

Abstract
To identify additional cell fusion genes in Saccharomyces cerevisiae, we performed a high-copy suppressor screen of fus2Δ. Higher dosage of three genes, BEM1, LRG1, and FUS1, partially suppressed the fus2Δ cell fusion defect. BEM1 and FUS1 were high-copy suppressors of many cell-fusion-defective mutations, whereas LRG1 suppressed only fus2Δ and rvs161Δ. Lrg1p contains a Rho-GAP homologous region. Complete deletion of LRG1, as well as deletion of the Rho-GAP coding region, caused decreased rates of cell fusion and diploid formation comparable to that of fus2Δ. Furthermore, lrg1Δ caused a more severe mating defect in combination with other cell fusion mutations. Consistent with an involvement in cell fusion, Lrg1p localized to the tip of the mating projection. Lrg1p-GAP domain strongly and specifically stimulated the GTPase activity of Rho1p, a regulator of β(1-3)-glucan synthase in vitro. β(1-3)-glucan deposition was increased in lrg1Δ strains and mislocalized to the tip of the mating projection in fus2Δ strains. High-copy LRG1 suppressed the mislocalization of β(1-3) glucan in fus2Δ strains. We conclude that Lrg1p is a Rho1p-GAP involved in cell fusion and speculate that it acts to locally inhibit cell wall synthesis to aid in the close apposition of the plasma membranes of mating cells.
CELL fusion is a widespread eukaryotic phenomenon that is necessary for fertilization (see reviews of Wassarman et al. 2001; Talbot et al. 2003) and developmental processes including muscle, placenta, and bone formation (see reviews of Shemer and Podbilewicz 2000; Taylor 2000; Abmayr et al. 2003; Shemer and Podbilewicz 2003). Cell fusion has also been implicated in the pathologies of diseases such as human immunodeficiency virus infection (Fais et al. 1997) and osteoporosis (Vignery 2000). Finally, cell fusion events appear to be one of the mechanisms by which adult stem cells regenerate certain tissues (Vassilopoulos and Russell 2003; Vassilopoulos et al. 2003; Wang et al. 2003).
The yeast Saccharomyces cerevisiae is a facile organism for the investigation of cell fusion during conjugation. Conjugation in yeast is the sequence of events during mating that culminates in the formation of a diploid zygote (for reviews see Sprague and Thorner 1992; Marsh and Rose 1997). S. cerevisiae has two haploid mating types, MATa and MATα, each of which secretes a specific mating pheromone (a-factor or α-factor) that binds receptors on the surface of the cell of the opposite mating type. Pheromone binding activates a MAP kinase signal transduction pathway, which leads to arrest in G1 of the cell cycle and transcriptional induction of genes required for conjugation (reviewed in Dohlman and Thorner 2001; Posas et al. 1998; Elion 2000). In response to pheromone gradients, haploid yeast cells direct their growth toward mating partners, a process called shmoo formation (Segall 1993). Mating-specific polarization involves cytoskeletal reorganization, asymmetric growth, and the deposition of proteins necessary for cell and nuclear fusion at or near the zone of contact between the mating cells.
Electron microscopy studies (Byers and Goetsch 1975; Gammie et al. 1998) and time lapse digital imaging (Maddox et al. 1999) have provided morphological and temporal descriptions of the events of cell and nuclear fusion. Mutants have also helped to define the identity of key components and the order of the events of mating (see review of Marsh and Rose 1997). Matings of cell-fusion defective mutants (Fus−) accumulate prezygotes that retain cell wall material at the contact region and typically have unfused nuclei.
FUS1 (McCaffrey et al. 1987; Trueheart et al. 1987; Trueheart and Fink 1989), FUS2 (Trueheart et al. 1987; Elion et al. 1995), RVS161 (Brizzio et al. 1998), FIG1, FIG2 (Erdman et al. 1998), and PRM1 (Heiman and Walter 2000) are thought to have specific functions during mating, because they are induced by pheromone and the proteins localize to the zone of cell fusion. Mutations in genes required for the establishment of cell polarity, including SPA2, PEA2, and BNI1, also cause pronounced defects in cell fusion (Dorer et al. 1997; Gammie et al. 1998). Finally, it is likely that cell fusion requires vesicular transport of necessary components to the region of cell contact because two membrane trafficking genes, CHS5 (Dorer et al. 1997; Santos and Snyder 1997; Santos et al. 1997) and TPM1 (Hasek et al. 1987; Liu and Bretscher 1992), are required for efficient cell fusion. At least two distinct signaling pathways regulate cell fusion, the pheromone response pathway (Elion et al. 1990, 1993; Fujimura 1992; Brizzio et al. 1996; Elia and Marsh 1996) and the PKC1 pathway (Philips and Herskowitz 1997).
Although a number of cell fusion genes have been discovered, it is likely that additional components remain unidentified. Genetic analysis showed that FUS2 is one of the most downstream genes involved in the process of cell fusion (Gammie et al. 1998). By performing a high-dosage suppressor screen of fus2Δ, we aimed to identify additional genes involved in cell fusion.
MATERIALS AND METHODS
Microbial and molecular techniques:
The yeast strains and plasmids used in this study are listed in Tables 1 and 2, respectively. Yeast techniques were conducted according to published procedures (Rose et al. 1990; Burke et al. 2000). Limited plate matings and assays for cytoplasmic mixing in zygotes have been previously described (Gammie et al. 1998; Gammie and Rose 2002). Quantitative microscopic matings using differential interference contrast (DIC) optics to visualize the zygote morphology and 4′,6-diamidino-2-phenylindole (DAPI) fluorescence to assess the position of the nuclear DNA were done as described previously (Gammie and Rose 2002). The Synthesizing and Sequencing Facility at Princeton University performed all sequencing reactions and synthesized the oligonucleotides for PCR and gene disruptions. Standard molecular biology manipulations were performed according to Ausubel (1994) and Sambrook et al. (1989).
TABLE 1.
Strains used in this study
| Strain | Genotype | Sourcea |
|---|---|---|
| MY427 | MATahis4-539 lys2-801 ura3-52 | This laboratory |
| MY1817 | MATafus2-Δ3 his4-34 leu2-1,112 ura3-52 | This laboratory |
| MY1894 | MATα trp1-Δ1 | This laboratory |
| MY3375 | MATaura3-52 leu2-Δ1 his3-Δ200 | This laboratory |
| MY3377 | MATaura3-52 leu2-Δ1 his3-Δ200 trp1-Δ63 | This laboratory |
| MY3492 | MATa/α lys2-801/+ trp1-Δ63/trp1-Δ63 ade2-101/+ his3-Δ200/ his3-Δ200 leu2-Δ1/leu2-11,112 ura3-52/ura3-52 | This laboratory |
| MY3608 | MATaspa2-964 leu2 ura3-52 | This laboratory |
| MY3773 | MATα spa2-964 lys2-Δ202 ura3-52 | This laboratory |
| MY3905 | MATα rvs161Δ::LEU2 his3-Δ200 leu2-Δ1 ura3-52 | This laboratory |
| MY3909 | MATarvs161Δ::LEU2 ura3-52 his3-Δ200 leu2-Δ1 trp1-Δ63 | This laboratory |
| MY4160 | MATafus1-Δ1 fus2-Δ3 lys2-801 ura3-52 | This laboratory |
| MY4164 | MATα fus1-Δ1 ura3-52 trp1-Δ1 | This laboratory |
| MY4177 | MATα fus2-410 ura3-52 lys2-Δ202 leu2 | This laboratory |
| MY4384 | MATα ura3-52 leu2-Δ1 trp1-Δ63 [PGAL-GFP LEU2 CEN] | This laboratory |
| MY4495 | MATα ura3-52 leu2-Δ1 lys2-Δ801 his3-Δ200 rvs161::LEU2 | This laboratory |
| MY4843 | MATα fus1-Δ1 fus2-Δ3 trp1-Δ1 ura3-52 cyh2 ρ° | This laboratory |
| MY4859 | MATafus2::URA3 rvs161::LEU2 ura3-52 his3-Δ200 leu2 trp1-Δ63 | This laboratory |
| MY5489 | MATafus2::URA3 ura3-52 trp1-Δ1 leu2-3,112 | This laboratory |
| MY5494 | MATalrg1Δ::HIS3 leu2-Δ1 his3-Δ200 ura3-52 trp1-Δ63 | This laboratory |
| MY5500 | MATα lrg1Δ::HIS3 trp1-Δ63 leu2 ura3-52 | This laboratory |
| MY5503 | MATα lrg1Δ::HIS3 lys2-801 trp1-Δ63 leu2 ura3-52 leu2-Δ1 his3-Δ200 | This laboratory |
| MY5641 | MATalrg1Δ::HA leu2-Δ1 his3-Δ200 ura3-52 trp1-Δ63 | This laboratory |
| MY5727 | MATalrg1Δ::HIS3 rvs161::LEU2 ura3-52 trp1-Δ63 | This laboratory |
| MY5728 | MATα lrg1Δ::HIS3 rvs161::LEU2 ura3-52 trp1-Δ6 | This laboratory |
| MY5730 | MATalrg1Δ::HIS3 fus2::URA3 leu2-Δ1 trp1-Δ63 | This laboratory |
| MY5791 | MATα lrg1Δ::HIS3 fus2::URA3 leu2 trp1-Δ63 [PGAL-GFP CEN LEU2] | This laboratory |
| MY5796 | MATaspa2Δ::URA3 lrg1::HIS3 leu2-3112 | This laboratory |
| MY5800 | MATα spa2Δ::URA3 lrg1::HIS3 leu2-3112 [PGAL-GFP CEN LEU2] | This laboratory |
| MY5805 | MATα lrg1Δ::HIS3 fus1-Δ1 trp1-Δ63 ura3-52 leu2 | This laboratory |
| MY5806 | MATalrg1Δ::HIS3 fus1-Δ1 leu2 his3-Δ200 ura3-52 trp1-Δ63 | This laboratory |
| MY5953 | MATα lrg1-ΔRhoGAP::HIS3 trp1-Δ63 leu2 ura3-52 | This laboratory |
| MY5960 | MATalrg1-ΔRhoGAP::HIS3 trp1-Δ63 leu2 ura3-52 | This laboratory |
| MY6003 | MATaspa2-964 ura3-52 leu2 [BEM1 2μ URA3] [PGAL-GFP CEN LEU2] | This laboratory |
| MS5208 | MATaspa2Δ::URA3 his3-Δ200 ura3-52 leu2-3 leu2-112 ade2-101 | This laboratory |
| JY424 | MATafus2-Δ3 his4-34 leu2-3 leu2-112 ura3-52 | G. Fink (Whitehead Institute) |
| JY427 | MATafus1-Δ1 leu2-3 leu2-112 ura3-52 | G. Fink (Whitehead Institute) |
| JY430 | MATα fus1-Δ1 trp1-Δ1 ura3-52 canr | G. Fink (Whitehead Institute) |
The MS strains from the Rose laboratory are congenic with S288C. The MY strains are also congenic with S288C, except the parent strains are from Fred Winston's laboratory (Harvard University, Cambridge, MA).
