

Abstract
The duplication of genes and genomes is believed to be a major force in the evolution of eukaryotic organisms. However, different models have been presented about how duplicated genes are preserved from elimination by purifying selection. Preservation of one of the gene copies due to rare mutational events that result in a new gene function (neofunctionalization) necessitates that the other gene copy retain its ancestral function. Alternatively, preservation of both gene copies due to rapid divergence of coding and noncoding regions such that neither retains the complete function of the ancestral gene (subfunctionalization) may result in a requirement for both gene copies for organismal survival. The duplication and divergence of the tandemly arrayed homeotic clusters have been studied in considerable detail and have provided evidence in support of the subfunctionalization model. However, the vast majority of duplicated genes are not clustered tandemly, but instead are dispersed in syntenic regions on different chromosomes, most likely as a result of genome-wide duplications and rearrangements. The Myb oncogene family provides an interesting opportunity to study a dispersed multigene family because invertebrates possess a single Myb gene, whereas all vertebrate genomes examined thus far contain three different Myb genes (A-Myb, B-Myb, and c-Myb). A-Myb and c-Myb appear to have arisen by a second round of gene duplication, which was preceded by the acquisition of a transcriptional activation domain in the ancestral A-Myb/c-Myb gene generated from the initial duplication of an ancestral B-Myb-like gene. B-Myb appears to be essential in all dividing cells, whereas A-Myb and c-Myb display tissue-specific requirements during spermatogenesis and hematopoiesis, respectively. We now report that the absence of Drosophila Myb (Dm-Myb) causes a failure of larval hemocyte proliferation and lymph gland development, while Dm-Myb−/− hemocytes from mosaic larvae reveal a phagocytosis defect. In addition, we show that vertebrate B-Myb, but neither vertebrate A-Myb nor c-Myb, can complement these hemocyte proliferation defects in Drosophila. Indeed, vertebrate A-Myb and c-Myb cause lethality in the presence or absence of endogenous Dm-Myb. These results are consistent with a neomorphic origin of an ancestral A-Myb/c-Myb gene from a duplicated B-Myb-like gene. In addition, our results suggest that B-Myb and Dm-Myb share essential conserved functions that are required for cell proliferation. Finally, these experiments demonstrate the utility of genetic complementation in Drosophila to explore the functional evolution of duplicated genes in vertebrates.
IT has been extensively reported that genome or large chromosomal regional duplications may be responsible for the structure and evolution of vertebrate genomes from preduplication invertebrate genomes (Abi-Rached et al. 2002; McLysaght et al. 2002; Panopoulou et al. 2003). For example, at least 15% of the known human genes are recognizable as duplicates (Li et al. 2001). While controversial, it has been proposed that vertebrate genome evolution has occurred through two whole-genome duplication events that are thought to have occurred early in vertebrate evolution ∼500 million years ago (Ohno 1999). Consistent with this model, many vertebrate multigene families are represented by a single homolog in modern invertebrate species such as the sea urchin, Drosophila, and Caenorhabditis elegans (Holland 1999; Meyer and Schartl 1999). Conclusive support for whole-genome duplication as a source for duplicate gene innovation has recently been shown for the yeast Saccharomyces cerevisiae. Analysis of the complete genome of a related yeast species, Kluyveromyces walti, has demonstrated that the genome of S. cerevisiae is a degenerate tetraploid that arose from an ancient whole-genome duplication after the divergence of the two species from a common ancestor (Wolfe and Shields 1997; Kellis et al. 2004).
Of considerable interest in the study of gene and genome evolution is the mechanism(s) by which duplicated genes are preserved in the face of constant selective pressure (reviewed in (Prince and Pickett 2002). Current theories propose three alternative fates for duplicated genes: (1) one copy is rendered nonfunctional by mutations or eliminated by genomic rearrangements (nonfunctionalization); (2) both copies are retained due to a rare mutational event in one copy that creates a selective advantage (neofunctionalization); and (3) both copies are retained due to complementary loss-of-function mutations that can occur at the level of regulatory regions well as protein structural domains (subfunctionalization). Duplicated genes can occur in tandem arrays (e.g., the Hom/Hox gene clusters), dispersed duplications residing on syntenic chromosomal regions (e.g., the three different immunoglobulin chain genes), or via recent whole-genome duplication (e.g., the polyploid nature of cereal crops such as maize). Increased ploidy has occurred in some vertebrate lineages, including the salmonid fish, zebrafish, Xenopus laevis, and the red viscacha rat, as a result of additional genome-wide duplications that appear to have occurred much more recently (Bailey et al. 1978; Hughes and Hughes 1993; Amores et al. 1998; Gallardo et al. 1999).
The subfunctionalization model was proposed in large part due to comparative studies of the Hox genes of mice and zebrafish (Force et al. 1999; Lynch and Force 2000). For example, complementary degenerative mutations in the cis-regulatory regions that control the putative ancestral expression of the mouse Hoxb1 gene have been subdivided between the present-day zebrafish hoxb1b and hoxb1a gene duplicates (McClintock et al. 2002). However, it remains unclear whether studies of unusually large tandemly arrayed duplicate genes such as those in the Hox clusters can be generalized to explain the preservation of many dispersed gene duplications that occurred during vertebrate evolution from a common ancestor shared with modern invertebrates.
We believe that a functional analysis of the vertebrate Myb gene family in Drosophila may provide another useful model system to understand the survival of duplicate genes during vertebrate evolution. Vertebrate genomes contain three different Myb genes (A-Myb, B-Myb, and c-Myb), which in humans and mice reside on three different chromosomes in regions that appear to be syntenic between human and mouse. Invertebrates such as Drosophila and invertebrate chordates such as sea urchin (Strongylocentrotus purpuratus) and sea squirt (Ciona intestinalis) contain a single member of the Myb gene family. Genes encoding closely related Myb domains have been identified in the cellular slime mold (Dictyostelium discoideum) and in all major plant lineages (Stober-Grasser et al. 1992; Braun and Grotewold 1999; Kranz et al. 2000). In fact, Myb repeat-containing proteins are highly represented in plants, with >200 Myb domain-encoding genes represented in maize and >100 present in Arabidopsis, demonstrating the utility of Myb gene duplication during the evolution of flowering plants (Rabinowicz et al. 1999; Riechmann et al. 2000; Stracke et al. 2001; Dias et al. 2003). Phylogenetic analysis of animal Myb proteins suggests that the vertebrate Myb genes may have arisen via the putative whole-genome duplication events mentioned above, in that A-Myb and c-Myb arose via a more recent gene duplication, with the common ancestor of A-Myb and c-Myb resulting from a duplication of a B-Myb/Drosophila Myb-like gene (Simon et al. 2002). The duplication that generated the ancestral A-Myb/c-Myb gene was followed by the insertion of a DNA sequence that encodes the highly conserved transcriptional activation domain present only in A-Myb and c-Myb, but not in B-Myb or Dm-Myb.
All three vertebrate Myb genes have been implicated in cell proliferation and malignancies (Ganter and Lipsick 1999). c-Myb expression is required for definitive hematopoiesis since targeted disruption of the c-Myb gene in mice results in late embryonic death due to a failure of fetal liver hematopoiesis (Mucenski et al. 1991). The expression of c-Myb has also been detected in immature epithelial cells in a variety of tissues including the colon, respiratory tract, skin, and retina (Queva et al. 1992; Sitzmann et al. 1995). The expression of the A-Myb gene also occurs in a tissue-specific manner, with the highest levels occurring in germinal center B-lymphocytes, mammary gland ductal epithelium, the testis, and the central nervous system (Sleeman 1993; Mettus et al. 1994; Trauth et al. 1994). Mice with a targeted disruption of the A-Myb gene are viable but exhibit a failure of spermatogenesis and of mammary gland proliferation in response to pregnancy (Toscani et al. 1997). In contrast to the tissue-specific expression of A-Myb and c-Myb, B-Myb is ubiquitously expressed throughout mouse development (Sitzmann et al. 1996). The B-Myb gene is thought to play a role in cell cycle progression and, similar to other S-phase genes, is directly regulated by the p16/cyclin D/Rb family/E2F pathway (Lam and Watson 1993; Zwicker et al. 1996). The B-Myb “knockout” mouse phenotype is early embryonic lethality at days 4.5–6.5 with apparent defects in inner cell mass formation (Tanaka et al. 1999).
Drosophila Myb (Dm-Myb) was initially implicated in cell cycle progression with a role in G2/M progression (Katzen and Bishop 1996; Katzen et al. 1998). These findings were supported by experiments demonstrating Dm-Myb-induced expression of cyclin B, an important regulator of the G2/M transition (Okada et al. 2002). Further experiments have demonstrated a role for Dm-Myb in the maintenance of genomic integrity and in the suppression of centrosome amplification (Fung et al. 2002), with ectopic expression of Dm-Myb promoting proliferation in diploid cells and suppressing endoreduplication in endocycling cells (Fitzpatrick et al. 2002). Analyses of Dm-Myb null larvae have suggested the primary cell cycle defect occurs in S- or G2-phase, resulting in a variety of M-phase abnormalities such as anomalous chromosomal condensation, increased aneuploidy and polyploidy, and spindle defects (Manak et al. 2002). Further supporting a role in S-phase, Dm-Myb is required for amplification of the chorion gene loci in Drosophila follicle cells, binding both in vitro and in vivo to site-specific DNA replication enhancer elements (Beall et al. 2002).
