclin D and Cdk4 was first suggested by yeast two-hybrid experiments (Finley et al. 1996; Sauer et al. 1996) and later confirmed by co-immunoprecipitation experiments (Meyer et al. 2000). If Cyclin D and Cdk4 function exclusively as a complex, double-mutant phenotypes would be predicted to correspond to the single-mutant phenotypes. In contrast, if Cyclin D and Cdk4 also provided some major functions independently, double mutants would be expected to have a more severe phenotype than single mutants. For a careful comparison of single and double mutants, we analyzed readily quantifiable aspects including viability, body weight, wing area, wing cell number, and size. Moreover, this experiment was designed such that all the different genotypes developed as siblings with the same maternal contribution under identical growth conditions. In addition, for the determination of the different sibling genotypes, we did not rely on adult visible phenotypes induced by dominant marker mutations on balancer chromosomes with potential side effects on the analyzed parameters. Genotypes were therefore determined with a PCR assay (Figure 1C) after completion of the phenotypic analyses. The comparison of single- and double-mutant phenotypes did not reveal major differences (Table 4). A second experiment gave very similar results (data not shown) although in this case the fraction of eclosing double mutants and their average weight was slightly lower compared to single mutants. In both experiments, however, all the differences between weight, wing size, and cell numbers of single and double mutants were not statistically significant (P-values obtained with t-test >0.05), while the differences between mutants and CycD+; Cdk4+ siblings, which also tended to eclose slightly faster than the mutants, were highly significant. Our findings therefore fail to provide evidence for independent functions of Cyclin D or Cdk4.
TABLE 4.
Comparison ofCycD,Cdk4 single and double mutants
| Weightc
|
Wing aread
|
||||||
|---|---|---|---|---|---|---|---|
| Genotypea CycD; Cdk4 |
Survivalb (%) | (mg) | (%) | (mm2) | (%) | Cell numberd (%) | Cell sized (%) |
| +; + | 100 | 0.85 ± 0.08 | 100 | 1.13 ± 0.04 | 100 | 100 | 100 |
| +; − | 87 | 0.69 ± 0.07 | 81 | 1.06 ± 0.02 | 94 | 88 | 107 |
| −; + | 100 | 0.71 ± 0.07 | 84 | 1.01 ± 0.03 | 89 | 88 | 108 |
| −; − | 91 | 0.71 ± 0.06 | 84 | 1.03 ± 0.06 | 91 | 83 | 111 |
For comparison of single- and double-mutant phenotypes, a cross was set up as illustrated in Figure 3C, resulting in male progeny hemizygous for either the CycD+ (indicated by +) or the CycD1 allele (indicated by −). Moreover, the males were either heterozygous (indicated by +) or homozygous (indicated by −) for the Cdk43 allele. The genotype of individual male progeny was determined with a PCR assay (Figure 1C) following measurement of fly weight, wing size, and wing-hair density.
For the comparison of the developmental fitness associated with the four different genotypes expected to segregate with equal frequency according to Mendelian rules, all 170 eclosing males from a cross were genotyped. The number of progeny males that survived to the adult stage is given relative to the number of +; + males, which was set as 100%.
All males were weighed individually before calculation of an average weight ± standard deviation. The average weight of +; + males was set as 100%.
At least five wings were analyzed for each genotype (see materials and methods). The values obtained with +; + males were set as 100%.