TABLE 2.
Plasmids used in this study
| Plasmid | Relevant markers | Source |
|---|---|---|
| pMR3397 | RVS161 URA3 2μ ampr | This laboratory |
| pMR3453 | PGAL-GFP CEN4 ARS1 LEU2 ampr | This laboratory |
| pMR3562 | BEM1 2μ URA3 ampr | This laboratory |
| pMR3725 | FUS2 TRP1 CEN4 ARS1 ampr | This laboratory |
| pMR3859 | LRG1 URA3 2μ ampr | This laboratory |
| pMR3860 | YDL241W URA3 2μ ampr | This laboratory |
| pMR4910 | Ptet-6xHN-CDC42 Cmr | This laboratory |
| pMR4911 | Ptet-6xHN-RHO1 Cmr | This laboratory |
| pMR4912 | Ptet-6xHN-RHO2 Cmr | This laboratory |
| pMR4913 | Ptet-6xHN-RHO3 Cmr | This laboratory |
| pMR4914 | Ptet-6xHN-RHO4 Cmr | This laboratory |
| pMR4915 | Ptet-6xHN-RHO5 Cmr | This laboratory |
| pMR4917 | Ptet-6xHN-LRG1-GAP Cmr | This laboratory |
| pPROTet.E133 | Ptet-6xHN Cmr | Clontech (Palo Alto, CA) |
| pRS414 | TRP1 CEN ampr | P. Heiter (University of British Columbia) |
| pRS416 | URA3 CEN ampr | P. Heiter (University of British Columbia) |
| pRS424 | TRP1 2μ ampr | P. Heiter (University of British Columbia) |
| pRS426 | URA3 2μ ampr | P. Heiter (University of British Columbia) |
| pSB257 | FUS2 2μ URA3 ampr | G. Fink (Whitehead Institute) |
| pSB273 | FUS1 2μ URA3 ampr | G. Fink (Whitehead Institute) |
| pTS595 | PGAL-GFP CEN ARS URA3 ampr | T. Stearns (Stanford University) |
| pCY362 | BEM1 2μ URA3 ampr | I. Herskowitz (University of California, San Francisco) |
| YEp24 | URA3 2μ ampr | Botstein laboratory (Princeton University) |
High-copy suppression of Fus− defects:
A YEp24 2μ-based yeast genomic DNA library (Carlson and Botstein 1982) was transformed into a MATa fus2Δ strain (JY424). Approximately 13,200 transformants were mated to a MATα fus1Δfus2Δ lawn (MY4843). Plasmids conferring suppressing activity were recovered from the cells (Burke et al. 2000), transformed into fus2Δ, and retested. Yeast colony PCR (Burke et al. 2000) was performed to identify and eliminate plasmids containing the FUS2 gene.
DNA sequencing was used to identify the genes carried on the suppressing plasmids. Six plasmids contained FUS1. Two candidate plasmids contained overlapping regions of chromosome IV, including two open reading frames, YDL241W and LRG1. To identify the suppression gene, YDL241W was cloned as a ClaI/SpeI fragment and LRG1 as a XhoI/HindIII fragment into pRS426, a URA3 2μ vector (Sikorski and Hieter 1989). Only the LRG1-expressing plasmid (pMR3859) suppressed the fus2Δ mating defect. Two candidate plasmids contained overlapping regions from chromosome II, which encoded BEM1. pCY362, harboring just the BEM1 gene on a 2μ vector (Ira Herskowitz, University of California, San Francisco) was shown to suppress the fus2Δ mating defect. One additional candidate plasmid contained NAB3, a high-copy suppressor of the CLN3/DAF1-1 mating defect (Sugimoto et al. 1995).
For analysis of the suppression of rvs161Δ, fus1Δ, and spa2-964 Fus− phenotypes, zygotes were prepared and were examined by the green fluorescent protein (GFP) cytoplasmic mixing assay and DAPI/DIC method (Gammie and Rose 2002). rvs161Δ, fus1Δ, and spa2-964 zygotes were examined after mating on YEPD at 30° for 2, 3, and 8 hr, respectively. rvs161Δ zygotes were from matings between rvs161Δ (MY3909) containing the 2μ vector (pRS426), 2μ LRG1 (pMR3859), 2μ RVS161 (pMR3397), or 2μ BEM1 (pMR3562), mated to MATα rvs161Δ (MY4495 + pTS595). Strains for the GFP mixing analysis testing a bilateral suppression of fus1Δ and spa2-964 were: WT × WT (MY3377 × MY4384), fus1Δ × fus1Δ (JY427 + pTS595 × JY430), fus1Δ [2μ BEM1] × fus1Δ [2μ BEM1] (JY427 + pMR3453 + pMR3562 × MY4164 + pMR3562), fus1Δ [2μ LRG1] × fus1Δ [2μ LRG1] (JY430 + pMR3859 × JY424 + pMR3453 + pMR3859), spa2-964 × spa2-964 (MY3608 + pTS595 × MY3773), spa2-964 [2μ BEM1] × spa2-964 [2μ BEM1] (MY6003 × MY3773 + pMR3562), and spa2-964 [2μ LRG1] × spa2-964 [2μ LRG1] (MY3608 + pMR3453 + pMR3859 × MY3773 + pMR3859). Strains to analyze the zygote morphology for fus1 and spa2 mutant matings were as detailed below. fus1Δ × fus1Δ were such that both partners contained the vector (JY427 + pTS595 × JY430), 2μ BEM1 (JY427 + pMR3453 + pMR3562 × MY4164 + pMR3562), or 2μ LRG1 (JY430 + pMR3859 × JY424 + pMR3453 + pMR3859). The spa2-964 × spa2-964 zygotes observed in matings were where both partners contained the vector (MY3608 + pTS595 × MY3773), 2μ BEM1 (MY6003 × MY3773 + pMR3562), or 2μ LRG1 (MY3608 + pMR3453 + pMR3859 × MY3773 + pMR3859).
Strain construction:
Generation of lrg1Δ and a truncated version of LRG1 that lacks the Rho-GAP domain, each marked with HIS3, were done by one step-gene replacement (Rothstein 1991). Primers used to create the lrg1Δ strain were as follows: LRG1-5′ (5′-TCT TCA AAG TAT GCC GGG TAT TGA TGG GCA CGG AAG ATG TCG TTT TAA GAG CTT GGT GAG) and LRG1-3′ (5′-ATA AGA ACG ACA AAC CTC GAA ATC TGA GGG GAA GGA GAA GAT CCG TCG AGT TCA AGA GAA). The primers used to create a truncated LRG1 strain lacking the Rho-GAP domain (deletion spanning codons 528–1017) were LRG1-3′ listed above and LRG1LIM (5′-CTC AAT TGA TGA TCC GGC CTC TGT GCC GGG TTT CAA ATT TCG TTT TAA GAG CTT GGT GAG). The PCR reaction products were transformed into wild-type haploid (MY3377) and diploid (MY3492) strains. Confirmation of both junctions of the lrg1Δ and the truncated LRG1 alleles was performed using PCR.
The fus1Δ lrg1Δ double mutant was made by two-step gene replacement (Scherer and Davis 1979) using pSB281 (Fink Laboratory, Whitehead Institute, Cambridge, MA) in lrg1Δ strains MY5494 and MY5500. The spa2Δ lrg1Δ, fus2Δ lrg1Δ, and rvs161Δ lrg1Δ double mutants were obtained from crosses between MY5500 (lrg1Δ) and MY4859 (fus2Δ rvs161Δ) to obtain fus2Δ lrg1Δ and rvs161Δ lrg1Δ and between MY5500 (lrg1Δ) and MS5208 (spa2Δ) to obtain spa2Δ lrg1Δ.
A PCR-based epitope-tagging procedure (Schneider et al. 1995) was employed to insert the hemagglutinin (HA) epitope into the LRG1 C-terminal coding region, generating a LRG1::HA fusion on the chromosome. Primers used were: LRG1HAU (5′-TCA AAG ATT CGA CCA CGG TCA TAC AAG GTG AAA TAA ACA AAA GGG AAC AAA AGC TGG) and LRG1HAD (5′-GAA AAA AAG GAA AAT GAG GGG AAA CTT ACA GTT TCT GAA TAT TAC TAT AGGAGCG AAT TGG). The PCR product was transformed into a wild-type MATa (MY3377) strain, and integration was verified by PCR and DNA sequencing of the fusion junction. The functionality of the LRG1::HA fusion was established by mating the strain (MY5641) to a fus1Δ fus2Δ lawn (MY4843 + pRS424).
Immunoblotting analysis of Lrg1::HA protein:
The LRG1::HA strain (MY5641) and an isogenic strain lacking the fusion (MY3377) were grown to midexponential phase and treated with 6 μm α-factor in MeOH or with MeOH alone for 90 min at 30°. Total protein extracts were obtained (Burke et al. 2000), fractionated by SDS polyacrylamide gel electrophoresis (SDS-PAGE), and immunoblotted according to standard procedures (Ausubel 1994). The membrane was probed with a mouse α-HA monoclonal antibody (12CA5) from ascites fluid (Princeton Monoclonal Facility) at a dilution of 1:1000. The secondary antibody was α-mouse IgG conjugated to horseradish peroxidase (HRP; Amersham Pharmacia Biotech, Piscataway, NJ) at a dilution of 1:2500. Lrg1p::HA was detected using the ECL protocol and reagents (Amersham Pharmacia Biotech).
Fluorescence microscopy:
Indirect immunofluorescence was done essentially as described previously (Santos and Snyder 1997; Gammie and Rose 2002) with the following specifications. Strain MY5641 and a negative control strain (MY3377) were grown overnight in YEPD pH 3.5 to early exponential phase and treated with 6 μm α-factor for 2 hr at 30°. Cells were fixed with 3.7% formaldehyde at room temperature for 1 hr and spheroplasted for ∼30 min. Both the primary antibody, mouse α-HA (12CA5), and the secondary antibody, CY3-conjugated goat α-mouse IgG (Sigma-Aldrich, St. Louis), were preabsorbed to fixed cells lacking the HA epitope. The primary antibody was added to the immobilized fixed pheromone-treated cells and incubated overnight at 4° and the secondary antibody was incubated for 2 hr. Cells were stained with DAPI and observed using DIC optics and fluorescence microscopy (Gammie and Rose 2002).
Aniline blue staining was carried out as described by Watanabe et al. (2001), with minor modifications. Briefly, after treatment with mating pheromone, cells were collected by low-speed centrifugation (3000 rpm for 5 min), washed twice with PBS, sonicated for 20 sec, and incubated in 0.5% aniline blue (Wako USA, Richmond, VA) for 5 min. Cells were examined by fluorescence microscopy using the Chroma 31016/Hydroxycoumarin filter set (excitation wavelength, 405 nm; emission wavelength, 460 nm; Chroma Technology, Brattleboro, VT).