We hypothesize that the gene duplication and domain acquisition events that gave rise to an A-Myb/c-Myb progenitor represent an example of neofunctionalization and facilitated the positive selection of the duplicated ancestral A-Myb/c-Myb gene. On the basis of the similar protein sequences but differing tissue-specific expression patterns of A-Myb and c-Myb, it is probable that subfunctionalization facilitated the preservation of these genes following a second duplication event. One prediction of this model is that B-Myb retains the ancestral function and is the true ortholog of Dm-Myb. We therefore investigated which, if any, of the vertebrate Myb genes would be able to complement Dm-Myb null mutations. In particular, to test whether Dm-Myb is required for a tissue-specific function similar to the requirement of c-Myb in mammalian hematopoiesis, we choose to investigate the functional evolution of the vertebrate Myb genes in lymph gland of Drosophila since recent evidence indicates a high degree of functional conservation between fly and mammalian hematopoiesis (Evans et al. 2003). The failure of all of the vertebrate Myb genes to complement Dm-Myb null mutations would indicate subfunctionalization and extreme divergence of all the vertebrate Myb genes as a result of the gene duplication events, such that none of the paralogs possesses a complete ancestral function. Complementation with B-Myb but neither A-Myb nor c-Myb would be consistent with the neofunctionalization of the ancestral A-Myb/c-Myb gene. Complementation of hemocyte defects with c-Myb but neither A-Myb nor B-Myb would support a conserved tissue-specific role for c-Myb that arose by subfunctionalization.
In Drosophila larvae the paired lymph gland lobes that flank the dorsal vessel are sites of hematopoiesis (Lanot et al. 2001). Differentiation of putative hematopoietic stem cells gives rise to three characterized hemocyte lineages: plasmatocytes/macrophages that phagocytose microorganisms and apoptotic cells; lamellocytes that encapsulate foreign material too large to be internalized; and crystal cells that initiate a melanotic response by lysis and release of enzymes at, or near, foreign bodies. Remarkably, the differentiation of prohemocytes in the Drosophila lymph gland requires the function of transcription factors with sequence identity to important regulators of mammalian hematopoiesis such as the GATA-related gene serpent (srp; Rehorn et al. 1996; Lebestky et al. 2000); the acute myeloid leukemia-1 (AML1)-related gene lozenge (lz; Lebestky et al. 2000); and signaling pathways such as those including Toll (reviewed in Dearolf 1998; Mathey-Prevot and Perrimon 1998), Janus kinase and signal transducers and activators of transcription (JAK/STAT), vascular endothelial growth factor (VEGF; Cho et al. 2002; Munier et al. 2002), and Notch (Duvic et al. 2002; Lebestky et al. 2003).
Here we present genetic rescue experiments that indicate vertebrate B-Myb and Dm-Myb are functional orthologs. We find that B-Myb, but neither A-Myb nor c-Myb, can restore normal levels of proliferation, correct the hematopoietic defects of Dm-Myb−/− larval lymph gland hemocytes, and rescue circulating crystal cells. Through use of mosaic analyses we show that Dm-Myb−/− hemocytes are defective in phagocytosis. Furthermore, Dm-Myb is required for the hyperproliferation of hemocytes that result from gain-of-function mutations in the Toll and JAK/STAT pathways. Taken together, these results indicate that Dm-Myb and B-Myb are generally required for proliferation while the paralogous genes, A-Myb and c-Myb, have acquired new functions.
MATERIALS AND METHODS
Drosophila stocks and genetics:
Control larvae were y, w67. Other stocks were: UAS-Dm-Myb transgene line y,w67,P{w[+mC]=UAS-Dm-Myb}; UAS-chicken B-Myb transgene line y,w67;+;P{w[+mC]=UAS-B-Myb}; UAS-chicken c-Myb transgene line y,w67;+;P{w[+mC]=UAS-c-Myb}; UAS-chicken A-Myb transgene line y,w67;+;P{w[+mC]=UAS-A-Myb}; and GAL4-e33c (P{w[+mW.hs]=GAL4}e33c); Dm-Myb null allele MH107 (Manak et al. 2002), hopTuml (Hanratty and Ryerse 1981), and lz null allele lzR15 (Daga et al. 1996) were balanced over FM7i, P{w+mC=ActGFP}JMR3 (Bloomington Stock Center) for identification of mutant larvae; mosaic analysis with a repressible cell marker (MARCM) line (P{neoFRT}19A, P{tubP-GAL80}LL1, P{hsFLP}1, w[*]; P{UAS-mCD8::GFP.L}) (Bloomington Stock Center); Toll10b (Tl8); and msnlacZ (P{PZ}msnP1707).
The Dm-Myb cDNA rescue line was created by recombining Dm-Myb null allele MH107 with a y,w67,P{w[+mC]=UAS-Dm-Myb} transgene on the X chromosome. In addition, the Dm-Myb null allele MH107 was recombined with the dominant gain-of-function mutant hopTuml. Successful recombinants were balanced over FM7i, P{w+mC=ActGFP}JMR3 (Bloomington Stock Center) for identification of mutant larvae.
To express transgenes in the lymph gland and hemocytes, flies carrying the target genes under UAS control (the GAL4 response element) were crossed with flies carrying the GAL4-e33c enhancer trap (Brand and Perrimon 1993). To perform the flp/FRT experiments, the Dm-Myb null allele MH107 was recombined with an X chromosome containing an FRT site at 19A. A stable line for cDNA expression in Dm-Myb mutant lymph glands was then generated through a two-generation cross that consisted of MH107, FRT19A, and GAL4-e33c; this line was balanced over FM7i, P{w+mC=ActGFP}JMR3 and TM3, P{ActGFP}JMR2, Ser[1] (Bloomington Stock Center).
To generate mosaic larvae via the MARCM method, heat-shock conditions were as follows: larvae were raised at 18° and early third instar larvae were placed twice at 37° for 3 hr at 24-hr intervals. Larvae were analyzed 48 hr after the second heat shock.
Stocks were cultured on standard cornmeal, molasses, yeast, agar medium and maintained at 25°. Late wandering third instar larvae were used for all experiments, as they display maximal numbers of hemocytes in the lymph gland and in the circulation.
Immunohistochemistry:
Lymph glands from wandering third instar larvae of the appropriate genotype were carefully dissected in Schneider's medium containing 10 mm ethylenediaminetetraacetic acid (EDTA) and a protease inhibitor cocktail (Complete, Mini, EDTA-free, Roche, Indianapolis). The lymph glands were fixed to poly-lysine-coated slides in PBS containing 4% formaldehyde for 15 min. After several washes with a buffer containing PBS and 0.2% Triton X-100 (PBST), the tissue was blocked for 1 hr in PBST and 1% BSA (PBSTB) at room temperature and then incubated in PBSTB overnight at 4° with the primary antibody at the appropriate dilution [mouse anti-Myb (5E) at 1:200 (Sleeman 1993); mouse anti-BrdU, Alexa Fluor 488 conjugate at 1:20 (Molecular Probes, Eugene, OR); rabbit anti-phosphohistone H3 (PH3) at 1:500 (Upstate Biotechnologies, Lake Placid, NY); mouse anti-β-gal at 1:2000 (Promega); wheat germ agglutinin (WGA) Alexa Fluor 633 conjugate at 1:500 (Molecular Probes)]. Several washes in PBST were followed by a 2-hr incubation in PBSTB with the appropriate secondary antibody at room temperature [goat anti-mouse IgG Alexa Fluor 594 or Alexa Fluor 488; goat anti-rabbit IgG Alexa Fluor 594 or Alexa Fluor 488 diluted to 1:500 (Molecular Probes)]. The tissue was washed several times in PBST and was treated with DNase-free RNase A (2 mg/ml). The lymph glands were finally mounted in Vectashield fluorescent mounting medium (Vector Laboratories, Burlingame, CA) containing the nucleic acid stains propidium iodide (PI) or TOTO-3 (Molecular Probes). Images were obtained with a Nikon PCM confocal microscope.
Statistical analysis:
Statistical analysis of PH3-positive lymph gland hemocytes was performed using the Microsoft Excel software. Lymph glands were analyzed for each genotype and the ratio of PH3-positive hemocytes to total number of hemocytes was evaluated using Student's t-test (two-sample assuming unequal variances), with values of P < 0.05 considered as statistically significant.
Hemocyte proliferation:
Late third instar larval lymph glands were dissected in Schneider's medium and the protocol for the ABSOLUTE-S SBIP Cell Proliferation assay kit (Molecular Probes) was followed as per manufacturer's instructions with the following exceptions. Briefly, lymph glands were incubated at room temperature for 40 min in 1 ml of Schneider's medium containing 2 μl of BrdU Photolyte stock solution. Dimethyl sulfoxide (DMSO) was added to the incubation to a final concentration of 2% (v/v) and 2 μl of photolyte enhancer. The lymph glands were incubated for an additional 20 min. The tissue was fixed to poly-lysine-coated slides and washed as described above. Slides were placed on the illuminating surface of a UV light box for 10 min. Following UV treatment the lymph glands were washed with the wash buffer provided. The DNA-labeling solution was prepared according to the kit instructions and the lymph glands were incubated for 60 min at 37°. The tissue was rinsed several times with the rinse buffer provided and the antibody staining was performed as described above.
Injection and bacterial experiments:
Approximately 50 nl of AlexaFluor594-labeled fluorescent heat-killed Escherichia coli (Molecular Probes) were injected into late third instar mosaic larvae generated via the MARCM system. Injections were performed using the Picospritzer III (Parker Hannifin, Richland, MI). For bacterial challenge experiments, third instar larvae were pricked with a thin needle inoculated with a concentrated bacterial pellet (4 × 1011 cells/ml) from an overnight culture of E. coli and Micrococcus luteus bacteria.
Hemocyte collection and visualization of hemocytes:
Circulating hemocytes were collected by rupturing the larval cuticle using a pair of fine forceps. Hemocytes were collected in ice-cold Schneider's medium containing 10 mm EDTA and a protease inhibitor cocktail (Complete, Mini, EDTA-free; Roche) to prevent cell aggregation and melanization and were kept on ice until incubation with fluorescence-activated cell-sorting (FACS) probes. Crystal cells were visualized by heating the larvae for 10 min at 70°, which results in a specific blackening of these cells. Heated larvae used for sectioning were fixed for 4 hr on ice in 4% formaldehyde in PBS. The fixed larvae were then paraffin embedded, sectioned, and stained with Wright/Giemsa stain.
