

Abstract
Epigenetic mutation, heritable developmental variation not based on a change in nucleotide sequence, is widely reported in plants. However, the developmental and evolutionary significance of such mutations remains enigmatic. On the basis of our studies of the endogenous Arabidopsis transposon CACTA, we propose that the inheritance of epigenetic gene silencing over generations can function as a transgenerational genome defense mechanism against deleterious movement of transposons. We previously reported that silent CACTA1 is mobilized by the DNA hypomethylation mutation ddm1 (decrease in DNA methylation). In this study, we report that CACTA activated by the ddm1 mutation remains mobile in the presence of the wild-type DDM1 gene, suggesting that de novo silencing is not efficient for the defense of the genome against CACTA movement. The defense depends on maintenance of transposon silencing over generations. In addition, we show that the activated CACTA1 element transposes throughout the genome in DDM1 plants, as reported previously for ddm1 backgrounds. Furthermore, the CACTA1 element integrated into both the ddm1-derived and the DDM1-derived chromosomal regions in the DDM1 wild-type plants, demonstrating that this class of transposons does not exhibit targeted integration into heterochromatin, despite its accumulation in the pericentromeric regions in natural populations. The possible contribution of natural selection as a mechanism for the accumulation of transposons and evolution of heterochromatin is discussed.
METHYLATION of cytosine, together with histone modifications, plays a central role in epigenetic gene regulation. Mutations affecting DNA methylation induce transcriptional perturbations and developmental defects in both vertebrates and plants (Li et al. 1992, 1995; Finnegan et al. 1996; Kakutani et al. 1996, 1997; Ronemus et al. 1996; Soppe et al. 2000; Stancheva and Meehan 2000; Stokes et al. 2002; Kankel et al. 2003; Kinoshita et al. 2004). Interestingly, some of the developmental abnormalities in plants induced by changes in DNA methylation and transcription are inherited over many generations (Jacobsen and Meyerowitz 1997; Soppe et al. 2000; Stokes et al. 2002). Similar epigenetic variations heritable over generations have also been reported in natural plant populations and in some mouse strains (Cubas et al. 1999; Whitelaw and Martin 2001; Rakyan et al. 2003). However, the developmental and evolutionary significance of the inheritance of epigenetic information over generations remain unclear.
In addition to the genes silenced at specific developmental stages, many eukaryotes have constitutive heterochromatic chromosomal regions, which are condensed and silent constitutively throughout the life cycle. Notably, the major components of such constitutive heterochromatin are often silent transposons and their derivatives. For example, pericentromeric heterochromatin regions of the flowering plant Arabidopsis thaliana contain many copies of a retroelement-related sequence, Athila, as well as several other classes of sequences related to retroelements and DNA-type transposons (Pelissier et al. 1996; Arabidopsis Genome Initiative 2000). Such transposon-rich regions have been found in the genomes of many plant species, although it is not well understood how the transposon-related sequences accumulate in those regions (Schmidt et al. 1995; Pearce et al. 1996; Miller et al. 1998; Presting et al. 1998; Miura et al. 2004).
The transposon-related sequences in plants, vertebrates, and some fungi are often marked by the DNA methylation, and it has been proposed that the primary function of DNA methylation may be to protect the genome from the deleterious effects of transposons (Yoder et al. 1997; Matzke et al. 1999; Selker et al. 2003). Consistent with such a “genome defense” hypothesis for the function of DNA methylation, some endogenous Arabidopsis transposons are mobilized by mutations affecting genomic DNA methylation (Miura et al. 2001; Singer et al. 2001; Kato et al. 2003). For example, the endogenous DNA-type transposon CACTA1 is silent in the wild-type background but transposes in mutants deficient in the DDM1 (decrease in DNA methylation) gene, which encodes a chromatin-remodeling factor (Jeddeloh et al. 1999; Brzeski and Jerzmanowski 2003). The ddm1 mutation affects DNA methylation, transcription, and transposition of these transposons, although it is unknown whether the ddm1 mutation affects the specificity of the transposon integration sites (Miura et al. 2001, 2004).
In this study, we demonstrated that the CACTA1 transposon activated by the ddm1 mutation maintains its mobility in the DDM1 wild-type background; in this sense, the activated CACTA1 behaves in a manner similar to epigenetic mutations. In addition, this system allowed us to directly examine the integration site specificity of CACTA1 in the DDM1 wild-type background. As the ddm1-derived pericentromeric regions still lost the epigenetic marks of heterochromatin, we were able to compare the integration preference for the ddm1-derived and wild-type-derived regions. No bias for preferential integration into the heterochromatic regions was detected. On the basis of these results, we discuss the biological meaning of epigenetic inheritance over generations and control of transposons.
MATERIALS AND METHODS
Plant materials and genotyping:
The isolation of the ddm1 mutants in Columbia (Col) background was previously reported by Vongs et al. (1993). The wild-type C24 plant was a gift from Jean Finnegan. The ddm1-1 mutant and the wild-type DDM1 alleles were distinguished by NsiI digestibility of the PCR product using primers 5′-ATTTGCTGATGACCAGGTCCT-3′ and 5′-CATAAACCAATCTCATGAGGC-3′.
