

Abstract
Histone acetylation and deacetylation activate or repress transcription, yet the physiological relevance of reversible changes in chromatin structure and gene expression is poorly understood. We have shown that disrupting the expression of AtHD1 that encodes a putative Arabidopsis thaliana histone deacetylase induces a variety of developmental abnormalities. However, causal effects of the AtHD1 disruption on chromatin structure and gene expression are unknown. Using Arabidopsis spotted oligo-gene microarray analysis, here we report that >7% of the transcriptome was up- or downregulated in A. thaliana plants containing a T-DNA insertion in AtHD1 (athd1-t1), indicating that AtHD1 provides positive and negative control of transcriptional regulation. Remarkably, genes involved in ionic homeostasis and protein synthesis were ectopically expressed, whereas genes in ionic homeostasis, protein transport, and plant hormonal regulation were repressed in athd1-t1 leaves or flowers, suggesting a role of AtHD1 in developmental and environmental regulation of gene expression. Moreover, defective AtHD1 induced site-specific and reversible acetylation changes in H3-Lys9, H4-Lys12, and H4 tetra-lysines (residues 5, 8, 12, and 16) in homozygous recessive and heterozygous plants. Transcriptional activation was locus specific and often associated with specific acetylation sites in the vicinity of promoters, whereas gene repression did not correlate with changes in histone acetylation or correlated directly with H3-Lys9 methylation but not with DNA methylation. The data suggest that histone acetylation and deacetylation are promoter dependent, locus specific, and genetically reversible, which provides a general mechanism for reversible gene regulation responsive to developmental and environmental changes.
ACETYLATION and deacetylation of lysine residues in the N termini of core histones are catalyzed by intrinsic histone acetyltransferases (HATs) and histone deacetylases (HDs; HDAs; HDACs), providing a mechanism for reversibly modulating chromatin structure and transcriptional regulation (Brownell and Allis 1996; Jenuwein and Allis 2001). Hyperacetylation relaxes chromatin structure and activates gene expression, whereas hypoacetylation induces chromatin compaction and gene repression. In contrast to “cemented” chromatin modifications such as DNA and histone methylation (Jenuwein and Allis 2001; Richards and Elgin 2002; Rabinowicz et al. 2003), histone acetylation and deacetylation are reversible and therefore play a unique role in transcriptional regulation associated with developmental programs and environmental conditions, including day length (Tian et al. 2003), flowering (He et al. 2003; Ausin et al. 2004; Kim et al. 2004), osmotic and oxidative stress (Brunet et al. 2004; De Nadal et al. 2004), and cell aging (Imai et al. 2000).
Arabidopsis has 18 members of a putative histone deacetylase family (Pandey et al. 2002). AtHD1, a RPD3 homolog in yeast, has four closely related members, namely, AtHD1 (or AtHDA19), AtHDA6, 7, and 9 (Tian et al. 2003). Other members of the gene family include eight RPD3/HDA1-like genes, two SIR2 homologs, and four plant-specific HD2 genes (Wu et al. 2003). Different members within a group might have evolved specific functions. Indeed, AtHDA6 is responsible for silencing transgenes, repetitive DNA, and rDNA loci (Murfett et al. 2001; Aufsatz et al. 2002; Lippman et al. 2003; Probst et al. 2004), whereas AtHD1 is a putative global transcriptional regulator throughout Arabidopsis development (Tian and Chen 2001; Tian et al. 2003). Disrupting AtHD1 by antisense AtHD1 expression (Tian and Chen 2001) or T-DNA insertion (athd1-t1; Tian et al. 2003) induces various developmental abnormalities and ectopic expression of tissue-specific genes such as SUPERMAN (Tian and Chen 2001). It is unclear whether and how AtHD1 directly affects chromatin structure of the target genes and plant development. Using spotted oligo-gene microarrays (Chen et al. 2004), we analyzed genome-wide changes in gene expression in athd1-t1 lines during vegetative growth and flower development. Moreover, we examined histone acetylation and gene expression changes in wild-type, heterozygous, and homozygous athd1-t1 plants using immuno-blot and chromatin immunoprecipitation (ChIP) assays. The data indicate that (1) AtHD1 both negatively and positively regulates the expression of different sets of genes during leaf and flower development; (2) disrupting AtHD1 induces site-specific and reversible changes in histone acetylation and irreversible changes in histone methylation; (3) gene activation is associated with increased levels of site-specific histone acetylation, whereas gene repression does not correlate with changes in histone acetylation or correlate with histone methylation; and (4) changes in gene expression and histone acetylation are locus specific, occur in the vicinity of the promoter, and are independent of DNA methylation. The data obtained in AtHD1-defective lines, together with previous findings of the involvement of histone deacetylases in stress response (Brunet et al. 2004; De Nadal et al. 2004), flower development (He et al. 2003; Tian et al. 2003; Ausin et al. 2004; Kim et al. 2004), and cell aging (Imai et al. 2000; Hekimi and Guarente 2003), suggest that reversible modifications of histone acetylation and deacetylation provide an active and dynamic mechanism for gene regulation responsive to changes in developmental programs and environmental cues.
MATERIALS AND METHODS
Plant materials:
Arabidopsis thaliana ecotype Ws (AtHD1/AtHD1, +/+), AtHD1/athd1-t1 (+/−), and athd1-t1/athd1-t1 (−/−) plants were produced as previously described (Tian et al. 2003). The heterozygous plants (+/−) were generated by backcrossing the homozygous plants (−/−) to Ws (+/+) for inheritance studies on changes in histone acetylation and methylation. The plants were grown in a growth chamber under growth conditions of 22°/18° (day/night) and 14 hr of illumination per day. DNA and RNA were isolated from tissues collected from a pool of 32 plants in each line. Rosette leaves were collected at prebolting stage (∼3 weeks) for DNA and RNA preparation and ChIP, while flower buds were harvested after the first flower bloomed.
