

Abstract
Epoxide hydrolases play an important role in the biodegradation of organic compounds and are potentially useful in enantioselective biocatalysis. An analysis of various genomic databases revealed that about 20% of sequenced organisms contain one or more putative epoxide hydrolase genes. They were found in all domains of life, and many fungi and actinobacteria contain several putative epoxide hydrolase-encoding genes. Multiple sequence alignments of epoxide hydrolases with other known and putative α/β-hydrolase fold enzymes that possess a nucleophilic aspartate revealed that these enzymes can be classified into eight phylogenetic groups that all contain putative epoxide hydrolases. To determine their catalytic activities, 10 putative bacterial epoxide hydrolase genes and 2 known bacterial epoxide hydrolase genes were cloned and overexpressed in Escherichia coli. The production of active enzyme was strongly improved by fusion to the maltose binding protein (MalE), which prevented inclusion body formation and facilitated protein purification. Eight of the 12 fusion proteins were active toward one or more of the 21 epoxides that were tested, and they converted both terminal and nonterminal epoxides. Four of the new epoxide hydrolases showed an uncommon enantiopreference for meso-epoxides and/or terminal aromatic epoxides, which made them suitable for the production of enantiopure (S,S)-diols and (R)-epoxides. The results show that the expression of epoxide hydrolase genes that are detected by analyses of genomic databases is a useful strategy for obtaining new biocatalysts.
Enantiopure epoxides and vicinal diols are valuable intermediates in the synthesis of a number of pharmaceutical compounds. Epoxide hydrolases (EC 3.3.2.3) catalyze the conversion of epoxides to the corresponding diols. If they are enantioselective, they can be used to produce enantiopure epoxides by means of kinetic resolution (5). In the past, when only epoxide hydrolases from mammalian sources were known (12), the use of epoxide hydrolases in biocatalysis was hampered by their poor availability and insufficient catalytic performance, such as a low turnover rate or poor enantioselectivity. The potential for biocatalytic application of epoxide hydrolases was significantly increased with the discovery of microbial epoxide hydrolases (41), which are easier to produce in large quantities. The cloning and overexpression of several enantioselective epoxide hydrolases, e.g., from Agrobacterium radiobacter (35), Aspergillus niger (3), and potato plants (40), not only facilitated large-scale production of these enzymes but also made it possible to improve their biocatalytic properties by site-directed or random mutagenesis (34, 36, 43).
Since many microbial genome sequences are available in the public domain, it is useful to screen these databases for genes that might encode new enzymes with interesting properties. Novel epoxide hydrolases can be identified by performing a BLAST search of the genomic databases, using amino acid sequences of known epoxide hydrolases as queries. This approach will result in putative epoxide hydrolases but also in amino acid sequences from structurally and mechanistically related enzymes, such as esterases and dehalogenases (33), which can be filtered out using conserved epoxide hydrolase sequence motifs that define the active site (Fig. 1). The putative epoxide hydrolase-encoding genes can subsequently be cloned and overexpressed in a host with no endogenous epoxide hydrolase activity, such as Escherichia coli, and the activity of the encoded proteins can be tested.
FIG. 1.

Structure and mechanism of α/β-hydrolase fold epoxide hydrolases. The active site shown in panels A and B is from the epoxide hydrolase from A. radiobacter AD1 (EchA) (31, 35, 36). (A) Tyrosine-assisted ring opening of the epoxide by the nucleophilic aspartate. (B) Hydrolysis of the alkyl-enzyme intermediate by an activated water molecule. (C) General topology and structure of α/β-hydrolase fold epoxide hydrolases. I, main domain with α/β-hydrolase fold; II, cap domain; 1, catalytic nucleophile (D107 in EchA); 2, histidine base (H275); 3, charge relay acid, which can be positioned at either 3a or 3b (D246 is at position 3b); 4, ring-opening tyrosines (Y152/Y215); 5, H-G-X-P motif (the X is usually an aromatic residue in epoxide hydrolases [W38]); 6, G-X-Sm-X-S/T motif. The region of helix α4a can be nonhelical or missing in some epoxide hydrolases. The length of the loop between helixes α6 and α7 can vary considerably. Extensions with additional domains of 40 to 300 amino acids can occur at both the N and C termini.
Most epoxide hydrolases for which sequence information is presently available are members of the α/β-hydrolase fold family, to which lipases, esterases, and haloalkane dehalogenases also belong. These enzymes consist of a main domain that is composed of a central β-sheet surrounded by α-helices and a variable cap domain positioned on top of the substrate binding site (Fig. 1) (33). They use a catalytic mechanism that involves an Asp/Ser/Cys-His-Asp/Glu (nucleophile-histidine-acid) catalytic triad located on top loops of the main domain. The positions of these residues are structurally conserved in the order nucleophile-histidine-acid (Fig. 1A and B) (33), but in the primary amino acid sequence the order is nucleophile-acid-histidine, with the acid located either in front of the cap domain (Fig. 1C, position 3a) or after the cap domain (Fig. 1C, position 3b).
In the case of α/β-hydrolase fold epoxide hydrolases, the catalytic triad nucleophile is an invariable aspartate that opens the epoxide ring by nucleophilic attack (Fig. 1A) (4, 35). The ring-opening reaction is assisted by two conserved tyrosines that are located in the cap domain (Fig. 1C) (6, 36). The resulting alkyl-enzyme intermediate is subsequently hydrolyzed by a water molecule that is activated by a histidine that functions as a proton acceptor and, in turn, is assisted by the acidic residue (Fig. 1B). The negative charge that develops on the carbonyl oxygen of the nucleophilic aspartate during hydrolysis of the alkyl enzyme intermediate is stabilized by two backbone amides that are contributed by the residue following the catalytic nucleophile and residue X in a conserved H-G-X-P motif (Fig. 1C). This motif is located between strand β3 and helix α1 (Fig. 1C, position 5) and is conserved in haloalkane dehalogenases and epoxide hydrolases. Residue X is usually an aromatic residue in epoxide hydrolases, whereas it is an asparagine or a glutamate in haloalkane dehalogenases. The side chain of the amino acid at this position lines the active site (31, 51). Between the H-G-X-P motif and the catalytic nucleophile, there is a conserved G-X-Sm-X-S/T motif of unknown function (Fig. 1C, position 6).
This paper describes the screening of various genomic databases for epoxide hydrolases of the α/β-hydrolase fold family. Based on phylogenetic analysis of the resulting sequences and comparison to other α/β-hydrolase fold enzymes that have a nucleophilic aspartate, the epoxide hydrolases were divided into different phylogenetic groups. Ten of these putative epoxide hydrolases, together with two known bacterial epoxide hydrolases, were cloned and overexpressed in E. coli and subsequently tested for their biocatalytic potential.
MATERIALS AND METHODS
Materials.
The epoxides that were used in this study are indicated in Fig. 2. The same numbering is used for the epoxides and the corresponding diols, with the extension “a” for the diol. Epoxides 2, 7, 9, 10, 20, and 25 (Fig. 2), racemic and enantiopure diols 20a and 23a, and l-epinephrine were obtained from Acros (Geel, Belgium). Epoxide 5 was obtained from Enzis (Groningen, The Netherlands), and epoxides 6, 11, 12, 15 to 17, 19, and 23 were obtained from Aldrich. Furthermore, epoxides 8 and 14 were obtained from Lancaster (Frankfurt am Main, Germany), epoxides 13, 21, and 24 were obtained from Fluka, epoxide 12 was obtained from Tokyo Kasei, and epoxide 22 was obtained from Sigma. Epoxides 1, 3, and 4 were synthesized from para-methylbenzaldehyde, para-chlorobenzaldehyde, and meta-chlorobenzaldehyde, respectively. Chiral high-performance liquid chromatography (HPLC) columns were obtained from Diacel. A G-TA capillary column was obtained from Altech, and a CP Chirasil Dex CB column was obtained from Chrompack. All DNA-modifying enzymes and ampicillin were obtained from Roche Molecular Biochemicals, except for Pwo DNA polymerase (GENAXIS GmbH) and Pfu DNA polymerase (Stratagene). Other medium components were obtained from Merck and Difco. The plasmid vector pMAL-c2x and amylose resin were obtained from New England Biolabs. E. coli strain TOP10 was obtained from Invitrogen Inc., and strain BL21(DE3)/pLysS Rosetta was obtained from Novagen. Oligonucleotides were obtained from Eurosequence or Sigma-Genosys, and IPTG (isopropyl-β-d-thiogalactopyranoside) was obtained from Eurogentec. The constructs were sequenced at the Biomedical Centre in Groningen, using the malE (forward) and M13/pUC (reverse) primers.
FIG. 2.