Analysis of transcription, transposition, and methylation of CACTA elements:
CACTA1 transcript was detected by RT-PCR, the CACTA mobility was examined by Southern analysis, and the cytosine methylation status in the CACTA1 5′ region was examined using the bisulfite-mediated sequencing method as described previously (Kato et al. 2003).
Characterization of the CACTA1 integration sites:
Identification of integration site:
Wild-type DDM1/DDM1 plants were selected from the F2 families from crosses between ddm1/ddm1 and DDM1/DDM1 plants. The genomic locations of the CACTA1 integration sites were determined by suppression PCR with modification of the conditions described previously (Miura et al. 2001). The genomic DNA was digested by EcoRV or HincII before adapter ligation. The CACTA1 flanking regions were amplified from the ligated product using the primer pairs 5′-GGATCCTAATACGACTCACTATAGGGC-3′ + 5′-CAGCGACAGATCTTAGCTTTTAGGTTG-3′ and, subsequently, 5′-AATAGGGCTCGAGCGGC-3′ + 5′-GGATTCGACAGATCTAAGGCA-3′. The PCR products obtained were separated by agarose gel and each fragment was directly sequenced with the primer 5′-AGTGTTGGCGCTGAAGTGAAT-3′. The integration sites of transposed CACTA1 were determined from flanking sequences using BLAST search in The Arabidopsis Information Resource (TAIR, http://www.arabidopsis.org/).
Detection of Col/C24 polymorphism around the integration site:
To detect polymorphism between Col and C24, ∼2-kb regions around each of the CACTA1 integration sites were sequenced for these ecotypes. The primers used for amplification and sequencing of these regions are shown in the supplementary data available at http://www.genetics.org/supplemental/.
Determination of the origin of the integration site:
The flanking sequences of both the 5′ and the 3′ sides of transposed CACTA1 were amplified using primers, one from the flanking region and the other from CACTA1 (shown as solid and open arrows, respectively, in Figure 4A). Finally, the polymorphic sites were sequenced directly to determine whether the integration site was ddm1 (Col) or wild type (C24) derived. For each of the integration sites, at least two polymorphic sites were examined.
Figure 4.—


Distribution of integration sites of the CACTA1 element in a wild-type background. (A) Procedures to characterize integration sites of CACTA1. (Top) Regions flanking the 5′ side of CACTA1 were sequenced by suppression PCR. Identified flanking regions are shown as solid. (Middle) The regions spanning 2 kb (1 kb upstream and 1 kb downstream) surrounding the integration sites were amplified from the Col and C24 genomes and sequenced. Polymorphism in the nucleotide sequence between Col and C24 is indicated by an “x.” (Bottom) Sequences flanking the 5′ side and 3′ side of transposed CACTA1 were amplified with primers near the integration site (solid arrow) and from the transposon (open arrows). The parental origin of the integration site was identified using Col/C24 polymorphism (indicated by an “x”). (B) Integration sites of transposed CACTA1 were examined in 11 wild-type DDM1 F2 plants derived from C24 WT × ddm1 (plants 1, 2, 3, 6, 7, 9, 11, 12, 13, 23, and 24 in Figure 2A). The integration sites of CACTA1 are shown by arrowheads. Open and solid arrowheads represent the integration into the Col (ddm1-derived) and C24 (wild-type-derived) genomes, respectively (shaded arrowheads: unclassified integration sites). For each of the integration sites, the number corresponds to the plant number in Figure 2A. The position of each integration site was estimated using theTAIR MapViewer (http://arabidopsis.org/servlets/mapper).
RESULTS
The ddm1-derived CACTA1 remains transcriptionally active in the wild-type background:
We first examined whether the CACTA1 transposon activated by the ddm1 DNA hypomethylation mutation remains active in the wild-type background. The ddm1/ddm1 mutants with an active CACTA1 element were crossed to wild-type DDM1/DDM1 plants of the C24 strain. The C24 strain does not contain sequence that hybridizes to the CACTA probe in Southern blot analysis, which enabled us to follow the ddm1-derived CACTA1 copies after segregation.
The CACTA1 transcript was detectable in F1 DDM1/ddm1 heterozygous plants produced by crosses between wild-type C24 plants and ddm1 mutants (Figure 1A). The transcript was not detectable in the control F1 hybrid plants from crossing wild-type C24 to wild-type Col plants. Essentially the same results were obtained from reciprocal crosses (Figure 1A). The transcript was also detectable after intrastrain crosses between Col and ddm1 (Figure S1 available at http://www.genetics.org/supplemental/). This was not due to heterozygosity of the DDM1 locus, because the transcript was undetectable in the DDM1/ddm1 heterozygote in which the CACTA1 locus had been replaced by repeated backcrossing to Col wild type (Figure 1A). In addition, the transcript was detectable in DDM1/DDM1 wild-type homozygotes generated by an additional backcross of the F1 plants to the wild-type plants (Figure 1B). These results indicate that CACTA1 activated by the ddm1 mutation remains transcriptionally active even in the wild-type DDM1 background. The active state was inherited through both male and female meiotic passages.