DNA and RNA analyses:
Total RNA was extracted using the Trizol reagent (Invitrogen, San Diego). DNA isolation and DNA or RNA blot analysis were performed as previously described (Tian et al. 2003). For microarray and RT-PCR analyses, the mRNA was isolated from 500 μg of total RNA with the FastTrack 2.0 mRNA isolation kit (Invitrogen). RT-PCR analysis was performed using ∼500 ng of mRNA mixed with 1 μl of oligo(dT) (Amersham, Buckinghamshire, UK) and 2 μg of random nonamer (Gene Link) in a total volume of 17 μl for denaturation and primer annealing using SuperScript II reverse transcriptase. The synthesized cDNA was purified by QIAquick kit (QIAGEN, Chatsworth, CA) and adjusted to a final concentration of 15 ng/μl. An aliquot of 0.5 μl was used for PCR reaction in the volume of 25 μl using the primers designed according to the 3′ end sequences (supplementary Tables 3 and 4 at http://www.genetics.org/supplemental/). The PCR reaction included 1 cycle of 94° for 2 min, 30 cycles of 94° for 30 sec, 52° for 30 sec, and 72° for 1 min. The ACTIN2/7 gene was amplified and served as a control (Tian et al. 2003). An aliquot of 5 μl RT-PCR products was used for agarose gel electrophoresis.
Microarray analysis:
Spotted oligo-gene microarrays were developed as previously described (Lee et al. 2004). Gene names and accession numbers of the 26,090 70-mer oligos can be found at http://oligos.qiagen.com/arrays/oligosets_arabidopsis.php. Each slide was printed with 27,648 features, including 26,090 70-mer oligos and controls (Chen et al. 2004). Slide hybridization, washing, and scanning were modified from a published protocol (Lee et al. 2004). Four repeated dye-swap experiments were performed in each comparison (e.g., Ws vs. athd1/athd1 in leaves) (supplementary Figure 1 at http://www.genetics.org/supplemental/). The data were normalized using a robust locally weighted regression (or lowess) (Cleveland 1979) and analyzed statistically using a linear model (Lee et al. 2004). We selected the genes found to be statistically significant (α = 0.05 with multiple comparison correction) using the per-gene variance assumption (supplementary Table 1 at http://www.genetics.org/supplemental/). Functional categories of up- and downregulated genes in leaves and flower buds of the athd1-t1 line were classified using PENDANT (http://mips.gsf.de/proj/thal/db/index.html) and compared using Venn diagrams.
Figure 1.—
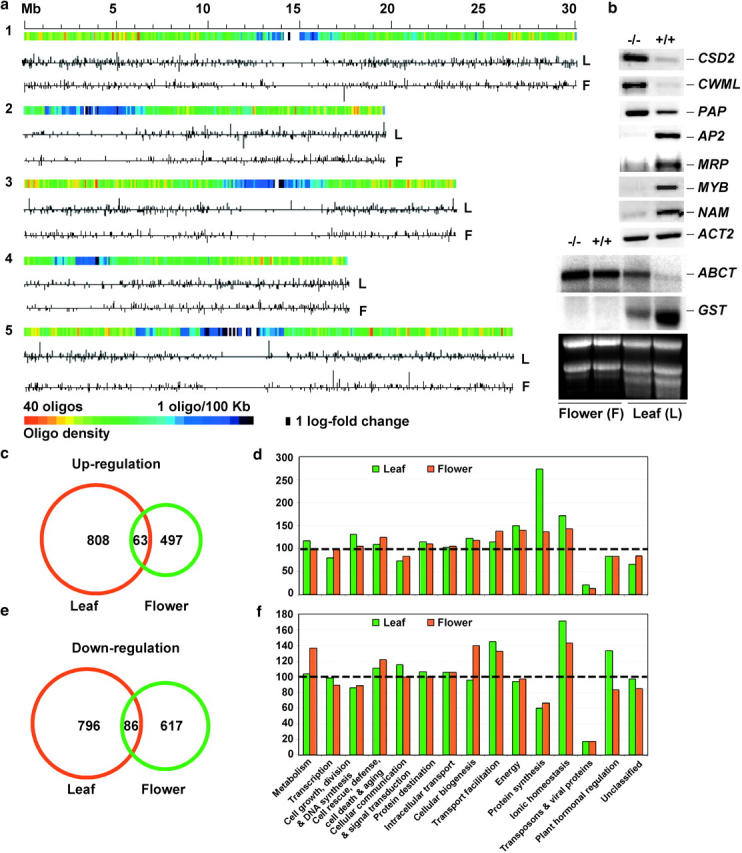
(a) Chromosomal distribution of differentially expressed genes in the athd1-t1 leaves (L) and flowers (F). The 70-mer oligos were mapped in five chromosomes using the annotated gene sequences correspondent to genomic coordinates, as shown in color gradients from high (red) to low (blue) gene density. The up- and downregulated genes in the athd1-t1 lines are indicated by the vertical lines above or below the horizontal line, respectively, the length of which is proportional to natural logarithm fold changes of gene expression between the athd1-t1 and Ws lines. (b) RT-PCR and Northern blot analyses of differentially expressed genes (supplementary Tables 3 and 4 at http://www.genetics.org/supplemental/) in leaves (L) and flowers (F) of Ws and athd1-t1 lines. (c) Only 4.6% of the upregulated genes detected in the leaves and flowers overlapped. (d) The relative ratio in the y-axis was calculated using the percentage of upregulated genes from each functional category detected in the athd1-t1 lines divided by the percentage of ∼26,000 annotated genes in the same category in the genome (Arabidopsis Genome Initiative 2000). The dashed line (at 100) indicates that the percentage of all annotated genes in this functional category is equal to the percentage of the genes that are upregulated in the athd1-1 lines. (e) Only 5.7% of the downregulated genes detected in the leaves and flowers overlapped. (f) The percentages of downregulated genes detected were analyzed using the same method as in d.