Flow cytometry probes:
Two probes were used for the purpose of gating hemocytes: monochlorobimane (MCB; Sigma, St. Louis) and PI (Molecular Probes). MCB is a cell-permeable, nonfluorescent probe that is conjugated to intracellular glutathione (GSH) by glutathione-S-transferases (GSTs), leading to the formation of fluorescent glutathione-S-bimane adducts (excited at 407 nm; measured with a 440/40-wavelength/bandpass-collection filter). Thus, MCB can be used as a specific probe to measure intracellular GSH levels within living cells (Herzenberg et al. 1997). PI (excited at 488 nm; measured with a 665/30 collection filter) is excluded from live cells but readily enters into dead cells through their permeable membrane and intercalates into their DNA to yield bright fluorescence. Cells were incubated with probes for 20 min at room temperature in the dark in 1 ml of staining medium consisting of RPMI 1640 medium with 2.5 mm probenecid (Sigma) at pH 7.4. Probenecid is used to inhibit efflux of glutathione-S-bimane adducts. Cells were washed once with 10 ml ice-cold staining medium and centrifuged for 5 min at 4° at 1500 rpm. Supernatant was discarded and the pellet was kept on ice and resuspended in staining medium with 2 μm PI, just before acquisition on the FACS.
Acquisition and analysis of FACS data:
Samples were acquired on a modified FACStarPlus (Becton Dickinson, Franklin Lakes, NJ) equipped with three lasers (krypton laser at 407 nm, argon laser at 488 nm, and dye laser at 595 nm) and connected to MoFlo electronics (Cytomation, Fort Collins, CO) supplemented with home-built electronics (Stanford Shared FACS Facility, Stanford, CA). Data were archived via FACS-Desk software and compensated, analyzed, and presented using FlowJo software (Tree Star, San Carlos, CA). The FACS instrument was standardized with fluorochrome-containing beads so that the fluorescence reading in each channel was automatically adjusted to a value that is maintained constant. This ensured that data obtained on different days were comparable.
Total RNA isolation and real-time PCR:
Five flies were snap frozen for each time point and total RNA was prepared according to the animal tissue isolation protocol using the RNeasy Mini kit (QIAGEN, Valencia, CA), with the addition of DNase I treatment to degrade the genomic DNA. RNA prepared from these flies was subjected to real-time RT-PCR analysis using published conditions (Schneider and Shahabuddin 2000). RT-PCR was conducted using a Bio-Rad (Richmond, CA) iCycler with TaqMan probes and rTth polymerase (Perkin-Elmer, Norwalk, CT) as per manufacturer's directions. The following primers were used: for diptericin, left primer (5′) ACC GCA GTA CCC ACT CAA TC (3′), right primer (5′) CCC AAG TGC TGT CCA TAT CC (3′), and Taqman probe 6FAM-TCC AGG GTC ACC AGA AGG TGT G-TAMRA; for drosomycin, left primer (5′) GTA CTT GTT CGC CCT CTT CG (3′), right primer (5′) CTT GCA CAC ACG ACG ACA G (3′), and Taqman probe 6FAM-TCC GGA AGA TAC AAG GGT CCC TGT G-TAMRA; and for ribosomal protein 15A, left primer (5′) TGG ACC ACG AGG AGG CTA GG (3′), right primer (5′) GTT GGT GCA TGG TCG GTG A (3′), and Taqman probe 6FAM-TGG GAG GCA AAA TTC TCG GCT TC-TAMRA.
RESULTS
Dm-Myb is required for hemocyte development and function:
Dm-Myb−/− larvae are completely devoid of visible crystal cells (Figure 1B). Crystal cells were detected by heating the larvae to 70° for 10 min, resulting in the premature melanization of the cells and making them easily visible through the cuticle of control larvae (Figure 1A) (Rizki and Rizki 1984). Visible crystal cells were also absent in Dm-Myb, Black Cell double-mutant larvae, which is of interest because the dominant Black Cell mutation causes aberrant melanization of crystal cells (data not shown). The crystal cell lineage development requires the transcription factor Lz (Lebestky et al. 2000); consistent with this requirement, lz null (lzR15) larvae are similarly devoid of visible crystal cells (Figure 1E). Sectioning through the Dm-Myb−/− larvae confirmed the complete absence of crystal cells and also revealed reduced plasmatocyte numbers compared to that of controls (Figure 1, F and G). Thus, Dm-Myb is required for the proliferation of larval hemocytes and differentiation of the crystal cell lineage.
Figure 1.—
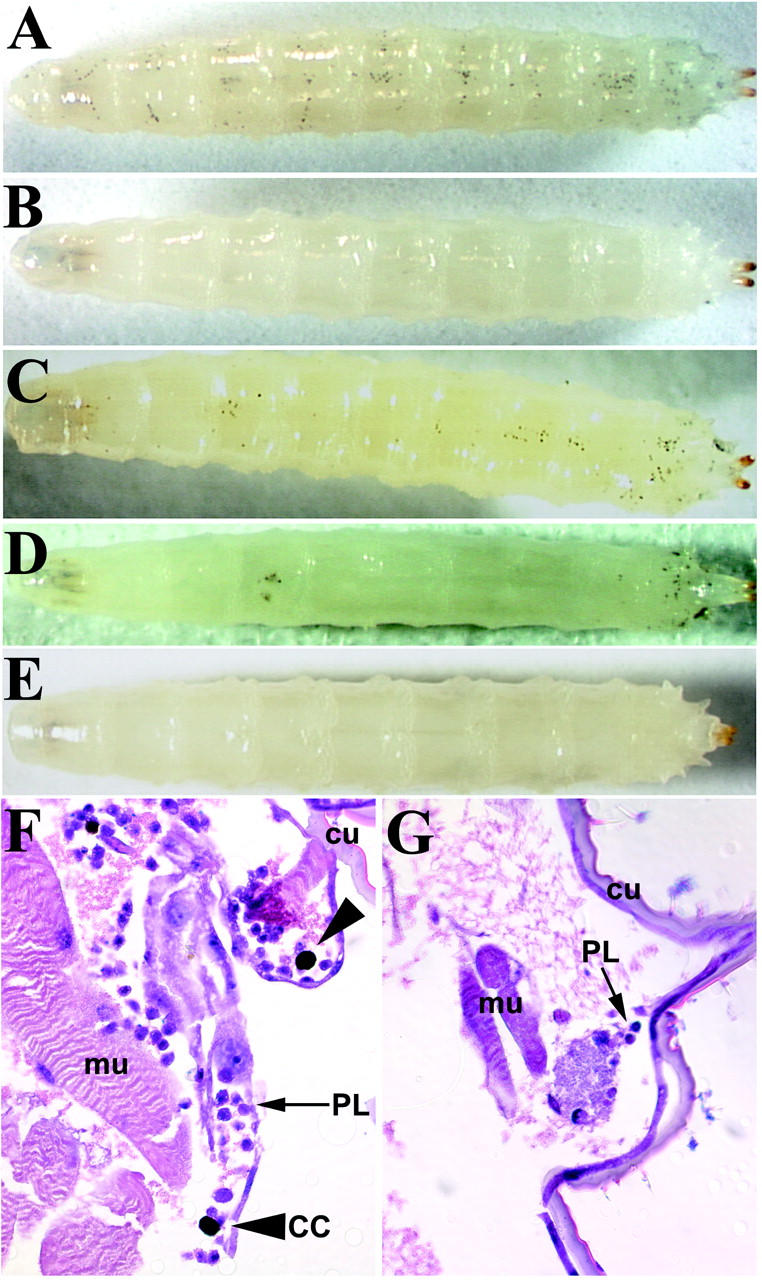
Dm-Myb−/− larvae lack crystal cells. (A–G) Third instar larvae were heated to 70° for 10 min to visualize crystal cells. (A) Control (y,w67). (B) Dm-Myb−/− (MH107). (C) Dm-Myb−/− rescue with Dm-Myb (MH107, UAS-Dm-Myb; GAL4-e33c). (D) Dm-Myb−/− rescue with vertebrate B-Myb (MH107; UAS-B-Myb/ GAL4-e33c). (E) Homozygous lozenge null (lzR15). (F and G) Heat-treated third instar larvae were paraffin embedded, sectioned, and stained with the Wright-Giemsa stain. (F) Control (y,w67); (G) Dm-Myb−/− (MH107). Large arrowheads (F) indicate darkened crystal cells; small arrows indicate (F and G) plasmatocytes. Note the decreased number of plasmatocytes in the Dm-Myb mutant (CC, crystal cell; cu, cuticle; mu, muscle; PL, plasmatocyte).
Dm-Myb−/− hemocytes demonstrate a phagocytosis defect:
Due to the severe proliferation defect of hemocytes both in the circulation and in the lymph gland (see Figure 3 below) of Dm-Myb−/− larvae, we generated mosaic animals using the flp/FRT system to further characterize the role of Dm-Myb in larval hematopoiesis. This system utilizes the flp recombinase to induce site-specific mitotic recombination at the FRT site during the G2 phase of the cell cycle leading to the generation of homozygous mutant cells (−/−) in an otherwise heterozygous animal (+/−) (Golic and Lindquist 1989; Xu and Rubin 1993). Application of this technique should facilitate the study of the Dm-Myb proliferation defect in the context of wild-type neighboring hematopoietic cells and might allow us to determine whether Dm-Myb−/− hemocytes adopt a limited repertoire of cell fates (i.e., a block in hemocyte differentiation) in the presence of a normal lymph gland environment. We choose to positively label the Dm-Myb−/− hemocytes with the fluorescent marker GFP via the MARCM system (Lee and Luo 1999) since this technique would facilitate analysis of mutant hemocytes by flow cytometry. This system has in trans to the mutant gene of interest a dominant repressor (GAL80) of GAL4-driven expression of a cell marker (UAS-mCD8a::GFP). Heat-shock-induced expression of the flp recombinase results in mitotic recombination events at FRT sites, generating homozygous mutant cells that are GFP+ due to loss of the GAL80 repressor of GAL4 activation. Through use of the MARCM system we were able to generate GFP-positive, Dm-Myb−/− hemocytes both in the lymph gland and in the hemolymph (Figure 2A). An analysis of Dm-Myb mosaic lymph glands with antibodies against the previously characterized Drosophila hemocyte markers including croquemort, a CD36-like protein involved in plasmatocyte phagocytosis (Franc et al. 1996, 1999); peroxidasin (Nelson et al. 1994); PDGF/VEGF receptor (Munier et al. 2002); and Notch did not reveal any difference between Dm-Myb+ and Dm-Myb−/− hemocytes (supplementary Figure S1 at http://www.genetics.org/supplemental/).