Epoxide substrates used in this study. 1, para-methylstyrene oxide; 2, styrene oxide; 3, para-chlorostyrene oxide; 4, meta-chlorostyrene oxide; 5, para-nitrostyrene oxide; 6, cis-stilbene oxide; 7, trans-stilbene oxide; 8, chalcone-α,β-epoxide; 9, phenyl glycidyl ether; 10, para-nitrophenyl glycidyl ether; 11, epichlorohydrin; 12, epibromohydrin; 13, vinyl oxirane; 14, tert-butyloxirane; 15, 1,2-epoxybutane; 16, 1,2-epoxypentane; 17, 1,2-epoxyhexane; 18, 1,2-epoxyheptane; 19, 1,2-epoxyoctane; 20, cis-2,3-epoxybutane; 21, trans-2,3-epoxybutane; 22, fosfomycin; 23, cyclohexene oxide; 24, limonene-1,2-epoxide; 25, 3,4-epoxytetrahydrofuran.
BLAST search, multiple sequence alignments, and phylogenetic analysis.
In order to obtain putative epoxide hydrolase-encoding sequences, we screened the Pedant (http://pedant.gsf.de), NCBI (http://www.ncbi.nlm.nih.gov), TIGR (http://www.tigr.org), and Sanger (http://www.sanger.ac.uk) sequence databases and the Rhodococcus RHA1 genome database (http://www.rhodococcus.ca). Epoxide hydrolases for which the activity and sequence are described in the literature served as query sequences. The resulting BLAST hits were manually screened for the presence of the putative epoxide hydrolase active-site residues and the motifs depicted in Fig. 1C. Sequences that contained the catalytic triad, at least one ring-opening tyrosine, and motifs 5 and 6 were annotated as putative epoxide hydrolases. All putative epoxide hydrolase sequences were aligned, together with the known epoxide hydrolase sequences and a number of known and putative haloalkane and haloacetate dehalogenases that also belong to the α/β-hydrolase fold family and possess an invariable aspartate as the catalytic nucleophile. The resulting phylogenetic tree was displayed using TreeView and edited. The multiple sequence alignments were displayed as bit scores for each position, using the WebLogo sequence generator (http://weblogo.berkeley.edu).
Cloning of epoxide hydrolase genes.
Genes encoding putative epoxide hydrolases were amplified from respective sources of genomic DNA (see below), using the primer pairs described in the supplemental material, and subsequently cloned into the pMAL-c2x plasmid vector. Upon bacterial expression of these constructs, epoxide hydrolases were obtained as fusion proteins with a maltose binding protein (MalE) domain linked to the N terminus of the epoxide hydrolase domain via a polyasparagine linker and a small decapeptide. The following DNA sources served as templates: whole-cell material from an overnight culture (Bsueh, Bfueh1, Npueh1, Npueh2, and Ppueh), genomic DNA added up to 0.05 ng μl−1 (Draeh, Rpaeh2, Scoeh6, and Tfueh), and plasmid DNA (AraEchA [43], Coreh [29], and MtuEphF). Primers were used at 0.4 nM in a reaction mixture with a 0.2 mM concentration of each deoxynucleoside triphosphate and 0.025 U μl−1 Pwo DNA polymerase or, in the case of DNAs with high G+C contents (see the supplemental material), 0.05 U μl−1 Pfu polymerase. The reactions with high-G+C-content DNA were performed in the presence of 5% (vol/vol) dimethyl sulfoxide (DMSO). For genes with no high G+C content, the temperature program used was 15 min at 94°C without polymerase, followed by 30 cycles of 60 s at 94°C, 45 s at 58°C, and 70 s at 72°C and a final step of 4 min at 72°C. For genes with high G+C content, the temperature program used was 15 min at 94°C without polymerase, followed by 30 cycles of 60 s at 95°C, 45 s at 68°C − 0.5°C cycle−1 (each cycle the temperature was lowered 0.5°C), and 120 s at 72°C and a final step of 4 min at 72°C. For PCR amplification of MtuEphF, the following program was used: 15 min at 94°C without polymerase, followed by 30 cycles of 60 s at 94°C, 45 s at 50°C, and 70 s at 68°C and a final step of 4 min at 68°C. The PCR products were digested with various restriction enzymes (see the supplemental material for details) and subsequently ligated into BamHI-HindIII-, EcoRI-HindIII-, EcoRI-PstI-, or XbaI-HindIII-digested pMAL-c2x plasmid DNA, using T4 DNA ligase. The ligation mixtures were transformed into E. coli TOP10 cells by electroporation. The transformants were plated on LB medium containing ampicillin. Colonies were checked for inserts by using PCR with Taq polymerase and with colony material as the template. Positive colonies were used to inoculate 5 ml of liquid LB medium and grown overnight at 37°C. Plasmid DNA was extracted using a plasmid purification kit from Roche, and the inserts were sequenced.
Protein expression and purification.
Induction conditions for the expression of fusion proteins were optimized by adding up to 0.4 mM IPTG to a 5-ml culture with an optical density at 600 nm (OD600) of ∼0.2, followed by overnight incubation at various temperatures ranging from 8 to 37°C. The cells were harvested and resuspended in 0.4 ml TEDANG buffer (20 mM Tris-HCl, pH 7.4, 1 mM EDTA, 1 mM dithiothreitol, 0.02% [wt/vol] NaN3, 200 mM NaCl, 10% [vol/vol] glycerol) at 4°C. After brief sonication, a cell extract (CFE) was obtained by centrifugation. Cell extracts were analyzed for expression by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. The levels of expression were determined from Coomassie-stained gels with the GelPro Analyzer program. Total protein contents were determined using the Bradford reagent. For Bsueh, expression was optimized further by transforming the pMAL-c2x-Bsueh construct into E. coli BL21(DE3)/pLysS Rosetta, which has elevated levels of rare tRNAs that could enhance expression.
Preparative-scale production of proteins was achieved by induction of 1- to 3-liter cultures at an OD600 of ∼0.2 with 0.4 mM IPTG, followed by overnight incubation at temperatures ranging from 8 to 30°C. Cells were harvested by centrifugation, washed, and resuspended in TEDANG buffer at 4°C. The cells were lysed by sonication, and cell extracts were obtained by centrifugation at 200,000 × g for 90 min.
Purification of proteins from CFE was achieved using amylose resin to selectively bind the fusion proteins. Resin with bound fusion protein was subsequently poured into a column, and after the column was washed with TEDANG buffer, the fusion protein was eluted with the same buffer containing 10 mM maltose. Fractions containing active fusion protein were pooled and concentrated. The protein content of the purified enzyme fractions was determined by A280 measurements. The extinction coefficient was calculated from the amino acid sequences of the MalE-EH fusion proteins, using Lasergene-Protean.
Spectrophotometric epoxide hydrolase assays.
All continuous spectrophotometric measurements were performed on a Kontron Uvikon 930 UV/VIS spectrophotometer. Epoxide hydrolase activities toward para-nitrostyrene oxide (pNSO; epoxide 5 in Fig. 2) and para-nitrophenyl glycidyl ether (pNPGE; epoxide 10) were determined in 100 mM Tris-SO4 (pH 7.5) as described previously (43). Errors in values of initial activities calculated from the decrease (pNSO, 310 nm) or increase (pNPGE, 350 nm) in absorbance were <10%, as indicated by duplicate measurements. The detection limit for these initial activities was 0.002 U mg−1 (units are defined in μmol min−1).
The steady-state kinetic parameters of the purified novel epoxide hydrolases were determined by measuring progress curves for the conversion of enantiopure pNSO (epoxide 5) and pNPGE (epoxide 10). The substrate was added at a concentration of up to 0.5 mM, with a final concentration of DMSO of <1% (vol/vol). A suitable amount of purified protein was added to start the reaction. The spectrophotometric conversion traces for the separate enantiomers of epoxides 5 and 10 were directly fitted to Michaelis-Menten kinetics, as described before, to obtain kcat, Km, and kcat/Km values (36, 43). In case the Km was much higher than the substrate concentration used, the spectrophotometric traces were fitted according to first-order kinetics, and the first-order rate constant equals kcat/Km. In this case, only lower limits of kcat and Km were obtained. More details on the fitting procedures used can be found in the supplemental material. Errors in values for kcat, Km, and kcat/Km were <10%, as indicated by duplicate measurements.