TABLE 1.
ChIP analysis using a subset of differentially expressed genes detected between Ws andathd1-t1 lines
| Acetylation or methylation profile
|
|||||||
|---|---|---|---|---|---|---|---|
| Locus ID | Description | H4- tetraAc |
H4- K12Ac |
H3- K9Ac |
H3- K9Me |
Tissue | Expression |
| At1g14970 | Putative auxin-independent promoter (AXI) | 0 | 0 | ++ | 0 | Leaf | ++ |
| At2g3691 | A. thaliana P glycoprotein (PGP1) | 0 | ++ | 0 | ND | Leaf | ++ |
| At2g2819 | Putative copper/zinc superoxide dismutase (CSD2) | 0 | ++ | 0 | 0 | Leaf | ++ |
| At3g23130 | Superman | 0 | 0 | 0 | ND | Leaf | ++ |
| At1g02920 | Glutathione S-transferase (GST11) | 0 | 0 | 0 | 0 | Leaf | — |
| At4g34590 | BZip transcription factor | 0 | ND | ++ | — | Flower | ++ |
| At1g01060 | Late elongated hypocotyls protein (LHY) | ++ | ND | ++ | ND | Flower | ++ |
| At2g02850 | Plantacyanin (ARPN) | ++ | ND | ++ | ND | Flower | ++ |
| At2g22980 | Putative serine carboxypeptide I (SCP) | 0 | ND | 0 | 0 | Flower | — |
0 indicates a small or no difference between Ws and athd1-t1; ++, increased level of histone acetylation or gene expression; —, decreased level of histone acetylation, methylation, or gene expression; ND, not determined.
ChIP assays:
ChIP was modified from a published protocol (Lawrence et al. 2004). Approximately 1 g of leaves or 0.5 g of flower buds was used for each ChIP assay. The fresh tissues were subjected to vacuum infiltration in a formaldehyde (1%) solution for crosslinking the chromatin proteins to DNA. Chromatin was extracted and sonicated (Fisher, model 60 sonicator) at half-maximal power for 5- × 10-sec pulses with chilling on ice for 3 min after each pulse. The average size of the resulting DNA fragments produced was ∼0.3–1.0 kb. An aliquot of chromatin solution (1/10 of total volume) was used to determine the DNA fragment sizes and serve as input DNA. The remaining chromatin solution was diluted 10-fold and divided into two aliquots. One aliquot was incubated by adding 10 μl of antibodies (anti-tetra-acetyl-histone H4, anti-acetyl-histone H4-K12, anti-dimethyl-histone H3-K9, or anti-acetyl-histone H3-K9, Upstate Biotechnology, Lake Placid, NY). The other aliquot was incubated without antibodies (as a control). After incubation at 4° with rotation for overnight, the solution was added to 40 μl of DNA/protein A agarose and incubated for another 2 hr. The immunocomplexes were eluted and crosslinks were reversed by incubation at 65° for 6 hr. Residual protein was degraded by proteinase K and DNA was extracted and dissolved in 50 μl of ddH2O.
ChIP and chop PCR:
An aliquot (1 μl) of ChIP DNA was used for semiquantitative PCR analysis to determine the amount of genomic DNA immunoprecipitated in the ChIP assays (Lawrence et al. 2004) using the primer pairs designed from promoter and/or coding regions of the genes (supplementary Tables 4–6 at http://www.genetics.org/supplemental/), which amplified ∼350-bp DNA fragments. The concentration of each ChIP DNA sample was adjusted empirically such that an equal amount of ACT2/7 was amplified (Tian et al. 2003). All PCR reactions were performed in 25 μl using 1.0 μl of immunoprecipitated DNA for 25–35 cycles of PCR amplification. For chop PCR, 1 μg of DNA was digested to completion using McrBC. PCR was performed using an aliquot of 50 ng of the digested DNA and the same primer pairs used in the ChIP assays in a 25-μl PCR reaction for 25 cycles of amplification.
RESULTS AND DISCUSSION
Genome-wide analysis of gene expression changes in athd1-t1 lines:
We analyzed transcriptome changes in the athd1-t1 line in leaves and flower buds, two important developmental stages. In each comparison, we performed four dye-swap experiments using two biological replicates (supplementary Figure 1 and supplementary Table 1 at http://www.genetics.org/supplemental/). The data were analyzed using a linear model and the results were adjusted for multiple comparisons (Lee et al. 2004) to test the null hypothesis of no differential gene expression between the wild type (Ws) and athd1-t1. The data were analyzed using two versions of the same statistical test (i.e., t-test). The first analysis was based on a t-test using a common variance assumption for all genes, while the second analysis acknowledged the per-gene variances for individual genes via the biological replicates. The genes found to be statistically significant (α = 0.05) under per-gene variances included those that had relatively small fold changes but that may be biologically relevant (Lee et al. 2004). We detected 2789 (or 10.7%) and 2010 (or 7.8%) genes that were significantly different between Ws and athd1-t1 in leaves and flower buds, respectively (supplementary Table 1 at http://www.genetics.org/supplemental/). For further analyses using chromosomal display and semiquantitative RT-PCR analysis, we selected the genes that were statistically significant and more than ±1.25-fold changes between the two lines in the leaves (1753 or 6.7%) or flowers (1263 or 4.8%). The arbitral fold cut (±1.25) was used because it is probably the fold-change limit that can be detected by other assays such as RT-PCR (Lee et al. 2004).