Calcofluor white staining for chitin was carried out according to methods developed in Pringle et al. (1991). Briefly, cells were fixed by addition of formaldehyde (3.7%), incubated for 30 min at room temperature, and washed twice with PBS. Fluorescent Brightener 28 (Sigma, St. Louis) was added to a final concentration of 0.17 mg/ml. After 10 min, cells were then washed three times with PBS, resuspended in 100 μl of PBS, and examined by fluorescent microscopy using DAPI filter sets.
Mating projection and cell cycle arrest analysis of lrg1Δ:
Pheromone sensitivity assays for both wild-type (MY3375) and lrg1Δ (MY5494) MATa strains were performed by spreading ∼105 cells onto YEPD plates; placing sterile filter disks on the agar; and administering different dilutions of α-factor pheromone (Princeton Syn/Seq Facility), 5 × 10−8 m, 5 × 10−7 m, and 5 × 10−6 m, diluted in methanol, to the disks. The plates were incubated at 23° for 2 days. The ability to form a mating projection was assessed by adding 6 μm of α-factor pheromone to early exponential growth phase MATa lrg1Δ (MY5494) and wild-type (MY3375) cultures for 0, 2, 4, and 6 hr. The numbers of unbudded, small-budded, and large-budded cells and cells with a mating projection were measured by microscopy using DIC optics (Gammie and Rose 2002).
GTPase assays:
The open reading frames of all six yeast Rho-GTPases, including CDC42, RHO1, RHO2, RHO3, RHO4, RHO5, and of the LRG1 GAP homology domain (representing amino acids 551–1018) were amplified from genomic DNA by PCR (Burke et al. 2000) and inserted into pPROTet.E133 vector (Clontech, Palo Alto, CA) to create in-frame fusions between six repeated His-Asn residues (6xHN) and the GTPases or Lrg1p Rho-GAP. The recombinant plasmids (listed in Table 2) were sequenced to confirm the fusion and integrity of the constructs.
The proteins were expressed in exponentially growing bacterial cells (DH5αPRO) with 100 ng/ml anhydrotetracycline (Clontech) for 4 hr. The GTPase proteins were purified using NTA spin columns (QIAGEN, Valencia, CA) and previously published conditions (Apanovitch et al. 1998). The Lrg1Rho-GAP protein was purified using the Talon resin batch (Clontech; Apanovitch et al. 1998). Cells containing the pPROTet.E133 vector were processed in parallel during each purification as a control for background GTPase activity or for Lrg1-GAP activity. Protein concentrations were determined using the Bradford protein assay (Bio-Rad Laboratories, Hercules, CA) with bovine serum albumin as the protein standard. Protein purity was assessed using SDS-PAGE and Coomassie staining (Ausubel 1994).
GTPase assays were developed using a modification of previous methods (Wagner et al. 1992; Apanovitch et al. 1998). Assay conditions were: 5 μl of the purified GTPase or a vector control sample, 5 μl of 2× GTPase buffer (50 mm HEPES, pH 7.6, 1 mm EDTA, 2 mm DTT, 20 μm GTP), and 4 μl [α-32P]GTP (800 Ci/mmol at 12.5 μm; New England Nuclear, Boston; Perkin Elmer, Norwalk, CT). Samples were incubated at room temperature for 30 min to allow for GTP binding and then added to a tube with 2.5 μl of 2× GTPase buffer, 2.5 μl of purified Lrg1-GAP or control vector extracts, and 1.1 μl of 0.1 m MgCl2. At appropriate times, 2-μl aliquots were added to 2 μl stop buffer (0.5% SDS, 5 mm EDTA, 50 mm GDP, 50 mm GMP, 50 mm GTP) and placed on ice. After heating at 70° for 2 min, the samples (1 μl) were spotted onto PEI cellulose plates (Sigma-Aldrich). The PEI cellulose was pretreated prior to use with 1 m NaCl for 30 min and washed with multiple changes of fresh dH2O before drying and sample application. The PEI cellulose plates were placed in a resolving tank with ∼200 ml of 0.6 m NaH2PO4 (pH 4.0). The PEI cellulose plates were dried after the liquid had migrated 75% of the length and the GDP and GTP levels were quantitated using a PhosphorImager and ImageQuant software (Molecular Dynamics, Sunnyvale, CA). For each sample values from vector extracts were subtracted to determine the level of hydrolyzed GTP above background. The fold induction of GTPase activity represents the activity found in the presence of purified Lrg1 divided by the activity in the absence of Lrg1 (vector extracts).
RESULTS
Isolation of LRG1 and BEM1 as high-copy suppressors of fus2Δ:
Fus2p was identified as one of the most downstream components, in genetic studies of the cell fusion pathway (Gammie et al. 1998). However, increased dosage of FUS1 partially suppressed fus2Δ, apparently by hyperactivating a second pathway required for efficient cell fusion (Trueheart et al. 1987; Gammie et al. 1998). To identify additional components required for cell fusion, we performed a screen to isolate additional high-copy suppressors of fus2Δ mutants. To accomplish this, a fus2Δ strain was transformed with a YEp24 2μ yeast genomic DNA library (Carlson and Botstein 1982) and the transformants were screened by mating to a fus1Δ fus2Δ lawn. Under these conditions, the efficiency of mating is strongly dependent on the mating ability of the fus2Δ parent. Candidate plasmids were isolated from transformants showing enhanced mating ability and retested, and the DNA inserts were characterized (see materials and methods). Four plasmids contained FUS2 and six contained FUS1. In two plasmids, the insert DNA contained BEM1 (bud emergence mediator), a gene implicated in polarization of the MAP kinase cascade during mating (Moskow et al. 2000). Two of the suppressing plasmids contained LRG1, a gene implicated in sporulation and mating (Muller et al. 1994; Watanabe et al. 2001). For both LRG1 and BEM1, the identity of the suppressing gene was confirmed using a plasmid containing only the single open reading frame (see materials and methods). Finally, NAB3, isolated once, was also previously identified as a high-copy suppressor of CLN3/DAF1-1 mating defects (Sugimoto et al. 1995) and codes for an RNA-processing protein (Wilson et al. 1994; Conrad et al. 2000; Steinmetz et al. 2001).
The ability of the plasmids to suppress the fus2Δ mating defect in plate mating assays is shown in Figure 1A. This experiment shows unilateral dosage suppression, in which only one partner contains the suppressing plasmid. In this case, the fus2Δ strain contains a high-copy vector plasmid either with no insert (the null control) or with FUS2 (the wild-type control), and FUS1, BEM1, or LRG1 each mated to a fus1Δ fus2Δ lawn. The results show that increased gene dosage of BEM1 or LRG1 suppressed the mating defect of fus2Δ as well as high-copy FUS1 (Figure 1A).
Figure 1.—
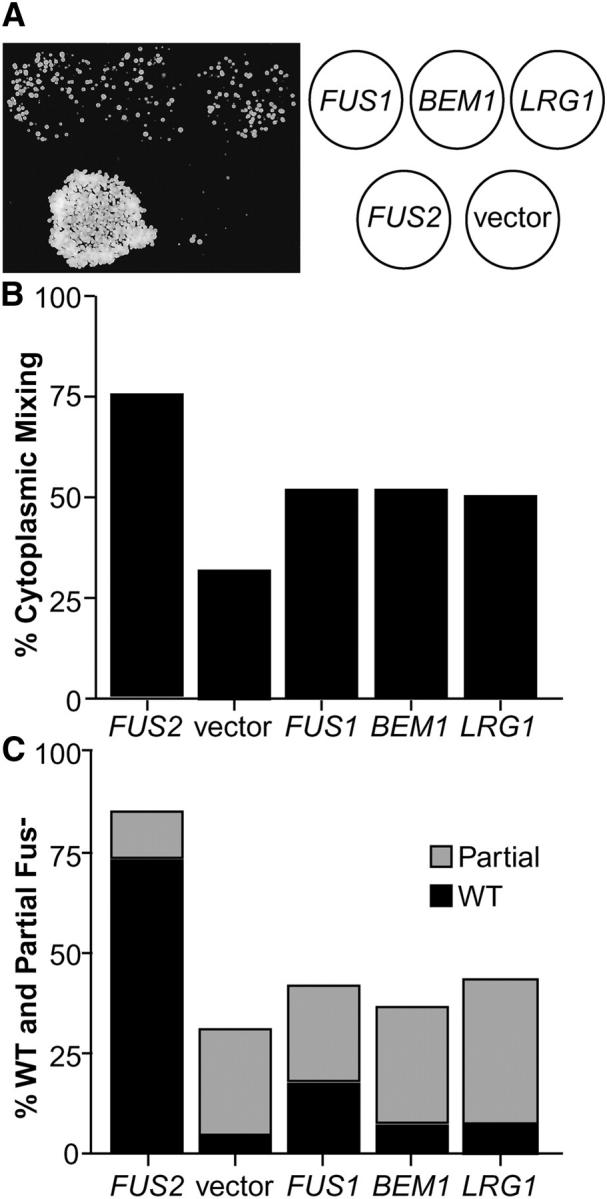
Unilateral dosage suppression of fus2Δ mating defects. (A) A limited plate-mating analysis of the fus2Δ strain containing suppressing plasmids identified in the screen. The fus2Δ strain (JY424) was transformed with FUS1 2μ (pSB273), BEM1 2μ (pCY362), LRG1 2μ (pMR3859), FUS2 2μ (pSB257), or a 2μ vector (pRS416). Each strain was patched onto a plate and mated to a fus1Δfus2Δ (MY4843) lawn on YEPD for 5 hr at 30°. The cells were transferred to the appropriate selective medium and grown at 30°. Diploid growth is shown in the photograph to the left with the legend for the patches to the right. (B) Soluble GFP assay of cytoplasmic mixing in zygotes. Failure of mating pairs to mix cytoplasms during conjugation is diagnostic of a complete block in cell fusion. The matings shown employed the same fus2Δ strains as described above mated to fus2Δ partners expressing soluble GFP (MY4177 + pMR3453). Cells were allowed to mate for 1.5 hr on YEP-GAL plates at 30°. Zygotes were scored for the distribution of GFP fluorescence. The “mixed” category consisted of wild-type zygotes (mixed cytoplasm with no intervening cell wall between partners) and partially defective zygotes (mixed cytoplasm but retained visible cell wall between partners). The “unmixed” category of zygotes retained an intact intervening cell wall where only one partner contained the soluble GFP. Only the mixed category is graphed. Approximately 100 zygotes were scored for each mating. (C) Microscopic analysis of zygotes to assess nuclear position and cell wall morphology. The strains described above were allowed to mate on YEPD plates for 3 hr at 30°. After fixation, the nuclei were visualized using DAPI and cell wall morphology was assessed by DIC optics. Zygotes were scored as wild type, partially defective, or completely defective (Gammie and Rose 2002). Wild-type zygotes have a fused nucleus and no visible septum. Partially defective zygotes have a fused nucleus and a partial septum. Completely defective zygotes have unfused nuclei and a visible intervening septum. Graphed are the percentages of wild-type zygotes (solid bars) and partially defective zygotes (shaded bars). Approximately 400 zygotes were scored for each mating.