DISCUSSION
D-type cyclin-cdk complexes are of crucial importance in human tumorigenesis. Since these complexes have been conserved in evolution, it is readily possible to use model organisms like D. melanogaster for functional characterizations. Here we extend our previous characterization of Drosophila Cdk4 mutants by phenotypic comparisons with CycD mutants. As previously observed for Cdk4 (Meyer et al. 2000), we find that Cyclin D is not required for progression through the G1 phase of the cell cycle. Some escapers develop to the adult stage even when both maternal and zygotic Cdk4+ or CycD+ function is abolished. Moreover, FACS analyses demonstrate that the cell-cycle profile of wing-imaginal disc cells homozygous for null mutations in Cdk4 (Meyer et al. 2000) or CycD (Figure 2B) is essentially indistinguishable from that of wild type. Our evidence therefore is not consistent with the prevailing idea that D-type cyclin-cdk complexes primarily regulate progression through the G1 phase. In cultured mammalian cells, where the most support for this suggestion has accumulated, D-type cyclin-cdk complexes have been shown to act in part by titrating CIP/KIP inhibitors away from CycE/Cdk2 complexes, which are thus freed to stimulate cell-cycle progression (Adams 2001; Ortega et al. 2002). In contrast, binding of Dacapo, the single known Drosophila CIP/KIP family member, to Drosophila Cyclin D-Cdk4 has not been detectable (Meyer et al. 2000). This provides a potential explanation for the apparent discrepancy. It should be noted, however, that the strong genetic interactions we report among CycD, CycE, and Cdk2 (Table 3), previous interaction tests performed with Rbf (Datar et al. 2000; Xin et al. 2002), and target gene analysis (B. Lynch, A. F. A. de la Cruz and B. A. Edgar, unpublished results) indicate that Drosophila CyclinD-Cdk4 complexes do play a significant, if redundant, activating role in the E2F/RBF network, just as described in mammals. In the CycD and Cdk4 mutants, CycE/Cdk2 complexes are presumably sufficient to perform this function.
While not revealing a specific role during G1, the Drosophila mutant phenotypes provide compelling evidence that Cyclin D-Cdk4 promotes cellular growth and thereby accelerates progression through all the cell-cycle phases proportionally. CycD and Cdk4 mutants develop into small but normally proportioned flies with an average weight of ∼20% less than that of wild-type siblings (Meyer et al. 2000; this work). Conversely, overexpression of Cyclin D and Cdk4 has the opposite effect, causing increased growth in organs such as the eye, wing, and salivary glands (Datar et al. 2000). Moreover, growth regulation by Cyclin D and Cdk4 is also clearly apparent at the cellular level. Clones of wing-imaginal disc cells either lacking one of the Cyclin D-Cdk4 complex partners or overexpressing the complex grow slower or faster, respectively, than wild-type clones (Datar et al. 2000; Meyer et al. 2000; this work).
In Drosophila, the growth-promoting function of Cyclin D-Cdk4 can be interrogated using genetic approaches. Initial results have so far argued that Cyclin D-Cdk4 is not part of one of the other pathways (insulin/TOR, ras, myc, bantam), which are known to control cellular and organismal growth rates (Frei and Edgar 2004). However, we have recently identified the Hif-1 prolyl hydroxylase as a key growth stimulator downstream of Cyclin D-Cdk4 (Frei and Edgar 2004). This raises the possibility that Cyclin D-Cdk4 is interconnected with metabolic pathways sensitive to oxygen levels (Lavista-Llanos et al. 2002; Frei and Edgar 2004). Future analyses might therefore reveal whether an involvement in oxygen-related metabolism represents the evolutionary conserved role of Cyclin D-Cdk4 in multicellular eukaryotes and throw a new light on its significance in human tumors, where oxygen limitation is a known and crucial challenge. Cyclin D-Cdk4 has also recently been implicated in the JAK-STAT pathway by an independent genetic approach in Drosophila (Chen et al. 2003).
Our comparison of CycD and Cdk4 mutant phenotypes is also of interest with regard to functions provided by these proteins independently. In particular, D-type cyclins have been proposed to regulate a number of transcription factors without a partner kinase (see Introduction). Moreover, overexpression of UAS-CycD alone or UAS-Cdk4 alone does often have phenotypic consequences that vary in extent with different GAL4 driver lines. For instance, ey-GAL4-driven UAS-CycD expression suppresses the inhibitory effects of simultaneous UAS-RBF1 expression dramatically (Datar et al. 2000). Similarly, da-GAL4-driven UAS-CycD expression during development of otherwise wild-type flies results in an increased adult fly weight (Table 1). However, in these experiments in Drosophila, the overexpressed Cyclin D might execute its effect in combination with excess Cdk4 expressed from the endogenous Cdk4 gene, as suggested by the finding that da-GAL4-driven UAS-CycD expression in Cdk4 mutants increases adult fly weight at most marginally (data not shown). Our findings that loss of CycD+ or Cdk4+ function, as well as simultaneous loss of both CycD+ and Cdk4+ function, results in essentially indistinguishable phenotypes and does not necessarily prevent development to the adult stage, demonstrate that neither Cyclin D nor Cdk4 provides essential functions in Drosophila independently of each other.