Substrate profiling of the various epoxide hydrolases was done using the adrenaline test (46). Initial testing of various vicinal diols in a reaction with IO4− and adrenaline revealed that adenochrome was rapidly produced with all diols that are formed from the epoxides shown in Fig. 2, except for diols derived from glycidyl ethers (epoxides 9 and 10), which do not react fast enough with IO4− (<50% conversion in 45 min). Substrates were added at concentrations of up to 1 mM from 50 mM stock solutions in water (epoxide 22) or DMSO (all other substrates) to 100 μl 100 mM sodium phosphate (pH 8.0) in microtiter plates. Purified protein or CFE was transferred to 100 mM sodium phosphate (pH 8.0) by desalting over an Econopack desalting column in order to remove components that can be oxidized by IO4−, such as glycerol and Tris. After 1 h of incubation of the enzyme with the substrate at room temperature, up to 1 mM NaIO4 was added from a 10 mM stock solution, and the mixture was incubated for 45 min, after which l-epinephrine was added to determine the amount of remaining NaIO4. The A490 value, which is a measure of the amount of adenochrome formed by the reaction with remaining IO4− (46), was determined for each well. After correction for incubations without substrate but with enzyme and visual inspection of the microtiter plates, the activities toward the different substrates were scored as high (++, 60 to 100% conversion), intermediate (+, 10 to 60% conversion), or low (−, <10% conversion).
Kinetic resolution.
The enantioselectivities of the enzymes for various epoxides (Fig. 2) were determined by performing a kinetic resolution of 0.5 to 2 mM of racemic substrate in 12 to 20 ml of 100 mM Tris-SO4, pH 7.5, at room temperature or 30°C. Sampling, chiral gas chromatography (GC), and HPLC analysis were done as described before (43). The conditions for chiral GC and HPLC analysis are described in the supplemental material. The E values were calculated from the resulting data, as described before (26, 43).
Conversion of meso-epoxides.
The activities of the expressed epoxide hydrolases toward meso-epoxides 20 and 23 were determined at a 2 mM substrate concentration in 20 ml 100 mM Tris-SO4, pH 7.5. The reaction was started by adding a suitable amount of CFE or purified enzyme, and samples were taken in time and extracted with hexane containing an internal standard. The samples were analyzed by GC (see the supplemental material for the conditions used). Errors in values of initial activities were <10%, as indicated by duplicate measurements. The detection limit for these conversions was 0.002 μmol min−1 mg−1.
The diastereomeric composition of the product resulting from the conversion of epoxide 20 to 2,3-butanediol (diol 20a) was determined by dimethoxypropane extraction of a 2-ml sample from an incubation of 5 mM of substrate with enzyme. The organic phase was incubated with Amberlite/H+ resin for 1 h and subsequently dried over a short column of anhydrous MgSO4. The same procedure was performed with commercially available reference compounds of the three different diastereomers of diol 20a. Analysis was done by chiral GC (see the supplemental material for GC methods).
The diastereomeric composition of 1,2-cyclohexanediol (diol 23a) was determined by extracting 2 ml of a mixture of 10 mM substrate and enzyme with diethyl ether. The organic phase was dried over a short column of anhydrous MgSO4. Commercially obtained samples of the three different diastereomers of diol 23a were used as reference compounds. Analysis was done by chiral GC (see the supplemental material for the conditions used).
The E values for the formation of (R,R)- and (S,S)-diols from meso-epoxides were calculated from the ee of the product (eep) by using equation 1, as follows:
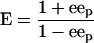 |
(1) |
RESULTS AND DISCUSSION
Phylogenetic analysis.
To identify the suitability of genomic sequences as a source of new biocatalytically useful epoxide hydrolases, we first made an inventory of potential targets for overexpression in E. coli by screening various eukaryotic and prokaryotic genome databases for the presence of putative epoxide hydrolase-encoding sequences. The amino acid sequences of epoxide hydrolases for which the activity is known were used as query sequences. In total, 438 sequenced organisms were screened for the presence of putative epoxide hydrolases (Table 1). The obtained hits were manually screened for the sequence motifs that are characteristic of epoxide hydrolases (Fig. 1C) in order to filter out false-positive results. All sequences that contained the catalytic triad, at least one ring-opening tyrosine, and the H-G-X-P and G-X-Sm-X-S/T motifs were annotated as putative epoxide hydrolases. In this way, 292 amino acid sequences of putative epoxide hydrolases were identified in 93 different organisms for which no epoxide hydrolase activity had previously been reported. These sequences were subsequently aligned with the amino acid sequences of other α/β-hydrolase fold enzymes that have the conserved nucleophilic aspartate, including 42 epoxide hydrolases with known activity, haloalkane dehalogenases, and fluoroacetate dehalogenases. Analysis of the multiple sequence alignment revealed that 18 hydrolases that were annotated in the databases as epoxide hydrolases most likely are fluoroacetate dehalogenases (see below). The remaining 274 putative epoxide hydrolases originated from 91 taxonomically different organisms (Table 1). From this set, 35 sequences were removed before further phylogenetic analysis since they were 100% identical to sequences from closely related organisms. A multiple sequence alignment was done with the remaining 239 putative epoxide hydrolases found in sequencing projects, 6 putative epoxide hydrolases from other sources, 42 known epoxide hydrolases, 19 (putative) fluoroacetate dehalogenases, and 8 (putative) haloalkane dehalogenases.
TABLE 1.
Epoxide hydrolases in various classes of sequenced organisms
| Domain | Class | No. of organisms sequenced | No. of organisms in which an EH(s) was detecteda | Avg no. of EHs per EH-containing strainb | Subclass(es) with many EHs | No. of EHs used in alignmentc |
|---|---|---|---|---|---|---|
| Archaea | 24 | 1 (4) | 1 (1) | 1 | ||
| Bacteria | Actinobacteria | 30 | 18 (60) | 5 (1-11) | Mycobacteria, Streptomycetes | 67 |
| Chlamydiae | 11 | |||||
| Cyanobacteria | 16 | 7 (44) | 1.6 (1-3) | 11 | ||
| Firmicutes | 103 | 2 (2) | 1 (1) | 2 | ||
| α-Proteobacteria | 39 | 12 (31) | 3.3 (1-12) | Rhizobiaceae | 41 | |
| β-Proteobacteria | 25 | 12 (48) | 3.3 (1-9) | Burkholderiaceae | 38 | |
| δ-Proteobacteria | 8 | 1 (13) | 1 (1) | 2 | ||
| ɛ-Proteobacteria | 6 | |||||
| γ-Proteobacteria | 91 | 8 (9) | 2.9 (1-7) | Pseudomonadaceae | 24 | |
| Spirochaetales | 6 | 1 (17) | 3 (3) | 3 | ||
| Others | 30 | 4 (13) | 1.5 (1-2) | 13 | ||
| Eukaryota | Fungi/metazoa | 37 | 21 (57) | 2.5 (1-10) | Ascomycota | 59 |
| Others | 10 | 4 (40) | 2.5 (1-4) | 26 |
The percentage of epoxide hydrolase-containing organisms within each class is indicated in parentheses.
The range of putative epoxide hydrolase genes per strain is indicated in parentheses.
Including known epoxide hydrolases.
The database search results showed that 20% of the organisms that have been sequenced thus far contain one or more putative epoxide hydrolases. However, in some phylogenetic (sub)classes, such as actinobacteria and fungi, >50% of the sequenced organisms contain at least one putative epoxide hydrolase, whereas in other (sub)classes, such as firmicutes and chlamydiae, they are hardly present (Table 1). Most microbial epoxide hydrolases described in the literature originate from actinobacteria (41) and fungi (39), and their epoxide hydrolase activity was determined with whole cells (39, 41). Since many of the sequenced actinobacteria and fungi appear to contain more than one putative epoxide hydrolase (Table 1), reported activities and enantioselectivities could well result from more than one enzyme.
The phylogenetic tree that resulted from the final multiple sequence alignment identified eight different phylogenetic groups (Fig. 3). On average, the α/β-hydrolase fold and cap domain sequences together (as indicated in Fig. 1C) were 290 to 310 amino acids long (Table 2). In some phylogenetic groups, the average total sequence length was significantly larger, which was, in most cases, the result of an N- or C-terminal extension. The nucleophilic aspartate, the histidine, and the H-G-X-P and G-X-Sm-X-S/T motifs aligned perfectly for all sequences. For all putative epoxide hydrolases, two ring-opening tyrosines were identified in the cap domain. The position of the first tyrosine (Y152 in EchA) (Fig. 1) especially varied considerably between the different groups, but it was conserved within each group. The charge relay acid was predominantly an aspartate, usually located at position b (Fig. 1; Table 2). The average amino acid sequence identity of the complete sequences was only 14%, or 19% if only sequences with known activity were included, so a low sequence identity appears to be normal for this class of enzymes. The relatively long nodes compared to the branches of the phylogenetic tree also indicate low overall identity, even within a group (20 to 35%) (Fig. 3). Apparently, only a few residues are crucial for proper performance of the various enzymes, and the overall α/β-hydrolase fold and active-site geometry allow significant sequence variation, as observed in previous studies on phylogenetic relationships between functionally different α/β-hydrolase fold enzymes (8, 9). This is most strikingly demonstrated by the fact that the amino acid sequences of the epoxide hydrolases from A. radiobacter AD1 (35) and A. niger (3) are <10% identical, but the α/β-hydrolase fold part of their X-ray structures and their active sites are virtually superimposable (31, 51). Most of the phylogenetic groups of epoxide hydrolases consist of homologs from evolutionarily very distant organisms. Since the overall identity within each group is rather low, and since the high identities that do occur are between enzymes that originated from closely related organisms, the majority of these genes are probably orthologs that were separated during speciation. Since bacteria are biased toward deleting genes that are no longer needed (28), several specialized microorganisms such as lactic acid bacteria probably lost their epoxide hydrolase genes during evolution. Indeed, most epoxide hydrolases occur in organisms with large genomes (>8 Mb), even though the size of their collective genomes is similar to that of the small-genome organisms (<2 Mb) (Fig. 4). Lateral transfer may play a role in some cases, as suggested by the situation encountered in Mycobacterium tuberculosis, which contains 11 α/β-fold hydrolases (3 dehalogenases and 8 epoxide hydrolases) in a 4-Mb genome that harbors less than 4,000 genes in total.