To demonstrate that the improved growth observed in plate mating assays was due to suppression of the cell fusion defect, we performed microscopic analyses of mating cells. We used a soluble GFP expressed in one fus2Δ parent as a marker for cytoplasmic mixing during mating (Gammie and Rose 2002) and examined mating to fus2Δ strains harboring the high-copy suppressor plasmids. FUS1, BEM1, and LRG1 on high-copy plasmids were able to suppress the fus2Δ cell fusion defect to the same degree (Figure 1B; 52, 52, and 50% of the cells showed mixed cytoplasms, respectively). In contrast, the wild-type control (fus2Δ strain containing FUS2 in high copy) showed 75% cytoplasmic mixing and the null mutant control (fus2Δ strain containing the vector) showed 32% mixing. Full suppression by wild-type FUS2 was not observed presumably because of prior plasmid loss from the parent strain containing the suppressing plasmid. Note that the cytoplasmic mixing assay detects all fusion events, including events that do not eventually form a diploid zygote.
We also examined the morphology of the zygotes and the positions of the nuclei (Figure 1C) in these matings. Zygotes with defects in cell fusion typically exhibit characteristic residual septa (Figure 4A and Gammie and Rose 2002), which may interfere with nuclear migration and fusion (Elion et al. 1995). In this experiment we examined unilateral suppression by the plasmid in a fus2Δ × fus2Δ mating. Three classes of zygotes were observed: wild type, partial Fus−, or full Fus− (Gammie and Rose 2002). Wild-type zygotes have a single fused nucleus and no discernible septum. Partial Fus− zygotes have a single fused nucleus and a remnant septum. Full Fus− zygotes have two unfused nuclei and a pronounced intervening septum. In high copy, FUS1 was the only gene that strongly suppressed the formation of remnant septa in the fus2Δ zygotes as evidenced by the increased percentage of wild-type zygotes containing single nuclei (Figure 1C). However, all three genes were able to partially suppress the fus2Δ cell fusion defect as revealed by the increased number of zygotes in which nuclear fusion had occurred (wild-type or partial Fus−) and the decreased numbers of full Fus− zygotes relative to the vector control (Figure 1C).
Figure 4.—
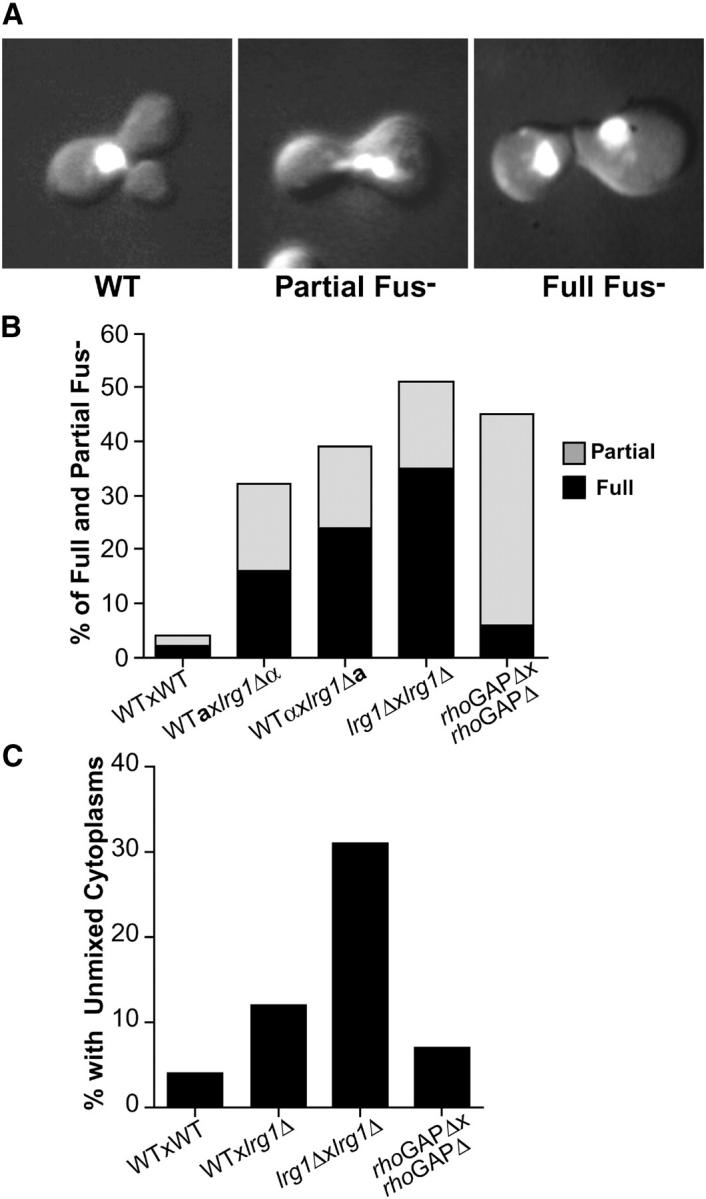
Quantitative analysis of lrg1Δ cell fusion defects. (A) Representative lrg1Δ zygotes from each of the three categories, wild type (WT), partial Fus−, and full Fus−, that were scored for morphology and nuclear staining analysis as described previously for Figure 1. The images are a combination of DAPI fluorescence and DIC optics. (B) Microscopic characterization of lrg1Δ cell fusion defects. The results from microscopic analysis of zygotes from wild-type matings (WT × WT), unilateral (lrg1Δ × WT) and bilateral (lrg1Δ × lrg1Δ) lrg1Δ matings, along with bilateral LRG1 GAP domain-deleted matings (rhoGAPΔ × rhoGAPΔ) are shown. Whether the strain is MATa cells (a) or MATα (α) is denoted for the bilateral matings. Graphed are the percentages of fully defective (solid bars) and partially defective (shaded bars) zygotes. (C) Cytoplasmic mixing assay in lrg1Δ zygotes. Scoring was done as previously described in Figure 1B. Only the percentage of unmixed cytoplasms category is graphed. Approximately 100 zygotes were scored for each mating. The strains used in both microscopic analyses were WT MATa (MY3377), WT MATα (MY4384), lrg1Δ MATa (MY5494), lrg1Δ MATα (MY5500 + pMR3453), rhoGAPΔ MATa (MY5960), and rhoGAPΔ MATα (MY5953 + pMR3453).
To further examine the suppression of the fus2Δ mating defect we measured diploid formation during mating to a fus1Δ fus2Δ strain using a quantitative filter-mating assay. In this assay, cells are allowed to mate for a brief period of time, diluted, and plated onto selective media, thereby preventing subsequent mating with adjacent cells. This is in contrast to the semiquantitative plate-mating assays (Figure 1A), where cells remain in proximity and may undergo additional rounds of matings until nutrients become limiting. In the filter-mating assay, the control FUS2 plasmid resulted in 31% diploids and the plasmid vector resulted in 0.15% diploids. Elevated copies of LRG1, FUS1, and BEM1 all partially suppressed the mating defect of fus2Δ to comparable extents, giving rise to diploids at frequencies 6- to 10-fold higher than that of the vector control (0.88, 0.90, and 1.6%, respectively). We conclude that in high copy, both LRG1 and BEM1, like FUS1, increase the efficiency of cell fusion in a fus2Δ mating.
LRG1 and BEM1 dosage suppression analyses of other cell-fusion-defective alleles:
Previous genetic data are consistent with the hypothesis that cell fusion involves multiple partially redundant pathways (Trueheart et al. 1987; Gammie et al. 1998); FUS2 and RVS161 define one pathway and SPA2 and FUS1 represent separate and distinct pathways (Gammie et al. 1998). To help place LRG1 and BEM1 in the cell fusion pathways, we tested the ability of increased dosage of LRG1 and BEM1 to suppress the mating defects of other Fus− mutants. On the basis of either cytoplasmic mixing or zygote morphology, increased dosage of LRG1 partially suppressed rvs161Δ, slightly suppressed spa2-964, but failed to suppress fus1Δ (Figure 2B). In contrast, high-copy BEM1 suppressed all of the tested cell fusion alleles, including rvs161Δ, fus1Δ, and spa2-964. Interestingly, increased dosage of BEM1 suppressed fus1Δ to the same extent as high-copy suppression by FUS2. In summary, our data show that LRG1 functions differently than either FUS1 or BEM1 in high-copy suppression assays, by exclusively impacting the Fus2p/Rvs161p pathway.
Figure 2.—

BEM1 and LRG1 dosage suppression analysis of rvs161Δ, fus1Δ, and spa2-964 cell fusion defects. The percentages of cytoplasmic mixing (left graphs) and cell fusion (right graphs) were assayed as described in Figure 1, B and C, respectively. − indicates vector-containing strains and + indicates (A) BEM1 2μ- or (B) LRG1 2μ-containing strains. Wild type (WT) is the average value obtained for three separate experiments. Strains for this analysis are detailed in materials and methods.
Phenotypic characterization of lrg1Δ cell fusion defects:
We next wanted to determine if LRG1 might have a direct role in cell fusion or act solely as a suppressor of cell fusion mutants. The LRG1 gene encodes a 1017-amino-acid polypeptide with a predicted molecular weight of 117 kD. On the basis of analyses performed by the Munich Information Center for Protein Sequences (MIPS), the LRG1 open reading frame is predicted to possess four LIM (lin-11, Isl-1, mec-3) domains, a potential transmembrane domain, and a Rho-GTPase-activating protein (Rho-GAP) homology domain (Figure 3A). The SWISS-PROT Protein Database (Boeckmann et al. 2003) also indicates the Rho-GAP homology domain and three of the four LIM domains (LIM1, LIM2, and LIM4).
Figure 3.—

Deletions of LRG1 result in mating defects. (A) Structural organization of Lrg1p. The 1017-amino-acid (aa) Lrg1p protein is depicted schematically. The Lrg1p structural predictions are from Munich Information Center for Protein Sequences (MIPS). Lrg1p's four LIM domains (LIM1, 27–89 aa; LIM2, 96–148 aa; LIM3, 155–184 aa; and LIM4, 417–474 aa), a putative transmembrane domain (TM, 348–368 aa), and a Rho GAP homology domain (755–910 aa) are represented by the open rectangles. The ranges of the complete deletion (lrg1Δ) and lrg1Δ lacking the Rho-GAP homology domain (rhoGAPΔ) are indicated by the arrows. SWISS-PROT Protein Database also predicts LIM1, LIM2, LIM4, and the Rho-GAP homology domain. (B) Limited plate-mating analysis of the lrg1Δ strains. Wild type (WT) (MY1894), Rho-GAPΔ (MY5953), and lrg1Δ (MY5500 + pRS426) were mated against a fus1Δfus2Δ (MY4160) lawn on YEPD for 2 hr at 30°. The cells were transferred to the appropriate selective medium and grown at 30°. Diploid growth is shown in the photograph.
Previous studies using a W303 strain background suggested that LRG1 plays a major role in mating and meiosis (Muller et al. 1994), although the nature of the mating defect was not reported. To confirm the phenotype, we created S288C strains containing either a complete deletion (lrg1Δ) or a deletion of the Rho-GAP homology domain (lrg1-rhoGAPΔ). Both lrg1Δ strains exhibited mating defects (Figure 3B); however, the observed defect was significantly less severe than the ∼1000-fold defect previously reported (Muller et al. 1994).
