

Abstract
Approximately 6000 DNA elements, totaling nearly 15 Mb, are coordinately excised from the developing somatic genome of Tetrahymena thermophila. An RNA interference (RNAi)-related mechanism has been implicated in the targeting of these germline-limited sequences for chromatin modification and subsequent DNA rearrangement. The excision of individual DNA segments can be inhibited if the homologous sequence is placed within the parental somatic nucleus, indicating that communication occurs between the parental and developing genomes. To determine how the DNA content of one nucleus is communicated to the other, we assessed DNA rearrangement occurring in wild-type cells that were mated to cells that contained the normally germline-limited M element within their somatic nuclei. M-element rearrangement was blocked in the wild-type cell even when no genetic exchange occurred between mating partners, a finding that is inconsistent with any genetic imprinting models. This inhibition by the parental somatic nucleus was rapidly established between 5 and 6 hr of conjugation, near or shortly after the time that zygotic nuclei are formed. M-element small RNAs (sRNAs) that are believed to direct its rearrangement were found to rapidly accumulate during the first few hours of conjugation before stabilizing to a low, steady-state level. The period between 5 and 6 hr during which sRNA levels stabilize correlates with the time after which the parental genome can block DNA rearrangement. These data lead us to suggest that homologous sRNAs serve as mediators to communicate sequence-specific information between the parental and developing genomes, thereby regulating genome-wide DNA rearrangement, and that these sRNAs can be effectively compared to the somatic genome of both parents.
EUKARYOTIC cells go to great lengths to ensure the integrity of their genomes. In addition to mechanisms that orchestrate the sequential events of chromosome replication and division, others exist that protect the genome from the spread of invading DNA elements. The existence of such genome defense mechanisms has been uncovered largely due to studies of diverse epigenetic phenomena such as transgene-induced silencing or cosuppression, described in both plants and animals (see Waterhouse et al. 2001), and repeat-induced point mutation or gene silencing in fungi (Goyon and Faugeron 1989; Selker 1990, 2002).
Studies from the last several years have revealed that the triggers for many such homology-dependent events are likely double-stranded RNAs (dsRNAs; reviewed in Hannon 2002). The epigenetic phenomena induced by dsRNA have been collectively called RNA interference (RNAi)-related processes. The dsRNA molecules are processed into 20–30 nucleotide effector species by an RNAse III-related protein called Dicer (Bernstein et al. 2001). Mutations in genes required for RNAi in Caenorhabditis elegans result in the mobilization of transposable elements, providing further evidence that RNAi appears to serve as a genome defense mechanism that silences invading DNA elements to limit their spread to other loci (Ketting et al. 1999; Tabara et al. 1999). Furthermore, the RNAi machinery of Schizosaccharomyces pombe is required for heterochromatin formation at the mating-type locus and for silencing of centromeric repeats (Hall et al. 2002; Volpe et al. 2002), which implicates this process in endogenous mechanisms important for chromosome stability (see Dernburg and Karpen 2002).
The ciliated protozoan Tetrahymena thermophila has emerged as a useful model to understand how eukaryotic cells can differentially regulate individual copies of homologous sequence. Tetrahymena, like other ciliated protozoa, are unusual in that these single-celled organisms contain two functionally distinct genomes, which serve the equivalent of germline and somatic roles (reviewed in Karrer 2000). The germline genome is contained within the transcriptionally silent micronucleus that primarily serves as the genetic repository to pass along an intact genome during sexual development. The somatic genome, which consists of a rearranged subset of the germline DNA, is found within the polyploid macronucleus and is the source for gene expression. Tetrahymena divide by binary fission during vegetative growth, and the micronucleus and macronucleus are propagated by mitotic and amitotic division, respectively. Upon conjugation, the germline micronuclei within mating pairs complete meiosis. One of four haploid products in each partner is selected to undergo a mitotic division followed by reciprocal exchange between cells and karyogamy of the resulting gametic nuclei to form the precursors of the new somatic and germline nuclei within the developing progeny. The parental macronuclei are then resorbed during the later stages of development.
During differentiation of the somatic genome, extensive DNA rearrangement results in the fragmentation of the five germline chromosomes into 200–300 “mini”-chromosomes (reviewed in Yao et al. 2002). Furthermore, DNA deletion eliminates ∼15% (totaling nearly 15 Mb) of the germline DNA from an estimated 6000 loci in the developing macronucleus (Yao and Gorovsky 1974; Yao et al. 1984). The eliminated sequences are both unique and repetitive, noncoding sequences as well as some that resemble transposable elements (Cherry and Blackburn 1985; Wuitschick et al. 2002; Fillingham et al. 2004). How such diverse sequences can be efficiently targeted for elimination during nuclear development has been a long-standing enigma. Recent studies indicate that homologous RNAs play a critical role in these DNA rearrangements (reviewed in Mochizuki and Gorovsky 2004b). Bidirectional transcription of germline-limited sequences has been detected in the early stages of conjugation (Chalker and Yao 2001). This transcription is the likely source of dsRNA that is presumably cleaved by a Dicer-like ribonuclease into ∼28-nt small RNAs (sRNAs; Mochizuki et al. 2002). These sRNAs, which have been called scan RNAs, are believed to target the methylation of lysine 9 of histone H3 (K9H3) on chromatin of the homologous sequence in the developing somatic nuclei (Taverna et al. 2002). Disruption of the genes encoding either an Argonaute/Piwi-related protein (TWI1) or a chromodomain-containing protein (PDD1) causes an inability for cells to accumulate sRNAs, target K9H3 methylation, and promote subsequent DNA elimination, which together provide strong evidence of a role for an RNAi-like mechanism targeting this chromatin modification (Coyne et al. 1999; Mochizuki et al. 2002; Taverna et al. 2002; Liu et al. 2004). Thus many steps in the process of DNA rearrangement are mechanistically similar to RNA-directed establishment of heterochromatin formation observed in S. pombe, which likely has parallels in the majority of eukaryotes. Injection of dsRNA complementary to genomic regions normally retained in the somatic nucleus into developing Tetrahymena cells induced elimination of the homologous sequence, conclusively demonstrating that DNA rearrangement is an RNA-directed process (Yao et al. 2003).
The use of sRNAs to target chromatin modification on a genome-wide scale is quite remarkable; however, what may be an even more striking step in this process is the involvement of the parental somatic genome in regulating the extent of rearrangement. It has been shown that a germline-limited sequence (i.e., the M- and R-deletion elements) placed into the somatic macronucleus can inhibit the normally efficient elimination of the homologous DNA sequence during nuclear differentiation (Chalker and Yao 1996). Similar homology-dependent regulation of DNA rearrangement has also been demonstrated in the ciliate, Paramecium tetraurelia (Duharcourt et al. 1995, 1998). These findings imply that, at least in part, genomic regions are targeted for elimination by comparing the DNA content of the germline genome with that of the parental somatic genome. To understand mechanistically the transfer of sequence-specific information between the parental and developing genomes, we used genetic manipulation to determine the requirements and timing of this step in DNA rearrangement. We found that the inhibition of DNA rearrangement by the parental genome requires no direct genetic exchange and must occur using factors free to travel through the cytoplasm of mating pairs. Results from comparing the timing of this inhibition by the parental genome and the accumulation of specific sRNAs support a model in which sRNAs, likely with associated proteins, are effector molecules performing this regulation. These sRNAs appear to be produced only from transcription of the germline genome, not from transcription of the homologous sequence in the somatic genome, and are rapidly compared to the parental genome of both mating partners to determine the sequences to be eliminated from the developing genome.
MATERIALS AND METHODS
Strains and growth conditions:
All growth and manipulation of T. thermophila cells were performed as previously described (Gorovsky et al. 1975; Asai and Forney 1999). For most experiments, 1× SPP (1% proteose peptone, 0.2% dextrose, 0.1% yeast extract, 0.003% sequestrine) was used as the growth medium. Cell populations were prestarved in 10 mm Tris-HCl (pH 7.4) prior to mixing to induce synchronous mating. Wild-type strains CU427 {chx1-1/chx1-1 [VI, cycloheximide sensitive (cy-s)]} and CU428 {mpr1-1/mpr1-1 [VII, 6-methylpurine sensitive (mp-s)]} were obtained from P. J. Bruns (Howard Hughes Medical Institute, Chevy Chase, MD). M+ strains HC76-M5B and HC81-M3A were previously described (Chalker and Yao 1996). Chloramphenicol-resistant (cm-r) strains arising from a spontaneous mutation of CU427 were selected by two rounds of growth of a starting inoculum of 3 × 106 cells into 100 ml of 1× SPP medium containing 200 μg/ml chloramphenicol for 4–5 days at 30°. After the second round, drug-resistant cells were subcloned into medium containing 250 μg/ml chloramphenicol, and four subclones were subsequently checked for the ability to confer drug resistance to their vegetative progeny through cytoplasmic inheritance, presumably due to inheritance of resistant mitochondria (Roberts and Orias 1973). Subclone HC27c-2 (chx1-1/chx1-1 [VI, cy-s, cm-r]) was used for the pair disruption experiments described below.