Figure 1.—

(A) Transcription of ddm1-derived CACTA1 after crosses to wild-type plants. Semiquantitative RT-PCR reactions corresponding to 25 ng and 4 ng of input total RNA are shown for each plant. C24 and Col represent wild-type (DDM1/DDM1) plants in those ecotypes. The DDM1/ddm1 heterozygote in lane 10 was derived from a ddm1/ddm1 Columbia plant, which was backcrossed six times to the wild-type Columbia plant in the heterozygous state (Kakutani et al. 1999). (B) Transcription of ddm1-derived CACTA1 after two successive backcrosses to C24 wild-type plants. Because C24 does not have CACTA1, plants with and without CACTA1 segregate after the second backcross (lanes 3, 5, and 6, and lanes 4 and 7, respectively). The plants in lanes 3 and 4 and lanes 5–7 are siblings. The constitutively expressed GapC gene was used as a control. Length of the predicted PCR product: CACTA1, 0.64 kb for cDNA and 0.72 kb for genomic DNA; GapC, 0.54 kb for cDNA and 0.82 kb for genomic DNA.
ddm1-derived CACTA1 remains mobile in the wild-type DDM1 background:
The ddm1 mutation induces not only transcription but also transposition of CACTA1 (Miura et al. 2001). We next examined the mobility of the ddm1-derived CACTA1 element in the wild-type DDM1 background. DDM1/DDM1 homozygotes were selected from self-pollinated progenies of the F1 DDM1/ddm1 (wild-type C24 × ddm1). In the Southern hybridization analysis, DDM1/DDM1 wild-type individuals in the F2 generation showed several new bands, which were undetectable in their direct parents (F1 DDM1/ddm1, plants A, B, C, and D in Figure 2A). These new bands in the F2 plants should reflect the transposition events in the F1 or F2 generation, suggesting that the CACTA1 activated by the ddm1 mutation remained mobile in the presence of the wild-type DDM1 allele. Furthermore, the CACTA1 continued to transpose in the next generation in the DDM1/DDM1 background; we examined self-pollinated progeny from some of the F2 DDM1/DDM1 wild-type homozygotes and found additional bands, which were undetectable in the previous generation (F2 wild-type DDM1/DDM1 parents; Figure 2B). Mobilization of CACTA1 was not due to the interstrain crosses between Col and C24, because transposition of CACTA1 was not detected in the F2 wild-type hybrid from crosses between two wild-type strains (Figure S2 at http://www.genetics.org/supplemental/). In addition, we were able to detect transposition in DDM1/DDM1 plants segregating from intrastrain crosses between ddm1 and wild-type Col plants (Figure S3 at http://www.genetics.org/supplemental/). These observations indicate that the CACTA1 element activated by the ddm1 mutation remained mobile for at least two generations in the presence of the wild-type DDM1 gene.
Figure 2.—
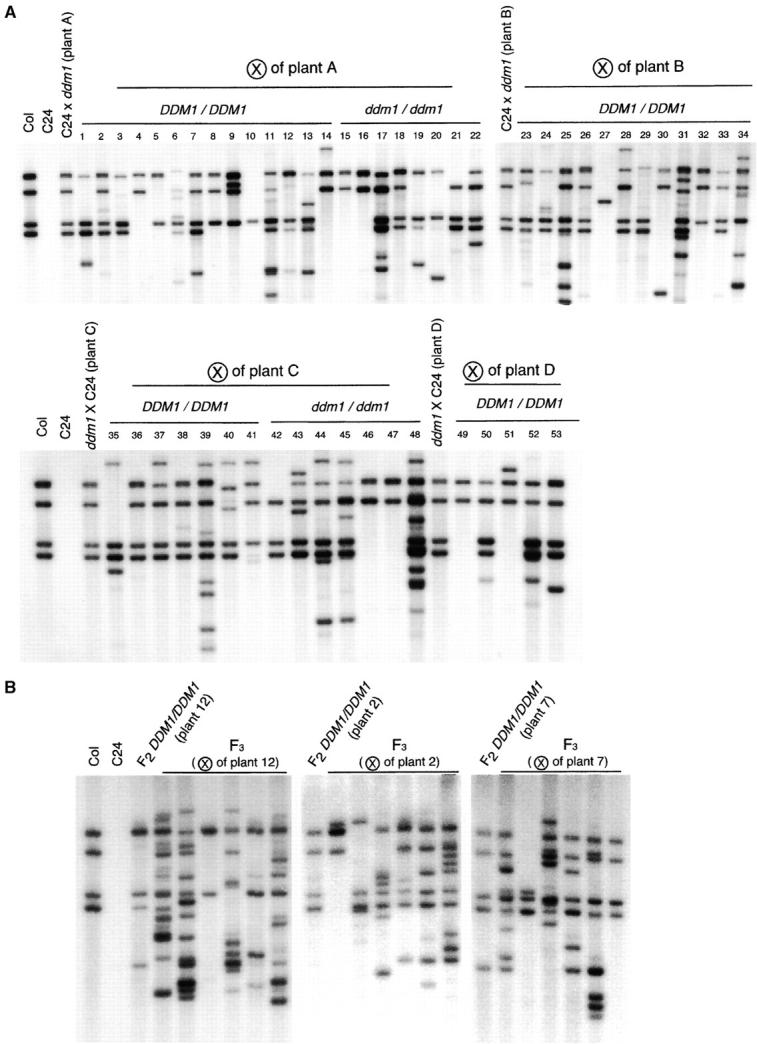
Transposition of ddm1-derived CACTA1 in a wild-type DDM1 background. Genomic DNAs were digested with EcoRV, which is insensitive to cytosine methylation, and examined by Southern analysis with a probe corresponding to the 5′ end of CACTA1 (probe B of Miura et al. 2001). C24 and Col represent wild-type (DDM1/DDM1) plants of those ecotypes. The C24 strain does not contain sequence that hybridizes to this probe. (A) The DDM1/ddm1 F1 plants (plants A–D) were derived from crosses between ddm1/ddm1 and wild-type (C24) DDM1/DDM1. From self-pollinated progeny of each of the F1 plants, DDM1/DDM1 and ddm1/ddm1 segregants were selected and their band patterns were compared to those of their direct parents. A circled X represents self-pollination. We initially examined five F1 plants, but one of them was not used for the F2 analysis, because it showed a strong new band, possibly reflecting transposition in the F0 ddm1/ddm1 generation. Cleavage by another enzyme, HindIII, suggests that plant C has a weak additional band possibly reflecting somatic transposition (not shown). (B) The wild-type DDM1/DDM1 plants in the F3 generation were derived by self-pollination of DDM1/DDM1 wild-type F2 plants, which correspond to plants 12, 2, and 7.