We examined lrg1Δ zygotes to establish whether the mating defect was at the cell fusion step. Zygotes from the lrg1Δ matings had the characteristic phenotypes indicative of defects in cell fusion, including a septum between the two mating cells, unfused nuclei, and an enlarged region of cell-cell contact (Figure 4A). Bilateral lrg1Δ matings (both partners defective) produced approximately twofold more full Fus− zygotes than a unilateral mating (one partner defective; Figure 4B). In unilateral matings, the cell fusion defect was visible when the mutation was present in either mating type, indicating lrg1Δ does not confer a cell-type-specific defect. In bilateral matings, both the complete deletion (lrg1Δ) and the partial deletion (lrg1-rhoGAPΔ) strains showed equivalently reduced levels of wild-type zygotes. However, in the lrg1-rhoGAPΔ matings, the majority of the defective zygotes showed only a partial Fus− defect (approximately threefold more frequent than lrg1Δ), suggesting that the defect is less severe.
Analysis of cytoplasmic mixing during mating produced similar results. In bilateral matings, soluble GFP was uniformly dispersed in lrg1-rhoGAPΔ zygotes almost as frequently as wild type (Figure 4C). In contrast, the cytoplasms remained unmixed in 32% of the zygotes in bilateral lrg1Δ matings. The severity of the lrg1Δ cytoplasmic mixing defect is comparable to that seen with fus2Δ matings (Gammie et al. 1998). In accord with the morphological analysis, lrg1Δ unilateral matings exhibited a defect intermediate between the bilateral mutant and wild-type matings.
Several cell fusion mutants exhibit significant cytoplasmic mixing, in spite of the presence of a residual septum between the mating cells (Gammie et al. 1998). Cytoplasmic mixing in these cases is accomplished presumably via a pore of inadequate proportions to form a viable diploid as indicated by the frequent presence of unfused nuclei. Although Lrg1p is clearly required for cell fusion, the more severe phenotype of the complete deletion suggests that the Rho-GAP domain is not exclusively responsible for Lrg1p's function. Possibly one or all of Lrg1p's LIM or transmembrane domains contribute to the efficiency of cell fusion.
lrg1Δ mutants respond normally to pheromone:
Reduced pheromone signaling can lead to defects in cell fusion (Brizzio et al. 1996). We therefore investigated whether lrg1Δ mutants exhibit pheromone response defects by examining cell cycle arrest and formation of mating projections in the presence of α-factor. Zones of growth inhibition for wild type and lrg1Δ were of equal diameter at each of four different α-factor concentrations, indicating that cell cycle arrest was normal (data not shown). Furthermore, the dose and time dependence of mating projection formation were identical to wild type (data not shown). Therefore, we conclude that the lrg1Δ strains are able to cell cycle arrest and polarize to form mating projections with wild-type proficiency in response to pheromone.
Genetic interactions between LRG1 and other cell fusion genes:
To genetically position LRG1 in the cell fusion pathways, we analyzed the phenotypes of double mutants constructed between lrg1Δ and deletions of several other cell fusion genes. Combining lrg1Δ with fus2Δ, rvs161Δ, fus1Δ, or spa2Δ resulted in a more severe cell fusion defect than any single deletion, as determined either by semiquantitative plate matings (our unpublished observations) or by the cytoplasmic mixing assay (Figure 5). Note that for the spa2Δ strain significantly longer times were required to see appreciable levels of cytoplasmic mixing. Taken together, this analysis suggests that LRG1 does not act exclusively in any of the previously defined pathways (Gammie et al. 1998). Interestingly, the combination of lrg1Δ with fus2Δ or rvs161Δ resulted in particularly dramatic increases in the cell fusion defect (Figure 5). This result suggests that the cells lacking the Fus2p/Rvs161p pathway are especially sensitive to the activity of the Lrg1p pathway, consistent with Lrg1p overexpression being a specific suppressor of fus2Δ and rvs161Δ.
Figure 5.—
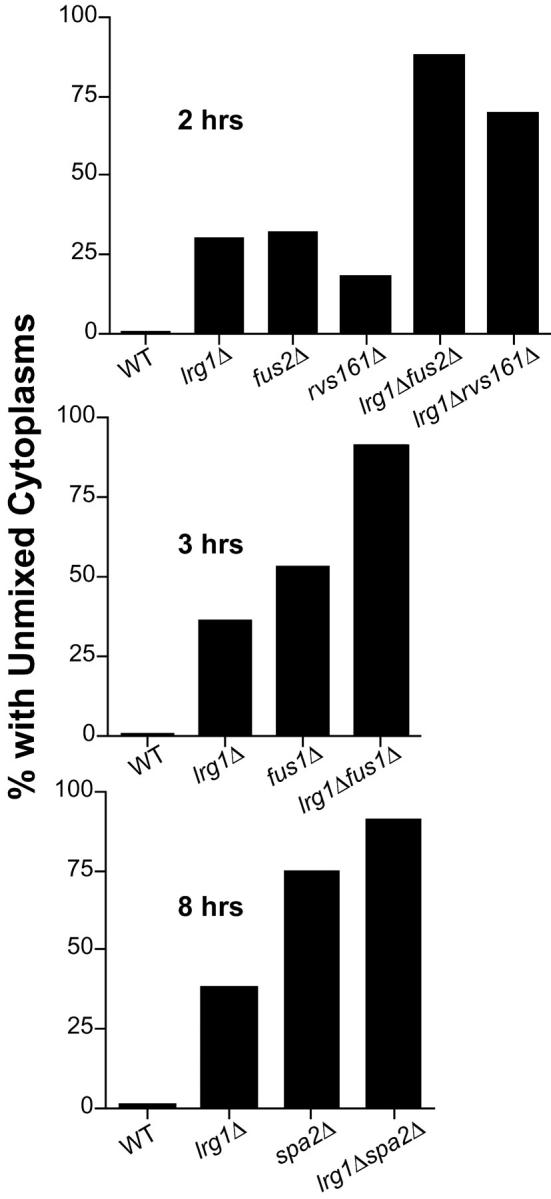
Microscopic analysis of Fus− double mutants. Scoring for all the analyses were performed as previously described in the Figure 1 legend. The percentages of zygotes without cytoplasmic mixing in the fus2Δlrg1Δ and rvs161Δ lrg1Δ analyses are shown in the top. Matings were done for 2 hr on YEPD. The strains used for these matings were WT × WT (MY3377 × MY4384), lrg1Δ × lrg1Δ (MY5494 × MY5500 pl pMR3453), fus2Δ × fus2Δ (JY424 × MY4177 + pMR3453), rvs161Δ × rvs161Δ (MY3909 + pTS595 × MY3905), fus2Δ lrg1Δ × fus2Δlrg1Δ (MY5730 × MY5791), and rvs161Δ lrg1Δ × rvs161Δlrg1Δ (MY5727 × MY5728 + pTS595). The percentages of zygotes with unmixed cytoplasms in the fus1Δlrg1Δ analysis are shown in the middle. Matings were done for 3 hr on YEPD. The strains used for these matings were WT × WT (MY3377 × MY4384), lrg1Δ × lrg1Δ (MY5494 × MY5500 + pMR3453), fus1Δ × fus1Δ (JY427 + pTS595 × JY430), and fus1Δlrg1Δ × fus1Δlrg1Δ (MY5806 + pTS595 × MY5805). The percentages of zygotes with unmixed cytoplasms in the spa2Δ lrg1Δ analysis are displayed in the bottom. Matings were done for 8 hr on YEP-GAL to maintain GFP expression. The strains used for these matings were WT × WT (MY3377 × MY4384), lrg1Δ × lrg1Δ (MY5494 × MY5500 + pMR3453), spa2Δ × spa2Δ (MY3608 + pTS595 × MY3773), and spa2Δlrg1Δ × spa2Δlrg1Δ (MY5796 × MY5800).
Expression and localization of Lrg1p during mating:
Many of the genes involved in cell fusion are induced by pheromone. To analyze the levels and localization of Lrg1p during mating we inserted three copies of the HA epitope into the carboxy-terminal coding region of chromosomal LRG1. The strain harboring the epitope-tagged LRG1 (LRG1::HA) mated like wild type, indicating that the fusion protein was fully functional. We examined the level of Lrg1p::HA with and without exposure to α-factor pheromone and found that Lrg1p, of the expected molecular weight, was expressed to the same level in mitotic and pheromone-treated cells (Figure 6A). These results have been confirmed at the transcriptional level by microarray analyses (Roberts et al. 2000). The constitutive expression of LRG1 suggests that Lrg1p has additional functions during vegetative growth consistent with previous observations (Lorberg et al. 2001; Watanabe et al. 2001).
Figure 6.—

Expression and Localization of Lrg1p::HA during pheromone treatment. (A) Immunoblot analysis of the expression levels of Lrg1p::HA with (+) and without (−) α-factor pheromone treatment for 90 min at 30°. Total protein extracts were blotted and probed with a mouse α-HA monoclonal antibody, 12CA5 as the primary antibody and α-mouse IgG-HRP as the secondary antibody. Strains used were LRG1::HA (MY5641) and the wild-type (WT) isogenic strain lacking the epitope (MY3377). Positions of molecular weight standards (kD) are indicated. (B–E) Immunofluorescence localization of Lrg1p in pheromone-treated cells. Pheromone-induced cells were fixed and prepared for indirect immunofluorescence using 12CA5 and CY3-conjugated goat α-mouse IgG antibodies. Visualization of Lrg1p::HA is shown in B and D. The position of the nuclei in the same cells was observed using DAPI staining of the DNA (C and E). B and C show cells containing LRG1::HA (MY5641). D and E show cells without the HA epitope (MY3377).
Not all cell fusion proteins are induced by pheromone; however, many localize to the tip of the mating projection (Trueheart et al. 1987; Gehrung and Snyder 1990; Elion et al. 1995; Valtz and Herskowitz 1996; Evangelista et al. 1997; Brizzio et al. 1998; Erdman et al. 1998; Philips and Herskowitz 1998). Accordingly, we used indirect immunofluorescence to determine the localization of Lrg1p::HA in pheromone-induced cells. After 2 hr of pheromone stimulation, Lrg1p::HA localized to a bright dot at the tip of the mating projection in 98% of cells (Figure 6B). No fluorescence was observed in cells that were not expressing the HA epitope (Figure 6D) or that had no primary antibody added (data not shown), showing that the observed fluorescence was specific to Lrg1::HA. Localization to the projection tip is consistent with the hypothesis that Lrg1p has a direct role in cell fusion.