Cytogamy:
At 4.5–5 hr after the mixing of cells, conjugating pairs were administered a hyperosmotic shock by adding 30% glucose to 1.4% final concentration and, after 45 min, diluted 10-fold with 10 mm Tris-HCl as previously described (Cole and Bruns 1992). Mating pairs were allowed to recover at least 30 min and then cloned into individual drops of medium. The progeny of cloned pairs were allowed to divide for 3 days at 30° and then replicated into 1× SPP medium containing either 25 μg/ml cycloheximide or 15 μg/ml 6-methylpurine.
Pair disruption:
To physically separate pairs prematurely, 1 ml of conjugating cells (∼1 × 105 pairs/ml) were vortexed in the presence of glass beads for 1–2 min as previously described (Virtue and Cole 1999). Disrupted pairs were allowed to recover by mixing with an equal volume of 1× SPP and incubating at 30° for at least 30 min. Individual cells were cloned into drops of medium and grown for 3 days prior to screening for drug resistance as described above. Cells were replicated to 1× SPP containing 250 μg/ml chloramphenicol to identify progeny derived from strain HC27c-2.
DNA analysis:
Southern blot analysis was performed by standard methods using a macronuclear M-element region probe as previously described (Austerberry and Yao 1987; Chalker and Yao 1996). Assessment of DNA rearrangement without DNA isolation was accomplished by lysing 300–400 cells in 30 μl of 0.5% Tween-20, 0.5% NP-40, 10 mm Tris-HCl (pH 8), 50 mm KCl, and 300 μg/ml proteinase K for 20 min at 65°, followed by incubation at 95° for 10 min. One-tenth of each lysate was then used as template in 30-μl PCR reactions. The assay utilizes the following three oligonucleotide primers to detect the unrearranged and rearranged forms, two that correspond to sequences flanking the eliminated region and a third that is complementary to sequences within the germline-limited region: MacL, 5′-AGC TTA AAC AAA TGC CAT ATT GAG-3′; MacR, 5′-GTG GGG AGG GAG AAG GAT TCA AC-3′; and MicL, 5′-ATA TTG TGT GGT ACA ATA GGT TGT CGT AG-3′.
RNA analysis:
Total RNA was isolated from Tetrahymena cells using RNAsol extraction (Fan et al. 1999). RNA (15 μg/lane) was electrophoresed in 1.2% agarose/1× MOPS/1% formaldehyde gels and transferred to nylon membranes (Osmonics, Minnetonka, MN) by downward capillary blotting. Filters were hybridized with single-stranded riboprobes specific to the 0.6-kbp germline-limited M-element synthesized from plasmids pMint2 or pMint7 as previously described (Chalker and Yao 2001). Strand specificity of riboprobes was examined by hybridization to in vitro transcribed RNA immobilized on membranes. Riboprobes routinely hybridized at least 100-fold more efficiently to antisense transcripts than to sense transcripts. For detection of small RNAs, samples (30 μg/lane) were separated on 15% polyacrylamide-urea-1× Tris/borate/EDTA (TBE) gels and stained with 0.2 μg/ml ethidium bromide for 15 min, followed by destaining for >30 min in 0.5× TBE. For hybridization, RNA was transferred in 0.5× TBE to 0.22-μm charged-nylon membranes (Osmonics) by semidry electroblotting using an Owl model HEP-1 apparatus for 45 min at 1.5–2 mA/cm2. Filters were hybridized to M-element riboprobes in 0.25 m sodium phosphate (pH 7.2)/7% SDS (Church and Gilbert 1984) overnight at 55° and washed in 0.5× SSC/1% SDS at 60° four times for >15 min each followed by exposure to X-ray film. To quantify loading, filters where rehybridized to an end-labeled antisense oligonucleotide specific to the 5S ribosomal RNA. Hybridization intensity was determined by phosphorimage analysis (Bio-Rad personal FX imager; Richmond, CA).
RESULTS
Macronuclear M-element copies in one partner block rearrangement of the homologous element in a wild-type mating partner without genetic exchange:
DNA rearrangement of the M-deletion element (Figure 1A) and other germline-limited sequences is normally extremely efficient (Austerberry et al. 1984; Yao et al. 1984; Austerberry and Yao 1988). Nevertheless, we previously found that placing copies of the M- or R-deletion element within the parental macronucleus caused failure of DNA rearrangement of the homologous element during macronuclear development (Chalker and Yao 1996). To investigate the mechanism by which the parental nucleus can communicate its genetic composition to the developing nucleus, we set out to determine how and when the “signal” that blocks DNA rearrangement is transferred between nuclear compartments. We took advantage of our earlier observation that copies of the M element in the parental macronucleus of one cell can block DNA rearrangement occurring in the nuclei of a wild-type mating partner (Chalker and Yao 1996) to ask whether the M+ partner must exchange genetic material with the wild-type conjugant to transfer the inhibitory signal. One possibility is that the parental nucleus communicates its sequence composition to the germline during vegetative growth when the micronucleus is positioned in a cleft of the macronucleus during most of the cell cycle, thereby establishing an imprint upon the micronucleus that determines sequences that should be protected from DNA elimination during the subsequent nuclear development. If this were the case, transfer of the imprint would require nuclear exchange between partners. To test this, we initiated crosses between “M+” strains, which carry the M element on a high-copy vector within their macronuclei, and wild-type strains, but blocked gametic nucleus exchange by administering a hyperosmotic shock (Hamilton et al. 1988; Cole and Bruns 1992; Figure 1B). A shock given to the cells between 4.5 and 5 hr of conjugation results in a significant percentage of mating pairs undergoing cytogamy, a developmental program in which each mating partner generates its new macronucleus exclusively from its own germline genome. Because each nucleus develops from uniparental genetic material, we were able to determine the identity of each progeny's parent using the presence of different genetic markers in the micronuclei of the M+ and wild-type strains. For example, in the cross of the M+ strain HC76M5B (VI, chx1-1/chx1-1) and the wild-type strain CU428 (VII, mpr1-1/mpr1-1), the progeny with nuclei that developed within the confines of the M+ strain were resistant to the antibiotic cycloheximide (cy-r), but sensitive to 6-methylpurine (mp-s), whereas those developing in the wild-type partner were conversely cy-s and 6-methylpurine resistant (mp-r). If genetic exchange had occurred, the progeny would be resistant to both drugs.