Hypomethylation in CACTA1 promoter region is inherited in the presence of the wild-type DDM1 allele:
We next examined whether the CACTA1 hypomethylated by ddm1 mutation remained hypomethylated in the wild-type background. We examined DNA methylation in ∼300 bp of the 5′ terminal region, which includes the entire upstream region from the transcriptional starting site (K. Watanabe and T. Kakutani, unpublished results). In wild-type Col, this region was heavily methylated, especially at the CpG sites. In contrast, DNA methylation was almost completely lost in this region in the ddm1 mutant (Figure 3; Figure S4 at http://www.genetics.org/supplemental/; Kato et al. 2003). The F1 DDM1/ddm1 plant (wild-type C24 × ddm1) has one CACTA1 copy derived from the ddm1 mutant parent. The CACTA1 copy in this F1 plant remained hypomethylated for both CpG and non-CG sites. In the F1 hybrid from a control interstrain cross (wild-type C24 × wild-type Col), the CACTA1 copy was normally methylated (Figure 3; Figure S4 at http://www.genetics.org/supplemental/. Essentially the same methylation patterns were obtained from reciprocal crosses (data not shown). These observations suggest that the ddm1-induced hypomethylation in this region was meiotically and mitotically transmitted even in the presence of the wild-type DDM1 copy. This observation is consistent with the inheritance of transposon mobility.
Figure 3.—

DNA methylation status of ddm1-derived CACTA1 in the presence of a wild-type DDM1 allele. The DNA methylation pattern of the bottom strand of the 5′ terminal 310-bp sequence of CACTA1 was determined by the bisulfite-mediated sequencing method. A total of 8–10 independent clones were examined for each plant. Distribution of the methylation in each clone is shown in Figure S4 at http://www.genetics.org/supplemental/.
Integration specificity of the CACTA transposon in the wild-type DDM1/DDM1 background:
DNA methylation is necessary for immobilization of CACTA and other transposons, consistent with the transcriptional activation of a variety of transposons and transposon-related sequences in DNA methylation mutants (Hirochika et al. 2000; Steimer et al. 2000; Lindroth et al. 2001; Miura et al. 2001; Singer et al. 2001; Johnson et al. 2002; Kato et al. 2003; Lippman et al. 2003). On the other hand, it is not known whether DNA methylation and the associated heterochromatin formation in the transposon integration site affect transposition efficiency. In other words, it is not known whether any of the Arabidopsis transposons preferentially integrate into heterochromatic regions.
We have previously shown that all five loci for CACTA transposons in the Col genome and 11 polymorphic integration sites of the related sequences in other ecotypes are localized near the centromeric heterochromatin (Miura et al. 2004). In contrast, they transpose throughout the genome in the ddm1 mutant background. The difference in integration sites between natural populations and ddm1 could be due to loss of pericentromeric heterochromatin mark(s) in the ddm1 mutant background (Miura et al. 2001, 2004). However, it is not clear whether the ddm1 mutation affects integration site specificity of the transposon.
We examined the integration sites of transposed CACTA1 in F2 DDM1/DDM1 wild-type plants derived from the crosses between ddm1 mutant and wild-type C24 plants. Self-pollinated progeny of F1 without strong additional bands (plants A and B in Figure 2A) was used to avoid detecting transposition in the F0 (ddm1) generation. The detected transposition should occur in the F1 (DDM1/ddm1) or F2 (DDM1/DDM1) generation. We confirmed each of the insertions by PCR using primers from flanking sequences and transposon sequences. The CACTA1 integration sites in the F2 DDM1/DDM1 background distributed to chromosomal arms as well as to pericentromeric regions (Figure 4B). This is in contrast to the pericentromeric distribution of CACTA-like sequences fixed in natural populations (Miura et al. 2004). In addition, although most of the CACTA-related sequences distribute in transposon-rich regions in natural populations, the integration sites for transposition induced in the laboratory did not show such a bias. Even in the DDM1 wild-type background, they were distributed in both transposon-rich and gene-rich regions (Table 1).