Figure 3.—
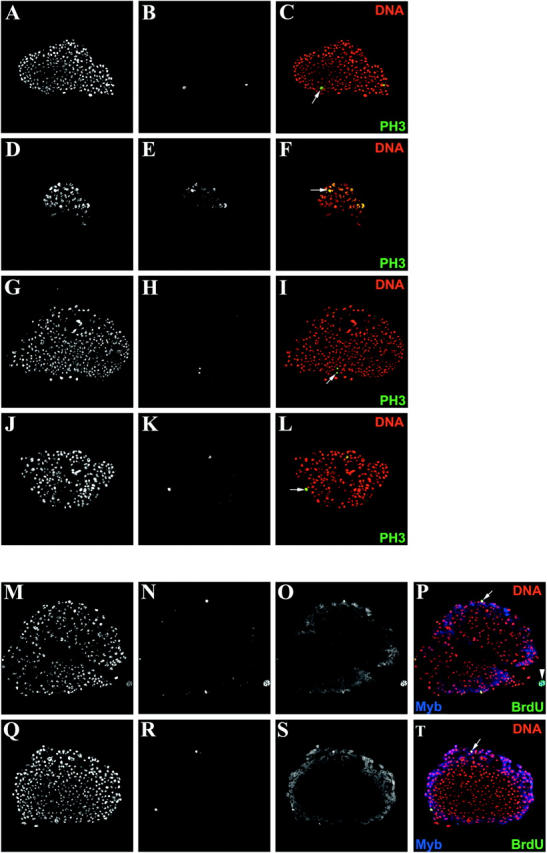
Dm-Myb and vertebrate B-Myb are able to rescue Dm-Myb−/− hematopoietic defects. (A–C) Similarly staged third instar control larvae (y,w67) lymph gland and (D–F) Dm-Myb−/− (MH107) lymph gland. (G–L) Rescue of normal numbers and morphology of PH3-positive cells in third instar larval lymph glands. (G–I) Dm-Myb−/− rescue with Dm-Myb (MH107, UAS-Dm-Myb; GAL4-e33c). (J–L) Dm-Myb−/− rescue with vertebrate B-Myb (MH107; UAS-B-Myb/GAL4-e33c). Primary third instar larval lymph gland lobes were stained with TOTO-3 for DNA (A, D, G, and J) and anti-PH3 antibody (B, E, H, and K). PH3+ cells are indicated by the arrows in the two-color composites (C, F, I, and L). (M–T) Restoration of proliferation as measured by BrdU-incorporating cells in rescued third instar lymph glands. (M–P) Dm-Myb−/− rescue with Dm-Myb (MH107, UAS-Dm-Myb; GAL4-e33c). (Q–T) Dm-Myb−/− rescue with vertebrate B-Myb (MH107; UAS-B-Myb/GAL4-e33c). Hemocytes were stained with TOTO-3 for DNA (M and Q), anti-BrdU antibody (N and R), and anti-Myb antibody (O and S). Large arrowhead (P) indicates pericardiocyte; small arrows (P and T) indicate BrdU+ and Dm-Myb+ and B-Myb+ hemocytes, respectively.
Figure 2.—

Dm-Myb−/− hemocytes are defective in phagocytosis. (A) A mosaic third instar larvae with the Dm-Myb−/− mutant circulating hemocytes labeled with mCD8::GFP fusion protein (arrow) using the MARCM system (MH107, FRT19A/hs-flp, tub-gal80, FRT19A; UASmCD8::GFP; GAL4-e33c). Note the GFP+ lymph glands indicated by the arrowhead. (B) A flow cytometry density plot with the measurement of heat-killed E. coli labeled with Alexa Fluor594 on the y-axis and GFP fluorescence on the x-axis. Hemocytes were collected from injected mosaic larvae generated using the MARCM system. The four populations of hemocytes correspond to Dm-Myb+ (GFP−) and Dm-Myb−/− (GFP+) with or without fluorescent E. coli. (C) To assay the phagocytic function of the two populations (GFP+ vs. GFP−), ratios of hemocytes positive for uptake of E. coli to hemocytes negative for uptake were generated for GFP+ hemocytes and for GFP− hemocytes. A comparison of these ratios between the two populations revealed a phagocytosis defect in the Dm-Myb−/− hemocytes in two independent experiments, demonstrating that Dm-Myb−/− hemocytes (green bars) are 57–68% defective in phagocytosis compared to that of the Dm-Myb+ hemocytes (red bars).
Using a flow cytometry staining strategy that facilitates the discrimination of Drosophila larval hemocytes from contaminating events acquired during data collection (Tirouvanziam et al. 2004), we were able to measure phagocytosis in living hemocytes. Phagocytosis was tested by the injection of controlled volumes of fluorescently labeled, heat-killed E. coli into Dm-Myb−/− mosaic larvae generated using the MARCM system. The flow cytometry staining strategy combined PI, a cell-impermeable DNA dye that selectively labels dead cells, and MCB, a cell-permeable probe for the tripeptide antioxidant GSH. Proper intracellular GSH levels are required for cell survival; thus, dead cells and debris lack GSH whereas live hemocytes are highly positive for MCB (GSHhi) and negative for PI (PI−). After the application of the GSHhi PI− gate, the two populations of hemocytes [Dm-Myb+ (GFP−) and Dm-Myb− (GFP+)] from MARCM mosaic larvae were compared in their ability to take up heat-killed E. coli labeled with the Alexa Fluor594 dye (Figure 2B). Calculating the ratio of hemocytes positive for uptake of E. coli to negative hemocytes between the two populations (GFP+ vs. GFP−) revealed a phagocytosis defect in the Dm-Myb−/− hemocytes. A comparison of these ratios reveals that Dm-Myb−/− hemocytes demonstrate a 57–68% decrease in phagocytosis compared to that of Dm-Myb+ cells as determined by two independent experiments (Figure 2C). Our estimation of hemocytes positive for phagocytosis of labeled E. coli may also include cells with bacteria bound to the cell surface but not internalized. Trypan blue is commonly used to quench the fluorescence of nonphagocytosed particles (Elrod-Erickson et al. 2000); however, this approach could not be used in these experiments since this dye interfered with other probes used in the FACS analysis.
Dm-Myb and vertebrate B-Myb expression restore lymph gland hemocyte proliferation and differentiation:
Consistent with the low number of plasmatocytes observed in whole larval sections (Figure 1G), the lymph glands of Dm-Myb−/− animals are undersized relative to those of control animals in similarly staged third instar larvae (Figure 3), the period of greatest lymph gland hemocyte proliferation (Lanot et al. 2001). Furthermore, the reduced number of hemocytes contained within the lymph glands of Dm-Myb−/− larvae have aberrant nuclei as revealed by DNA staining (Figure 3D). Interestingly, use of an antibody directed against condensed chromatin, PH3, revealed a substantial increase of Dm-Myb−/− hemocytes positive for this marker of mitosis (Figure 3, E and F). Elevated PH3 staining is consistent with previous experiments in which loss of Dm-Myb resulted in abnormal mitoses and increased PH3 staining in larval brain and imaginal discs (Manak et al. 2002). The undersized lymph glands of Dm-Myb mutant larvae in combination with elevated levels of PH3 staining in Dm-Myb−/− hemocytes suggest a cell cycle block and indicate that Dm-Myb is required for larval lymph gland hemocyte proliferation.
To determine the functional redundancy between the vertebrate Myb genes and Dm-Myb, rescue of Dm-Myb−/− larval lymph glands was performed via GAL4-mediated expression of a transgenic cDNA placed under the control of a UAS element. Expression of a transgene was accomplished using a previously characterized lymph gland and pericardiocyte enhancer trap-generated GAL4 driver (GAL4-e33c; Harrison et al. 1995), and expression of Dm-Myb resulted in the rescue of circulating crystal cells in Dm-Myb−/− larvae (Figure 1C). Expression of vertebrate B-Myb using the GAL4-e33c driver also restored circulating crystal cells in Dm-Myb−/− larvae (Figure 1D). Furthermore, lymph gland crystal cells were restored in Dm-Myb−/− larvae as detected by Lz immunolocalization in UAS-Dm-Myb rescued and in UAS-B-Myb rescued lymph glands (supplementary Figure S2 at http://www.genetics.org/supplemental/). However, expression of vertebrate A-Myb or c-Myb via the GAL4-e33c driver failed to rescue circulating crystal cells. Indeed, expression of these transgenes with the same driver was not compatible with viability. This is consistent with expression of vertebrate Myb transgenes in the eye of Drosophila; A-Myb and c-Myb expression leads to an aberrant eye phenotype whereas expression of B-Myb and Dm-Myb shows no visible phenotype (data not shown).
Directed expression of UAS-Dm-Myb (Figure 3, H and I) and UAS-B-Myb (Figure 3, K and L) with the GAL4-e33c driver in Dm-Myb−/− lymph glands restored normal levels of PH3 staining in all examined lymph glands (compare to PH3 staining of control lymph glands in Figure 3B and to Dm-Myb−/− lymph glands in Figure 3E). No statistical difference was found in the mean ratio of PH3-positive hemocytes to total number of hemocytes between control lymph glands (1.9 ± 0.4%, n = 8) and UAS-Dm-Myb and UAS-B-Myb rescued lymph glands (2.2 ± 0.9%, n = 7 and 1.1 ± 0.8%, n = 5, respectively). The mean ratio of PH3-positive hemocytes to total number of hemocytes was found to be significantly higher (0.02 > P > 0.01) between Dm-Myb−/− mutant lymph glands (22.4 ± 10.8%, n = 5) and control lymph glands, UAS-Dm-Myb rescued lymph glands, and UAS-B-Myb rescued lymph glands.