Biochemical assays confirm that Lrg1p is the GAP for Rho1p:
Given Lrg1p's homology with Rho-GAP proteins, we determined which Rho-GTPase is activated by Lrg1p. Six Rho-type GTPases are identified in the yeast genome (Rho1p, Rho2p, Rho3p, Rho4p, Rho5p, and Cdc42p). To identify the relevant protein, we expressed each of the known yeast Rho-GTPases in Escherichia coli and examined the GTPase activity of each alone or in combination with the GAP domain of Lrg1p (Lrg1p-GAP). Each protein was expressed in E. coli as a fusion protein with six His-Asn repeats (6xHN) at its amino terminus. The fusion proteins were then purified using metal-ion chromatography (see materials and methods).
In vitro GTPase assays (Figure 7) were performed for all six Rho-GTPases. As a control, parallel GTPase assays were performed using extracts from identical purifications using cells containing the bacterial expression vector expressing only the 6xHN polypeptide. From each assay, the very low levels of contaminating E. coli GTPase activity have been subtracted. The GTPase assays were performed in the presence of either purified Lrg1p-GAP or equivalent extracts from cells expressing only the 6xHN polypeptide. These results unequivocally demonstrated that the GTPase activity of Rho1p was greatly stimulated by the presence of Lrg1p-GAP (∼36-fold). None of the other GTPases tested exhibited any change in activity. These data are consistent with the observation that activated Rho1p interacted with Lrg1p in a yeast two-hybrid assay (Watanabe et al. 2001). In sum, Lrg1p is a GAP for Rho1p, implicating Rho1p in the regulation of cell fusion.
Figure 7.—

Lrg1p stimulation of Rho1p GTPase Activity. The assay used purified Rho-GTPase proteins Rho1p, Rho2p, Rho3p, Rho4p, Rho5p, or Cdc42p in the presence or absence of purified Lrg1p GAP protein. The reactions were allowed to proceed for 0 sec, 2 min, 4 min, 8 min, and 16 min. The data plotted for each Rho-GTPase are a reflection of the fold stimulation in GTPase activity in the presence (+) of purified Lrg1 divided by the activity in the absence (−) of Lrg1 (purified vector extract added).
Localization of β(1-3)-glucan is abnormal in fus2 and lrg1 mutants:
Rho1p has multiple functions including serving as a regulatory subunit of β(1-3)-glucan synthase (Drgonova et al. 1996; Mazur and Baginsky 1996; Qadota et al. 1996). Therefore, we determined whether the lrg1 and fus2 mutants have defects in the formation and/or localization of β(1-3)-glucan during mating, using a specific fluorescent dye, aniline blue (Figure 8A). Wild-type shmoos showed pronounced staining with aniline blue along the sides and base of the shmoo projection, consistent with increased levels of β(1-3)-glucan. However, the tip of the shmoo was not stained with the dye, indicating a local decrease in β(1-3)-glucan deposition. In the lrg1 and fus2 mutants, the entire surface of the shmoo projection was stained, including the tip. In the lrg1 mutant, overall staining of the cell was distinctly brighter than that of the wild-type strain, consistent with a role for Lrg1p as a negative regulator of Rho1p. Remarkably, overexpression of Lrg1p from the 2μ plasmid restored the normal pattern of aniline blue staining to the fus2 mutant. The introduction of wild-type FUS2 also restored the normal staining pattern, whereas the empty vector had no effect. As a control for the specificity of the defect on cell wall components, we examined the pattern of chitin deposition using the fluorescent dye, calcofluor white (Figure 8B). In wild-type strains, chitin is concentrated along the base and sides of the shmoo projection, similar to the localization of β(1-3)-glucan. The wild-type pattern of chitin localization was observed in both the fus2 and the lrg1 (data not shown) mutant strains. The normal levels of chitin suggest that Lrg1p is not acting through the recently identified role of Rho1 as a regulator of chitin synthase III transport (Valdivia and Schekman 2003). Taken together these data suggest that loss of Fus2p (and Lrg1p) leads to unregulated β(1-3)-glucan synthase at the shmoo tip, which can be suppressed by increased levels of Lrg1p.
Figure 8.—

The localization of β(1-3)-glucan is correlated with Fus2p and Lrg1p function. (A) Shmoos were stained with aniline blue and examined by fluorescence microscopy for β(1-3) glucan distribution. Wild-type (MY427) cells show strong staining at the neck, but not the tip of shmoos. Both fus2 (MY5489) and lrg1 (MY5503) strains showed strong staining throughout the mating projection including the shmoo tip. fus2 [FUS2] shows staining of a fus2Δ strain (MY5489) containing a centromere-based FUS2 plasmid (pMR3725). fus2 [LRG12μ] shows staining of a fus2Δ strain (MY1817) containing LRG1 2μ (pMR3859). fus2 [vector] shows staining of a fus2Δ strain (MY5489) containing a centromere-based vector control plasmid (pRS414). (B) Chitin localization in shmoos from the indicated strains was determined by staining with calcofluor white and examination by fluorescence microscopy. All strains showed the wild-type staining pattern.
DISCUSSION
Summary:
LRG1 and BEM1 were identified in a screen for high-copy suppressors of fus2Δ. High-copy BEM1 was similar to FUS1 and FUS2 in that it suppressed cell fusion mutations affecting several pathways. We speculate that increased dosage of Bem1p suppresses the defects by increasing the concentration of key signaling components at the site of cell fusion (Leeuw et al. 1995; Lyons et al. 1996; Moskow et al. 2000) and thereby compensates for an array of cell fusion deficiencies.
In contrast to the broad suppression capabilities of increased BEM1, high-copy LRG1 suppression was specific to the FUS2-RVS161 cell fusion pathway. We confirmed that Lrg1p is part of the network of proteins required for the formation of the diploid zygote by showing that deletions of LRG1 result in cell fusion deficiencies, and that LRG1 mutations exhibit genetic interactions with known cell fusion genes. In addition, Lrg1p localizes to a highly focused point at the tip of the mating projection, a feature of many cell fusion components.
We speculated that the role of Lrg1p in mating is dependent upon its GAP domain. The GAP domain of Lrg1p was required for mating and the Lrg1p GAP specifically stimulated the GTPase activity of Rho1p in vitro. Moreover, loss of Lrg1p leads to increased β(1-3) glucan and high-copy LRG1 leads to a reduction of excess β(1-3)-glucan at the mating projection in fus2Δ mutants. Taken together, these results suggest that Lrg1p functions in mating by locally inhibiting β(1-3)-glucan synthase activity via Rho1p at the site of cell fusion.
The roles of Lrg1p's structural domains in cell fusion:
LRG1 encodes a protein sequence with a putative transmembrane domain, three to four putative LIM domains, and a Rho-GAP homology domain. The reality of the transmembrane domain is less certain, as not all protein structure programs predict the transmembrane domain. If functional, the transmembrane region would be positioned such that three of the four putative LIM domains would be external to the cell, assuming that the Rho-GAP domain is cytoplasmic. Of the Lrg1p structural elements, the Rho-GAP domain clearly plays an important role in mating as deletion of the region caused a defect in cell fusion. However, the less severe defect of the GAP domain deletion relative to a complete deletion of the gene suggests that the other domains may also contribute to cell fusion. Of these, the putative LIM domains could be of functional significance. LIM domain-containing proteins have diverse roles in a wide range of organisms from yeast to humans (Dawid et al. 1998). LIM domains are thought to mediate zinc binding and protein-protein interactions important for cellular localization (Michelsen et al. 1993; Dawid et al. 1998; Khurana et al. 2002). Possibly, the Lrg1p LIM domains mediate interactions with other components at the tip of the mating projection and may be important for the localization of proteins such as Rho1p at the site of cell fusion.
Lrg1p regulated Rho1p during cell fusion:
Rho1p has been reported to have multiple functions in vegetative cells. These include a role in cell polarization, by the activation and localization of the formin Bni1p (Kohno et al. 1996; Dong et al. 2003) and by localization of the secretory exocyst complex (Guo et al. 2001). Rho1p is also required for cell wall maintenance, serving as an upstream regulator of the Pkc1p pathway (Nonaka et al. 1995; Drgonova et al. 1996; Kamada et al. 1996) and as the regulatory subunit of β(1-3)-glucan synthase (Mazur and Baginsky 1996; Qadota et al. 1996).
In principle, alterations in any of these Rho1p targets could lead to defects in cell fusion. Loss of cell polarization, as observed for bni1 and spa2 mutants, is known to cause defects in cell fusion (Dorer et al. 1997; Gammie et al. 1998). In addition, the Pkc1-mediated stress response impacts a myriad of functions involving cell fusion (Philips and Herskowitz 1997, 1998), including the localization (Kohno et al. 1996) and activation (Dong et al. 2003) of Bni1p. Finally, Rho1p, acting as a regulatory subunit of β(1-3)-glucan synthase, could regulate cell wall synthesis between fusing cells. We favor this final mode of action for Lrg1p regulation of Rho1p during cell fusion for the reasons detailed below.
As a Rho1-GTPase-activating protein, Lrg1p increases the rate of conversion of active Rho1p-GTP into inactive Rho1p-GDP. Therefore, deletion of LRG1 should increase the activity of Rho1p and elevated dosage of LRG1 should decrease the activity of Rho1p. That is, Lrg1p is expected to be a negative regulator of Rho1p. With this in mind, we suggest that Rho1p's general involvement in polarization during vegetative growth is not likely to be relevant to the specific Lrg1p-regulated role of Rho1p in cell fusion during mating. First, the defects in Bni1p and exocyst polarization observed in rho1 mutants are a consequence of the loss of Rho1p activity (Guo et al. 2001; Dong et al. 2003). Thus, increased activity of Rho1p, caused by a lack of Lrg1p, would not be expected to cause significant defects in cell polarization. Consistent with this prediction, we observed that neither the loss of Lrg1p nor the overexpression of Lrg1p caused observable changes in cell polarization in response to mating pheromone. Second, Rho1p's activation of Bni1p has been shown to be a temperature-dependent, stress response mediated by Pkc1p (Dong et al. 2003) and is not likely to occur during standard mating conditions. Finally, Bni1p localization is strongly dependent upon Spa2p (Fujiwara et al. 1999); therefore, if the localization of Bni1p were the main function of Lrg1p-regulated Rho1p, then the double-mutant phenotype of a spa2Δ lrg1Δ should have been no worse than either of the single-mutant phenotypes. Instead, we observed a more severe phenotype in the double-mutant matings. Taken together, we conclude that the role of Lrg1p-modulated Rho1p is not likely to be due to significant alterations in polarization during mating.
It is more difficult to rule out a model in which Lrg1p regulates Rho1p's effects on the Pkc1p pathway during mating. Philips and Herskowitz showed that hyperactivated Pkc1p leads to cell fusion defects (Philips and Herskowitz 1997), and in the absence of Lrg1p, increased activity of Rho1p would be expected to activate the Pkc1p pathway. Conflicting reports either exclude (Watanabe et al. 2001) or include (Lorberg et al. 2001) Lrg1p in the Pkc1p pathway. Watanabe et al. (2001) provide evidence that loss of Lrg1p does not lead to changes in the phosphorylation of downstream proteins or in the transcription of genes regulated by Pkc1p in mitotic cells; whereas Lorberg et al. (2001) show the converse. While the role of Lrg1p is still controversial, it has been confirmed that Rho1p has distinct and genetically separable effects on the Pkc1p pathway and β(1-3)-glucan synthase activity (Drgonova et al. 1996; Roh et al. 2002).