TABLE 1.
Distribution of the integration sites ofCACTA1 fixed in natural populations or induced in the laboratory inddm1 orDDM1
| Gene-rich regionsa |
Transposon-rich regionsb | Total | |
|---|---|---|---|
| Natural populationsc | 1 | 10 | 11 |
| Laboratory | |||
| ddm1d | 11 | 6 | 17 |
| DDM1e | 9 | 13 | 22 |
| To ddm1-derived regions |
5 | 8 | 13 |
| To DDM1-derived regions |
3 | 4 | 7 |
| Unclassified | 1 | 1 | 2 |
By chi-square test, the difference between natural populations and ddm1 is significant (P < 0.004). Although the difference between natural populations and DDM1 is not significant (P = 0.06), the difference is significant (P = 0.007) if 100-kb regions with only one sequence annotated to be transposon related are classified as gene rich rather than transposon rich.
Insertion into regions without sequences annotated to be transposon, transposase, or reverse transcriptase related within 50-kb + 50-kb window.
Insertion into regions other than those mentioned in footnote a.
This work.
The pericentromeric heterochromatic regions of Arabidopsis are detectable by DAPI staining as condensed structures and show a high degree of methylation in cytosine and lysine 9 (K9) of histone H3, which depend on the DDM1 gene; the ddm1 mutation abolishes DNA and H3K9 methylation and reduces the DAPI-stained chromocenter size (Soppe et al. 2002). Interestingly, all these effects of the ddm1 mutation on the pericentromeric regions are heritable; in the F1 hybrid between ddm1 and wild type, half of the chromocenters show decondensation and reduced methylation of DNA and H3K9, while the other half are indistinguishable from the normal wild-type chromocenter (Soppe et al. 2002). This observation suggests that the ddm1-induced loss of pericentromeric heterochromatin, associated with loss of DNA and H3K9 methylations, is heritable as an epigenetic imprint in the presence of wild-type DDM1 copy (Soppe et al. 2002). This offers a useful system to examine whether CACTA1 has integration preference between the heterochromatic and euchromatic regions.
We examined whether CACTA1 integrates preferentially into the wild-type-derived pericentromeric region. The wild-type-derived and the ddm1-derived chromosomal regions were distinguished using nucleotide sequence polymorphism between the two parental ecotypes, Col and C24 (detailed procedures are shown in Figure 4A and materials and methods). Instead of using molecular markers already available, we surveyed Col/C24 polymorphisms by sequencing 2-kb regions around each integration site to minimize possible ambiguity due to meiotic recombination with linked markers. Within the 22 CACTA1 integration sites, we could identify the Col/C24 polymorphisms in 20 loci. We examined the origin of alleles for each of these sites. A total of 13 insertions were on ddm1-derived chromosomes, while 7 were on wild-type-derived chromosomes (Figure 4B; Table 1). For each of the integration sites, identity was confirmed by examining more than two polymorphic sites. These results are not biased by segregation, because the ddm1- and wild-type-derived centromeric regions segregated randomly in the F2 generation (Table S1 at http://www.genetics.org/supplemental/). Taken together, these results demonstrate that CACTA1 integration is not biased toward wild-type-derived heterochromatic regions.
DISCUSSION
In the wild-type Col ecotype, the CACTA1 is localized within a pericentromeric region and remain silent. However, it is mobilized by mutations abolishing genomic DNA methylation (Miura et al. 2001; Kato et al. 2003). We showed in this study that the CACTA1 transposon mobilized by the DNA hypomethylation mutation ddm1 remained mobile in DDM1 wild-type backgrounds. The results are striking considering that CACTA1 is completely silent in original wild-type Col plants. We have never found movement of this transposon in >100 wild-type Col plants examined (A. Miura, M. Kato and T. Kakutani, unpublished results). In addition, six Col accessions, Col-0, -1, -2, -3, -4, and -5, all showed the same band pattern of CACTA (Miura et al. 2004). CACTA did not transpose in the F2 generation from a cross between Col and C24 (Figure S2 at http://www.genetics.org/supplemental/). In recombinant inbred lines between Col and Landsberg erecta (Lister and Dean 1993), only bands corresponding to the parental ecotypes segregated, suggesting that CACTA did not transpose during the initial interstrain cross nor the subsequent repeated self-pollinations (A. Miura and T. Kakutani, unpublished results).
One possible explanation for the mobility of the ddm1-derived CACTA1 could be that the CACTA1 transposed into a euchromatic arm region in the ddm1 mutant background, and it therefore remained mobile. However, even CACTA1 in the original pericentromeric region remained mobile in the DDM1 wild-type background; DDM1/DDM1 plants without any additional band to the original Col pattern also generated progeny with new bands (plant 2 and its progeny in Figure 2B). These results suggest that the ddm1 mutation induces change in a heritable epigenetic mark(s), which is critical for transposon mobility.
The inheritance of epigenetic change of transcription over generations has been reported in several plant genes (Jacobsen and Meyerowitz 1997; Soppe et al. 2000; Cubas et al. 2001; Stokes et al. 2002). Two of them, FWA and BAL, are due to ectopic transcription, which can be induced by the ddm1 or met1 mutation (Soppe et al. 2000; Stokes et al. 2002; Kankel et al. 2003). CACTA1 behaved in a similar manner. It has recently been shown that other transposons (or transposon-like sequences) are transcribed in the ddm1 mutant and continue to be transcribed after a cross to a wild-type plant (Lippman et al. 2003).