Yeast RPD3 is a transcriptional regulator (Bernstein et al. 2000; Vogelauer et al. 2000) that affects many genes located near telomeres. To determine whether AtHD1 has specific chromosomal targets, we mapped the differentially expressed genes in five Arabidopsis chromosomes (Figure 1a), generating transcription maps in leaves and flowers in the athd1-t1 lines. The differentially expressed genes were randomly distributed relative to the oligo-gene density across five chromosomes. There was no obvious cluster of up- or downregulated genes in a specific region. Moreover, relatively equal numbers of genes were up- or downregulated in the athd1-t1 line, suggesting that AtHD1 is a negative and positive regulator of gene expression.
A subset of the genes encoding transcription factors and homeotic proteins important to plant development detected in microarray analysis was verified by semiquantitative RT-PCR or RNA blot analysis (Figure 1b and supplementary Tables 3 and 4 at http://www.genetics.org/supplemental/). Notably, NO APICAL MERISTEM (NAM) gene was repressed in the athd1-t1 leaves, which may correlate with defective shoot apical meristems as previously observed in Arabidopsis (Tian et al. 2003). A gene encoding ABC transporter (ABCT) that displayed a flower-specific expression pattern was ectopically expressed in the athd1-t1 leaves, whereas a glutathione transferase (GST) gene was repressed in the athd1-t1 leaves. Furthermore, of the differentially expressed genes detected, 871 (49.7%) were upregulated and 882 (50.3%) were downregulated in the leaves, whereas 560 (44.3%) genes were upregulated and 703 (55.7%) genes were repressed in the flowers (Figure 1, c and e). There is a little overlap between the upregulated (χ2 = 1006, P ≈ 0) or downregulated (χ2 = 1056, P ≈ 0) genes in leaves and flowers, indicating that athd1-t1 induces silencing or ectopic expression of different sets of genes in two developmental stages. When the data were compared with the genes exhibiting tissue-specific expression patterns, 85 “leaf-specific” genes were ectopically expressed in the flower buds and 360 “flower-specific” genes were reactivated in the leaves of the athd1-t1 line. The data suggest that AtHD1 plays a role in developmentally regulated gene expression.
The differentially expressed genes detected in the athd1-t1 leaves and flowers were classified into 15 functional and 1 unclassified category (Arabidopsis Genome Initiative 2000; Figure 1, d and f). Remarkably, the number of genes involved in ionic homeostasis and transport facilitation affected by athd1-t1 in both leaves and flowers was 40–85% higher than a genome-wide average, indicating a general role of AtHD1 in response to various growth conditions such as osmotic and oxidative stress and cell aging as observed in yeast and mammalian cells (Imai et al. 2000; Brunet et al. 2004; De Nadal et al. 2004). The number of differentially expressed genes in plant hormonal regulation and protein synthesis was 40–150% higher in the leaves than in the flowers, suggesting an important role of AtHD1 in phytohormone-dependent gene regulation during vegetative growth. The genes in metabolism and cellular biogenesis were affected by athd1-t1 more in the flowers than in the leaves, suggesting a role of AtHD1 in rapid cell divisions and cellular growth during flower development (Meyerowitz 1996). Notably, transposons were underrepresented in the athd1-t1 lines, suggesting that in contrast to AtHDA6 (Murfett et al. 2001; Aufsatz et al. 2002; Lippman et al. 2003; Probst et al. 2004), AtHD1 generally does not affect repetitive DNA.
Changes in histone acetylation, methylation, and gene expression in the wild-type, heterozygous, and homozygous athd1-t1 plants:
To study inheritance of changes in histone acetylation and methylation, the heterozygous plants (+/−) were generated by backcrossing the homozygous athd1-t1 plants (−/−) to wild-type plants (+/+). The overall levels of H4 tetra-lysine, H4-K12, and H3-K9 acetylation were increased 1.5- to 4-fold in the leaves of athd1-t1 homozygous (−/−) plants (Figure 2), whereas H4 K5 acetylation levels were not affected (data not shown). Both wild-type (Ws, +/+) and heterozygous (+/−) plants had similarly low levels of acetylated histones, suggesting that histone acetylation and deacetylation are reversible. However, H3-K9 methylation was decreased in both homozygous recessive and heterozygous plants, suggesting that histone methylation is irreversible and that some residual methylation remains in the heterozygous plants. In the athd1-t1 line the decreased levels of H3-K9-dimethyl correlated with the increased levels of H3-K9 acetylation, indicating a mutually exclusive competition between acetylation and methylation for the specific lysine residue (Jenuwein and Allis 2001). Moreover, histone methylation appears to be suppressed in the homozygous plants to the same extent as in the heterozygous plants, even though the phenotypic effects are observed only when homozygous for the knockout mutation.
Figure 2.—

Effects of athd1-t1 on histone acetylation, methylation, and gene expression. (a) H3-K9Ac, H4-tetraAc, and K12Ac were hyperacetylated and H3-K9 was hypomethylated in athd1-t1 lines. The ratios indicate relative abundance of proteins in the athd-t1 homozygous (−/−) and heterozygous (+/−) plants compared to Ws (+/+). C.H., purified core histones. (b) ChIP assays show a correlation of histone acetylation and methylation with gene activation and silencing, respectively. Semiquantitative PCR was used to estimate relative enrichment of DNA fragments in the chromatin immunoprecipitates by specific antibodies as shown. ACT2 was amplified as a control for DNA quantification. Input (I) and mock (M) indicate PCR products amplified using the DNA recovered from the chromatin complexes before ChIP analysis and without antibodies, respectively. Fold changes in the heterozygous and homozygous plants relative to the wild type are shown under each lane, except for no changes (ACT2) or a missing sample (BZIP11, H3-K9Me). ACT2 was slightly hyperacetylated in the athd1-t1 lines.