In addition, cell proliferation was restored as measured by the presence of BrdU-incorporating cells in UAS-Dm-Myb rescued lymph glands (PFigure 3, N and P) and UAS-B-Myb rescued lymph glands (Figure 3, R and T). Furthermore, colocalization of Dm-Myb and B-Myb proteins with BrdU was observed in mitotically cycling hemocytes (Figure 3, N–P and R–T). In addition, Dm-Myb protein and BrdU also colocalize in pericardiocytes (Figure 3, Q–S), large polyploid cells whose nuclei undergo endoreduplication without mitosis. These cells flank the dorsal vessel and are interspersed among the lymph gland lobes (Kambysellis and Wheeler 1972). Together, these results implicate Dm-Myb in a functional role during or shortly after S-phase in both mitotic and endocycling cells, consistent with previous results in larval brain cells and endocycling larval fat body cells (Manak et al. 2002). What's more, B-Myb can restore normal proliferation of hemocytes and differentiation of the crystal cell lineage in Dm-Myb−/− larvae. Note that rescue of hemocytes in the lymph gland Dm-Myb mutant larvae by GAL4-e33c-driven expression of UAS-Dm-Myb and UAS-B-Myb results in a restoration of normal nuclear morphology as determined by nuclear DNA staining (Figure 3, G, J, M, and Q). Thus, the differentiation of crystal cell lineage in the larval lymph gland and in circulation requires normal proliferation, which is Dm-Myb dependent, and vertebrate B-Myb can complement this function of Dm-Myb. The lethality associated with the expression of A-Myb and c-Myb indicates that little functional overlap between these genes and B-Myb and Dm-Myb is likely and supports the neofunctionalization of an ancestral A-Myb/c-Myb gene following the first duplication.
Dm-Myb is required for JAK/STAT pathway-mediated hemocyte hyperproliferation:
To establish whether Dm-Myb is generally required for proliferation of hemocytes, epistasis experiments were conducted using a Dm-Myb null mutation and dominant substitution mutations of the Toll receptor (Tl10b) and the Jak kinase, hopscotch (hopTuml); dominant gain-of-function mutations in these genes result in hyperactivation of their respective pathways, leading to hemocyte overproliferation and abnormal lamellocyte differentiation (Harrison et al. 1995; Luo et al. 1995; Qiu et al. 1998). The experiments shown above (Figure 1, B and C) indicate that proper differentiation of normal crystal cells requires Dm-Myb-dependent proliferation. To determine whether Dm-Myb is required for the dysregulated overproliferation and differentiation phenotypes of Tl10b and hopTuml mutants, double-mutant larvae lacking Dm-Myb in conjunction with these dominant alleles of Toll and hopscotch were generated.
We found that, in addition to an overproliferation of plasmatocytes in the primary lymph gland lobes, the secondary lymph gland lobe hemocytes aberrantly differentiate into lamellocytes in hopTuml mutants (Figure 4, E–H). It is thought that the normally smaller secondary lymph gland lobes serve as a reservoir of undifferentiated prohemocytes (Lanot et al. 2001); however, in hopTuml larvae the secondary lobes enlarge with concomitant abnormal differentiation of lamellocytes. Hemocytes in the secondary lobes of control (yw67; Figure 4, A–D) and Dm-Myb−/− (Figure 4, I–L) larvae do not demonstrate abnormal lamellocyte differentiation as determined by the absence of an increased expression of lamellocyte enhancer-trap marker (msnlacZ) over the background staining seen in the control. While hemocytes in the secondary lymph gland lobes of hopTuml, Dm-Myb−/− double mutants show an increased expression of the lamellocyte enhancer-trap marker (msnlacZ), these β-gal-positive cells fail to overproliferate and do not adopt the flattened shape characteristic of differentiated lamellocytes (Figure 4, M–P). The lamellocyte enhancer-trap marker (msnlacZ) is a lacZ reporter insertion in the misshapen gene, a component of the JNK kinase pathway. These results suggest that this stress pathway may be upregulated in Dm-Myb−/− hemocytes in the absence of lamellocyte differentiation.
Figure 4.—

Dm-Myb mutation is epistatic to the hopTuml mutation. Comparative analysis of lymph gland hemocyte proliferation and lamellocyte differentiation in secondary lymph gland lobes of third instar larvae is shown. (A–D) Control with lamellocyte enhancer-trap (y,w67; msnlacZ). (E–H) hopTuml with lamellocyte enhancer-trap (hopTuml; msnlacZ). Arrows (H) indicate β-gal-positive lamellocytes. (I–L) Dm-Myb mutant with lamellocyte enhancer-trap (MH107; msnlacZ). (M–P) hopTuml, Dm-Myb−/− double mutants with lamellocyte enhancer-trap (hopTuml, MH107; msnlacZ). Arrows (P) indicate β-gal-positive hemocytes. Cells were stained with TOTO-3 for DNA (A, E, I, and M), anti-β-gal antibody (B, F, J, and N), and anti-PH3 antibody (C, G, K, and O).
Furthermore, the double-mutant (hopTuml, MH107) hemocytes exhibit amorphous nuclear DNA and PH3 staining similar to that of the Dm-Myb single mutant (compare Figure 4, M and O, with Figure 4, I and K), suggesting that Dm-Myb is epistatic to hopTuml. The mean ratios of PH3-positive hemocytes to total number of hemocytes between control secondary lymph glands (1.9 ± 0.6%, n = 6) and hopTuml secondary lobes (1.9 ± 0.1%, n = 7) were statistically indistinguishable. Similarly, there was no significant difference between the mean ratio of PH3-positive hemocytes to total number of hemocytes in the secondary lymph gland lobes of the Dm-Myb−/− single mutant (24.2 ± 10.5%, n = 5) and the hopTuml, Dm-Myb−/− double mutants (16.5 ± 5.7%, n = 12). However, there was highly significant difference (P < 0.001) in the mean ratio of PH3-positive hemocytes to total number of hemocytes in the secondary lymph gland between hopTuml, Dm-Myb−/− double mutants and both control and hopTuml single mutants. In summary, an activated JAK/STAT pathway cannot drive the proliferation of hemocytes in the absence of Dm-Myb.
Dm-Myb is required for proliferation driven by the Toll pathway:
Through the use of a dominant mutation in the Toll receptor (Tl10b) and a lamellocyte enhancer-trap marker (msnlacZ; Braun et al. 1997), we demonstrated that WGA binding identifies abnormal lamellocyte differentiation in the secondary lymph gland lobes of Tl10b larvae (Figure 5, F–H) but not in control (Figure 5, B–D), Dm-Myb mutant (Figure 5 J–L), or Tl10b, Dm- Myb−/− double mutants (PFigure 5, O and P). In Tl10b larvae the secondary lobes dramatically enlarge with concomitant abnormal differentiation of lamellocytes. The binding of WGA has previously been shown to be a marker of differentiation and activation of lamellocytes in Drosophila (Nappi and Silvers 1984). Interestingly, the lymph gland hemocytes of Tl10b, Dm-Myb−/− double mutants fail to overproliferate and instead exhibit the aberrant PH3 staining characteristic of Dm-Myb−/− animals, suggesting that Dm-Myb is epistatic to Tl10b (Figure 5, M and N). Note that Dm-Myb−/− hemocytes express increased levels of the lamellocyte enhancer-trap marker (msnlacZ; Figure 5J); however, these high-expressing cells do not demonstrate increased levels of WGA binding (Figure 5K). Together these results demonstrate that an activated Toll pathway cannot drive the proliferation of hemocytes in the absence of Dm-Myb.
Figure 5.—
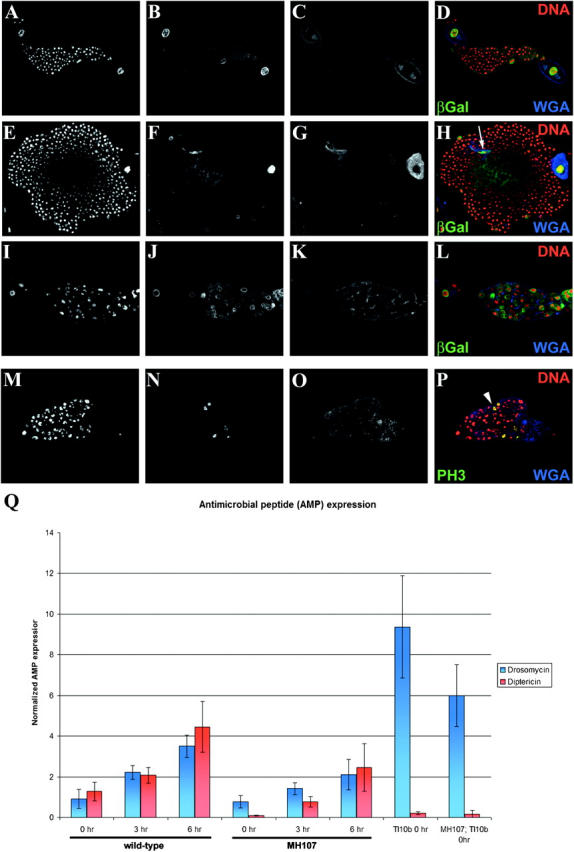
Toll10b, Dm-Myb−/− double-mutant secondary lymph gland hemocytes fail to overproliferate and abnormally differentiate into lamellocytes. (A–D) Control with lamellocyte enhancer-trap (y,w67; msnlacZ). (E–H) Toll10b mutant with lamellocyte enhancer-trap (Tl10b/msnlacZ). WGA+ and enhancer-trap (msnlacZ)-positive lamellocytes are indicated by the arrow (H). (I–L) Dm-Myb mutant with lamellocyte enhancer-trap (MH107; msnlacZ). (M–P) Toll10b, Dm-Myb−/− double mutant (MH107; Tl10b). Increased PH3-labeled hemocytes are indicated with an arrowhead (P) in the Toll10b, Dm-Myb−/− double mutant. Third instar secondary larval lymph gland lobes were stained with PI (A, E, I, and M), wheat germ agglutinin (WGA; C, G, K, and O) and anti-β-gal antibody (B, F, and J) or anti-PH3 antibody (N). (Q) Drosomycin and Diptericin are expressed in Dm-Myb mutant larvae in response to bacterial challenge. A needle inoculated with E. coli and M. luteus was used to prick late third instar larvae and the induction of Drosomycin (blue bars) and Diptericin (red bars) expression was measured by real-time RT-PCR analysis. Drosomycin and Diptericin transcript levels were normalized to the expression of the ribosomal protein 15a transcript in each sample. (Columns 1–3) Control (yw67) Drosomycin and Diptericin expression over 0, 3, and 6 hr of bacterial challenge. (Columns 4–6) Dm-Myb mutant (MH107) Drosomycin and Diptericin expression over 0, 3, and 6 hr of bacterial challenge. (Column 7) Unchallenged Toll10b dominant mutant Drosomycin and Diptericin expression. (Column 8) Unchallenged Toll10b, Dm-Myb−/− double-mutant (MH107; Tl10b) Drosomycin and Diptericin expression.