Figure 1.—
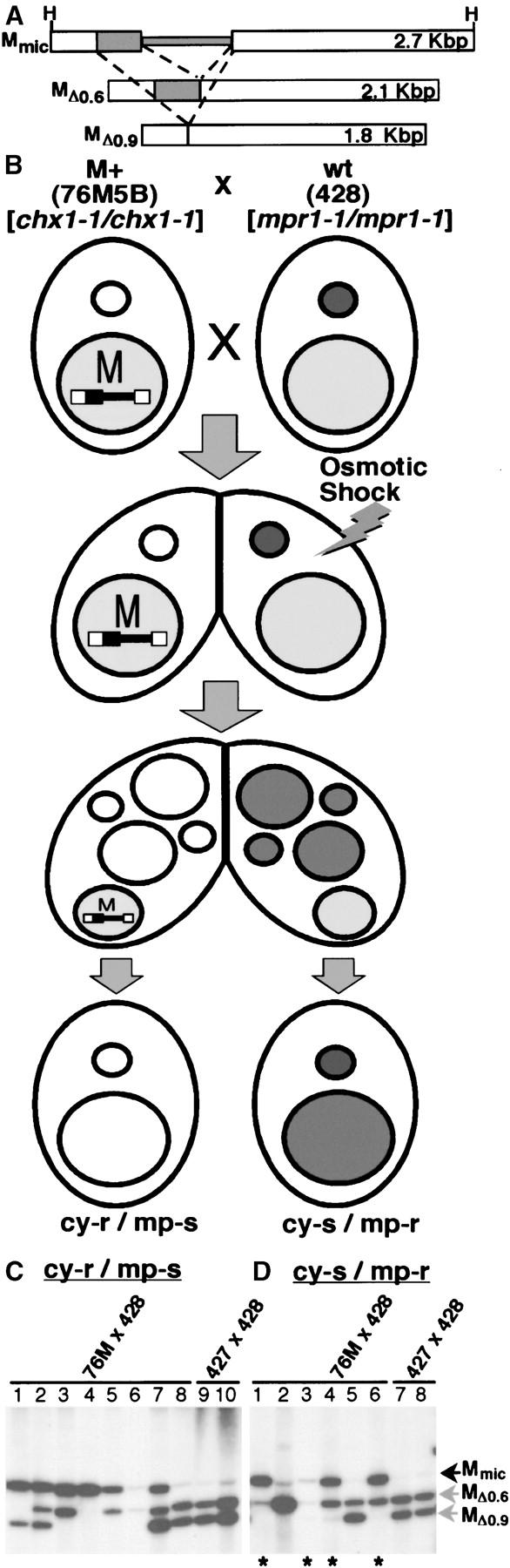
Inhibition of M-element rearrangement in a wild-type cell occurs without nuclear exchange with an M+ partner. (A) Diagram depicting the M element shows the micronuclear unrearranged element (Mmic) and the two alternative rearranged macronuclear forms, MΔ0.6 or MΔ0.9, resulting from excision of a 0.6-kbp segment (narrow shaded box) or an additional 0.3-kbp segment (wide shaded box), respectively. The wide, open boxes represent the macronucleus-retained flanking regions. The size (in kilobase pairs) of the genomic HindIII (H) fragment corresponding to each form as detected in Southern blot analysis at the bottom is given to the right. As depicted in B, an osmotic shock was administered to conjugating pairs either 4.5 or 5 hr after mixing prestarved cell populations. This treatment blocks exchange of gametic nuclei and induces cytogamy, a developmental program in which the new macronuclei generated in each cell (two develop in each mating partner) are formed using the genome of its own micronucleus. The M+ micronuclei (small open circles) are homozygous for the chx1-1 marker conferring resistance to cycloheximide when brought into expression in the progeny; the wild-type micronuclei (small solid circles) are homozygous for the mpr1-1 marker conferring 6-methylpurine resistance. The progeny cells whose new macronuclei developed from the micronuclei of the M+ cell are cy-r/mp-s, whereas the progeny whose macronuclei derive solely from the wild-type micronuclear genome are cy-s/mp-r. (C and D) Southern blot analysis of total genomic DNA isolated from the progeny whose macronuclei developed from the micronuclear genome of the M+ or the wild-type partner was used to examine the degree of M-element rearrangement. The bands corresponding to the sizes given in A for the unrearranged or rearranged forms are indicated. Asterisks indicate DNA of progeny derived from cytogamy of the wild-type partner showing unrearranged copies of the M element remaining in the macronuclear chromosomes. The last two lanes show the DNA of a control cross of two wild-type strains induce to undergo cytogamy.
We assessed M-element rearrangement in cells that had undergone cytogamy, and as expected, the majority of progeny derived from the M+ cell exhibited failure of M-element rearrangement (Figure 1; Table 1). The more revealing observation was that the majority of progeny that developed from the wild-type partner and solely from that parent's micronuclear genome also had failed to eliminate the M element (Figure 1; Table 1). Therefore, inhibition of M-element rearrangement in the wild-type mating partner requires no transfer of genetic material from the M+ cell. We performed this experiment by crossing two different M+ strains, HC76M5B and HC81M3A, to wild-type strains CU428 and CU427, respectively, and obtained comparable results (Table 1). Overall, 90% (18/20) of progeny derived from the M+ strain and 67% (10/15) from the wild-type partner showed partial or full failure of M-element rearrangement. It should be noted that the vector-borne M element is lost from the cells along with the parental macronucleus during conjugation and any remaining copies resulted from blocked DNA rearrangement of the M-element locus in the developing macronuclei. Four to eight copies of each chromosome undergo independent rearrangement in each developing macronucleus, and this inhibitory action imposed by the parental macronucleus typically affected only a subset of these. Even so, the failure of rearrangement induced by the presence of the M element is quite dramatic.
TABLE 1.
M-element inhibition upon induced cytogamy
| Crossa | Progeny phenotypeb | Failed excision (%)c |
|---|---|---|
| 427 × 428 | mp-r (wt) | 0/4 (0) |
| cy-r (wt) | 0/5 (0) | |
| cy-r, mp-r | 0/2 (0) | |
| 428 × 76M | mp-r (wt) | 6/9 (67) |
| cy-r (M+) | 10/10 (100) | |
| cy-r, mp-r | 2/2 (100) | |
| cy-r, mp-rd | 2/2 (100) | |
| 427 × 81M | mp-r (M+) | 8/10 (80) |
| cy-r (wt) | 4/6 (67) | |
| cy-r, mp-r | 4/4 (100) | |
| cy-r, mp-rd | 2/2 (100) |
Wild-type strains used were CU427 (427) and CU428 (428); M+ strains used were HC76M5B (76M) and HC81M3A (81M).
For the resistance expected upon cytogamy, the wild-type (wt) or M+ partner (M+) giving rise to each is indicated in parentheses. mp-r, 6-methylpurine resistant; cy-r, cycloheximide resistant. Double resistance (cy-r, mp-r) indicates genetic exchange between partners.
The fraction of pairs (x/n, where n is the total analyzed) showing detectable failure of M-element deletion for each class is shown. The percentage of pairs showing failure is given in parentheses.
Progeny of control crosses not subjected to osmotic shock.
To control for the possibility that the hyperosmotic shock used to block nuclear exchange might have impaired DNA rearrangement in the wild-type cell, we subjected a cross of two wild-type strains to the identical treatment and found that M-element rearrangement was unperturbed (Figure 1; Table 1). Thus, we find no support for the establishment of a germline imprint, but instead this study indicates that inhibition of DNA rearrangement by the parental macronuclear DNA is signaled through the cytoplasm of the mating partners during conjugation.
Inhibition of rearrangement by the parental macronucleus is rapidly enforced during conjugation:
We felt that we could gain insight into the nature of the signal being communicated by determining the period of development during which the parental nucleus can block DNA rearrangement. To facilitate this study, we first developed a PCR assay that allowed assessment of M-element rearrangement for large numbers of individual progeny lines without DNA isolation (Figure 2; see materials and methods for details). The Δ0.6- and Δ0.9-kbp rearranged forms (MΔ0.6 and MΔ0.9) of the M element are detected as 601- and 284-bp PCR products, respectively, whereas the unrearranged element (Mmic) is evident as a 386-bp product. When we used this assay to assess the state of M-element rearrangement, a very low level of the 386-bp product, corresponding to the unrearranged elements remaining within micronuclei, was observed in the progeny of wild-type cells (Figure 2, lanes 1–3) while abundant quantities of this PCR-generated fragment were detected in the progeny of M+ strains (lanes 4–11), indicative of failed DNA rearrangement. We also isolated DNA from these same progeny lines and compared the PCR results to Southern blot analysis. This comparison showed that the levels of PCR products corresponding to the unrearranged form and the 0.9-kbp deletion provide a good assessment of the quantities of these two chromosomal forms in macronuclei, but the levels of the largest product representing the 0.6-kbp deletion were typically underrepresented relative to the actual quantity, likely due to a PCR bias toward smaller products. Nevertheless, this assay easily distinguished cells that failed to rearrange copies of the M element from those in which rearrangement was complete.