The transposition of CACTA1 in the DDM1 wild-type background allowed us to directly examine its integration specificity in the wild-type background. We have previously shown that all CACTA transposons in the Col ecotype and most of the related sequences in other ecotypes tend to be localized in pericentromeric heterochromatin (Miura et al. 2004). This genomic distribution could be due to selective integration near the centromere. In the present study, we showed that CACTA1 did not transpose preferentially into pericentromeric regions even in the DDM1 background. In addition, CACTA1 did not show preferential integration into wild-type-derived chromosomal regions compared to ddm1-derived regions (Figure 4B). These observations suggest that CACTA1 does not preferentially integrate into heterochromatic regions.
The reason that CACTA elements tend to be localized near the centromere rather than near the chromosomal-arm regions in natural populations remains unknown. One possible mechanism to account for the accumulation of CACTA1 in pericentromeric regions is natural selection; even though CACTA1 integrates into chromosome arm regions, the resultant chromosome may be eliminated from the natural population by purifying selection. It is possible that CACTA insertions into gene-rich regions sometimes reduce host fitness by direct gene disruption. In fact, Tc transposon insertions fixed in the Caenorhabditis elegans genome show a bias against insertion into coding regions, while the high-frequency insertions induced in the laboratory in the mut-7 mutant background show a more random distribution (Rizzon et al. 2003). This observation suggests natural selection against gene disruption. In addition to the direct gene disruption by insertion, transposon insertion sometimes disturbs the proper function of nearby host genes by affecting their transcription (Fedoroff 1996; Martienssen 1996). These mechanisms may also contribute to differentiation of gene-rich and transposon-rich regions in the plant genome. An alternative explanation is that the transposition is random, but the low frequency of transposon excision in the heterochromatic region results in net accumulation of “cut-and-paste” type transposons. Meiotic recombination rates tend to be low in the heterochromatin region, which would interfere with recombination-based mechanisms to remove transposons. However, recent analysis revealed that transposon abundance does not generally correlate with the low meiotic recombination rate in the Arabidopsis genome (Wright et al. 2003).
Another important question is whether integration of other classes of transposons is controlled in a similar manner. Although accumulation near the centromere is conserved among many classes of transposons, the underlying mechanisms might differ. With regard to transcriptional activation, different transposons respond differently to mutations affecting DNA and histone modifications (Johnson et al. 2002; Lippman et al. 2003). Despite the extensive investigation of transcription of many endogenous Arabidopsis transposon families, epigenetic control of integration specificity has been examined in only two of them: CACTA (this work and Miura et al. 2001) and AtMu (Singer et al. 2001). AtMu integrates throughout the genome, as is the case in CACTA, at least in the ddm1 mutant background (Singer et al. 2001).
Inheritance of differential epigenetic states over generations is an enigmatic phenomenon found in plants. Similar inheritance of epigenetic variations has also been reported for some alleles of mammalian genes (Whitelaw and Martin 2001; Rakyan et al. 2003). Interestingly, the affected mammalian alleles have transposon insertions compared to the wild-type allele. We propose that inheritance of epigenetic silencing is used, at least for some sequences, for transgenerational genome defense against deleterious genome rearrangement induced by transposon movement. It has long been known that maize transposons sometimes switch between active and inactive states (McClintock 1958). Such active or inactive states are also often inherited over multiple generations. Correlation between reversible transposon activity and DNA methylation has been found on a variety of transposons (Chandler and Walbot 1986; Schwartz and Dennis 1986; Banks et al. 1988; Martienssen et al. 1990). If one of the major functions of DNA methylation and the epigenetic modification system is a defense against the deleterious effects of transposons and other invasive elements (Yoder et al. 1997; Matzke et al. 1999), the heritable property would be advantageous, because the silencing is maintained at every stage of development, including early development before de novo silencing is established.
Acknowledgments
We thank Akiko Terui for technical assistance, Asuka Miura for sharing plant strains and DNA, and Eric Richards for comments on the manuscript. This work was supported by a Grant-in-Aid for Creative Scientific Research (no. 14GS0321) from the Japan Society for the Promotion of Science.