FIG. 3.

Phylogenetic tree of α/β-hydrolase fold enzymes with a nucleophilic aspartate. Symbols: •, enzymes for which an epoxide hydrolase function is described in the literature or databases; ○, enzymes identified in this study and active with one or more epoxides; □, putative epoxide hydrolases for which no epoxide substrate was found. The black nodes are (putative) epoxide hydrolases, red nodes are (putative) haloacetate dehalogenases, green nodes are (putative) haloalkane dehalogenases, and orange nodes in group 3 are putative epoxide hydrolases with a C-terminal part that is homologous to short-chain dehydrogenase/reductase family proteins (SDR proteins). Groups 1 to 8 are described in Table 2.
TABLE 2.
Phylogenetic groups of α/β-hydrolase fold enzymes with a nucleophilic aspartate
| Groupa | Source(s) | Activity (putative) | No. of members (known, new) | Example(s) of members with known activity | Charge relay acid(s) (positionb) (frequency [%]) | Avg size (amino acids) |
|---|---|---|---|---|---|---|
| 1 | Bacterial, fungal | Epoxide hydrolase | 22 (1, 0) | BD8876 (49) | D (b) (100) | 300 |
| 1A | Bacterial | Epoxide hydrolase | 12 (0, 0) | D (b) (100) | 344 | |
| 2 | Bacterial, archaeal | Epoxide hydrolase | 26 (0, 3) | Bsueh, Draeh, Npueh 1 | D (b) (100) | 295 |
| Nematode | 4 (0, 0) | 367 | ||||
| 3 | Fungal | Epoxide hydrolase | 11 (0, 0) | D (b) (100) | 342 | |
| Bacterial | Epoxide hydrolase | 12 (0, 0) | 313 | |||
| Epoxide hydrolase-SDR | 12 (0, 0) | 591 | ||||
| 4 | Bacterial | Epoxide hydrolase | 27 (0, 1) | Npueh2, Ppueh | D (b) (100) | 308 |
| Fungal | Epoxide hydrolase | 6 (0, 0) | 295 | |||
| Bacterial | Haloalkane dehalogenase | 8 (5, 0) | Xanthobacter autotrophicus DhlA (23) | D (b) (50), E (a) (50) | 299 | |
| 5 | Animal | Epoxide hydrolase | 19 (13, 0) | Human micosomal EH (22), Manduca sexta juvenile hormone EH (18) | D (b) (25), E (b) (75) | 459 |
| Fungal | 22 (6, 0) | A. niger EH (3), R. glutinis EH (45) | 418 | |||
| Bacterial | 44 (4, 1) | BD9883 (49), Bfueh1 | 399 | |||
| 6 | Bacterial, fungal | Epoxide hydrolase | 14 (2, 0) | Corynebacterium sp. strain C12 EH (29), BD8877 (49) | D (a) (64), E (a) (36) | 293 |
| Bacterial | Haloacetate dehalogenase | 19 (1, 0) | Moraxella sp. DehL (24) | D (a) (100) | 297 | |
| 7 | Bacterial | Epoxide hydrolase | 4 (2, 0) | Agrobacterium radiobacter AD1 EH (35), BD10721 (49) | D (b) (100) | 292 |
| 8 | Animal | Epoxide hydrolase | 4 (3, 0) | Human soluble EH (10) | D (b) (100) | 549 |
| Plant | 15 (11, 0) | Potato EH (40) | 337 | |||
| Bacterial | 26 (0, 1) | Rpaeh2 | 329 |
FIG. 4.
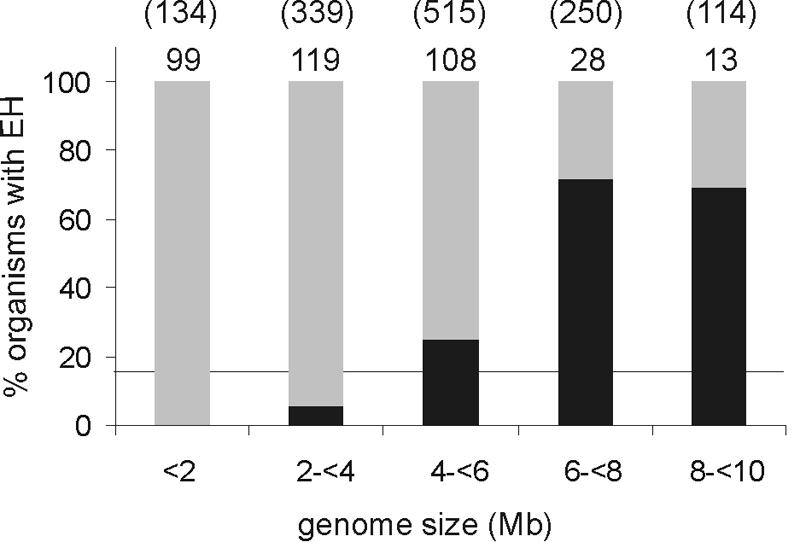
Occurrence of putative epoxide hydrolases in prokaryotes and archaea in relation to genome size. The number of organisms with a given genome size is indicated above each percentage bar, with the size of the collective genomes in this category shown in parentheses (in Mb). The percentages of organisms that have at least one putative epoxide hydrolase are indicated in black. The horizontal line indicates the average number of epoxide hydrolase-containing organisms (17.5%).
In order to investigate whether the phylogenetic study performed here was biased by the available sequenced genomes, a BLAST search with the same 42 query sequences of epoxide hydrolases with known activity was done on the open reading frames that were identified in the Sargasso Sea environmental genome sequencing project (44). In total, 61 complete putative epoxide hydrolase sequences were retrieved, which were all different from the 42 known epoxide hydrolases. A multiple sequence alignment including the 61 environmental sequences yielded the same eight phylogenetic groups. Most of the environmental sequences clustered either with other environmental sequences or with putative epoxide hydrolases from Burkholderia spp., which are very abundant in the Sargasso Sea (44). Since many of the sequences retrieved from the Sargasso Sea probably are missing part of their N and/or C terminus, they were not included in further phylogenetic studies.
Characteristics of phylogenetic groups.
The members of phylogenetic group 1 epoxide hydrolases originated predominantly from proteobacteria (>70% of the group members). This group includes an epoxide hydrolase that was recently identified in an environmental gene library (49). The members of subgroup 1A are, on average, 40 amino acids longer than the other members of this group. Using SignalP software (http://www.cbs.dtu.dk/services/SignalP), these extra amino acids were identified as an N-terminal signal peptide for protein excretion. A more thorough analysis of the various characteristic signal peptide regions suggested that these proteins are secreted via a twin arginine translocation pathway (1). This pathway usually translocates completely folded proteins, including cofactor-dependent enzymes, across cellular membranes (14).
Group 2 contains epoxide hydrolases from bacterial, archaeal, and eukaryotic origins. Some of the eukaryotic epoxide hydrolases originated from multicellular organisms. Thus far, only two classes of eukaryotic epoxide hydrolases from multicellular organisms are known, which are the microsomal/juvenile hormone epoxide hydrolases (group 5) and the cytosolic/plant epoxide hydrolases (group 8). The putative eukaryotic epoxide hydrolases that cluster in group 2 have an N-terminal extension of unknown function and are therefore somewhat longer than the enzymes in this group that are of prokaryotic origin (Table 2). Group 2 contains a relatively large number of putative epoxide hydrolases that originated from cyanobacteria (20%).