Figure 2.—
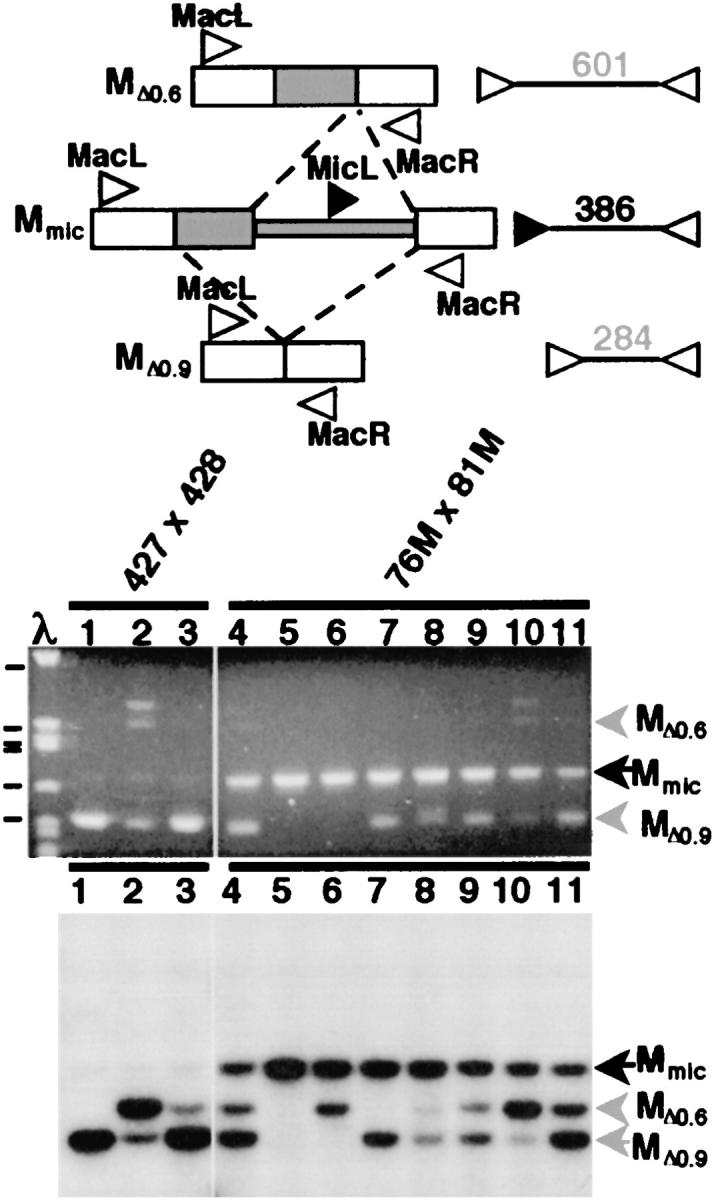
PCR-based detection of failed M-element rearrangement. The micronuclear, unrearranged M element (Mmic) and the two alternative rearranged macronuclear forms (MΔ0.6 or MΔ0.9) are depicted at the top as described in Figure 1. The relative positions of oligonucleotides complementary to DNA sequences in the right (MacR) and left flanking regions (MacL; open arrowheads) and the micronucleus-limited region (MicL; solid arrowhead) used for assessment of M-element rearrangement by PCR are also shown. The expected size of the PCR product generated by these primers indicating each form of the M element is given to the right. PCR (middle) and Southern blot (bottom) analysis examining the efficiency of M-element rearrangement detects significant levels of failed rearrangement in the progeny derived from M+ crosses (HC76M5B × HC81M3A, lanes 4–11), but not from wild-type crosses (CU427 × CU428, lanes 1–3). 76M, HC76M5B; 81M, HC81M3A; 427, CU427; and 428, CU428. The bands corresponding to the unrearranged and rearranged forms are indicated. The size markers in the leftmost lane are fragments of lambda-phage (λ) DNA digested with PstI. The indicated fragments in descending order are 805, 514, 468, 448, 339, and 264 bp, respectively. The low level of unrearranged M element detected in the progeny of wild-type crosses represents the two copies remaining in the micronucleus, as compared to the 50 copies found in the mature macronucleus.
Virtue and Cole (1999) showed that mating pairs could be physically disrupted as early as 1.5 hr after pairing and yet the individual partners could still go on to complete nuclear development. Thus by mating wild-type cells with M+ cells and disrupting these pairs at various times during conjugation, we believed that we could determine the time period during which the wild-type cell must be paired to an M+ cell to have its M-element rearrangement influenced by the DNA sequences in its partner's macronucleus (Figure 3). Pairs disrupted before gametic nuclear exchange that successfully complete development do so from uniparental genetic material, as in the induced cytogamy experiments describe above, and thus progeny derived from the M+ or wild-type partner could be identified by their single-drug-resistant phenotypes. Pairs disrupted after formation of the zygotic nucleus had their drug-resistant phenotype derived from both parents. When we first performed this experiment, we relied upon these drug-resistant phenotypes to determine which progeny cells were the direct descendents of the wild-type partners. Regardless of the time of pair disruption, at least 87% of the progeny whose macronuclei developed within the M+ cell, which, of course, retained continued presence of macronuclear M-element copies, exhibited failure of DNA rearrangement (Table 2). In contrast, prior to gametic nuclear exchange and karyogamy, which occurs between 4.5 and 5 hr after pairing (see Figure 5), we observed little failure of M-element excision (∼2%, 2/93) in the nuclei developing within the wild-type cell (Table 2). After karyogamy, we could no longer determine the parent of origin; however, we found that the percentage of cells that showed failure of M-element excision increased significantly from 67% at 5 hr to 93% by 6–7 hr (Table 2). If we can assume approximately equal recovery of progeny for each partner, these data indicate that the block to M-element rearrangement is rapidly communicated to the wild-type partner between 5 and 7 hr of conjugation. By 7 hr, the majority of the nuclei developing in the wild-type cells had been fully affected, and the continued presence of the M element in the parental macronucleus was no longer needed to block the excision events that would have occurred between 11 and 14 hr (Austerberry et al. 1984; Duharcourt and Yao 2002).
Figure 3.—
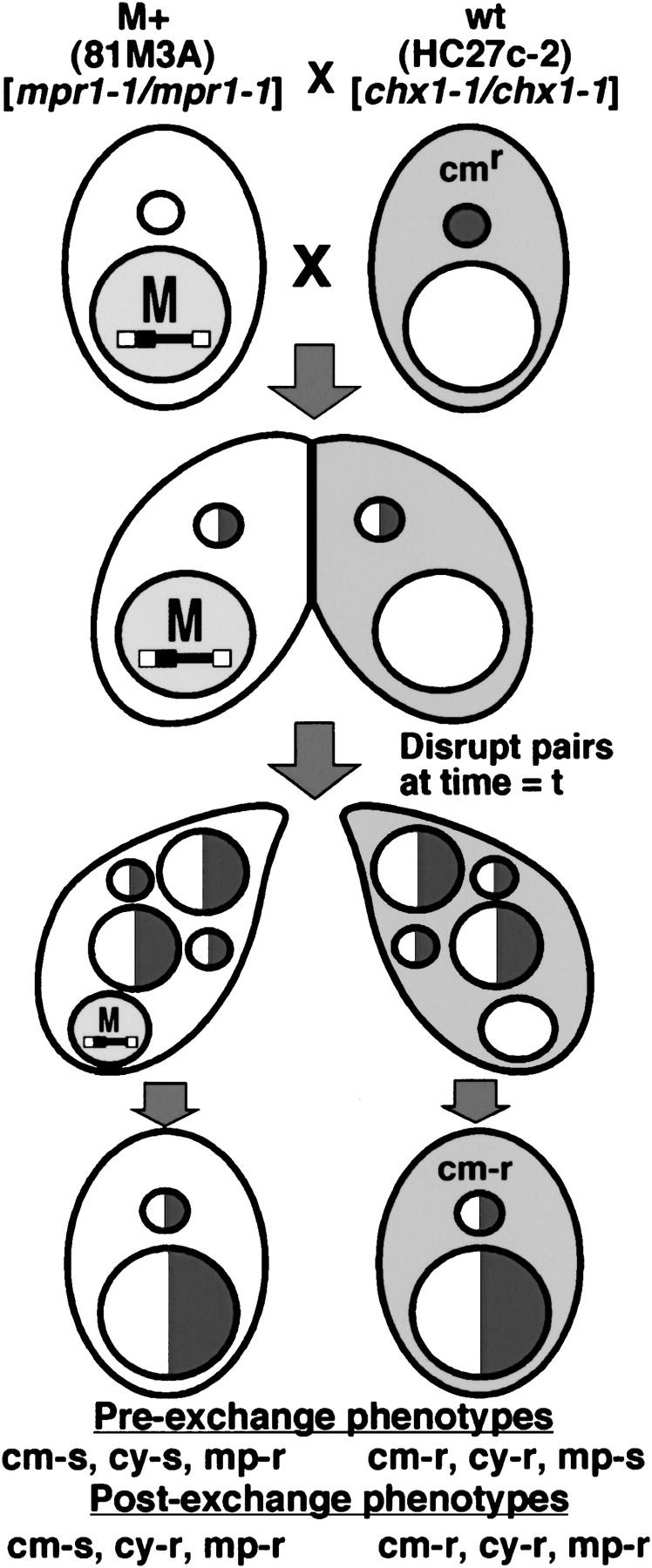
Strategy to determine developmental timing of parental macronuclear influence on M-element rearrangement. M+ strain HC81M3A was crossed to a chloramphenicol-resistant (cytoplasmically propagated as indicated by the shading) wild-type strain HC27c-2. Conjugating pairs were disrupted at various time points and individual cells were recovered and subsequently tested for their drug-resistance phenotype. The expected phenotype of progeny cells whose macronuclei had developed within the M+ or the wild-type partner, if separation occurred either before or after pronuclear exchange, is shown below each progeny cell. The half-open/half-shaded nuclei indicate nuclear exchange had occurred, producing heterozygous genomes.