ChIP assay (Lawrence et al. 2004) was used to investigate the role of a specific histone modification in the expression of target gene in Ws, heterozygous, and homozygous athd1-t1 plants (Figure 2b; Table 1; supplementary Table 5 at http://www.genetics.org/supplemental/). Gene activation was related to hyperacetylation of specific lysine residues with a few exeptions. H4-tetra and H3-K9 acetylation levels correlated with upregulation of LHY and APRN and the H4-K12 acetylation level with activation of PGP1 and CSD2. Upregulation of BZIP11 was associated with H3-K9 acetylation but not with H4-tetra acetylation, suggesting specificity for gene activation. Furthermore, BZIP11 activation also correlated with lower levels of H3-K9-dimethyl, providing evidence that histone H3-Lys9 is the site for acetylation and methylation competition, leading to gene activation or repression. It is notable that the acetylation level of a few genes was increased in the heterozygous plants, suggesting that potential epigenetic lesions may be induced by athd1-t1 via changes in “cemented” modifications such as histone methylation.
Of 11 genes tested, 8 (Figures 2 and 3a; Table 1) were associated with acetylation and/or methylation changes in at least one specific lysine residue. SUP was upregulated in athd1-t1 plants and in plants overexpressing antisense AtHD1 (Tian and Chen 2001); however, no changes in acetylation were detected. Two genes were repressed but their acetylation levels remained unchanged in the sites examined. The data suggest that disrupting AtHD1 expression also indirectly affects a set of downstream genes by activating transcriptional activators or repressors, as observed in yeast rpd3 deletion mutants (Bernstein et al. 2000; Robyr et al. 2002). Alternatively, other specific untested lysine residues (Jenuwein and Allis 2001) may be responsible.
Figure 3.—

The effects of athd1-t1 on histone acetylation in chromosomal domains and within a locus. (a) An ∼30-kb segment of chromosome 2 contains the At2g36910 locus that was upregulated in the leaves. The arrows and boxes indicate the directions and levels of gene expression. ChIP assays showed that locus At2g36910 was hyperacetylated whereas two neighboring loci, At2g36890 and At2g36920, were not affected in the athd1-t1 line. (b) ChIP analysis of an ∼28-kb segment of chromosome 4. The two loci (At4g34590 and 34620) were hyperacetylated and upregulated in the flowers. Acetylated levels of the loci flanking or between the two upregulated genes remained unaffected. Input (I) and mock (M) controls are shown for At4g34620. Fold changes in +/− and −/− lines relative to the wild type are shown below each lane. (c) Histone acetylation in the promoter region is associated with gene activation. The diagram shows the genomic sequence of LHY (At1g016060), including the start codon (ATG, +1), exons (boxes), and introns (lines). ChIP was performed using antibodies against H4-tetraAc and H3-K9Ac. Solid lines below the genomic diagram indicate the location of amplified PCR fragments. The amount of PCR products relative to the ACT2 control was quantified and is shown as a histogram. (d) The same ChIP analysis in c performed for ARPN (At2g02850).
Changes in histone acetylation and gene expression are locus specific:
Although histone acetylation directly affects the expression of target genes, it is unclear whether the altered acetylation status is localized or diffusible. To address this, we randomly selected two regions (∼30 kb each) containing upregulated and unaffected genes in chromosomes 2 and 4. ChIP analysis was performed for the selected loci and their neighboring genes (Table 1 and supplementary Tables 3–5 at http://www.genetics.org/supplemental/). Upregulation of At2g36910 in the leaves and of At4g34590 and At4g34620 in the flowers was associated with increased levels of H4-K12Ac and H3-K9Ac, respectively (Figure 3, a and b). Their neighboring genes located within the vicinity did not exhibit changes in acetylation levels. The data are reminiscent of results obtained from ChIP analysis of 88 genes in tobacco (Chua et al. 2004). Histone H4 acetylation occurs in the 300- to 600-bp sequences of the promoters or coding regions for one-third of the genes. Together, the data suggest that, unlike histone methylation, which may spread into neighboring heterochromatin regions (Noma et al. 2001), histone acetylation targets specific loci and does not affect the adjacent chromosomal domains examined.
Changes in histone acetylation are detected within the vicinity of promoters:
If histone acetylation is localized in a specific locus, it may affect promoters exclusively or other coding and noncoding sequences. To distinguish these scenarios, we mapped acetylation profiles in two loci, LHY (At1g01060) and ARPN (At2g02850), using primers designed to amplify individual DNA fragments spanning the promoters and coding sequences (Figure 3, c and d). As expected, both H4-tetra and H3-K9 acetylation levels were dramatically increased within the promoter region (∼500 bp) of the genes. No acetylation changes were detected beyond 500 bp upstream of the start codon or downstream after the first exon. Thus, acetylation is localized to a relatively small region from ∼500 bp of the promoter to the first exon, suggesting that AtHD1 modifies the chromatin structure within the promoter to prevent transcriptional initiation.