References
- Arabidopsis Genome Initiative, 2000. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408: 796–815. [DOI] [PubMed] [Google Scholar]
- Banks, J. A., P. Masson and N. Fedoroff, 1988. Molecular mechanisms in the developmental regulation of the maize Suppressor-mutator transposable element. Genes Dev. 2: 1364–1380. [DOI] [PubMed] [Google Scholar]
- Brzeski, J., and A. Jerzmanowski, 2003. Deficient in DNA methylation 1 (DDM1) defines a novel family of chromatin-remodeling factors. J. Biol. Chem. 278: 823–828. [DOI] [PubMed] [Google Scholar]
- Chandler, V. L., and V. Walbot, 1986. DNA modification of a maize transposable element correlates with loss of activity. Proc. Natl. Acad. Sci. USA 83: 1767–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cubas, P., C. Vincent and E. Coen, 1999. An epigenetic mutation responsible for natural variation in floral symmetry. Nature 401: 157–161. [DOI] [PubMed] [Google Scholar]
- Fedoroff, N., 1996 Epigenetic regulation of Spm, pp. 575–592 in Epigenetic Mechanisms of Gene Regulation, edited by A. Riggs, R. Martienssen and V. Russo. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Finnegan, E. J., W. J. Peacock and E. S. Dennis, 1996. Reduced DNA methylation in Arabidopsis thaliana results in abnormal plant development. Proc. Natl. Acad. Sci. USA 93: 8449–8454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirochika, H., H. Okamoto and T. Kakutani, 2000. Silencing of retrotransposons in Arabidopsis and reactivation by the ddm1 mutation. Plant Cell 12: 357–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen, S. E., and E. M. Meyerowitz, 1997. Hypermethylated SUPERMAN epigenetic alleles in Arabidopsis. Science 277: 1100–1103. [DOI] [PubMed] [Google Scholar]
- Jeddeloh, J. A., T. L. Stokes and E. J. Richards, 1999. Maintenance of genomic methylation requires a SWI2/SNF2-like protein. Nat. Genet. 22: 94–97. [DOI] [PubMed] [Google Scholar]
- Johnson, L., X. Cao and S. Jacobsen, 2002. Interplay between two epigenetic marks. DNA methylation and histone H3 lysine 9 methylation. Curr. Biol. 12: 1360–1367. [DOI] [PubMed] [Google Scholar]
- Kakutani, T., 1997. Genetic characterization of late-flowering traits induced by DNA hypomethylation mutation in Arabidopsis thaliana. Plant J. 12: 1447–1451. [DOI] [PubMed] [Google Scholar]
- Kakutani, T., J. A. Jeddeloh, S. K. Flowers, K. Munakata and E. J. Richards, 1996. Developmental abnormalities and epimutations associated with DNA hypomethylation mutations. Proc. Natl. Acad. Sci. USA 93: 12406–12411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakutani, T., K. Munakata, E. J. Richards and H. Hirochika, 1999. Meiotically and mitotically stable inheritance of DNA hypomethylation induced by ddm1 mutation of Arabidopsis thaliana. Genetics 151: 831–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kankel, M. W., D. E. Ramsey, T. L. Stokes, S. K. Flowers, J. R. Haag et al., 2003. Arabidopsis MET1 cytosine methyltransferase mutants. Genetics 163: 1109–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato, M., A. Miura, J. Bender, S. E. Jacobsen and T. Kakutani, 2003. Role of CG and non-CG methylation in immobilization of transposons in Arabidopsis. Curr. Biol. 13: 421–426. [DOI] [PubMed] [Google Scholar]
- Kinoshita, T., A. Miura, Y. Choi, Y. Kinoshita, X. Cao et al., 2004. One-way control of FWA imprinting in Arabidopsis endosperm by DNA methylation. Science 303: 521–523. [DOI] [PubMed] [Google Scholar]
- Li, E., T. H. Bestor and R. Jaenisch, 1992. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 69: 915–926. [DOI] [PubMed] [Google Scholar]
- Li, E., C. Beard and R. Jaenisch, 1995. Role for DNA methylation in genomic imprinting. Nature 366: 362–365. [DOI] [PubMed] [Google Scholar]
- Lindroth, A. M., X. Cao, J. P. Jackson, D. Zilberman, C. M. Mccallum et al., 2001. Requirement of CHROMOMETHYLASE3 for maintenance of CpXpG methylation. Science 292: 2077–2080. [DOI] [PubMed] [Google Scholar]
- Lippman, Z., B. May, C. Yordan, T. Singer and R. Martienssen, 2003. Distinct mechanisms determine transposon inheritance and methylation via small interfering RNA and histone modification. PLoS Biol. 1: E67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister, C., and C. Dean, 1993. Recombinant inbred lines for mapping RFLP and phenotypic markers in Arabidopsis thaliana. Plant J. 4: 745–750. [DOI] [PubMed] [Google Scholar]
- Martienssen, R., 1996 Epigenetic silencing of Mu transposable elements in maize, pp. 593–608 in Epigenetic Mechanisms of Gene Regulation, edited by A. Riggs, R. Martienssen and V. Russo. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Martienssen, R., A. Barkan, W. C. Taylor and M. Freeling, 1990. Somatically heritable switches in the DNA modification of Mu transposable elements monitored with a suppressible mutant in maize. Genes Dev. 4: 331–343. [DOI] [PubMed] [Google Scholar]
- Matzke, M. A., M. F. Mette, W. Aufsatz, J. Jakowitsch and A. J. Matzke, 1999. Host defenses to parasitic sequences and the evolution of epigenetic control mechanisms. Genetica 107: 271–287. [PubMed] [Google Scholar]