TABLE 2.
M-element rearrangement upon disruption of mating pairs
| Failed excision (%)b
|
|||
|---|---|---|---|
| Disruption time (hr)a |
M+ | wt | Postexchange |
| 2–4 | 15/16 (87) | 2/57 (4) | NA |
| 5 | 7/8 (88) | 0/25 (0) | 12/18 (67) |
| 6–7 | 10/10 (100) | 0/11 (0) | 52/55 (93) |
Prestarved cell populations were mixed at t = 0 hr and pairs were physically disrupted at the indicated time period.
The fraction (x/n, where n is the total analyzed) of single mated cells showing detectable failure of M-element deletion for each class is shown. The percentage showing failure is given in parentheses. Progeny cells showing single-drug resistance that was expected for nuclei developed within the wild-type (wt) or M+ partner disrupted prior to nuclear exchange are indicated in the second and third columns. Those showing double resistance indicating postgenetic exchange that could not be assigned a parent of origin are given in the fourth column. NA, not applicable.
Figure 5.—

Inhibition of DNA rearrangement correlates with declining sRNA levels. The events of conjugation starting with cell pairing complete in ∼14 hr, and the approximate time periods, indicated by the solid bars, for several of these are indicated. Above this time line is shown a plot of the accumulation/reduction cycle of M-element sRNAs and the period when the macronucleus can regulate DNA rearrangement. Note that inhibition of DNA rearrangement by the somatic genome coincides with the time that sRNA quantities reach a low, steady-state level.
These data appeared to show that the communication of the M+ state to the wild-type cells occurs rapidly after karyogamy. However, this interpretation required that there had been no bias in the survival of the M+ partner's vegetative progeny relative to that of the wild-type partner's. To more closely examine the timing of the DNA rearrangement block, we repeated this experiment using a wild-type strain that contained mitochondria that conferred resistance to the antibiotic chloramphenicol (Figure 3). The chloramphenicol-resistant phenotype is not passed to a mating partner during conjugation (Roberts and Orias 1973); therefore the progeny bearing nuclei that developed within this wild-type partner will be resistant to this drug while the progeny of the M+ partner will be sensitive, thereby allowing us to determine the parent of origin even after genetic exchange and karyogamy. M+ strain HC81M3A was crossed to chloramphenicol-resistant strain HC27c-2 and mating pairs were disrupted at 5, 5.5, or 6 hr after mixing. The drug-resistant phenotype expected for the progeny derived from either the M+ or the wild-type partner, disrupted either before or after nuclear exchange, could be clearly determined and is listed in Figure 3. The progeny recovered from pair disruption prior to pronuclear exchange showed 100% correspondence between the transmission of nuclear markers (conferring cy-r or mp-r) and the cytoplasmic marker (conferring cm-r; Table 3) proving the utility of the cytoplasmic marker to determine parental origin of the progeny.
TABLE 3.
Pair disruption reveals timing of macronuclear regulation of DNA rearrangement
| Failed excision (%)d
|
|||||
|---|---|---|---|---|---|
| Disruption time (hr)a |
Progeny phenotypeb |
Totalc | M+ (cm-s) | wt (cm-r) | ND |
| 5 | mp-r | 16 | 15/16 (94) | ||
| 5 | cy-r | 19 | 0/19 (0) | ||
| 5 | cy-r, mp-r | 54 | 29/30 (97) | 7/21 (33) | 3 |
| 5.5 | mp-r | 12 | 12 | ||
| 5.5 | cy-r | 18 | 18 | ||
| 5.5 | cy-r, mp-r | 32 | 8/15 (53) | 17 | |
| 6 | mp-r | 6 | 5/6 (83) | ||
| 6 | cy-r | 13 | 0/13 (0) | ||
| 6 | cy-r, mp-r | 142 | 46/53 (87) | 39/64 (61) | 25 |
| >10e | cy-r, mp-r | 138 | 25/57 (44) | 47/76 (62) | 5 |
Prestarved cell populations were mixed at t = 0 hr and pairs were physically disrupted at the indicated time.
mp-r, 6-methylpurine resistant; cy-r, cycloheximide resistant. Single resistance indicates disruption prior to nuclear exchange; double resistance indicates genetic exchange between partners.
Number of individual cells recovered giving the indicated drug resistance.
The fraction (x/n, where n is the total analyzed) of single mated cells showing detectable failure of M-element deletion for each class is shown. The percentage showing failure is given in parentheses. The progeny derived from the wild-type (wt) or M+ partner in each cross was determined by resistance or sensitivity to chloramphenicol (cm-r or cm-s), respectively. ND, state of M-element rearrangement not determined.
Pairs were allowed to separate normally without physical disruption and individuals were isolated into growth medium >20 hr after mixing.
We examined the rate of failed M-element excision in the progeny obtained after pair disruption using our PCR-based assay (Figure 2). We again found that >80% of the progeny of the M+ parent exhibited failed excision. We examined the DNA of 32 progeny derived from the wild-type partner that had been separated before pronuclear exchange (19 disrupted at 5 hr and 13 disrupted at 6 hr after mixing) and observed no failure of DNA rearrangement. This is consistent with our previous result showing a very low transmission of the block if cells are paired for <5 hr. Because pairing is not perfectly synchronous, these pre-exchange pairs likely are those for which pairing was delayed relative to the progeny that progressed past the point of karyogamy. In contrast, we found that the progeny of the wild-type partner derived from pairs that had undergone karyogamy, but were disrupted at 5, 5.5, or 6 hr showed an increasing rate of failed excision, from 33 to 61%, during this time period (Table 3). These data again show that the block to M-element excision is rapidly communicated to the wild-type partner starting at ∼5 hr of conjugation.
M-element transcripts synthesized in micronuclei, but not in macronuclei, are rapidly processed in small RNAs:
While it was apparent that the inhibition of M-element excision imposed by the homologous sequence within the parental macronucleus increased rapidly after exchange of gametic nuclei (Table 3), it is not the exchange per se that communicates the interfering signal (Figure 1). Factors that are free to move through the cytoplasm must mediate this form of epigenetic regulation. A likely mediator of this sequence-specific inhibition is RNA, especially given the fact that germline-specific transcription and homologous sRNAs have been implicated in the normal mechanism of DNA rearrangement (Chalker and Yao 2001; Mochizuki et al. 2002). Therefore, we examined the timing of M-element transcription and any processing into smaller species. RNA was isolated from both conjugating wild-type cells (CU427 × CU428) and M+ cells (HC76M5B × HC81M3A) and Northern blot analysis was employed using strand-specific M-element probes (Figure 4). In wild-type cells, the M element is transcribed bidirectionally in the micronucleus starting before 2 hr of conjugation and reaches a peak accumulation at ∼6 hr after mixing as described previously (Chalker and Yao 2001). In conjugating M+ strains, the M-element transcripts detected were extremely abundant, presumably due to the transcription of the high copy number of M elements within the macronucleus (Figure 4A). This observation opens the possibility that the M-element transcripts produced from the parental macronucleus act to inhibit M-element rearrangement. Such transcripts could freely travel through the cytoplasm and would be able to specifically block M-element rearrangement and would not be able to block the rearrangement of other elements (Chalker and Yao 1996).