Acknowledgments
We are indebted to Henning Jacobs for informing us about the isolation of KG04817 by the Berkeley Drosophila Genome Project and to Hugo Bellen for sending us this line. We thank Sandra Szameit for help during the initial isolation and characterization of the CycD alleles. Work in the laboratory of C.F.L. was supported by grants from the Deutsche Forschungsgemeinschaft (DFG Le 987/1-3 and 2-1) and the Fonds der Chemischen Industrie. Work in the laboratory of B.A.E. was supported by National Institutes of Health grant R01 GM61805.
References
- Adams, P. D., 2001. Regulation of the retinoblastoma tumor suppressor protein by cyclin/cdks. Biochim. Biophys. Acta 1471: M123–M133. [DOI] [PubMed] [Google Scholar]
- Bienvenu, F., H. Gascan and O. Coqueret, 2001. Cyclin D1 represses STAT3 activation through a Cdk4-independent mechanism. J. Biol. Chem. 276: 16840–16847. [DOI] [PubMed] [Google Scholar]
- Boxem, M., and S. van den Heuvel, 2001. lin-35 Rb and cki-1 Cip/Kip cooperate in developmental regulation of G1 progression in C. elegans. Development 128: 4349–4359. [DOI] [PubMed] [Google Scholar]
- Chen, X., S. W. Oh, Z. Zheng, H. W. Chen, H. H. Shin et al., 2003. Cyclin D-Cdk4 and cyclin E-Cdk2 regulate the Jak/STAT signal transduction pathway in Drosophila. Dev. Cell 4: 179–190. [DOI] [PubMed] [Google Scholar]
- Coqueret, O., 2002. Linking cyclins to transcriptional control. Gene 299: 35–55. [DOI] [PubMed] [Google Scholar]
- Datar, S. A., H. W. Jacobs, A. F. de la Cruz, C. F. Lehner and B. A. Edgar, 2000. The Drosophila cyclin D-Cdk4 complex promotes cellular growth. EMBO J. 19: 4543–4554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimova, D. K., O. Stevaux, M. V. Frolov and N. J. Dyson, 2003. Cell cycle-dependent and cell cycle-independent control of transcription by the Drosophila E2F/RB pathway. Genes Dev. 17: 2308–2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duronio, R. J., and P. H. O'Farrell, 1995. Developmental control of the G1 to S transition in Drosophila—cyclin E is a limiting downstream target of E2F. Genes Dev. 9: 1456–1468. [DOI] [PubMed] [Google Scholar]
- Duronio, R. J., P. H. O'Farrell, J. E. Xie, A. Brook and N. Dyson, 1995. The transcription factor E2f is required for S phase during Drosophila embryogenesis. Genes Dev. 9: 1445–1455. [DOI] [PubMed] [Google Scholar]
- Finley, Jr., R. L., B. J. Thomas, S. L. Zipursky and R. Brent, 1996. Isolation of Drosophila cyclin D, a protein expressed in the morphogenetic furrow before entry into S phase. Proc. Natl. Acad. Sci. USA 93: 3011–3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frei, C., and B. A. Edgar, 2004. Drosophila Cyclin D/Cdk4 requires Hif-1 prolyl hydroxylase (Hph) to drive cell growth. Dev. Cell 6: 241–251. [DOI] [PubMed] [Google Scholar]
- Ganter, B., S. Fu and J. S. Lipsick, 1998. D-type cyclins repress transcriptional activation by the v-Myb but not the c-Myb DNA-binding domain. EMBO J. 17: 255–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng, Y., W. Whoriskey, M. Y. Park, R. T. Bronson, R. H. Medema et al., 1999. Rescue of cyclin D1 deficiency by knockin cyclin E. Cell 97: 767–777. [DOI] [PubMed] [Google Scholar]