No DNA methylation changes were detected in a subset of genes that display expression changes in athd1-t1 lines:
Histone deacetylases are in the MeCP2 (Nan et al. 1998) and Dnmt1 (Fuks et al. 2000) complexes, suggesting that disrupting histone deacetylation may reduce DNA methylation levels, although changes in DNA methylation profiles in the centromeres and rDNA loci were not detected (Tian and Chen 2001). A chop assay without ChIP (Lawrence et al. 2004) using McrBC-digested DNA was used to determine whether the genes activated by histone acetylation are correlated with reduced levels of DNA methylation in the same loci (Figures 2b and 4a). None of the nine loci tested showed changes in DNA methylation between the Ws and athd1-t1 lines, although they displayed increased acetylation levels in at least one site (Table 2). As a control, the centromeres were demethylated in ddm1 but not in athd1-t1 plants.
Figure 4.—

DNA methylation in athd1-t1 lines and a model for the role of histone acetylation and deacetylation in plant development. (a) DNA methylation was not affected in genes that were up- or downregulated by histone acetylation or methylation. As a control, centromeric repeats were demethylated in the ddm1 mutant but not in the athd1-t1 line. (b) A simple model indicates that reversible modifications of histone acetylation and deacetylation provide a flexible regulatory system for changes in gene expression in response to developmental programs and environmental cues. The developmental and environmental signals are perceived by signal molecules, transcriptional activators, and repressors that recruit HATs (e.g., GCN5) and HDs (e.g., AtHD1, RPD3), respectively, resulting in changes in histone acetylation or deacetylation (e.g., H3-K9), which lead to transcriptional activation or repression. These changes are reversible when the signals are absent, although changes in histone acetylation/deacetylation are coupled with other cemented changes in histone or DNA methylation as previously reviewed (Jenuwein and Allis 2001; Richards and Elgin 2002), which may induce other epigenetic lesions. Thick, dashed, and thin arrows/lines indicate the interactions that are supported by the results from this report, this and other studies, and previous work, respectively (see text).
TABLE 2.
The number of differentially expressed genes detected between Ws andathd1-t1 lines using microarray data that are analyzed using a linear model and a false-discovery-rate multiple comparison correction
| FDR (α = 0.05)
|
||||
|---|---|---|---|---|
| Microarray experiment | Common variance | Per-gene variance | Shareda | Per-gene variance with a fold cut (±1.25)b |
| Leaves (Ws vs. athd1-t1) | 477 | 2789 | 260 | 1753 |
| Flower buds (Ws vs. athd1-t1) | 359 | 2010 | 87 | 1263 |
FDR, false discovery rate.
Shared sets of genes the expression level differences of which were found to be statistically significant using both common variance and per-gene variance.
The number of differentially expressed genes using an arbitrarily cut fold change (±1.25) from genes selected on the basis of per-gene variance.
The data suggest a mechanistic role of reversible histone acetylation and deacetylation in the transcriptional control of gene expression responsive to developmental programs and environmental cues (Figure 4b). Consistent with this model, AtHD1 acts primarily on euchromatic regions but not on transposons and repetitive DNA, providing new evidence that AtHD1 and AtHDA6 may have diverged functions. It is believed that core histones are acetylated in cytoplasm before they are transported into cell nuclei and further incorporated into chromatin during assembly (Brownell and Allis 1996; Kuo and Allis 1998). Histone deacetylases are recruited by transcriptional repressors, such as pRB (Brehm et al. 1998; Luo et al. 1998), YY1 (Yang et al. 1996), NcoR (Alland et al. 1997), and Ume6 (Kadosh and Struhl 1997), to fine-tune the acetylated sites so that the genes are turned “off.” The genes remain active if hypoacetylated histones are acetylated by HATs or if the repressors are absent. The reversible acetylation and deacetylation process may provide a means to turn gene expression “on” or “off” during development. Indeed, hyperacetylation of FLC that encodes a flowering repressor is controlled by FLD (He et al. 2003), a homolog of a mammalian protein found in histone deacetylase complexes. FVE, a gene involved in the autonomous pathway of flowering time, encodes a homolog of retinoblastoma-associated protein (Ausin et al. 2004). FVE has dual roles in regulating FLC and cold responses (Kim et al. 2004). Surprisingly, FLC expression and acetylation levels were not affected in the athd1-t1 lines that delayed flowering for 3 days (Tian et al. 2003). It is likely that disrupting AtHD1 activates some other genes in the flowering pathways that normally repress flower transition. Alternatively, other histone deacetylase associated proteins may be involved (He et al. 2003).
There is evidence that gene expression on stress is induced by the interaction of MAPK Hog1 with the osmotic responsive promoters through recruitment of RPD3-SIN3 histone deacetylase (De Nadal et al. 2004). Mammalian cells lacking the histone deacetylase complex are sensitive to osmotic stress. The data indicate that MAPK Hog1 recruits RPD3-SIN3 and targets to the promoter of osmotic responsive genes leading to histone deacetylation, which activates gene expression. Although the actual link between histone deacetylation and gene activation is unknown, the responses to osmotic stress-responsive genes are dependent on RPD3-SIN3, not on other histone deacetylases. In plants, the mutation in FVE induces cold tolerance and late flowering (Kim et al. 2004). Moreover, Arabidopsis homologs of GCN5 and ADA transcription factors interact with CBF1, a transcriptional activator involved in cold-responsive gene expression (Stockinger et al. 2001). Transgenic plants overexpressing antisense AtHD1 display pleiotropic developmental abnormalities including early senescence (Tian and Chen 2001). SIR2, an NAD-dependent histone deacetylase, is associated with yeast cell aging (Imai et al. 2000; Smith et al. 2000; Hekimi and Guarente 2003). SIRT1, a mammalian Sir2 homolog, appears to control the cellular response to oxidative stress by regulating the FOXO family of Forkhead transcription factors (Brunet et al. 2004). It is plausible that reversible reactions of histone acetylation and deacetylation are responsible for perceiving environmental and developmental signals (Figure 4b). These environmental signals are exerted by interacting with transcriptional activators (e.g., FOXO; Brunet et al. 2004), repressors (e.g., FVE; Ausin et al. 2004), and/or molecules involved in signal transduction (e.g., MAPK; De Nadal et al. 2004). These molecules recruit histone deacetylases (e.g., RPD3, AtHD1) to the promoters of the environmental or developmental responsive genes, which in turn remodel the chromatin and activate or repress transcription. The process is reversible so that the elevated levels of transcription may return to a “normal” state when the signals are removed or absent. However, if chromatin modifications involve histone or DNA methylation, the process is cemented and irreversible (Jenuwein and Allis 2001; Richards and Elgin 2002) and some residual effects may remain (Stokes et al. 2002; Stokes and Richards 2002). To avoid this, some specific histone deacetylases such as AtHD1 and RPD3 (De Nadal et al. 2004) and a histone acetyl transferase such as GCN5 (Stockinger et al. 2001) may be involved in the modification of some specific lysine residues (e.g., H3-K9) to induce dynamic and reversible changes in gene regulation (Figure 4b). The available data collectively suggest that histone acetylation and deacetylation play an active and reversible role in the modulation of gene expression in response to changes in developmental programs and environmental cues.