Dm-Myb is not required for Toll pathway-induced antimicrobial peptide gene expression:
A functional Toll pathway is required for the inducible expression of the antimicrobial peptide gene, Drosomycin, which is also constitutively expressed in Toll10b mutant larvae in the absence of immune challenge (Lemaitre et al. 1996). Through the use of real-time PCR analysis, the kinetics of Drosomycin and Diptericin expression were found to be similar in bacterially challenged control and Dm-Myb−/− larvae (Figure 5Q). Furthermore, Toll10b, Dm-Myb−/− double-mutant larvae constitutively express Drosomycin at comparable levels to those of Toll10b mutant animals. The differences in absolute levels of transcriptional response between Dm-Myb−/− and control larvae are likely due to the late third instar larval/prepupal lethality and underdeveloped fat body (the primary site of antimicrobial peptide synthesis) of the Dm-Myb mutants. Nevertheless, a functional Drosomycin transcriptional response in both Dm-Myb null mutant and Tl10b, Dm-Myb−/− double mutant larvae demonstrates that Dm-Myb is not required as a transcription factor for expression of this intensively studied downstream target of Toll pathway signaling. Rather, Dm-Myb appears to act as a more general factor in a separate, possibly nontranscriptional, pathway required for hemocyte overproliferation and the subsequent lamellocyte differentiation in response to hyperactivated Tl10b and hopTuml signaling.
DISCUSSION
The results shown here illustrate the functional conservation of B-Myb and Dm-Myb in restoring both the proliferation and differentiation capacity to Dm-Myb−/− larval lymph gland hemocytes. Phylogenetic analysis of the three vertebrate Myb proteins indicates that A-Myb and c-Myb arose via a more recent gene duplication event with the common ancestor of A-Myb and c-Myb resulting from the duplication of a B-Myb/Dm-Myb-like gene (Ganter and Lipsick 1999; Simon et al. 2002). In accordance with the classical model of duplicate gene preservation, the ancestral A-Myb/c-Myb gene likely acquired alterations in the coding region that led to a novel gene function, thus preserving the duplicate gene from purifying selection. Incorporating the phylogenetic data and the complementation experiments, we propose that the lethality due to the expression of A-Myb or c-Myb in this system indicates the ancestral A-Myb/c-Myb gene acquired a new function (neofunctionalization) subsequent to initial duplication from an ancestral B-Myb-like gene (Figure 6). Consistent with a neofunctional origin, both A-Myb and c-Myb possess a transcriptional activation domain not present in B-Myb or Dm-Myb. Analysis of vertebrate A-, B- and c-Myb sequences using a maximum-likelihood-based method for identifying evolutionarily constrained regions identified the transcriptional activation domain as a region under constraint in A- and c-Myb proteins but not B-Myb (Simon et al. 2002). Consistent with a duplicate gene under new and distinct selective constraints, the evolutionary constraint of this region of A- and c-Myb proteins suggests this region is functionally important in these paralogs, whereas this region is less important in B-Myb and the invertebrate Myb proteins.
Figure 6.—

Evidence for neofunctionalization following gene duplication within the vertebrate Myb gene family. Left, a consensus tree illustrating the phylogenetic relationship of animal Myb proteins; invertebrate Myb sequences are shown in black, the B-Myb clade is shown in yellow, the A-Myb clade is shown in cyan, and the c-Myb clade is shown in magenta. The pink circles indicate putative gene duplication events. Center, a cartoon representing the domain structure of the animal Myb proteins; the repeated Myb DNA-binding domain is shown in blue, the negative regulatory domain is shown in green, and the conserved transcriptional activation domain is shown in red. The ancestral A-Myb/c-Myb gene acquired the transcriptional activation domain (red, center) and likely a new function (neofunctionalization) between the two gene duplication events (pink circles, left). Right, the ability of each paralog to rescue Dm-Myb−/− hemocyte defects.
We propose that B-Myb and Dm-Myb do not function directly as traditional transcriptional activators but share conserved and essential roles in S-phase that facilitate the proliferation and subsequent differentiation of larval hemocytes. The co-immunolocalization of nuclear Dm-Myb and B-Myb proteins with BrdU was observed in rescue experiments in both mitotically dividing hemocytes and endocycling pericardiocytes for Dm-Myb. The absence of crystal cells in the Dm-Myb−/− larvae is consistent with a general requirement of Dm-Myb for proliferation. The crystal cell population may be particularly sensitive to perturbations in mitotic cell proliferation since these cells constitute a minor population of hemocytes (typically <5% of the total hemocyte population; Lanot et al. 2001). It seems likely that differentiation of crystal cells requires normal levels of proliferation and that larval hemocyte proliferation is Dm-Myb dependent, with vertebrate B-Myb functionally complementing this requirement for Dm-Myb.
In addition to a requirement for Dm-Myb in normal hemocyte proliferation and differentiation, epistasis experiments with dominant gain-of-function mutations in the Toll and JAK/STAT pathway suggest a general function for Dm-Myb and by implication B-Myb during the innate immune response. Dm-Myb is required for the phenotypes of hemocyte overproliferation and abnormal differentiation into lamellocytes that result from the hyperactivation of both the Toll and the JAK-STAT pathways. In support of a nontranscriptional role for Dm-Myb, the expression of Drosomycin, a known downstream target of Toll pathway signaling, in bacterially challenged Dm-Myb null mutant and in unchallenged Tl10b, Dm-Myb−/− double-mutant larvae argues against Dm-Myb acting as a transcriptional activator in this aspect of Toll pathway signaling. However, a transcriptional role for Dm-Myb in the expression of other unknown downstream targets of Toll signaling remains possible.
The phagocytosis defect uncovered using the MARCM approach is also consistent with a role for Dm-Myb in hemocyte proliferation. Through use of the MARCM system we have demonstrated that Dm-Myb−/− hemocytes exhibit a cell-autonomous defect in phagocytosis in a wild-type hemolymph environment. This result suggests that Dm-Myb−/− hemocytes are not properly developed and may be blocked in their differentiation. Our finding that Dm-Myb−/− hemocytes are impaired in phagocytosis in vivo is consistent with an in vitro assay using cultured Drosophila S2 cells, in which Dm-Myb was identified among 34 genes in an RNAi-based screen for defective phagocytosis (Ramet et al. 2002). The phagocytosis defect observed in the MARCM-generated Dm-Myb−/− hemocytes may be due to the inability of these cells to progress appropriately through the cell cycle. For example, appropriate progression through the cell cycle is thought to be required for proper differentiation of bipotent progenitor CD4+ helper T-cells in mammals (Mullen et al. 2001). The expression of lineage-restricted genes and silencing of alternative differentiated states has been shown to be cell cycle dependent in these cells. In particular, progenitor cells that are unable to cycle remain undifferentiated and bipotent even when exposed to maturation signals. A similar cell cycle-dependent differentiation mechanism has been observed in the slime mold Dictyostelium discoideum, where the tendency of single amoebae to differentiate into either prespore or prestalk cells occurs via a cell-autonomous mechanism dependent on the position of each cell within the cycle when exposed to an extrinsic developmental cue (Gomer and Firtel 1987).
The rescue of the crystal cell lineage and the restoration of proliferation and appropriate levels of PH3-positive hemocytes in the lymph gland of Dm-Myb−/− larvae by the expression of vertebrate B-Myb, but neither A-Myb nor c-Myb, indicates that Dm-Myb and B-Myb are functional orthologs whereas A-Myb and c-Myb have acquired new functions. Application of Drosophila genetics to screen for Drosophila genes that modify the phenotype of A-Myb and c-Myb expression in other tissues such as the eye would facilitate the identification of the genetic and biochemical pathways that resulted in the positive selection of the ancestral A-Myb/c-Myb gene in a preduplicate genome. We propose that duplication of a B-Myb-like ancestral gene was followed by the acquisition of the central activation domain in one gene copy, thereby imparting a neomorphic function to an A-Myb/c-Myb-like gene (Figure 6). The latter gene then duplicated, resulting in the genesis of the closely related A-Myb and c-Myb genes that presumably were retained due to further mutation and subfunctionalization. Although difficult to test in Drosophila, the differing tissue-specific expression patterns but conserved function as transcriptional activators suggests the extant A-Myb and c-Myb genes may have been preserved due to the partitioning of expression domains of the two genes subsequent to the second gene duplication event. It would therefore be of interest to test the functional redundancy of A-Myb and c-Myb experimentally in a mouse model.
While subfunctionalization of the ancestral A- and c-Myb gene likely contributed to the survival of these duplicate genes, it is tempting to speculate about the relationship between the role of B-Myb in proliferation and that of A-Myb and c-Myb in transcriptional regulation. Both B-Myb and c-Myb mRNA and protein expression begin in late G1 and continue into S-phase and like other S-phase genes such as cdc2, cyclinA, thymidylate synthetase, ribonucleotide reductase, and E2F-1, the B-Myb and c-Myb promoters contain binding sites for E2F, a family of transcription factors responsible for negatively regulating gene expression in G0 and early G1 (Torelli et al. 1985; Lipsick and Boyle 1987; Lam et al. 1992; Lam and Watson 1993; Zwicker et al. 1996; Campanero et al. 1999). Potentially the cell cycle regulation of both these genes is an important vestigial remnant of the ancestral function of Myb proteins, with the acquisition of the transcriptional activation domain leading to neofunctionalization of the ancestral A-Myb/c-Myb and a role as transcriptional activator. Consistent with a bricolage model of evolution, both A-Myb and c-Myb are expressed highly in and may be required for maintaining the proliferation of immature transiently amplifying cells prior to differentiation in a variety of tissues and cells such as the skin epithelium, the colon, testis, and during hematopoiesis (Queva et al. 1992; Sleeman 1993; Amaravadi and King 1994; Mettus et al. 1994; Trauth et al. 1994; Sitzmann et al. 1995). The findings reported here support the establishment of Drosophila as model for studying the evolution and functional divergence of other dispersed multigene families that arose during vertebrate evolution. In particular, many vertebrate transcription factor families appear to have arisen by gene duplication and divergence that resulted in highly conserved DNA-binding domains fused to much more rapidly evolving regulatory domains, as is the case in the Myb gene family.