Group 3 consists predominantly of putative epoxide hydrolases from actinobacteria, β-proteobacteria, and fungi. About one-third of the group members consist of an epoxide hydrolase domain (N-terminal) and a C-terminal domain that is homologous to SDR proteins (Table 2). The epoxide hydrolase domain of the fungal epoxide hydrolase sequences is, on average, 34 amino acids longer than that of the other group members.
In group 4, both epoxide hydrolases and haloalkane dehalogenases appear to be present (Fig. 3; Table 2). These structurally related enzymes, which also have similar mechanisms (2), can be distinguished at the amino acid level by the presence of the ring-opening tyrosines, which are missing in the haloalkane dehalogenases. Since these tyrosines do not always align perfectly, the H-G-X-P motif, which is located between sheet β3 and helix α1 (Fig. 1), is more suitable for distinguishing epoxide hydrolases from haloalkane dehalogenases. The main chain amide of the X residue is part of the oxyanion hole in both enzyme classes (Fig. 1B). The side chain is part of the wall of the active site and can therefore interact with the substrate (31, 51). In epoxide hydrolases, the X residue is always an aromatic amino acid, whereas in haloalkane dehalogenases it is a more hydrophilic or even charged residue such as asparagine or glutamate (Fig. 5A). Although the exact function of the motif is unknown, it can possibly be used to distinguish between epoxide hydrolases and haloalkane dehalogenases. The fungal enzymes present in this group consist predominantly of homologous epoxide hydrolase sequences of various Saccharomyces spp. (Fig. 3).
FIG. 5.

Multiple sequence alignments of conserved regions in functionally different α/β-hydrolase fold enzymes, represented as bit scores for each position, with the total height indicating the degree of conservation (maximum level, 4.32) and the relative heights of different symbols at the same position indicating the frequencies of the amino acids at that position. (A) H-G-X-P region in epoxide hydrolases and haloalkane dehalogenases. (B) Region around the nucleophilic aspartate in epoxide hydrolases and fluoroacetate dehalogenases.
More than half of the 42 known epoxide hydrolases that were included in the multiple sequence alignment clustered in group 5 (Fig. 3; Table 2). This group includes the well-known mammalian microsomal epoxide hydrolases (22) and the juvenile hormone epoxide hydrolases that are present in many insects (18). Group 5 is the best-defined group in the phylogenetic tree (Fig. 3). Its members are, on average, 100 (bacterial), 120 (fungal), and 150 (higher eukaryotes) amino acids longer than most other epoxide hydrolases (Table 2), which is mainly due to a large N-terminal extension. The mammalian and insect epoxide hydrolases of this group are membrane bound by an N-terminal membrane anchor (20). The fungal and bacterial epoxide hydrolases of this group do not contain this membrane anchor and are therefore probably soluble. The N-terminal extension is important for the phylogenetic distinction of the group 5 epoxide hydrolases from all others, but the differences are also clearly present in the α/β-hydrolase main domain. In contrast to the case for most epoxide hydrolases, the charge relay acid in this group of epoxide hydrolases is a glutamate in 75% of the cases instead of an aspartate (Table 2).
Group 6 contains both fluoroacetate dehalogenases and epoxide hydrolases. For all members of this group, the charge relay aspartate is located at position a instead of the more common position b (Fig. 1; Table 2). The putative fluoroacetate dehalogenase sequences all grouped together with the fluoroacetate dehalogenase from Moraxella sp. (24) (Fig. 3). These sequences contain three consecutive arginines distal from the nucleophilic aspartate in a DRXXRXXXR motif, whereas epoxide hydrolases usually have a conserved aromatic residue distally flanking the catalytic nucleophile (Fig. 5B). It was proposed that one of these arginines plays a role in the binding of the acid part of the fluoroacetate substrate (25). Furthermore, all of these sequences have the fluoroacetate dehalogenase from Moraxella sp. (24) as their highest scoring BLAST hit for proteins that have known activity. These putative haloacetate dehalogenases have often erroneously been annotated as epoxide hydrolases, including in a recent phylogenetic study of epoxide hydrolases (9). The presence of a conserved tyrosine in the cap domains of haloacetate dehalogenases that aligns perfectly with one of the ring-opening tyrosines of epoxide hydrolases contributes to the confusion. The epoxide hydrolase sequences in group 7 are similar to those in group 6. The first conserved ring-opening tyrosine and the charge relay aspartate, however, are located at different positions (Fig. 1; Table 2).
Group 8 contains a large number of known epoxide hydrolases from plants and mammals, including the well-known mammalian cytosolic epoxide hydrolases (Fig. 3; Table 2). There are no sequences originating from other, lower eukaryotic organisms, such as fungi, in this group. The plant epoxide hydrolases occur only in this group. The cap domain of this group of epoxide hydrolases has a 30-amino-acid excursion between helixes α6 and α7 (Fig. 1), resulting in a larger size of the epoxide hydrolase segment (Table 2).
Overexpression and purification of putative epoxide hydrolases.
Ten putative epoxide hydrolase-encoding genes from different phylogenetic groups and the bacterial epoxide hydrolases from A. radiobacter (35) (AraEchA) and Corynebacterium sp. strain C12 (29) (Coreh) were selected to be cloned and overexpressed in E. coli (Table 3). Since initial attempts to overexpress the epoxide hydrolases from Mycobacterium tuberculosis H37v (MtuEphF) and Pseudomonas putida KT2440 (Ppueh) in E. coli using the pGEF system (38) resulted in inclusion bodies (data not shown), it was decided to overexpress the enzymes as maltose binding protein-epoxide hydrolase fusion proteins (MalE-EH). The MalE tag was expected to facilitate proper folding of the epoxide hydrolased in E. coli (37). Moreover, it serves as a convenient purification tag. The MalE-EH fusion proteins were produced in E. coli at levels of 10 to 60% of the total soluble protein in CFEs, including the enzymes for which expression yielded inclusion bodies when no MalE was fused to the epoxide hydrolase (Table 3). The CFE prepared from an expression culture of the MalE-Bsueh (originating from Bacillus subtilis) construct in E. coli TOP10 cells resulted in rather low specific activities (0.03 μmol min−1 mg−1), possibly due to poor expression levels caused by the large number of rare codons present in the Bsueh gene. Expression of the MalE-Bsueh protein in E. coli BL21(DE3)/pLysS Rosetta, an E. coli variant that has increased levels of rare tRNAs, resulted in a twofold improvement in specific activity in the cell extract.
TABLE 3.
Properties of MalE-EH fusion proteins
| Enzyme | Origin | Groupg | Length (amino acids) | Molecular mass (kDa)a | Putative active-site residue
|
Expression level (% of total protein) | pNSO (compound 5) parameters
|
pNPGE (compound 10) parameters
|
|||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Nucleophile | Histidine | Acid | Tyrosine(s) | Activity (U mg−1) | Ed,f | Activity (U mg−1) | Ed,f | ||||||
| AraEchAb | Agrobacterium radiobacter AD1 | 7 | 294 | 34.1 | D107 | H275 | D246 | Y152, Y215 | 41 | 1.0 | 56 (R) | 4.0 | 3.4 (S) |
| Bsuehc | Bacillus subtilis | 2 | 286 | 32.8 | D103 | H265 | D237 | Y150, Y210 | 17 | <0.002e | — | 0.15 | 22 (R) |
| Bfueh1b | Burkholderia fungorum LB400 | 5 | 396 | 44.2 | D182 | H360 | E333 | Y239, Y308 | 29 | 0.16 | 45 (R) | 0.80 | 5.3 (R) |
| Corehc | Corynebacterium sp. strain C12 | 6 | 286 | 32.1 | D99 | H264 | D123 | Y167, Y209 | 19 | 0.67 | 24 (R) | <0.002 | — |
| Draehc | Deinococcus radiodurans R1 | 2 | 278 | 31.4 | D104 | H259 | D231 | Y150, Y205 | 36 | 0.11 | 21 (S) | 0.75 | 1.8 (R) |
| MtuEphFb | Mycobacterium tuberculosis H37v | 300 | 33.8 | D108 | H274 | D246 | Y153, Y213 | 13 | <0.002 | — | <0.002 | — | |
| Npuch1c | Nostoc punctiforme | 2 | 289 | 33.4 | D104 | H267 | D239 | Y150, Y211 | 55 | 1.3 | 20 (S) | 1.3 | 16 (R) |
| Npueh2b | Nostoc punctiforme | 4 | 300 | 34.7 | D112 | H273 | D246 | Y154, Y217 | 39 | <0.002 | — | 0.003 | — |
| Ppuehb | Pseudomonas putida KT2440 | 4 | 299 | 34.3 | D107 | H267 | D239 | Y213 | 12 | <0.002 | — | <0.002 | — |
| Rpach2b | Rhodopseudomonas palustris CGA009 | 8 | 316 | 34.7 | D100 | H294 | D261 | Y148, Y231 | 49 | 1.1 | 2.6 (R) | 36 | 2.2 (S) |
| Scoeh6b | Streptomyces coelicolor A3(2) | 6 | 292 | 32.1 | D103 | H270 | E127 | Y158, Y215 | 57 | <0.002 | — | <0.002 | — |
| Tfuehb | Thermobifida fusca | 5 | 393 | 43.3 | D183 | H366 | D341 | Y245, Y316 | 11 | <0.002 | — | <0.002 | — |
Molecular masses are for the EH segment of the fusion protein.