- Horstmann, S., S. Ferrari and K. H. Klempnauer, 2000. Regulation of B-Myb activity by cyclin D1. Oncogene 19: 298–306. [DOI] [PubMed] [Google Scholar]
- Inoue, K., and C. J. Sherr, 1998. Gene expression and cell cycle arrest mediated by transcription factor DMP1 is antagonized by D-type cyclins through a cyclin-dependent-kinase-independent mechanism. Mol. Cell. Biol. 18: 1590–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs, H. W., J. A. Knoblich and C. F. Lehner, 1998. Drosophila Cyclin B3 is required for female fertility and is dispensable for mitosis like Cyclin B. Genes Dev. 12: 3741–3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knirr, S., N. Azpiazu and M. Frasch, 1999. The role of the NK-homeobox gene slouch (S59) in somatic muscle patterning. Development 126: 4525–4535. [DOI] [PubMed] [Google Scholar]
- Knoblich, J. A., K. Sauer, L. Jones, H. Richardson, R. Saint et al., 1994. Cyclin E controls S phase progression and its downregulation during Drosophila embryogenesis is required for the arrest of cell proliferation. Cell 77: 107–120. [DOI] [PubMed] [Google Scholar]
- Knudsen, K. E., W. K. Cavenee and K. C. Arden, 1999. D-type cyclins complex with the androgen receptor and inhibit its transcriptional transactivation ability. Cancer Res. 59: 2297–2301. [PubMed] [Google Scholar]
- Lane, M. E., M. Elend, D. Heidmann, A. Herr, S. Marzodko et al., 2000. A screen for modifiers of cyclin E function in Drosophila melanogaster identifies Cdk2 mutations, revealing the insignificance of putative phosphorylation sites in Cdk2. Genetics 155: 233–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavista-Llanos, S., L. Centanin, M. Irisarri, D. M. Russo, J. M. Gleadle et al., 2002. Control of the hypoxic response in Drosophila melanogaster by the basic helix-loop-helix PAS protein similar. Mol. Cell. Biol. 22: 6842–6853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, H. M., L. Zhao and S. Y. Cheng, 2002. Cyclin D1 is a ligand-independent co-repressor for thyroid hormone receptors. J. Biol. Chem. 277: 28733–28741. [DOI] [PubMed] [Google Scholar]
- Meyer, C. A., H. W. Jacobs, S. A. Datar, W. Du, B. A. Edgar et al., 2000. Drosophila Cdk4 is required for normal growth and is dispensable for cell cycle progression. EMBO J. 19: 4533–4542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer, C. A., H. W. Jacobs and C. F. Lehner, 2002. Cyclin D-Cdk4 is not a master regulator of cell multiplication in Drosophila embryos. Curr. Biol. 12: 661–666. [DOI] [PubMed] [Google Scholar]
- Neufeld, T. P., A. F. de la Cruz, L. A. Johnston and B. A. Edgar, 1998. Coordination of growth and cell division in the Drosophila wing. Cell 93: 1183–1193. [DOI] [PubMed] [Google Scholar]
- O'Kane, C. J., 1998 Enhancer traps, pp. 131–178 in Drosophila: A Practical Approach, edited by D. B. Roberts. Oxford University Press, Oxford.