Acknowledgments
We thank Gary E. Hart for critical reading of the manuscript. The work was supported in part by grants from the National Institutes of Health (GM067015) to Z.J.C. and the National Science Foundation (DBI0077774) to R.D.W. and Z.J.C.
References
- Alland, L., R. Muhle, H. Hou, Jr., J. Potes, L. Chin et al., 1997. Role for N-CoR and histone deacetylase in Sin3-mediated transcriptional repression. Nature 387: 49–55. [DOI] [PubMed] [Google Scholar]
- Arabidopsis Genome Initiative, 2000. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408: 796–815. [DOI] [PubMed] [Google Scholar]
- Aufsatz, W., M. F. Mette, J. van der Winden, M. Matzke and A. J. Matzke, 2002. HDA6, a putative histone deacetylase needed to enhance DNA methylation induced by double-stranded RNA. EMBO J. 21: 6832–6841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ausin, I., C. Alonso-Blanco, J. A. Jarillo, L. Ruiz-Garcia and J. M. Martinez-Zapater, 2004. Regulation of flowering time by FVE, a retinoblastoma-associated protein. Nat. Genet. 36: 162–166. [DOI] [PubMed] [Google Scholar]
- Bernstein, B. E., J. K. Tong and S. L. Schreiber, 2000. Genome-wide studies of histone deacetylase function in yeast. Proc. Natl. Acad. Sci. USA 97: 13708–13713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brehm, A., E. A. Miska, D. J. McCance, J. L. Reid, A. J. Bannister et al., 1998. Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature 391: 597–601. [DOI] [PubMed] [Google Scholar]
- Brownell, J. E., and C. D. Allis, 1996. Special HATs for special occasions: linking histone acetylation to chromatin assembly and gene activation. Curr. Opin. Genet. Dev. 6: 176–184. [DOI] [PubMed] [Google Scholar]
- Brunet, A., L. B. Sweeney, J. F. Sturgill, K. F. Chua, P. L. Greer et al., 2004. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 303: 2011–2015. [DOI] [PubMed] [Google Scholar]
- Chen, Z. J., J. Wang, L. Tian, H.-S. Lee, J. J. Wang et al., 2004. The development of an Arabidopsis model system for genome-wide analysis of polyploidy effects. Biol. J. Linn. Soc. Lond. 82: 689–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua, Y. L., E. Mott, A. P. Brown, D. MacLean and J. C. Gray, 2004. Microarray analysis of chromatin-immunoprecipitated DNA identifies specific regions of tobacco genes associated with acetylated histones. Plant J. 37: 789–800. [DOI] [PubMed] [Google Scholar]
- Cleveland, W. S., 1979. Robust locally weighted regression and smoothing scatterplots. J. Am. Stat. Assoc. 74: 829–836. [Google Scholar]
- De Nadal, E., M. Zapater, P. M. Alepuz, L. Sumoy, G. Mas et al., 2004. The MAPK Hog1 recruits Rpd3 histone deacetylase to activate osmoresponsive genes. Nature 427: 370–374. [DOI] [PubMed] [Google Scholar]
- Fuks, F., W. A. Burgers, A. Brehm, L. Hughes-Davies and T. Kouzarides, 2000. DNA methyltransferase Dnmt1 associates with histone deacetylase activity. Nat. Genet. 24: 88–91. [DOI] [PubMed] [Google Scholar]
- He, Y., S. D. Michaels and R. M. Amasino, 2003. Regulation of flowering time by histone acetylation in Arabidopsis. Science 302: 1751–1754. [DOI] [PubMed] [Google Scholar]
- Hekimi, S., and L. Guarente, 2003. Genetics and the specificity of the aging process. Science 299: 1351–1354. [DOI] [PubMed] [Google Scholar]
- Imai, S., C. M. Armstrong, M. Kaeberlein and L. Guarente, 2000. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403: 795–800. [DOI] [PubMed] [Google Scholar]
- Jenuwein, T., and C. D. Allis, 2001. Translating the histone code. Science 293: 1074–1080. [DOI] [PubMed] [Google Scholar]
- Kadosh, D., and K. Struhl, 1997. Repression by Ume6 involves recruitment of a complex containing Sin3 corepressor and Rpd3 histone deacetylase to target promoters. Cell 89: 365–371. [DOI] [PubMed] [Google Scholar]
- Kim, H. J., Y. Hyun, J. Y. Park, M. J. Park, M. K. Park et al., 2004. A genetic link between cold responses and flowering time through FVE in Arabidopsis thaliana. Nat. Genet. 36: 167–171. [DOI] [PubMed] [Google Scholar]
- Kuo, M. H., and C. D. Allis, 1998. Roles of histone acetyltransferases and deacetylases in gene regulation. BioEssays 20: 615–626. [DOI] [PubMed] [Google Scholar]
- Lawrence, R. J., K. Earley, O. Pontes, M. Silva, Z. J. Chen et al., 2004. A concerted DNA methylation/histone methylation switch regulates rRNA gene dosage control and nucleolar dominance. Mol. Cell 13: 599–609. [DOI] [PubMed] [Google Scholar]