Acknowledgments
We thank David Schneider for instruction and use of the bacterial injection setup and real-time PCR analysis of antimicrobial peptide transcript levels, members of the Lipsick laboratory and Ranjiv Khush for fruitful discussions, and Chao Kung Chen for initiating studies of larval hematopoiesis in Dm-Myb mutants. We also thank Marilyn Masek (Stanford Histology Research Laboratory) for the sectioning and staining of the larvae; the Bloomington Stock Center for numerous Drosophila strains; and M. Meister, U. Banerjee, and K. Anderson for stocks and antibodies. This work was supported by National Institutes of Health grants R01 CA56509 (J.S.L.), R01 CA90307 (J.S.L.), and EB000231 (R.T. and L.A.H.).
References
- Abi-Rached, L., A. Gilles, T. Shiina, P. Pontarotti and H. Inoko, 2002. Evidence of en bloc duplication in vertebrate genomes. Nat. Genet. 31: 100–105. [DOI] [PubMed] [Google Scholar]
- Amaravadi, L., and M. W. King, 1994. Characterization and expression of the Xenopus c-Myb homolog. Oncogene 9: 971–974. [PubMed] [Google Scholar]
- Amores, A., A. Force, Y. L. Yan, L. Joly, C. Amemiya et al., 1998. Zebrafish hox clusters and vertebrate genome evolution. Science 282: 1711–1714. [DOI] [PubMed] [Google Scholar]
- Bailey, G. S., R. T. Poulter and P. A. Stockwell, 1978. Gene duplication in tetraploid fish: model for gene silencing at unlinked duplicated loci. Proc. Natl. Acad. Sci. USA 75: 5575–5579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beall, E. L., J. R. Manak, S. Zhou, M. Bell, J. S. Lipsick et al., 2002. Role for a Drosophila Myb-containing protein complex in site-specific DNA replication. Nature 420: 833–837. [DOI] [PubMed] [Google Scholar]
- Brand, A., and N. Perrimon, 1993. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118: 401–415. [DOI] [PubMed] [Google Scholar]
- Braun, E. L., and E. Grotewold, 1999. Newly discovered plant c-myb-like genes rewrite the evolution of the plant myb gene family. Plant Physiol. 121: 21–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun, A., B. Lemaitre, R. Lanot, D. Zachary and M. Meister, 1997. Drosophila immunity: analysis of larval hemocytes by P-element-mediated enhancer trap. Genetics 147: 623–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campanero, M. R., M. Armstrong and E. Flemington, 1999. Distinct cellular factors regulate the c-myb promoter through its E2F element. Mol. Cell. Biol. 19(12): 8442–8450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho, N. K., L. Keyes, E. Johnson, J. Heller, L. Ryner et al., 2002. Developmental control of blood cell migration by the Drosophila VEGF pathway. Cell 108: 865–876. [DOI] [PubMed] [Google Scholar]
- Daga, A., C. A. Karlovich, K. Dumstrei and U. Banerjee, 1996. Patterning of cells in the Drosophila eye by Lozenge, which shares homologous domains with AML1. Genes Dev. 10: 1194–1205. [DOI] [PubMed] [Google Scholar]
- Dearolf, C. R., 1998. Fruit fly “leukemia.” Biochim. Biophys. Acta 1377: M13–M23. [DOI] [PubMed] [Google Scholar]
- Dias, A. P., E. L. Braun, M. D. McMullen and E. Grotewold, 2003. Recently duplicated maize R2R3 Myb genes provide evidence for distinct mechanisms of evolutionary divergence after duplication. Plant Physiol. 131: 610–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvic, B., J. A. Hoffmann, M. Meister and J. Royet, 2002. Notch signaling controls lineage specification during Drosophila larval hematopoiesis. Curr. Biol. 12: 1923–1927. [DOI] [PubMed] [Google Scholar]
- Elrod-Erickson, M., S. Mishra and D. Schneider, 2000. Interactions between the cellular and humoral immune responses in Drosophila. Curr. Biol. 10: 781–784. [DOI] [PubMed] [Google Scholar]
- Evans, C. J., V. Hartenstein and U. Banerjee, 2003. Thicker than blood: conserved mechanisms in Drosophila and vertebrate hematopoiesis. Dev. Cell 5: 673–690. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick, C. A., N. V. Sharkov, G. Ramsay and A. L. Katzen, 2002. Drosophila myb exerts opposing effects on S phase, promoting proliferation and suppressing endoreduplication. Development 129: 4497–4507. [DOI] [PubMed] [Google Scholar]
- Force, A., M. Lynch, F. B. Pickett, A. Amores, Y. L. Yan et al., 1999. Preservation of duplicate genes by complementary, degenerative mutations. Genetics 151: 1531–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franc, N. C., J. L. Dimarcq, M. Lagueux, J. Hoffmann and R. A. Ezekowitz, 1996. Croquemort, a novel Drosophila hemocyte/macrophage receptor that recognizes apoptotic cells. Immunity 4: 431–443. [DOI] [PubMed] [Google Scholar]
- Franc, N. C., P. Heitzler, R. A. Ezekowitz and K. White, 1999. Requirement for croquemort in phagocytosis of apoptotic cells in Drosophila. Science 284: 1991–1994. [DOI] [PubMed] [Google Scholar]
- Fung, S. M., G. Ramsay and A. L. Katzen, 2002. Mutations in Drosophila myb lead to centrosome amplification and genomic instability. Development 129: 347–359. [DOI] [PubMed] [Google Scholar]
- Gallardo, M. H., J. W. Bickham, R. L. Honeycutt, R. A. Ojeda and N. Kohler, 1999. Discovery of tetraploidy in a mammal. Nature 401: 341. [DOI] [PubMed] [Google Scholar]
- Ganter, B., and J. S. Lipsick, 1999. Myb and oncogenesis. Adv. Cancer Res. 76: 21–60. [DOI] [PubMed] [Google Scholar]
- Golic, K. G., and S. Lindquist, 1989. The FLP recombinase of yeast catalyzes site-specific recombination in the Drosophila genome. Cell 59: 499–509. [DOI] [PubMed] [Google Scholar]
- Gomer, R. H., and R. A. Firtel, 1987. Cell-autonomous determination of cell-type choice in Dictyostelium development by cell-cycle phase. Science 237: 758–762. [DOI] [PubMed] [Google Scholar]
- Hanratty, W. P., and J. S. Ryerse, 1981. A genetic melanotic neoplasm of Drosophila melanogaster. Dev. Biol. 83: 238–249. [DOI] [PubMed] [Google Scholar]
- Harrison, D. A., R. Binari, T. S. Nahreini, M. Gilman and N. Perrimon, 1995. Activation of a Drosophila Janus kinase (JAK) causes hematopoietic neoplasia and developmental defects. EMBO J. 14: 2857–2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzenberg, L. A., S. C. De Rosa, J. G. Dubs, M. Roederer, M. T. Anderson et al., 1997. Glutathione deficiency is associated with impaired survival in HIV disease. Proc. Natl. Acad. Sci. USA 94: 1967–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland, P. W., 1999. Gene duplication: past, present and future. Semin. Cell Dev. Biol. 10: 541–547. [DOI] [PubMed] [Google Scholar]
- Hughes, M. K., and A. L. Hughes, 1993. Evolution of duplicate genes in a tetraploid animal, Xenopus laevis. Mol. Biol. Evol. 10: 1360–1369. [DOI] [PubMed] [Google Scholar]
- Kambysellis, M. P., and M. R. Wheeler, 1972. Banded polytene chromosomes in pericardial cells of Drosophila. J. Hered. 63: 214–215. [DOI] [PubMed] [Google Scholar]
- Katzen, A. L., and J. M. Bishop, 1996. myb provides an essential function during Drosophila development. Proc. Natl. Acad. Sci. USA 93: 13955–13960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzen, A. L., J. Jackson, B. P. Harmon, S. M. Fung, G. Ramsay et al., 1998. Drosophila myb is required for the G2/M transition and maintenance of diploidy. Genes Dev. 12: 831–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellis, M., B. W. Birren and E. S. Lander, 2004. Proof and evolutionary analysis of ancient genome duplication in the yeast Saccharomyces cerevisiae. Nature 428: 617–624. [DOI] [PubMed] [Google Scholar]
- Kranz, H., K. Scholz and B. Weisshaar, 2000. c-MYB oncogene-like genes encoding three MYB repeats occur in all major plant lineages. Plant J. 21: 231–235. [DOI] [PubMed] [Google Scholar]
- Lam, E. W., and R. J. Watson, 1993. An E2F-binding site mediates cell-cycle regulated repression of mouse B-myb transcription. EMBO J. 12: 2705–2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam, E. W., C. Robinson and R. J. Watson, 1992. Characterization and cell cycle-regulated expression of mouse B-myb. Oncogene 7: 1885–1890. [PubMed] [Google Scholar]
- Lanot, R., D. Zachary, F. Holder and M. Meister, 2001. Postembryonic hematopoiesis in Drosophila. Dev. Biol. 230: 243–257. [DOI] [PubMed] [Google Scholar]
- Lebestky, T., T. Chang, V. Hartenstein and U. Banerjee, 2000. Specification of Drosophila hematopoietic lineage by conserved transcription factors. Science 288: 146–149. [DOI] [PubMed] [Google Scholar]
- Lebestky, T., S. H. Jung and U. Banerjee, 2003. A Serrate-expressing signaling center controls Drosophila hematopoiesis. Genes Dev. 17: 348–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, T., and L. Luo, 1999. Mosaic analysis with a repressible neurotechnique cell marker for studies of gene function in neuronal morphogenesis. Neuron 22: 451–461. [DOI] [PubMed] [Google Scholar]