- Ortega, S., M. Malumbres and M. Barbacid, 2002. Cyclin D-dependent kinases, INK4 inhibitors and cancer. Biochim. Biophys. Acta 1602: 73–87. [DOI] [PubMed] [Google Scholar]
- Park, M., and M. W. Krause, 1999. Regulation of postembryonic G1 cell cycle progression in Caenorhabditis elegans by a cyclin D/CDK-like complex. Development 126: 4849–4860. [DOI] [PubMed] [Google Scholar]
- Ratineau, C., M. W. Petry, H. Mutoh and A. B. Leiter, 2002. Cyclin D1 represses the basic helix-loop-helix transcription factor, BETA2/NeuroD. J. Biol. Chem. 277: 8847–8853. [DOI] [PubMed] [Google Scholar]
- Reutens, A. T., M. Fu, C. Wang, C. Albanese, M. J. McPhaul et al., 2001. Cyclin D1 binds the androgen receptor and regulates hormone-dependent signaling in a p300/CBP-associated factor (P/CAF)-dependent manner. Mol. Endocrinol. 15: 797–811. [DOI] [PubMed] [Google Scholar]
- Royzman, I., A. J. Whittaker and T. L. Orr-Weaver, 1997. Mutations in Drosophila DP and E2F distinguish G1-S progression from an associated transcriptional program. Genes Dev. 11: 1999–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer, K., K. Weigmann, S. Sigrist and C. F. Lehner, 1996. Novel members of the cdc2-related kinase family in Drosophila: cdk4/6, cdk5, PFTAIRE, and PITSLRE kinase. Mol. Biol. Cell 7: 1759–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skapek, S. X., J. Rhee, D. B. Spicer and A. B. Lassar, 1995. Inhibition of myogenic differentiation in proliferating myoblasts by cyclin D1-dependent kinase. Science 267: 1022–1024. [DOI] [PubMed] [Google Scholar]
- Skapek, S. X., J. Rhee, P. S. Kim, B. G. Novitch and A. B. Lassar, 1996. Cyclin-mediated inhibition of muscle gene expression via a mechanism that is independent of pRB hyperphosphorylation. Mol. Cell. Biol. 16: 7043–7053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, A. V., and T. L. Orr-Weaver, 1991. The regulation of the cell cycle during Drosophila embryogenesis—the transition to polyteny. Development 112: 997–1008. [DOI] [PubMed] [Google Scholar]
- Stern, B., G. Ried, N. J. Clegg, T. A. Grigliatti and C. F. Lehner, 1993. Genetic analysis of the Drosophila cdc2 homolog. Development 117: 219–232. [DOI] [PubMed] [Google Scholar]
- Stevaux, O., and N. J. Dyson, 2002. A revised picture of the E2F transcriptional network and RB function. Curr. Opin. Cell Biol. 14: 684–691. [DOI] [PubMed] [Google Scholar]
- White, R. A. H., 1998 Immunolabelling of Drosophila, pp. 215–240 in Drosophila: A Practical Approach, edited by D. B. Roberts. Oxford University Press, Oxford.
- Wodarz, A., F. Grawe and E. Knust, 1993. Crumbs is involved in the control of apical protein targeting during Drosophila epithelial development. Mech. Dev. 44: 175–187. [DOI] [PubMed] [Google Scholar]
- Xin, S., L. Weng, J. Xu and W. Du, 2002. The role of RBF in developmentally regulated cell proliferation in the eye disc and in Cyclin D/Cdk4 induced cellular growth. Development 129: 1345–1356. [DOI] [PubMed] [Google Scholar]
- Xu, T., and G. M. Rubin, 1993. Analysis of genetic mosaics in developing and adult Drosophila tissues. Development 117: 1223–1237. [DOI] [PubMed] [Google Scholar]
- Zhang, J. M., Q. Wei, X. Zhao and B. M. Paterson, 1999. Coupling of the cell cycle and myogenesis through the cyclin D1-dependent interaction of MyoD with cdk4. EMBO J. 18: 926–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwijsen, R. M., E. Wientjens, R. Klompmaker, J. van der Sman, R. Bernards et al., 1997. CDK-independent activation of estrogen receptor by cyclin D1. Cell 88: 405–415. [DOI] [PubMed] [Google Scholar]
- Zwijsen, R. M., R. S. Buckle, E. M. Hijmans, C. J. Loomans and R. Bernards, 1998. Ligand-independent recruitment of steroid receptor coactivators to estrogen receptor by cyclin D1. Genes Dev. 12: 3488–3498. [DOI] [PMC free article] [PubMed] [Google Scholar]