Without directly excluding a role for the Pkc1p pathway, our data suggest that modulating β(1-3)-glucan synthase activity via Rho1p at the mating projection is likely to be the function of Lrg1p in mating. We found that both lrg1Δ and fus2Δ caused increased deposition of β(1-3)-glucan at the shmoo tip, and that the fus2Δ phenotype was suppressed by high-copy LRG1. In addition, examination of rho1 alleles revealed that rho1 mutations that specifically affect β(1-3)-glucan synthase, but not actin localization, were able to partially suppress lrg1Δ's mating defects (our unpublished observations). In light of these results, we speculate that the most plausible role for Lrg1p is to negatively regulate β(1-3)-glucan synthase via Rho1p. This inference does not diminish the possible relevance of other functions of Rho1p during mating, which would be independent of Lrg1p.
Lrg1p is not likely to be the only factor controlling the cell wall at the zone of cell fusion. Previous genetic studies and data presented here implicate FUS2 and RVS161 in cell wall breakdown. In high copy, Lrg1p can compensate for defects in Fus2p and Rvs161p functioning; however, Lrg1p does not appear to act in precisely the same pathway because the double mutants exhibit a more severe phenotype. We speculate that Lrg1p is important for the decreased synthesis of new cell wall material at the zone of cell fusion, whereas Fus2p and Rvs161p may be important for the delivery or activity of glucanases at the zone of cell fusion. This hypothesis is consistent with our previous observation that fus2 and rvs161 mutant zygotes accumulate vesicles at the zone of cell fusion (Gammie et al. 1998). Defects in both pathways would lead to accumulation of β(1-3)-glucan at the mating projection, but by different means.
Overall maintenance of the cell wall integrity is critical to cell viability. As such, the strict localization of cell wall removal is likely to be crucial for allowing cell fusion to occur while preserving the integrity of the zygote. The localization of Lrg1p at the eventual site of cell fusion is likely to be a significant aspect of this process. If cell polarity were disrupted, then Lrg1p would not be sufficiently localized to inhibit synthesis of β(1-3)-glucan at the site of cell fusion. Along these lines, high-copy Lrg1p was able to partially suppress defects in those cell fusion mutants that have properly polarized cellular components (e.g., fus2 and rvs161), but was unable to suppress defects in mutants that have failed to adequately polarize (e.g., spa2 and fus1).
The mechanism of cell wall breakdown during mating is likely to involve the concerted function of several separate activities including the polarization of key components and the localized breakdown of existing cell wall structure via secreted exo- and endoglucanases. In addition, we are proposing that the localized inhibition of β(1-3)-glucan synthesis at the zone of cell fusion represents a new regulatory component. Maintenance of the cell wall in regions of growth must involve a careful balance between deposition and degradation of the cell wall. Cell wall breakdown may then arise from a change in the balance between these two antagonistic activities. Only when the synthesis of the cell wall is negatively regulated at the site of cell fusion and there is a focused release of cell wall-degrading enzymes from polarized vesicles would a cell wall fusion pore of sufficient dimensions be formed efficiently. If any of the pathways were disrupted then cell fusion would be less effective. If two of the pathways were blocked, then cell fusion would be severely impacted. In keeping with this, cell fusion mutations in combination often result in much more severe blocks to fusion than does either mutation alone (Trueheart et al. 1987; Gammie et al. 1998; Philips and Herskowitz 1998). Additional work will be required to further unravel the multiple mechanisms contributing to efficient cell fusion during mating.
Acknowledgments
We are grateful to the laboratories of G. Fink, I. Herskowitz, P. Hieter, T. Stearns, and F. Winston for generously supplying plasmids and strains. This work was supported by a National Institutes of Health grant (GM37739) awarded to M. Rose.
References
- Abmayr, S. M., L. Balagopalan, B. J. Galletta and S. J. Hong, 2003. Cell and molecular biology of myoblast fusion. Int. Rev. Cytol. 225: 33–89. [DOI] [PubMed] [Google Scholar]
- Apanovitch, D. M., K. C. Slep, P. B. Sigler and H. G. Dohlman, 1998. Sst2 is a GTPase-activating protein for Gpa1: purification and characterization of a cognate RGS-Galpha protein pair in yeast. Biochemistry 37: 4815–4822. [DOI] [PubMed] [Google Scholar]
- Ausubel, F. M., 1994 Current Protocols in Molecular Biology. John Wiley & Sons, New York.
- Boeckmann, B., A. Bairoch, R. Apweiler, M. C. Blatter, A. Estreicher et al., 2003. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 31: 365–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brizzio, V., A. E. Gammie, G. Nijbroek, S. Michaelis and M. D. Rose, 1996. Cell fusion during yeast mating requires high levels of a-factor mating pheromone. J. Cell Biol. 135: 1727–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brizzio, V., A. E. Gammie and M. D. Rose, 1998. Rvs161p interacts with Fus2p to promote cell fusion in Saccharomyces cerevisiae. J. Cell Biol. 141: 567–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke, D., D. Dawson and T. Stearns, 2000 Methods in Yeast Genetics: A Laboratory Course Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Byers, B., and L. Goetsch, 1975. Behavior of spindles and spindle plaques in the cell cycle and conjugation of Saccharomyces cerevisiae. J. Bacteriol. 124: 511–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson, M., and D. Botstein, 1982. Two differentially regulated mRNAs with different 5′ ends encode secreted with intracellular forms of yeast invertase. Cell 28: 145–154. [DOI] [PubMed] [Google Scholar]
- Conrad, N. K., S. M. Wilson, E. J. Steinmetz, M. Patturajan, D. A. Brow et al., 2000. A yeast heterogeneous nuclear ribonucleoprotein complex associated with RNA polymerase II. Genetics 154: 557–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawid, I. B., J. J. Breen and R. Toyama, 1998. LIM domains: multiple roles as adapters and functional modifiers in protein interactions. Trends Genet. 14: 156–162. [DOI] [PubMed] [Google Scholar]
- Dohlman, H. G., and J. W. Thorner, 2001. Regulation of G protein-initiated signal transduction in yeast: paradigms and principles. Annu. Rev. Biochem. 70: 703–754. [DOI] [PubMed] [Google Scholar]
- Dong, Y., D. Pruyne and A. Bretscher, 2003. Formin-dependent actin assembly is regulated by distinct modes of Rho signaling in yeast. J. Cell Biol. 161: 1081–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorer, R., C. Boone, T. Kimbrough, J. Kim and L. H. Hartwell, 1997. Genetic analysis of default mating behavior in Saccharomyces cerevisiae. Genetics 146: 39–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drgonova, J., T. Drgon, K. Tanaka, R. Kollar, G. C. Chen et al., 1996. Rho1p, a yeast protein at the interface between cell polarization and morphogenesis. Science 272: 277–279. [DOI] [PubMed] [Google Scholar]
- Elia, L., and L. Marsh, 1996. Role of the ABC transporter Ste6 in cell fusion during yeast conjugation. J. Cell Biol. 135: 741–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elion, E. A., 2000. Pheromone response, mating and cell biology. Curr. Opin. Microbiol. 3: 573–581. [DOI] [PubMed] [Google Scholar]
- Elion, E. A., P. L. Grisafi and G. R. Fink, 1990. FUS3 encodes a cdc2/CDC28-related kinase required for the transition from mitosis into conjugation. Cell 60: 649–664. [DOI] [PubMed] [Google Scholar]
- Elion, E. A., B. Satterberg and J. E. Kranz, 1993. FUS3 phosphorylates multiple components of the mating signal transduction cascade: evidence for STE12 and FAR1. Mol. Biol. Cell 4: 495–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elion, E. A., J. Trueheart and G. R. Fink, 1995. Fus2 localizes near the site of cell fusion and is required for both cell fusion and nuclear alignment during zygote formation. J. Cell Biol. 130: 1283–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdman, S., L. Lin, M. Malczynski and M. Snyder, 1998. Pheromone-regulated genes required for yeast mating differentiation. J. Cell Biol. 140: 461–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evangelista, M., K. Blundell, M. S. Longtine, C. J. Chow, N. Adames et al., 1997. Bni1p, a yeast formin linking cdc42p and the actin cytoskeleton during polarized morphogenesis. Science 276: 118–122. [DOI] [PubMed] [Google Scholar]
- Fais, S., V. L. Burgio, M. R. Capobianchi, S. Gessani, F. Pallone et al., 1997. The biological relevance of polykaryons in the immune response. Immunol. Today 18: 522–527. [DOI] [PubMed] [Google Scholar]
- Fujimura, H. A., 1992. The DAC2/FUS3 protein kinase is not essential for transcriptional activation of the mating pheromone response pathway in Saccharomyces cerevisiae. Mol. Gen. Genet. 235: 450–452. [DOI] [PubMed] [Google Scholar]
- Fujiwara, T., K. Tanaka, E. Inoue, M. Kikyo and Y. Takai, 1999. Bni1p regulates microtubule-dependent nuclear migration through the actin cytoskeleton in Saccharomyces cerevisiae. Mol. Cell. Biol. 19: 8016–8027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gammie, A. E., and M. D. Rose, 2002. Assays of cell and nuclear fusion. Methods Enzymol. 351: 477–498. [DOI] [PubMed] [Google Scholar]
- Gammie, A. E., V. Brizzio and M. D. Rose, 1998. Distinct morphological phenotypes of cell fusion mutants. Mol. Biol. Cell 9: 1395–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehrung, S., and M. Snyder, 1990. The SPA2 gene of Saccharomyces cerevisiae is important for pheromone-induced morphogenesis and efficient mating. J. Cell Biol. 111: 1451–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, W., F. Tamanoi and P. Novick, 2001. Spatial regulation of the exocyst complex by Rho1 GTPase. Nat. Cell Biol. 3: 353–360. [DOI] [PubMed] [Google Scholar]
- Hasek, J., I. Rupes, J. Svobodova and E. Streiblova, 1987. Tubulin and actin topology during zygote formation of Saccharomyces cerevisiae. J. Gen. Microbiol. 133: 3355–3363. [DOI] [PubMed] [Google Scholar]
- Heiman, M. G., and P. Walter, 2000. Prm1p, a pheromone-regulated multispanning membrane protein, facilitates plasma membrane fusion during yeast mating. J. Cell Biol. 151: 719–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamada, Y., H. Qadota, C. P. Python, Y. Anraku, Y. Ohya et al., 1996. Activation of yeast protein kinase C by Rho1 GTPase. J. Biol. Chem. 271: 9193–9196. [DOI] [PubMed] [Google Scholar]