Initial activities were determined spectrophotometrically at a 0.5 mM substrate concentration with CFEs.
Initial activities were determined spectrophotometrically with (partially) purified protein.
The enantiomer that is preferentially hydrolyzed is indicated in parentheses.
Activity below the detection limit (0.002 U mg−1).
—, Activity too low for determination of an accurate E value.
See Fig. 3.
Amylose resin purification was first tested with the MalE-Coreh construct, resulting in 10 mg of 83% pure fusion protein from a 1-liter culture at an OD600 of ∼1.5, with a total activity yield of 84%. The MalE-EH fusion proteins of the three epoxide hydrolases from phylogenetic group 2 (Fig. 3; Table 2) were also produced at a preparative scale and conveniently purified from cell extracts using the MalE tag. The enzymes from Deinococcus radiodurans (Draeh) and Nostoc punctiforme (Npueh1) were obtained at >95% purity with good yields. The B. subtilis enzyme could only be obtained at 50 to 70% purity with a single round of amylose resin purification. The enzyme was unstable under the purification conditions used and was therefore not purified further (see the supplemental material for details on enzyme purifications).
Activity of MalE-epoxide hydrolase fusion proteins.
CFEs and/or purified samples of the MalE-EH fusion proteins were tested for activity towards a range of epoxides, using either a colorimetric assay (substrates 5 and 10) (Fig. 2) or an indirect assay that measures periodate depletion by its reaction with a diol (substrates 1, 2, 4, 7, 8, 11 to 14, 16, 17, and 19 to 25). In the latter assay, the diol that is formed by epoxide hydrolase activity reacts with periodate, and the amount of remaining periodate is measured by incubating it with adrenaline to yield a colored product (46). Using these assays, it appeared that of the 12 epoxide hydrolases that were tested, 8 were active with one or more epoxides (Table 4). The substrate ranges of the enzymes were generally very broad and included styrene oxide derivatives, phenyl glycidyl ethers, and most terminal aliphatic epoxides. Only a few enzymes showed activity with the sterically more demanding α,β-disubstituted epoxides (compounds 6 to 8, 20, 23, and 24). Among the proteins for which no activity was detected were MtuEphF and Ppueh. It is likely that the inactive proteins were correctly folded since they were expressed as soluble proteins. Activity was detected with proteins from different groups, and in combination with data in the literature, it was concluded that epoxide hydrolases occur in at least seven of the phylogenetic groups described above (Fig. 3; Table 2). The fact that Npueh2 is active toward pNPGE (compound 10) (Tables 3 and 4) shows that group 4 contains both haloalkane dehalogenases and epoxide hydrolases (Fig. 3). The nature of residue X in the H-G-X-P motif (motif 5 in Fig. 1), as described above, could indeed be used for distinction between the two functional classes (Fig. 5A).
TABLE 4.
Substrate ranges of cloned bacterial epoxide hydrolases
| Enzyme | Activity toward indicated epoxide substrate numbera:
|
||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Aromatic substrates
|
Aliphatic substrates
|
Cyclic substrates
|
|||||||||||||||||||
| 1 | 2 | 4 | 5 | 6 | 7 | 8 | 10 | 11 | 12 | 13 | 14 | 16 | 17 | 19 | 20 | 21 | 22 | 23 | 24 | 25 | |
| AraEchAc | ++ | ++ | ++ | ++ | − | − | − | ++ | ++ | ++ | ++ | ++ | ++ | ++ | ++ | + | − | − | + | − | − |
| Bsuehb | − | − | − | − | − | − | − | ++ | − | − | − | − | − | − | ++ | − | − | − | − | − | − |
| Bfuehlc | ++ | ++ | ++ | ++ | − | − | − | ++ | ++ | ++ | ++ | ++ | ++ | ++ | ++ | − | − | − | + | − | − |
| Corehb | ++ | ++ | ++ | ++ | ++ | − | − | − | + | + | + | ++ | + | ++ | ++ | − | − | − | ++ | + | − |
| Draehb | ++ | ++ | ++ | ++ | − | + | ++ | ++ | ++ | ++ | − | ++ | ++ | ++ | ++ | − | − | − | − | − | − |
| MtuEphFc | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − |
| Npueh1b | ++ | ++ | ++ | ++ | − | + | − | ++ | ++ | ++ | − | ++ | ++ | ++ | ++ | − | − | − | + | − | − |
| Npueh2c | − | − | − | − | − | − | − | + | − | − | − | − | − | − | − | − | − | − | − | − | − |
| Ppuehc | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − |
| Rpaeh2c | ++ | ++ | ++ | ++ | − | ++ | ++ | ++ | ++ | ++ | + | + | ++ | ++ | ++ | + | − | − | − | − | − |
| Scoeh6c | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − |
| Tfuehc | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − |
All assays were done with the adrenaline test (46), with the exception of that for epoxide 10, for which conversion was monitored spectroscopically. Activities were scored as follows: −, <3 nmol mg−1 min−1; +, >3 nmol mg−1 min−1; and ++, >10 nmol mg−1 min−1. Epoxide structures are shown in Fig. 2.
Measurements were performed with purified protein.
Measurements were performed with desalted cell extracts.
The epoxide hydrolase from Rhodopseudomonas palustris (Rpaeh2), which belongs to group 8 (Table 2), was active toward trans-stilbene oxide (compound 7) but not toward cis-stilbene oxide (compound 6) (Table 4). Both the mammalian and the plant soluble epoxide hydrolases, which also belong to phylogenetic group 8, have the same characteristic (11, 19, 30). Thus, this particular cis versus trans specificity is clearly linked to phylogenetic group 8. The three-dimensional structure of mammalian sEH, a group 8 member, revealed an L-shaped substrate access tunnel with the catalytic nucleophile sitting right at the kink (7). Compared to the active-site geometry of other EH structures, where the catalytic nucleophile sits at the very end of the substrate access tunnel, the particular active-site architecture of sEH, and possibly of the other group 8 members, does indeed favor the turnover of trans-1,2-disubstituted epoxides.
An analysis of the genes carried in the vicinity of the six putative epoxide hydrolase genes for which activity was found did not clearly reveal a function for the enzymes. However, the genes encoding the epoxide hydrolases from B. subtilis, Burkholderia fungorum (Bfueh1), and R. palustris are located near genes encoding putative enzymes that may be involved in the detoxification of xenobiotics or antibiotics. The genes surrounding the epoxide hydrolase genes from D. radiodurans and N. punctiforme are of unknown or unrelated function. Thus, the biological function of the epoxide hydrolases identified here is uncertain.
Enantioselectivity and unusual enantiopreference of group 2 enzymes.
The enantioselectivity of the active epoxide hydrolase fusion proteins was tested with various chiral substrates. The enantioselectivities with styrene epoxides and phenyl glycidyl ethers were generally low (E < 10) to modest (10 < E < 50) (Tables 3 and 5). This could possibly be influenced by the fact that the tested enzymes were fusion proteins, but the MalE-AraEchA enzyme showed no reduction in stereospecificity compared to the native enzyme (43), so in this case the MalE protein did not influence the catalytic properties of the enzyme. The highest E values were found for para-nitrostyrene oxide, using the D. radiodurans enzyme (Draeh), and for para-nitrophenyl glycidyl ether, using the B. subtilis epoxide hydrolase (Bsueh).
TABLE 5.