Figure 4.—
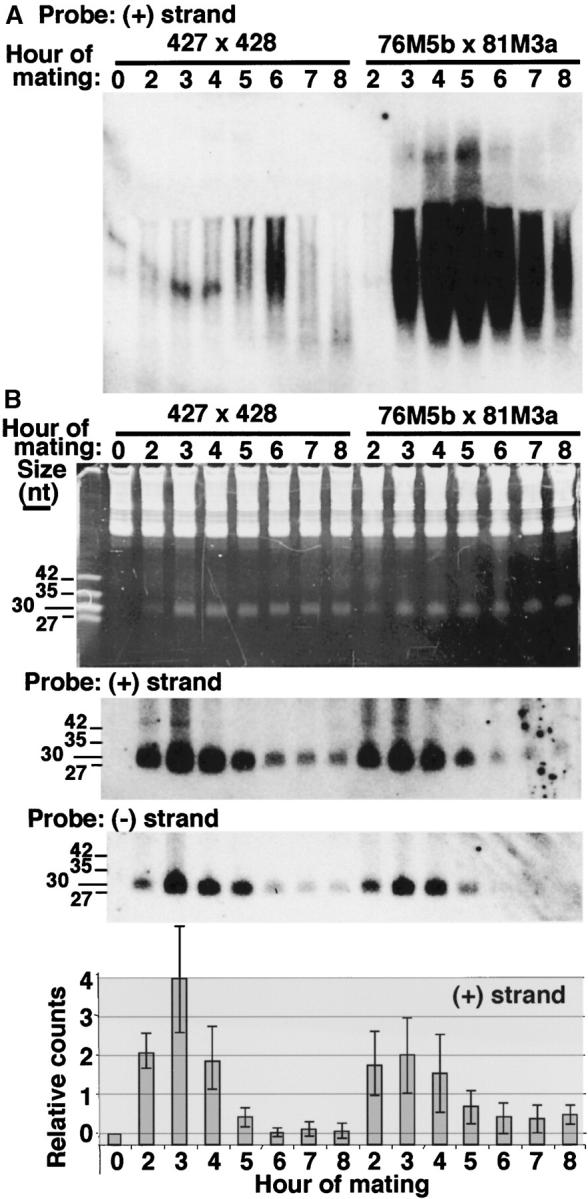
M-element transcript levels, but not sRNA levels, are altered in conjugating M+ strains. RNA isolated at the indicated times of conjugation was separated by electrophoresis on (A) 1.2% agarose-formaldehyde gels or (B) 15% polyacrylamide-urea gels, transferred to membranes, and hybridized to plus- or minus-strand-specific M-element riboprobes as indicated. Equal amounts of RNA from wild-type (427 × 428) or M+ (76M5B × 81M3A) mating populations were examined at each time point. (A) Heterogeneous M-element transcripts are much more abundant in conjugating M+ cells. (B) A population of sRNAs is observed on the ethidium bromide-stained gel (top) in relatively equal quantities by 3 hr of conjugation. The lengths of DNA oligonucleotide size markers are indicated (leftmost lane). Hybridization of the same samples (plus-strand probe) or duplicate samples (minus-strand probe) is shown below image of the stained gel. Quantification of the relative levels of plus-strand-specific M-element sRNAs at each time point is given at the bottom. Quantification of each sample was normalized to 5S rRNA hybridization, background hybridization of starved cell samples (lane 1) was subtracted, and the combined data sets of three individual RNA isolations and analyses were averaged. Error bars represent one standard deviation.
To determine whether M-element transcripts were processed into sRNA species, RNA isolated from conjugating cells was electrophoresed on 15% denaturing polyacrylamide gels and again hybridized to M-element-specific probes. M-element-specific sRNAs were found to accumulate between 2 and 4 hr of conjugation. This is the first detection of sRNAs to a particular eliminated sequence. The abundance of M-element sRNAs rapidly decreased between 4 and 6 hr to a low steady-state level that persisted until at least 12 hr of conjugation (Figure 4B and data not shown). Thus a large pool of specific sRNAs are produced, only a fraction of which are stabilized. If these sRNAs promote DNA rearrangement during nuclear differentiation by targeting H3-K9 methylation to the homologous sequence as has been proposed (Mochizuki et al. 2002; reviewed in Mochizuki and Gorovsky 2004b), this stable pool must contain the active targeting population, since this modification is not observed until developing macronuclei first appear, nearly 6 hr into conjugation (Taverna et al. 2002).
The dynamic accumulation and reduction was not evident in the overall population of sRNAs observed by staining the gel with ethidium bromide (Figure 4B). This may simply indicate that sRNAs homologous to many of the micronucleus-limited sequences do not show the dramatic accumulation/reduction cycle observed for the M element or that different micronucleus-limited sequences produce sRNAs at varying times during development. It also raises the possibility that the sRNAs observed on the stained gel represent multiple classes of developmentally produced sRNAs, not all of which need necessarily be involved in DNA rearrangement, and these may obscure the visualization of sRNAs dedicated to rearrangement.
To investigate how the parental macronuclear genome might influence accumulation of specific sRNAs, we compared the M-element sRNAs in conjugating wild-type cells to that of M+ cells and could detect no obvious difference in sRNA levels between these different cell populations at any developmental stage (Figure 4B). Given that M-element sRNAs accumulate normally in conjugating M+ cells, copies of this element in the parental macronucleus appear to inhibit DNA rearrangement by a means other than simply destabilizing the homologous sRNAs as has been suggested (Mochizuki et al. 2002), possibly by sequestering them in the parental macronucleus (see discussion).
One factor potentially complicating this analysis is that the M element appears to contain some moderately repetitive sequences as multiple DNA fragments hybridize to a probe specific for the 0.6-kbp micronucleus-limited region in Southern blot analysis of genomic DNA (D. L. Chalker, unpublished data). Even so, only the rearrangement of the one M-element locus is blocked by the homologous sequence placed in the macronucleus (Chalker and Yao 1996; D. L. Chalker, unpublished data). We can detect M-element-specific sRNAs in RNA isolated from crosses of nulli 4 strains that are missing the M-element-containing chromosome 4 from their micronuclei, demonstrating that other loci contribute to the production of this abundant pool of sRNAs (data not shown). Nevertheless, we believe we are detecting the specific sRNAs that are able to target M-element rearrangement because: (1) increasing the stringency of our last hybridization wash from 60° to 65° and 70° uniformly decreases our signal in RNA isolated from both wild-type and nulli 4 mating cells; (2) using probes specific to only the right or left half of the 0.6-kbp region detects similar levels of sRNAs; and (3) hybridization with probes to macronucleus-retained sequences of similar A + T content (the M element is >75% A + T) does not detect small RNAs (data not shown). Regardless of the copy number of M-element-related sequences in the micronucleus, if sequences in the macronucleus destabilize homologous sRNAs we should have observed a substantial decrease in any sRNAs that had sufficient homology to hybridize to our specific M-element probes, given that thousands of copies of the M element reside in the parental macronuclei of our M+ strains.
Another striking observation is that the M-element transcripts produced from the macronuclear copies of the M element are not being processed into sRNAs. If this were the case, we should have detected a significant increase of M-element sRNAs in the RNA isolated from M+ strains relative to that of the wild type, given the huge excess of M-element RNAs detected by conventional Northern blot analysis (Figure 4A). This finding indicates that the transcripts produced from the micronucleus can be distinguished from those produced from the macronucleus and it is only the micronucleus-derived M-element transcripts that are efficiently processed into sRNAs.
DISCUSSION
“Cross-talk” between the parental and developing macronuclei is an intriguing phenomenon illustrating that sequence-specific information can flow between nuclei through epigenetic mechanisms. DNA sequences within the parental macronucleus of one cell can interfere with the normally efficient programmed elimination of the homologous sequence from new macronuclei, even those nuclei developing within a wild-type conjugation partner. We have found that this cross-talk occurs without direct transfer of genetic material to the partner and must involve factors free to travel through the cells' cytoplasm (Figure 1). By physically disrupting conjugating pairs, we have shown that the inhibition of M-element rearrangement by the copies within the parental macronucleus is rapidly enforced between 5 and 6 hr of conjugation (Tables 2 and 3). We had previously reported that bidirectional M-element transcripts accumulate around this time of development (Chalker and Yao 2001). Here we have shown that M-element transcripts are processed into 28- to 30-nt sRNAs. These sRNAs rapidly accumulate in the first few hours of conjugation before their levels decline to generate a small, but stable pool (Figure 4B). From the summary of these data in Figure 5, it is apparent that the parental macronucleus' influence is enforced during the same time period that the M-element sRNA levels plateau. Together, these observations, along with other studies discussed below, lead us to conclude that sRNAs are the likely mediators of sequence-specific information between the germline, parental, and developing macronuclear genomes.