- Lemaitre, B., E. Nicolas, L. Michaut, J. M. Reichhart and J. A. Hoffmann, 1996. The dorsoventral regulatory gene cassette spatzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell 86: 973–983. [DOI] [PubMed] [Google Scholar]
- Li, W. H., Z. Gu, H. Wang and A. Nekrutenko, 2001. Evolutionary analyses of the human genome. Nature 409: 847–849. [DOI] [PubMed] [Google Scholar]
- Lipsick, J. S., and W. J. Boyle, 1987. c-myb protein expression is a late event during T-lymphocyte activation. Mol. Cell. Biol. 7: 3358–3360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, H., W. P. Hanratty and C. R. Dearolf, 1995. An amino acid substitution in the Drosophila hopTum-l Jak kinase causes leukemia-like hematopoietic defects. EMBO J. 14: 1412–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch, M., and A. Force, 2000. The probability of duplicate gene preservation by subfunctionalization. Genetics 154: 459–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manak, J. R., N. Mitiku and J. S. Lipsick, 2002. Mutation of the Drosophila homologue of the Myb protooncogene causes genomic instability. Proc. Natl. Acad. Sci. USA 99: 7438–7443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathey-Prevot, B., and N. Perrimon, 1998. Mammalian and Drosophila blood: JAK of all trades? Cell 92: 697–700. [DOI] [PubMed] [Google Scholar]
- McClintock, J. M., M. A. Kheirbek and V. E. Prince, 2002. Knockdown of duplicated zebrafish hoxb1 genes reveals distinct roles in hindbrain patterning and a novel mechanism of duplicate gene retention. Development 129: 2339–2354. [DOI] [PubMed] [Google Scholar]
- McLysaght, A., K. Hokamp and K. H. Wolfe, 2002. Extensive genomic duplication during early chordate evolution. Nat. Genet. 31: 200–204. [DOI] [PubMed] [Google Scholar]
- Mettus, R. V., J. Litvin, A. Wali, A. Toscani, K. Latham et al., 1994. Murine A-myb: evidence for differential splicing and tissue-specific expression. Oncogene 9: 3077–3086. [PubMed] [Google Scholar]
- Meyer, A., and M. Schartl, 1999. Gene and genome duplications in vertebrates: the one-to-four (-to-eight in fish) rule and the evolution of novel gene functions. Curr. Opin. Cell Biol. 11: 699–704. [DOI] [PubMed] [Google Scholar]
- Mucenski, M. L., K. McLain, A. B. Kier, S. H. Swerdlow, C. M. Schreiner et al., 1991. A functional c-myb gene is required for normal murine fetal hepatic hematopoiesis. Cell 65: 677–689. [DOI] [PubMed] [Google Scholar]
- Mullen, A. C., A. S. Hutchins, A. V. Villarino, H. W. Lee, F. A. High et al., 2001. Cell cycle controlling the silencing and functioning of mammalian activators. Curr. Biol. 11: 1695–1699. [DOI] [PubMed] [Google Scholar]
- Munier, A. I., D. Doucet, E. Perrodou, D. Zachary, M. Meister et al., 2002. PVF2, a PDGF/VEGF-like growth factor, induces hemocyte proliferation in Drosophila larvae. EMBO Rep. 3: 1195–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nappi, A. J., and M. Silvers, 1984. Cell surface changes associated with cellular immune reactions in Drosophila. Science 225: 1166–1168. [DOI] [PubMed] [Google Scholar]
- Nelson, R. E., L. I. Fessler, Y. Takagi, B. Blumberg, D. R. Keene et al., 1994. Peroxidasin: a novel enzyme-matrix protein of Drosophila development. EMBO J. 13: 3438–3447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno, S., 1999. Gene duplication and the uniqueness of vertebrate genomes circa 1970–1999. Semin. Cell Dev. Biol. 10: 517–522. [DOI] [PubMed] [Google Scholar]
- Okada, M., H. Akimaru, D. X. Hou, T. Takahashi and S. Ishii, 2002. Myb controls G(2)/M progression by inducing cyclin B expression in the Drosophila eye imaginal disc. EMBO J. 21: 675–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panopoulou, G., S. Hennig, D. Groth, A. Krause, A. J. Poustka et al., 2003. New evidence for genome-wide duplications at the origin of vertebrates using an amphioxus gene set and completed animal genomes. Genome Res. 13: 1056–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prince, V. E., and F. B. Pickett, 2002. Splitting pairs: the diverging fates of duplicated genes. Nat. Rev. Genet. 3: 827–837. [DOI] [PubMed] [Google Scholar]
- Qiu, P., P. C. Pan and S. Govind, 1998. A role for the Drosophila Toll/Cactus pathway in larval hematopoiesis. Development 125: 1909–1920. [DOI] [PubMed] [Google Scholar]
- Queva, C., S. A. Ness, F. A. Grasser, T. Graf, B. Vandenbunder et al., 1992. Expression patterns of c-myb and of v-myb induced myeloid-1 (mim-1) gene during the development of the chick embryo. Development 114: 125–133. [DOI] [PubMed] [Google Scholar]
- Rabinowicz, P. D., E. L. Braun, A. D. Wolfe, B. Bowen and E. Grotewold, 1999. Maize R2R3 myb genes: sequence analysis reveals amplification in the higher plants. Genetics 153: 427–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramet, M., P. Manfruelli, A. Pearson, B. Mathey-Prevot and R. A. Ezekowitz, 2002. Functional genomic analysis of phagocytosis and identification of a Drosophila receptor for E. coli. Nature 416: 644–648. [DOI] [PubMed] [Google Scholar]
- Rehorn, K. P., H. Thelen, A. M. Michelson and R. Reuter, 1996. A molecular aspect of hematopoiesis and endoderm development common to vertebrates and Drosophila. Development 122: 4023–4031. [DOI] [PubMed] [Google Scholar]
- Riechmann, J. L., J. Heard, G. Martin, L. Reuber, C. Jiang et al., 2000. Arabidopsis transcription factors: genome-wide comparative analysis among eukaryotes. Science 290: 2105–2110. [DOI] [PubMed] [Google Scholar]
- Rizki, T. M., and R. M. Rizki, 1984 The cellular defense system of Drosophila melanogaster, pp. 579–604 in Insect Ultrastructure, edited by R. C. King and H. Akai. Plenum, New York.
- Schneider, D., and M. Shahabuddin, 2000. Malaria parasite development in a Drosophila model. Science 288: 2376–2379. [DOI] [PubMed] [Google Scholar]
- Simon, A. L., E. A. Stone and A. Sidow, 2002. Inference of functional regions in proteins by quantification of evolutionary constraints. Proc. Natl. Acad. Sci. USA 99: 2912–2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitzmann, J., K. Noben-Trauth and K. H. Klempnauer, 1995. Expression of mouse c-myb during embryonic development. Oncogene 11: 2273–2279. [PubMed] [Google Scholar]
- Sitzmann, J., K. Noben-Trauth, H. Kamano and K. H. Klempnauer, 1996. Expression of B-Myb during mouse embryogenesis. Oncogene 12: 1889–1894. [PubMed] [Google Scholar]
- Sleeman, J. P., 1993. Xenopus A-myb is expressed during early spermatogenesis. Oncogene 8: 1931–1941. [PubMed] [Google Scholar]
- Stober-Grasser, U., B. Brydolf, X. Bin, F. Grasser, R. A. Firtel et al., 1992. The Myb DNA-binding domain is highly conserved in Dictyostelium discoideum. Oncogene 7: 589–596. [PubMed] [Google Scholar]
- Stracke, R., M. Werber and B. Weisshaar, 2001. The R2R3-MYB gene family in Arabidopsis thaliana. Curr. Opin. Plant Biol. 4: 447–456. [DOI] [PubMed] [Google Scholar]
- Tanaka, Y., N. P. Patestos, T. Maekawa and S. Ishii, 1999. B-myb is required for inner cell mass formation at an early stage of development. J. Biol. Chem. 274: 28067–28070. [DOI] [PubMed] [Google Scholar]
- Tirouvanziam, R., C. J. Davidson, J. S. Lipsick and L. A. Herzenberg, 2004. Fluorescence-activated cell sorting (FACS) of Drosophila hemocytes reveals important functional similarities to mammalian leukocytes. Proc. Natl. Acad. Sci. USA 101: 2912–2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torelli, G., L. Selleri, A. Donelli, S. Ferrari, G. Emilia et al., 1985. Activation of c-myb expression by phytohemagglutinin stimulation in normal human T lymphocytes. Mol. Cell. Biol. 5: 2874–2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toscani, A., R. V. Mettus, R. Coupland, H. Simpkins, J. Litvin et al., 1997. Arrest of spermatogenesis and defective breast development in mice lacking A-myb. Nature 386: 713–717. [DOI] [PubMed] [Google Scholar]
- Trauth, K., B. Mutschler, N. A. Jenkins, D. J. Gilbert, N. G. Copeland et al., 1994. Mouse A-myb encodes a trans-activator and is expressed in mitotically active cells of the developing central nervous system, adult testis and B lymphocytes. EMBO J. 13: 5994–6005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe, K. H., and D. C. Shields, 1997. Molecular evidence for an ancient duplication of the entire yeast genome. Nature 387: 708–713. [DOI] [PubMed] [Google Scholar]
- Xu, T., and G. M. Rubin, 1993. Analysis of genetic mosaics in developing and adult Drosophila tissues. Development 117: 1223–1237. [DOI] [PubMed] [Google Scholar]
- Zwicker, J., N. Liu, K. Engeland, F. C. Lucibello and R. Muller, 1996. Cell cycle regulation of E2F site occupation in vivo. Science 271: 1595–1597. [DOI] [PubMed] [Google Scholar]