Activities and enantioselectivities of novel group 2 epoxide hydrolases for terminal epoxides
| Enzyme and parameter | Activity (μmol min−1 mg−1) or enantioselectivitya with indicated epoxide substrate numberc:
|
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Aromatic substrates
|
Aliphatic substrates
|
||||||||||||
| 1 | 2 | 3 | 4 | 5 | 9 | 10 | 11 | 15 | 16 | 17 | 18 | 19 | |
| Bsueh | |||||||||||||
| Activity | <0.003 | <0.003 | <0.003 | <0.003 | <0.003 | 0.01 | 0.15 | <0.003 | <0.003 | <0.003 | <0.003 | 0.11 | 0.91 |
| Enantioselectivity | —b | — | — | — | — | — | 22 (R) | — | — | — | — | 1.2 (R) | 2.5 (R) |
| Draeh | |||||||||||||
| Activity | 0.42 | 0.06 | 0.68 | 0.19 | 0.11 | 0.85 | 0.75 | 0.07 | 0.04 | 0.31 | 4.3 | 13.8 | 25.6 |
| Enantioselectivity | 4.2 (S) | 5.2 (S) | 2.3 (S) | 6.4 (S) | 21 (S) | 1.4 (R) | 1.9 (R) | 1.3 (S) | 2.9 (R) | 3.5 (R) | 6.5 (R) | 4.6 (R) | 7.0 (R) |
| Npueh1 | |||||||||||||
| Activity | 0.37 | 0.42 | 2.1 | 0.74 | 1.3 | 1.3 | 1.3 | 0.21 | 0.33 | 1.2 | 1.2 | 1.7 | 2.4 |
| Enantioselectivity | 2.3 (S) | 1.9 (S) | 4.6 (S) | 1 | 17 (S) | 3.7 (R) | 16 (R) | 1.3 (R) | 1.5 (R) | 1.2 (R) | 1.7 (R) | 1.7 (R) | 1.8 (R) |
The preferentially hydrolyzed enantiomer is indicated in parentheses.
—, activity was too low to determine an accurate E value.
See Fig. 2 for epoxide structures.
Most well-described epoxide hydrolases, such as the ones from A. radiobacter AD1 (43), A. niger (16, 32), and Rhodothorula glutinis (47) and the mammalian microsomal epoxide hydrolase (13), are (R) specific toward styrene oxides and (S) specific toward phenyl glycidyl ethers. In contrast, Bsueh, Draeh, and Npueh1 (from N. punctiforme) had an opposite enantiopreference towards these compounds. The enantioselectivities varied from low (E < 10) to intermediate (10 < E < 50) and were generally higher for the aromatic substrates (epoxides 1 to 5, 9, and 10). All three of these epoxide hydrolases belong to phylogenetic group 2 (Fig. 3; Table 2). Of these three proteins, the B. subtilis enzyme has the most restricted substrate range. The enzyme had reasonable activities toward the terminal epoxides pNPGE (compound 10) and 1,2-epoxyoctane (compound 19), but with substrates that are only a little smaller (compounds 9 and 18), the activity dropped up to 10-fold. No activity was detectable with epoxides that have only a short substituent (compounds 11 to 17) and with most styrene oxides (Tables 4 and 5). In contrast, the epoxide hydrolase from D. radiodurans (Draeh) converted almost all terminal epoxides that were tested, but of the 1,2-disubstituted epoxides, only trans-stilbene oxide (compound 7) and trans-1,2-disubstituted chalcone oxide (compound 8) were converted (Tables 4 and 5). The enzyme prefers terminal epoxides that have a long substituent, especially unbranched 1,2-epoxyalkanes with a long chain length (epoxides 18 and 19). An increase of activity toward unbranched 1,2-epoxyalkanes with increasing lengths of the side group has also been found with epoxide hydrolases from R. glutinis and different Rhodothorula and Trichosporon spp. (15, 48). The epoxide hydrolase from N. punctiforme (Npueh1) was active not only with all terminal epoxides that were tested but also with the 1,2-disubstituted epoxides trans-stilbene oxide (compound 7) and cyclohexene oxide (compound 23) (Tables 4 and 5). Thus, the activity of Npueh1 is less influenced by the substituent on the epoxide ring than that of the Draeh enzyme.
Determination of the steady-state kinetic parameters for pNSO (compound 5) and pNPGE (compound 10) (Table 6) revealed that Npueh1 has a rather low Km for either one of their enantiomers (<0.2 mM). Indeed, the kinetic resolution data for this enzyme for most substrates (Table 5) could only be fitted when the Km values for the preferred enantiomers were set lower than 0.2 mM, indicating that this enzyme has a low Km for the preferred enantiomer with most terminal epoxides. In contrast, with the Draeh enzyme, the Km was far above the substrate concentration used for pNPGE and pNSO (>0.5 mM), and the shapes of the kinetic resolution curves indicate that this is also the case for most other epoxides. These high Km values indicate that the natural substrate for Draeh is probably not among the epoxides tested.
TABLE 6.
Steady-state kinetic parametersa for Draeh and Npueh1 acting on each enantiomer of pNSO (substrate 5) and pNPGE (substrate 10)
| Enzyme and parameter | Value for indicated enantiomer
|
|||
|---|---|---|---|---|
| (R)-pNSO | (S)-pNSO | (R)-pNPGE | (S)-pNPGE | |
| Draeh | ||||
| kcat (s−1) | >0.007 | >0.17 | >0.7 | >0.5 |
| Km (mM) | >0.5 | >0.5 | >0.3 | >0.3 |
| kcat/Km (mM−1 s−1) | 0.020 | 0.42 | 2.8 | 2.1 |
| E valueb | 21 (21) | 21 (21) | 1.3 (1.8) | 1.3 (1.8) |
| Npueh1 | ||||
| kcat (s−1) | >0.2 | 3.4 | >2.6 | 0.42 |
| Km (mM) | >0.5 | 0.20 | >0.025c | 0.028 |
| kcat/Km (mM−1 s−1) | 0.60 | 17 | 174 | 15 |
| E valueb | 28 (17) | 28 (17) | 12 (15) | 12 (15) |
Since for some enzymes the Km value exceeded substrate solubility, only lower limits could be determined for kcat and Km, and kcat/Km values were determined as first-order rate constants.
Calculated from the kcat/Km ratios of the two enantiomers. The E value calculated from a kinetic resolution experiment is given in parentheses.
At higher substrate concentrations, substrate inhibition occurred; therefore, kcat/Km could be determined only as a first-order rate constant at a low substrate concentration.
The three enzymes from group 2 were found to be (R) selective with aliphatic terminal epoxides and phenyl glycidyl ethers but (S) selective toward terminal epoxides with an aromatic substituent (Table 5), which makes them the first bacterial epoxide hydrolases that have this uncommon selectivity for which the genes have been cloned. Also, (S) selectivity toward meta-chlorostyrene oxide (compound 4) and pNSO (compound 5) and (R) selectivity toward pNPGE (compound 10) have not been well documented, and the uncommon enantiopreference of these enzymes widens the scope of bacterial epoxide hydrolases for the biocatalytic preparation of enantiopure epoxides and diols.
Conversion of meso-epoxides.
Five of the 12 recombinant epoxide hydrolases were active toward the meso-epoxides 20 and/or 23 (Table 4). The regioselective conversion of these (R,S)-configured meso compounds will result in optically enriched vicinal diols due to inversion of configuration at one of the two stereocenters. The activities and enantioselectivities of the MalE-EH fusion proteins toward meso-epoxides 20 and 23 were determined by following substrate depletion over time and measuring the enantiomeric composition of the resulting diol product (Table 7). The activities were rather low for most substrates (<0.05 μmol min−1 mg−1), but the enantiomeric excess of the product (eep) was in some cases very high (>95%). With Npueh1, the formation of (1S,2S)-1,2-cyclohexanediol from cyclohexene oxide occurred with an eep of >95%, which is a significant improvement over the 58% eep that was reported for an epoxide hydrolase (BD10090) obtained from an environmental gene library (49). Epoxide hydrolases with excellent enantioselectivities toward meso-epoxides could thus be obtained from genomic databases.
TABLE 7.
Activities of different fusion proteins for meso-epoxides
| Enzyme | Parameter value for indicated substrate
|
|||||
|---|---|---|---|---|---|---|
|
cis-2,3-Epoxybutane (substrate 20)
|
Cyclohexene oxide (substrate 23)
|
|||||
| Activity (μmol min−1 mg−1) | eep (%) | E valued | Activity (μmol min−1 mg−1) | eep (%) | E valued | |
| AraEchAa | 0.003 | 85 (2R,3R) | 12 | 0.005 | >99 (R,R) | >200 |
| Bfueh1a | <0.002 | —c | — | 0.035 | 89 (S,S) | 17 |
| Corehb | <0.002 | — | — | 1.4 | 30 (R,R) | 1.9 |
| Npueh1b | <0.002 | — | — | 0.016 | 97 (S,S) | 66 |
| Rpaeh2a | 0.019 | 58 (2R,3R) | 3.8 | <0.002 | — | — |
Initial activities were determined at a 2 mM substrate concentration with cell extracts.
Activities were measured with (partially) purified protein.
—, activity was too low to determine the enantiomeric excess of the product (eep).
The E value was calculated from the eep according to equation 1.
General conclusions.