The accumulation of M-element bidirectional transcripts and sRNAs is incongruous. While sRNA levels peak at 3 hr, the transcripts continue to accumulate until 6 hr of conjugation. These observations support a simple interpretation that M-element transcripts cease being processed around 3 hr, thus allowing for an increase in the steady-state transcript level and a decline in sRNAs. Possible explanations for a cessation of processing include a temporal change in the localization/activity of a predicted dicer-like processing enzyme or the export efficiency of the transcripts, if, for instance, processing occurs in the cytoplasm. Our finding that M-element transcripts synthesized in parental macronuclei are not efficiently processed into sRNAs indicates that they are not generally accessible to a dicer-like protein, a fact that argues that processing is compartmentalized and that only transcripts derived from the germline micronucleus are processed. Between 3 and 4 hr of conjugation, micronuclei complete meiosis (see Figure 5), which could represent the temporal signal that ends sRNA production. While we are not yet certain where M-element sRNAs are generated, a dicer-like protein should be localized in the cytoplasm at later stages of conjugation given that sense and antisense RNA injected together into the cytoplasm of mating cells (at least 4.5 hr into conjugation) can induce the elimination of the homologous sequence, assuming that sRNAs are essential for such induced DNA rearrangements (Yao et al. 2003).
At least for the M element, it appears that an excess of sRNAs are initially generated, only a fraction of which are stable. We suggest that the stabilization of sRNAs requires association with a limiting RNA-induced silencing complex-like protein machinery (Hammond et al. 2000) that would include the Piwi/Argonaute family member Twi1p (Mochizuki et al. 2002). This idea is supported by the observation that, in TWI1 knockout cells, M-element sRNAs accumulate during the first 4 hr of conjugation, but fail to be stabilized and cannot be detected by 6 hr of conjugation (D. L. Chalker, unpublished data).
How might DNA sequences within the parental macronucleus interfere with the elimination of their homologous element? Given that M-element sRNAs accumulate prior to the time that the block to rearrangement is communicated through the cytoplasm, they clearly have the potential to act as mediators of sequence-specific information between the germline micronuclei and the parental macronuclei. The idea that sRNAs produced from germline-derived transcripts are compared to the parental macronuclear genome was postulated to explain the influence of the parental macronucleus as part of the “scan RNA model” of DNA elimination and was supported by the observation that the Twi1p, which is initially cytoplasmic, localizes to parental macronuclei before developing macronuclei are formed (Mochizuki et al. 2002). It was suggested that sRNAs that encounter a homologous sequence in the parental macronucleus are degraded, removing them from the pool that targets DNA sequences for elimination in the developing macronucleus. However, our finding that the levels of M-element sRNAs produced and stabilized were not reduced in conjugating M+ cells indicates that sequences in the parental macronucleus do not simply promote the degradation of the homologous sRNAs. Instead we suggest that sRNAs that encounter their homologous sequence in the parental macronucleus, such as the M element in M+ strains, are sequestered and/or their associated proteins are specifically inactivated. That M-element copies need be present in the macronuclei of only one mating partner to block rearrangement of the homologous element in both partners indicates that the sRNAs produced in each cell must be effectively compared to the somatic genomes of both parents.
As we suggested above, the population of M-element sRNAs available to target excision is generated by 3–4 hr into conjugation and must assemble with Twi1p-containing complexes to be stabilized. To explain the timing of influence of the parental macronucleus, we suggest that sRNA-containing complexes are rapidly transported to the parental macronuclei and compared to the sequences residing within. Those that pair with macronuclear sequences are disassembled, but the proteins are free to return to the cytoplasm and pick up additional sRNAs from the existing pool. By 5 hr of conjugation, the sRNAs that were generated either must have been assembled into complexes or will have been degraded outside the parental macronucleus. Thus the inhibition of the parental macronucleus is not enforced until 5 hr of conjugation when the pool of free sRNAs is depleted (see Figure 5). The comparison to the parental genome continues until the developing macronuclei appear at ∼6 hr of conjugation, when the remaining sRNA-containing complexes relocalize there to direct DNA rearrangement. This interpretation of our data is also consistent with recent data showing that Twi1p-associated sRNAs are gradually enriched in germline-limited sequences as conjugation proceeds (Mochizuki and Gorovsky 2004a), findings that together support the designation of Tetrahymena sRNAs as scan RNAs (Mochizuki et al. 2002).
We must consider the possibility that M-element sRNAs may not interact directly with the macronuclear M-element DNA sequence, but rather may interact with homologous RNA transcripts. This is an important distinction that has major implications for understanding of the mechanism of RNAi-related processes, as sRNAs have not been shown to directly interact with DNA. Our observation that the extensive bidirectional transcription that occurs from M-element copies in the parental macronucleus does not lead to a corresponding increase in sRNA quantities indicates that the cell can distinguish between transcripts produced in the micronucleus and those that are produced in the macronucleus. This finding, along with other data presented, leads us to speculate that macronucleus-derived M-element transcripts are the factors that inhibit the targeting of the specific sRNAs to the developing macronucleus. It may be that the macronuclear-derived transcripts never leave the nucleus and thus are able to serve as a molecular sponge, absorbing the M-element sRNAs with which they interact. Alternately, these RNAs may be transported to the cytoplasm where they could serve a similar inhibitory role; although this is a less attractive possibility given that dsRNA injected in the cytoplasm can promote DNA elimination (Yao et al. 2003). Homologous RNA transcripts may also be required in the developing macronucleus for a sequence to be targeted by an sRNA-containing complex. This would explain the need for M-element transcription to continue even after the transcripts appear to cease being processed into sRNAs.
One important function of comparing the sRNA population to the parental genome would be to provide a means to ensure that sequences present in the somatic genome in the previous sexual generation are retained in the progeny. This would also serve as an elegant means of defense for the germline genome from the spread of invading transposable elements (Mochizuki et al. 2002; Yao et al. 2003). An element's transcription and, thus, its mobility in the silent micronucleus would be normally suppressed. It should rarely, if ever, be able to escape to the transcriptionally active macronucleus in subsequent generations, due to the cell's ability to recognize DNA segments present in the micronucleus, but not in the macronucleus and to excise those sequences. For such a genome surveillance system to be effective, it must be sensitive to rather low copy numbers of the invading sequence. In support of this, Yao et al. (2003) have shown that single-copy transgenes in the germline can be eliminated in a similar means as endogenous micronucleus-limited sequences. Such surveillance must also be able to efficiently determine the sequence content of the parental genome. Our data showing that a DNA sequence present in the parental genome of one mating partner can be rapidly scanned and inhibit DNA elimination indicates effective, dynamic interaction between the parental and developing genomes.
Acknowledgments
We thank Deborah Frank for comments on the manuscript and X. Zhen and C. Malone for technical assistance. This work was supported by National Science Foundation research grant MCB-0131421 to D.L.C. and by National Institutes of Health research grant GM-26210 to M.-C.Y.
References
- Asai, D. J., and J. D. Forney (Editors), 1999 Tetrahymena thermophila. Academic Press, San Diego.