- McClintock, B., 1958. The Suppressor-mutator system of control of gene action in maize. Carnegie Inst. Wash. Year Book 57: 415–429. [Google Scholar]
- Miller, J. T., F. Dong, S. A. Jackson, J. Song and J. Jiang, 1998. Retrotransposon-related DNA sequences in the centromeres of grass chromosomes. Genetics 150: 1615–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura, A., S. Yonebayashi, K. Watanabe, T. Toyama, H. Shimada et al., 2001. Mobilization of transposons by a mutation abolishing full DNA methylation in Arabidopsis. Nature 411: 212–214. [DOI] [PubMed] [Google Scholar]
- Miura, A., M. Kato, K. Watanabe, A. Kawabe, H. Kotani et al., 2004. Genomic localization of endogenous mobile CACTA family transposons in natural variants of Arabidopsis thaliana. Mol. Genet. Genomics 270: 524–532. [DOI] [PubMed] [Google Scholar]
- Pearce, S. R., U. Pich, G. Harrison, A. J. Flavell, J. S. Heslop-Harrison et al., 1996. The Ty1-copia group retrotransposons of Allium cepa are distributed throughout the chromosomes but are enriched in the terminal heterochromatin. Chromosome Res. 4: 357–364. [DOI] [PubMed] [Google Scholar]
- Pelissier, T., S. Tutois, S. Tourmente, J. M. Deragon and G. Picard, 1996. DNA regions flanking the major Arabidopsis thaliana satellite are principally enriched in Athila retroelement sequences. Genetica 97: 141–151. [DOI] [PubMed] [Google Scholar]
- Presting, G. G., L. Malysheva, J. Fuchs and I. Schubert, 1998. A Ty3/gypsy retrotransposon-like sequence localizes to the centromeric regions of cereal chromosomes. Plant J. 16: 721–728. [DOI] [PubMed] [Google Scholar]
- Rakyan, V. K., S. Chong, M. E. Champ, P. C. Cuthbert, H. D. Morgan et al., 2003. Transgenerational inheritance of epigenetic states at the murine Axin(Fu) allele occurs after maternal and paternal transmission. Proc. Natl. Acad. Sci. USA 100: 2538–2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzon, C., E. Martin, G. Marais, L. Duret, L. Segalat et al., 2003. Patterns of selection against transposons inferred from the distribution of Tc1, Tc3 and Tc5 insertions in the mut-7 line of the nematode Caenorhabditis elegans. Genetics 165: 1127–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronemus, M. J., M. Galbiati, C. Ticknor, J. Chen and S. L. Dellaporta, 1996. Demethylation-induced developmental pleiotropy in Arabidopsis. Science 273: 654–657. [DOI] [PubMed] [Google Scholar]
- Schmidt, T., S. Kubis and J. S. Heslop-Harrison, 1995. Analysis and chromosomal localization of retrotransposons in sugar beet (Beta vulgaris L.): LINEs and Ty1-copia-like elements as major components of the genome. Chromosome Res. 3: 335–345. [DOI] [PubMed] [Google Scholar]
- Schwartz, D., and E. Dennis, 1986. Transposase activity of the Ac controlling element in maize is regulated by its degree of methylation. Mol. Gen. Genet. 205: 476–482. [Google Scholar]
- Selker, E. U., N. A. Tountas, S. H. Cross, B. S. Margolin, J. G. Murphy et al., 2003. The methylated component of the Neurospora crassa genome. Nature 422: 893–897. [DOI] [PubMed] [Google Scholar]
- Singer, T., C. Yordan and R. A. Martienssen, 2001. Robertson's Mutator transposons in A. thaliana are regulated by the chromatin-remodeling gene decrease in DNA methylation (DDM1). Genes Dev. 15: 591–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soppe, W. J., S. E. Jacobsen, C. Alonso-Blanco, J. P. Jackson, T. Kakutani et al., 2000. The late flowering phenotype of fwa mutants is caused by gain-of-function epigenetic alleles of a homeodomain gene. Mol. Cell 6: 791–802. [DOI] [PubMed] [Google Scholar]
- Soppe, W. J., Z. Jasencakova, A. Houben, T. Kakutani, A. Meister et al., 2002. DNA methylation controls histone H3 lysine 9 methylation and heterochromatin assembly in Arabidopsis. EMBO J. 21: 6549–6559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stancheva, I., and R. R. Meehan, 2000. Transient depletion of xDnmt1 leads to premature gene activation in Xenopus embryos. Genes Dev. 14: 313–327. [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Steimer, A., P. Amedeo, K. Afsar, P. Fransz, O. M. Scheid et al., 2000. Endogenous targets of transcriptional gene silencing in Arabidopsis. Plant Cell 12: 1165–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokes, T. L., B. N. Kunkel and E. J. Richards, 2002. Epigenetic variation in Arabidopsis disease resistance. Genes Dev. 16: 171–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vongs, A., T. Kakutani, R. A. Martienssen and E. J. Richards, 1993. Arabidopsis thaliana DNA methylation mutants. Science 260: 1926–1928. [DOI] [PubMed] [Google Scholar]
- Whitelaw, E., and D. I. Martin, 2001. Retrotransposons as epigenetic mediators of phenotypic variation in mammals. Nat. Genet. 27: 361–365. [DOI] [PubMed] [Google Scholar]
- Wright, S. I., N. Agrawal and T. E. Bureau, 2003. Effects of recombination rate and gene density on transposable element distributions in Arabidopsis thaliana. Genome Res. 13: 1897–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoder, J. A., C. P. Walsh and T. H. Bestor, 1997. Cytosine methylation and the ecology of intragenomic parasites. Trends Genet. 13: 335–340. [DOI] [PubMed] [Google Scholar]