- Lee, H. S., J. Wang, L. Tian, H. Jiang, M. A. Black et al., 2004. Sensitivity of 70-mer oligonucleotides and cDNAs for microarray analysis of gene expression in Arabidopsis and Brasssica. Plant Biotechnol. J. 2: 45–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippman, Z., B. May, C. Yordan, T. Singer and R. Martienssen, 2003. Distinct mechanisms determine transposon inheritance and methylation via small interfering RNA and histone modification. PLoS Biol. 1: E67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, R. X., A. A. Postigo and D. C. Dean, 1998. Rb interacts with histone deacetylase to repress transcription. Cell 92: 463–473. [DOI] [PubMed] [Google Scholar]
- Meyerowitz, E. M., 1996. Plant development: local control, global patterning. Curr. Opin. Genet. Dev. 6: 475–479. [DOI] [PubMed] [Google Scholar]
- Murfett, J., X. J. Wang, G. Hagen and T. J. Guilfoyle, 2001. Identification of Arabidopsis histone deacetylase hda6 mutants that affect transgene expression. Plant Cell 13: 1047–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nan, X., H. H. Ng, C. A. Johnson, C. D. Laherty, B. M. Turner et al., 1998. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 393: 386–389. [DOI] [PubMed] [Google Scholar]
- Noma, K., C. D. Allis and S. I. Grewal, 2001. Transitions in distinct histone H3 methylation patterns at the heterochromatin domain boundaries. Science 293: 1150–1155. [DOI] [PubMed] [Google Scholar]
- Pandey, R., A. Muller, C. A. Napoli, D. A. Selinger, C. S. Pikaard et al., 2002. Analysis of histone acetyltransferase and histone deacetylase families of Arabidopsis thaliana suggests functional diversification of chromatin modification among multicellular eukaryotes. Nucleic Acids Res. 30: 5036–5055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Probst, A. V., M. Fagard, F. Proux, P. Mourrain, S. Boutet et al., 2004. Arabidopsis histone deacetylase HDA6 is required for maintenance of transcriptional gene silencing and determines nuclear organization of rDNA repeats. Plant Cell 16: 1021–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinowicz, P. D., W. R. McCombie and R. A. Martienssen, 2003. Gene enrichment in plant genomic shotgun libraries. Curr. Opin. Plant Biol. 6: 150–156. [DOI] [PubMed] [Google Scholar]
- Richards, E. J., and S. C. Elgin, 2002. Epigenetic codes for heterochromatin formation and silencing: rounding up the usual suspects. Cell 108: 489–500. [DOI] [PubMed] [Google Scholar]
- Robyr, D., Y. Suka, I. Xenarios, S. K. Kurdistani, A. Wang et al., 2002. Microarray deacetylation maps determine genome-wide functions for yeast histone deacetylases. Cell 109: 437–446. [DOI] [PubMed] [Google Scholar]
- Smith, J. S., C. B. Brachmann, I. Celic, M. A. Kenna, S. Muhammad et al., 2000. A phylogenetically conserved NAD+-dependent protein deacetylase activity in the Sir2 protein family. Proc. Natl. Acad. Sci. USA 97: 6658–6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockinger, E. J., Y. Mao, M. K. Regier, S. J. Triezenberg and M. F. Thomashow, 2001. Transcriptional adaptor and histone acetyltransferase proteins in Arabidopsis and their interactions with CBF1, a transcriptional activator involved in cold-regulated gene expression. Nucleic Acids Res. 29: 1524–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokes, T. L., and E. J. Richards, 2002. Induced instability of two Arabidopsis constitutive pathogen-response alleles. Proc. Natl. Acad. Sci. USA 99: 7792–7796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokes, T. L., B. N. Kunkel and E. J. Richards, 2002. Epigenetic variation in Arabidopsis disease resistance. Genes Dev. 16: 171–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian, L., and Z. J. Chen, 2001. Blocking histone deacetylation in Arabidopsis induces pleiotropic effects on plant gene regulation and development. Proc. Natl. Acad. Sci. USA 98: 200–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian, L., J. Wang, M. P. Fong, M. Chen, H. Cao et al., 2003. Genetic control of developmental changes induced by disruption of Arabidopsis histone deacetylase 1 (AtHD1) expression. Genetics 165: 399–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelauer, M., J. Wu, N. Suka and M. Grunstein, 2000. Global histone acetylation and deacetylation in yeast. Nature 408: 495–498. [DOI] [PubMed] [Google Scholar]
- Wu, K., L. Tian, C. Zhou, D. Brown and B. Miki, 2003. Repression of gene expression by Arabidopsis HD2 histone deacetylases. Plant J. 34: 241–247. [DOI] [PubMed] [Google Scholar]
- Yang, W. M., C. Inouye, Y. Zeng, D. Bearss and E. Seto, 1996. Transcriptional repression by YY1 is mediated by interaction with a mammalian homolog of the yeast global regulator RPD3. Proc. Natl. Acad. Sci. USA 93: 12845–12850. [DOI] [PMC free article] [PubMed] [Google Scholar]