- Khurana, T., B. Khurana and A. A. Noegel, 2002. LIM proteins: association with the actin cytoskeleton. Protoplasma 219: 1–12. [DOI] [PubMed] [Google Scholar]
- Kohno, H., K. Tanaka, A. Mino, M. Umikawa, H. Imamura et al., 1996. Bni1p implicated in cytoskeletal control is a putative target of Rho1p small GTP binding protein in Saccharomyces cerevisiae. EMBO J. 15: 6060–6068. [PMC free article] [PubMed] [Google Scholar]
- Leeuw, T., A. Fourest-Lieuvin, C. Wu, J. Chenevert, K. Clark et al., 1995. Pheromone response in yeast: association of Bem1p with proteins of the MAP kinase cascade and actin. Science 270: 1210–1213. [DOI] [PubMed] [Google Scholar]
- Liu, H., and A. Bretscher, 1992. Characterization of TPM1 disrupted yeast cells indicates an involvement of tropomyosin in directed vesicular transport. J. Cell Biol. 118: 285–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorberg, A., H. P. Schmitz, J. J. Jacoby and J. J. Heinisch, 2001. Lrg1p functions as a putative GTPase-activating protein in the Pkc1p-mediated cell integrity pathway in Saccharomyces cerevisiae. Mol. Genet. Genomics 266: 514–526. [DOI] [PubMed] [Google Scholar]
- Lyons, D. M., S. K. Mahanty, K. Y. Choi, M. Manandhar and E. A. Elion, 1996. The SH3-domain protein Bem1 coordinates mitogen-activated protein kinase cascade activation with cell cycle control in Saccharomyces cerevisiae. Mol. Cell. Biol. 16: 4095–4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddox, P., E. Chin, A. Mallavarapu, E. Yeh, E. D. Salmon et al., 1999. Microtubule dynamics from mating through the first zygotic division in the budding yeast Saccharomyces cerevisiae. J. Cell Biol. 144: 977–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh, L., and M. D. Rose, 1997 The pathway of cell and nuclear fusion during mating in S. cerevisiae, pp. 827–888 in The Molecular and Cellular Biology of the Yeast Saccharomyces: Cell Cycle and Cell Biology, edited by J. R. Pringle, J. R. Broach and E. W. Jones. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Mazur, P., and W. Baginsky, 1996. In vitro activity of 1,3-beta-D-glucan synthase requires the GTP-binding protein Rho1. J. Biol. Chem. 271: 14604–14609. [DOI] [PubMed] [Google Scholar]
- McCaffrey, G., F. J. Clay, K. Kelsay and G. F. Sprague, Jr., 1987. Identification and regulation of a gene required for cell fusion during mating of the yeast Saccharomyces cerevisiae. Mol. Cell. Biol. 7: 2680–2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelsen, J. W., K. L. Schmeichel, M. C. Beckerle and D. R. Winge, 1993. The LIM motif defines a specific zinc-binding protein domain. Proc. Natl. Acad. Sci. USA 90: 4404–4408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskow, J. J., A. S. Gladfelter, R. E. Lamson, P. M. Pryciak and D. J. Lew, 2000. Role of Cdc42p in pheromone-stimulated signal transduction in Saccharomyces cerevisiae. Mol. Cell. Biol. 20: 7559–7571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller, L., G. Xu, R. Wells, C. P. Hollenberg and W. Piepersberg, 1994. LRG1 is expressed during sporulation in Saccharomyces cerevisiae and contains motifs similar to LIM and rho/racGAP domains. Nucleic Acids Res. 22: 3151–3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonaka, H., K. Tanaka, H. Hirano, T. Fujiwara, H. Kohno et al., 1995. A downstream target of RHO1 small GTP-binding protein is PKC1, a homolog of protein kinase C, which leads to activation of the MAP kinase cascade in Saccharomyces cerevisiae. EMBO J. 14: 5931–5938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philips, J., and I. Herskowitz, 1997. Osmotic balance regulates cell fusion during mating in Saccharomyces cerevisiae. J. Cell Biol. 138: 961–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philips, J., and I. Herskowitz, 1998. Identification of Kel1p, a kelch domain-containing protein involved in cell fusion and morphology in Saccharomyces cerevisiae. J. Cell Biol. 143: 375–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posas, F., M. Takekawa and H. Saito, 1998. Signal transduction by MAP kinase cascades in budding yeast. Curr. Opin. Microbiol. 1: 175–182. [DOI] [PubMed] [Google Scholar]
- Pringle, J. R., A. E. Adams, D. G. Drubin and B. K. Haarer, 1991. Immunofluorescence methods for yeast. Methods Enzymol. 194: 565–602. [DOI] [PubMed] [Google Scholar]
- Qadota, H., C. P. Python, S. B. Inoue, M. Arisawa, Y. Anraku et al., 1996. Identification of yeast Rho1p GTPase as a regulatory subunit of 1,3-beta-glucan synthase. Science 272: 279–281. [DOI] [PubMed] [Google Scholar]
- Roberts, C. J., B. Nelson, M. J. Marton, R. Stoughton, M. R. Meyer et al., 2000. Signaling and circuitry of multiple MAPK pathways revealed by a matrix of global gene expression profiles. Science 287: 873–880. [DOI] [PubMed] [Google Scholar]
- Roh, D. H., B. Bowers, H. Riezman and E. Cabib, 2002. Rho1p mutations specific for regulation of beta(1→3)glucan synthesis and the order of assembly of the yeast cell wall. Mol. Microbiol. 44: 1167–1183. [DOI] [PubMed] [Google Scholar]
- Rose, M. D., F. Winston and P. Hieter, 1990 Methods in Yeast Genetics: A Laboratory Course Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Rothstein, R., 1991. Targeting, disruption, replacement, and allele rescue: integrative DNA transformation in yeast. Methods Enzymol. 194: 281–301. [DOI] [PubMed] [Google Scholar]
- Sambrook, J., E. F. Fritsch and T. Maniatis, 1989 Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Santos, B., and M. Snyder, 1997. Targeting of chitin synthase 3 to polarized growth sites in yeast requires Chs5p and Myo2p. J. Cell Biol. 136: 95–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos, B., A. Duran and M. H. Valdivieso, 1997. CHS5, a gene involved in chitin synthesis and mating in Saccharomyces cerevisiae. Mol. Cell. Biol. 17: 2485–2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherer, S., and R. W. Davis, 1979. Replacement of chromosome segments with altered DNA sequences constructed in vitro. Proc. Natl. Acad. Sci. USA 76: 4951–4955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider, B. L., W. Seufert, B. Steiner, Q. H. Yang and A. B. Futcher, 1995. Use of polymerase chain reaction epitope tagging for protein tagging in Saccharomyces cerevisiae. Yeast 11: 1265–1274. [DOI] [PubMed] [Google Scholar]
- Segall, J. E., 1993. Polarization of yeast cells in spatial gradients of alpha mating factor. Proc. Natl. Acad. Sci. USA 90: 8332–8336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shemer, G., and B. Podbilewicz, 2000. Fusomorphogenesis: cell fusion in organ formation. Dev. Dyn. 218: 30–51. [DOI] [PubMed] [Google Scholar]
- Shemer, G., and B. Podbilewicz, 2003. The story of cell fusion: big lessons from little worms. BioEssays 25: 672–682. [DOI] [PubMed] [Google Scholar]
- Sikorski, R., and P. Hieter, 1989. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122: 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprague, G. F., and J. W. Thorner, 1992 Pheromone response and signal transduction during the mating process of Saccharomyces cerevisiae, pp. 657–744 in The Molecular and Cellular Biology of the Yeast Saccharomyces: Gene Expression, edited by E. W. Jones, J. R. Pringle and J. R. Broach. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Steinmetz, E. J., N. K. Conrad, D. A. Brow and J. L. Corden, 2001. RNA-binding protein Nrd1 directs poly(A)-independent 3′-end formation of RNA polymerase II transcripts. Nature 413: 327–331. [DOI] [PubMed] [Google Scholar]
- Sugimoto, K., K. Matsumoto, R. D. Kornberg, S. I. Reed and C. Wittenberg, 1995. Dosage suppressors of the dominant G1 cyclin mutant CLN3–2: identification of a yeast gene encoding a putative RNA/ssDNA binding protein. Mol. Gen. Genet. 248: 712–718. [DOI] [PubMed] [Google Scholar]
- Talbot, P., B. D. Shur and D. G. Myles, 2003. Cell adhesion and fertilization: steps in oocyte transport, sperm-zona pellucida interactions, and sperm-egg fusion. Biol. Reprod. 68: 1–9. [DOI] [PubMed] [Google Scholar]
- Taylor, M. V., 2000. Muscle development: molecules of myoblast fusion. Curr. Biol. 10: R646–R648. [DOI] [PubMed] [Google Scholar]
- Trueheart, J., and G. R. Fink, 1989. The yeast cell fusion protein FUS1 is O-glycosylated and spans the plasma membrane. Proc. Natl. Acad. Sci. USA 86: 9916–9920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trueheart, J., J. D. Boeke and G. R. Fink, 1987. Two genes required for cell fusion during yeast conjugation: evidence for a pheromone-induced surface protein. Mol. Cell. Biol. 7: 2316–2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdivia, R. H., and R. Schekman, 2003. The yeasts Rho1p and Pkc1p regulate the transport of chitin synthase III (Chs3p) from internal stores to the plasma membrane. Proc. Natl. Acad. Sci. USA 100: 10287–10292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valtz, N., and I. Herskowitz, 1996. Pea2 protein of yeast is localized to sites of polarized growth and is required for efficient mating and bipolar budding. J. Cell Biol. 135: 725–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilopoulos, G., and D. W. Russell, 2003. Cell fusion: an alternative to stem cell plasticity and its therapeutic implications. Curr. Opin. Genet. Dev. 13: 480–485. [DOI] [PubMed] [Google Scholar]
- Vassilopoulos, G., P. R. Wang and D. W. Russell, 2003. Transplanted bone marrow regenerates liver by cell fusion. Nature 422: 901–904. [DOI] [PubMed] [Google Scholar]
- Vignery, A., 2000. Osteoclasts and giant cells: macrophage-macrophage fusion mechanism. Int. J. Exp. Pathol. 81: 291–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner, P., L. Hengst and D. Gallwitz, 1992. Ypt proteins in yeast. Methods Enzymol. 219: 369–387. [DOI] [PubMed] [Google Scholar]
- Wang, X., H. Willenbring, Y. Akkari, Y. Torimaru, M. Foster et al., 2003. Cell fusion is the principal source of bone-marrow-derived hepatocytes. Nature 422: 897–901. [DOI] [PubMed] [Google Scholar]
- Wassarman, P. M., L. Jovine and E. S. Litscher, 2001. A profile of fertilization in mammals. Nat. Cell Biol. 3: E59–E64. [DOI] [PubMed] [Google Scholar]
- Watanabe, D., M. Abe and Y. Ohya, 2001. Yeast Lrg1p acts as a specialized RhoGAP regulating 1,3-beta-glucan synthesis. Yeast 18: 943–951. [DOI] [PubMed] [Google Scholar]
- Wilson, S. M., K. V. Datar, M. R. Paddy, J. R. Swedlow and M. S. Swanson, 1994. Characterization of nuclear polyadenylated RNA-binding proteins in Saccharomyces cerevisiae. J. Cell Biol. 127: 1173–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]