- Austerberry, C. F., and M. C. Yao, 1987. Nucleotide sequence structure and consistency of a developmentally regulated DNA deletion in Tetrahymena thermophila. Mol. Cell. Biol. 7: 435–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austerberry, C. F., and M. C. Yao, 1988. Sequence structures of two developmentally regulated, alternative DNA deletion junctions in Tetrahymena thermophila. Mol. Cell. Biol. 8: 3947–3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austerberry, C. F., C. D. Allis and M. C. Yao, 1984. Specific DNA rearrangements in synchronously developing nuclei of Tetrahymena. Proc. Natl. Acad. Sci. USA 81: 7383–7387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein, E., A. A. Caudy, S. M. Hammond and G. J. Hannon, 2001. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 409: 363–366. [DOI] [PubMed] [Google Scholar]
- Chalker, D. L., and M.-C. Yao, 1996. Non-Mendelian, heritable blocks to DNA rearrangement are induced by loading the somatic nucleus of Tetrahymena thermophila with germ line limited DNA. Mol. Cell. Biol. 16: 3658–3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalker, D. L., and M. C. Yao, 2001. Nongenic, bidirectional transcription precedes and may promote developmental DNA deletion in Tetrahymena thermophila. Genes Dev. 15: 1287–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry, J. M., and E. H. Blackburn, 1985. The internally located telomeric sequences in the germline chromosomes of Tetrahymena are at the conserved ends of transposon-like elements. Cell 43: 747–758. [DOI] [PubMed] [Google Scholar]
- Church, G. M., and W. Gilbert, 1984. Genomic sequencing. Proc. Natl. Acad. Sci. USA 81: 1991–1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole, E., and P. Bruns, 1992. Uniparental cytogamy: a novel method for bringing micronuclear mutations of Tetrahymena into homozygous macronuclear expression with precocious sexual maturity. Genetics 132: 1017–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyne, R. S., M. A. Nikiforov, J. F. Smothers, C. D. Allis and M. C. Yao, 1999. Parental expression of the chromodomain protein Pdd1p is required for completion of programmed DNA elimination and nuclear differentiation. Mol. Cell 4: 865–872. [DOI] [PubMed] [Google Scholar]
- Dernburg, A. F., and G. H. Karpen, 2002. A chromosome RNAissance. Cell 111: 159–162. [DOI] [PubMed] [Google Scholar]
- Duharcourt, S., and M. C. Yao, 2002. Role of histone deacetylation in developmentally programmed DNA rearrangements in Tetrahymena thermophila. Eukaryot. Cell 1: 293–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duharcourt, S., A. Butler and E. Meyer, 1995. Epigenetic self-regulation of developmental excision of an internal eliminated sequence in Paramecium tetraurelia. Genes Dev. 9: 2065–2077. [DOI] [PubMed] [Google Scholar]
- Duharcourt, S., A. Keller and E. Meyer, 1998. Homology-dependent maternal inhibition of developmental excision of internal eliminated sequences in Paramecium tetraurelia. Mol. Cell. Biol. 18: 7075–7085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan, Q., R. Sweeney and M.-C. Yao, 1999 Creation and use of antisense ribosomes in Tetrahymena thermophila, pp. 533–547 in Tetrahymena thermophila, edited by D. J. Asai and J. D. Forney. Academic Press, New York. [DOI] [PubMed]
- Fillingham, J. S., T. A. Thing, N. Vythilingum, A. Keuroghlian, D. Bruno et al., 2004. A non-long terminal repeat retrotransposon family is restricted to the germ line micronucleus of the ciliated protozoan Tetrahymena thermophila. Eukaryot. Cell 3: 157–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorovsky, M. A., M. C. Yao, J. B. Keevert and G. L. Pleger, 1975. Isolation of micro- and macronuclei of Tetrahymena thermophila. Methods Cell Biol. 9: 311–327. [DOI] [PubMed] [Google Scholar]
- Goyon, C., and G. Faugeron, 1989. Targeted transformation of Ascobolus immersus and de novo methylation of the resulting duplicated DNA sequences. Mol. Cell. Biol. 9: 2818–2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall, I. M., G. D. Shankaranarayana, K. Noma, N. Ayoub, A. Cohen et al., 2002. Establishment and maintenance of a heterochromatin domain. Science 297: 2232–2237. [DOI] [PubMed] [Google Scholar]
- Hamilton, E. P., J. P. Suhr and E. Orias, 1988. Pronuclear fusion failure: an alternate conjugational pathway in Tetrahymena thermophila, induced by vinblastine. Genetics 118: 627–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond, S. M., E. Bernstein, D. Beach and G. J. Hannon, 2000. An RNA-directed nuclease mediates post-transcriptional gene silencing in Drosophila cells. Nature 404: 293–296. [DOI] [PubMed] [Google Scholar]
- Hannon, G. J., 2002. RNA interference. Nature 418: 244–251. [DOI] [PubMed] [Google Scholar]
- Karrer, K. M., 2000. Tetrahymena genetics: two nuclei are better than one. Methods Cell Biol. 62: 127–186. [DOI] [PubMed] [Google Scholar]
- Ketting, R. F., T. H. Haverkamp, H. G. van Luenen and R. H. Plasterk, 1999. Mut-7 of C. elegans, required for transposon silencing and RNA interference, is a homolog of Werner syndrome helicase and RNaseD. Cell 99: 133–141. [DOI] [PubMed] [Google Scholar]
- Liu, Y., K. Mochizuki and M. A. Gorovsky, 2004. Histone H3 lysine 9 methylation is required for DNA elimination in developing macronuclei in Tetrahymena. Proc. Natl. Acad. Sci. USA 101: 1679–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki, K., and M. A. Gorovsky, 2004. a Conjugation-specific small RNAs in Tetrahymena have predicted properties of scan (scn) RNAs involved in genome rearrangement. Genes Dev. 18: 2068–2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki, K., and M. A. Gorovsky, 2004. b Small RNAs in genome rearrangement in Tetrahymena. Curr. Opin. Genet. Dev. 14: 181–187. [DOI] [PubMed] [Google Scholar]
- Mochizuki, K., N. A. Fine, T. Fujisawa and M. A. Gorovsky, 2002. Analysis of a piwi-related gene implicates small RNAs in genome rearrangement in Tetrahymena. Cell 110: 689–699. [DOI] [PubMed] [Google Scholar]
- Roberts, C. T., Jr., and E. Orias, 1973. Cytoplasmic inheritance of chloramphenicol resistance in Tetrahymena. Genetics 73: 259–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selker, E. U., 1990. Premeiotic instability of repeated sequences in Neurospora crassa. Annu. Rev. Genet. 24: 579–613. [DOI] [PubMed] [Google Scholar]
- Selker, E. U., 2002. Repeat-induced gene silencing in fungi. Adv. Genet. 46: 439–450. [DOI] [PubMed] [Google Scholar]
- Tabara, H., M. Sarkissian, W. G. Kelly, J. Fleenor, A. Grishok et al., 1999. The rde-1 gene, RNA interference, and transposon silencing in C. elegans. Cell 99: 123–132. [DOI] [PubMed] [Google Scholar]
- Taverna, S. D., R. S. Coyne and C. D. Allis, 2002. Methylation of histone h3 at lysine 9 targets programmed DNA elimination in Tetrahymena. Cell 110: 701–711. [DOI] [PubMed] [Google Scholar]
- Virtue, M. A., and E. S. Cole, 1999. A cytogenetic study of development in mechanically disrupted pairs of Tetrahymena thermophila. J. Eukaryot. Microbiol. 46: 597–605. [DOI] [PubMed] [Google Scholar]
- Volpe, T. A., C. Kidner, I. M. Hall, G. Teng, S. I. Grewal et al., 2002. Regulation of heterochromatic silencing and histone H3 lysine-9 methylation by RNAi. Science 297: 1833–1837. [DOI] [PubMed] [Google Scholar]
- Waterhouse, P. M., M. B. Wang and T. Lough, 2001. Gene silencing as an adaptive defence against viruses. Nature 411: 834–842. [DOI] [PubMed] [Google Scholar]
- Wuitschick, J. D., J. A. Gershan, A. J. Lochowicz, S. Li and K. M. Karrer, 2002. A novel family of mobile genetic elements is limited to the germline genome in Tetrahymena thermophila. Nucleic Acids Res. 30: 2524–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao, M. C., and M. A. Gorovsky, 1974. Comparison of the sequences of macro- and micronuclear DNA of Tetrahymena thermophila. Chromosoma 48: 1–18. [DOI] [PubMed] [Google Scholar]
- Yao, M. C., J. Choi, S. Yokoyama, C. F. Austerberry and C. H. Yao, 1984. DNA elimination in Tetrahymena: a developmental process involving extensive breakage and rejoining of DNA at defined sites. Cell 36: 433–440. [DOI] [PubMed] [Google Scholar]
- Yao, M. C., S. Duharcourt and D. L. Chalker, 2002 Genome-wide rearrangements of DNA in ciliates, pp. 730–758 in Mobile DNA II, edited by N. Craig, R. Craigie, M. Gellert and A. Lambowitz. Academic Press, New York.
- Yao, M. C., P. Fuller and X. Xi, 2003. Programmed DNA deletion as an RNA-guided system of genome defense. Science 300: 1581–1584. [DOI] [PubMed] [Google Scholar]