The results reported here indicate that epoxide hydrolase genes that encode active proteins are widely present in microbial genomes. In about 20% of the organisms for which the whole genome sequence is known, putative epoxide hydrolase genes are present, and the expression of a number of these genes and testing with model substrates identified activity for 60% of them. Since only a restricted number of substrates was used, the percentage of organisms that carry active enzymes is probably larger. Thus far, the functions of most of these epoxide hydrolases remain unclear.
The various phylogenetic groups of α/β-hydrolase epoxide hydrolases harbor orthologs from phylogenetically very different organisms. Since the level of sequence identity within the groups is low, this is probably not caused by recent lateral gene transfer. A more likely explanation is that the common ancestor of epoxide hydrolases was present early in evolution and widespread among the various species. As a result of speciation, enzymes that cluster in a multiple sequence alignment are now present in phylogenetically unrelated organisms.
Active recombinant epoxide hydrolases could rapidly be obtained by cloning the genes as fusions to the C-terminal part of the maltose binding protein (MalE). This facilitated expression, prevented the formation of insoluble inclusion bodies, and served as a convenient purification tag. Preliminary experiments showed that the production of soluble protein failed for only 2 of the 12 enzymes that were synthesized as fusion proteins. This approach for obtaining new enzymes for biocatalysis is attractive since the gene of a positive hit is immediately available for further improvement by site-directed mutagenesis or directed evolution.
The cloning and characterization of the putative epoxide hydrolases led to several new active enzymes that can be evaluated as biocatalysts for enantioselective conversion. Three enzymes were (S) selective toward aromatic substrates, which is rather uncommon and has been observed only for a few other epoxide hydrolases (21, 27, 42, 50). Furthermore, the enzymes from B. fungorum (Bfueh1) and N. punctiforme (Npueh1) converted the prochiral substrate cyclohexene oxide to optically enriched (1S,2S)-1,2-cyclohexanediol with much higher enantioselectivities than previously reported (Table 7) (49). Thus, new epoxide hydrolases with interesting biocatalytic activities can be obtained using genomic databases, which defines genome analysis as a promising strategy to widen the scope of biocatalysts, as previously shown for carbohydrate-converting enzymes (17).
Supplementary Material
Acknowledgments
The various bacterial strains and/or their DNA material was kindly provided by James Tiedje (Michigan State University; B. fungorum), David Leak (Imperial College London, United Kingdom; Corynebacterium sp.), Theo Sonke (DSM Research, Geleen, The Netherlands; D. radiodurans R1), John C. Meeks (University of California; N. punctiforme), Stuart Levy (Tufts University; Pseudomonas fluorescens Pfo-1), Juan L. Ramos (Consejo Superior de Investigaciones Cientificas, Granada, Spain; P. putida KT2440), Caroline S. Harwood (University of Iowa; R. palustris CGA009), Stewart Cole (Institut Pasteur, Paris, France; M. tuberculosis), and Diana Irwin (Cornell University; Thermobifida fusca). We thank Birgit Schulze and Theo Sonke at DSM Research for valuable discussions.
The work of B.V.L. was supported by a grant from DSM to the University of Groningen.
Footnotes
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1.Albers, S.-V., and A. J. M. Driessen. 2002. Signal peptides of secreted proteins of the archaeon Sulfolobus solfataricus: a genomic survey. Arch. Microbiol. 177:209-216. [DOI] [PubMed] [Google Scholar]
- 2.Arand, M., D. F. Grant, J. K. Beetham, T. Friedberg, F. Oesch, and B. D. Hammock. 1994. Sequence similarity of mammalian epoxide hydrolases to the bacterial haloalkane dehalogenase and other related proteins. Implication for the potential catalytic mechanism of enzymatic epoxide hydrolysis. FEBS Lett. 338:251-256. [DOI] [PubMed] [Google Scholar]
- 3.Arand, M., H. Hemmer, H. Durk, J. Baratti, A. Archelas, R. Furstoss, and F. Oesch. 1999. Cloning and molecular characterization of a soluble epoxide hydrolase from Aspergillus niger that is related to mammalian microsomal epoxide hydrolase. Biochem. J. 344:273-280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arand, M., H. Wagner, and F. Oesch. 1996. Asp333, Asp495, and His523 form the catalytic triad of rat soluble epoxide hydrolase. J. Biol. Chem. 271:4223-4229. [DOI] [PubMed] [Google Scholar]
- 5.Archelas, A., and R. Furstoss. 2001. Synthetic applications of epoxide hydrolases. Curr. Opin. Chem. Biol. 5:112-119. [DOI] [PubMed] [Google Scholar]
- 6.Argiriadi, M. A., C. Morisseau, M. H. Goodrow, D. L. Dowdy, B. D. Hammock, and D. W. Christianson. 2000. Binding of alkylurea inhibitors to epoxide hydrolase implicates active site tyrosines in substrate activation. J. Biol. Chem. 275:15265-15270. [DOI] [PubMed] [Google Scholar]
- 7.Argiriadi, M. A., C. Morisseau, B. D. Hammock, and D. W. Christianson. 1999. Detoxification of environmental mutagens and carcinogens: structure, mechanism, and evolution of liver epoxide hydrolase. Proc. Natl. Acad. Sci. USA 96:10637-10642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barth, S., M. Fischer, R. D. Schmid, and J. Pleiss. 2004. The database of epoxide hydrolases and haloalkane dehalogenases: one structure, many functions. Bioinformatics 20:2845-2847. [DOI] [PubMed] [Google Scholar]
- 9.Barth, S., M. Fischer, R. D. Schmid, and J. Pleiss. 2004. Sequence and structure of epoxide hydrolases: a systematic analysis. Proteins 55:846-855. [DOI] [PubMed] [Google Scholar]
- 10.Beetham, J. K., T. Tian, and B. D. Hammock. 1993. cDNA cloning and expression of a soluble epoxide hydrolase from human liver. Arch. Biochem. Biophys. 305:197-201. [DOI] [PubMed] [Google Scholar]
- 11.Bellevik, S., J. Zhang, and J. Meijer. 2002. Brassica napus soluble epoxide hydrolase (BNSEH1). Eur. J. Biochem. 269:5295-5302. [DOI] [PubMed] [Google Scholar]
- 12.Bellucci, G., G. Berti, G. Ingrosso, and E. Mastrorilli. 1980. Stereoselectivity in the epoxide hydrase catalyzed hydrolysis of the stereoisomeric 4-tert-butyl-1,2-epoxycyclohexanes. J. Org. Chem. 45:299-303. [Google Scholar]
- 13.Bellucci, G., C. Chiappe, A. Cordoni, and F. Marioni. 1994. Different enantioselectivity and regioselectivity of the cytosolic and microsomal epoxide hydrolase catalyzed hydrolysis of simple phenyl substituted epoxides. Tetrahedron Lett. 35:4219-4222. [Google Scholar]
- 14.Berks, B. C., F. Sargent, and T. Palmer. 2000. The Tat protein export pathway. Mol. Microbiol. 35:260-274. [DOI] [PubMed] [Google Scholar]
- 15.Botes, A. L., C. A. G. M. Weijers, P. J. Botes, and M. S. van Dyk. 1999. Enantioselectivities of yeast epoxide hydrolase for 1,2-epoxides. Tetrahedron 10:3327-3336. [Google Scholar]
- 16.Choi, W. J., E. C. Huh, H. J. Park, E. Y. Lee, and C. Y. Choi. 1998. Kinetic resolution for optically active epoxides by microbial enantioselective hydrolysis. Biotechnol. Tech. 12:225-228. [Google Scholar]
- 17.Daines, A. M., B. A. Maltman, and S. L. Flitsch. 2004. Synthesis and modifications of carbohydrates, using biotransformations. Curr. Opin. Chem. Biol. 8:106-113. [DOI] [PubMed] [Google Scholar]
- 18.Debernard, S., C. Morisseau, T. F. Severson, L. Feng, H. Wojtasek, G. D. Prestwich, and B. D. Hammock. 1998. Expression and characterization of the recombinant juvenile hormone epoxide hydrolase (JHEH) from Manduca sexta. Insect Biochem. Mol. Biol. 28:409-419. [DOI] [PubMed] [Google Scholar]
- 19.Fretland, A. J., and C. J. Omiecinski. 2000. Epoxide hydrolases: biochemistry and molecular biology. Chem.-Biol. Interact. 129:41-59. [DOI] [PubMed] [Google Scholar]
- 20.Friedberg, T., B. Lollmann, R. Becker, R. Holler, and F. Oesch. 1994. The microsomal epoxide hydrolase has a single membrane signal anchor sequence which is dispensable for the catalytic activity of this protein. Biochem. J. 303:967-972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grogan, G., C. Rippé, and A. Willets. 1997. Biohydrolysis of substituted styrene oxides by Beauveria densa CMC 3240. J. Mol. Catal. B 3:253-257. [Google Scholar]
- 22.Jackson, M. R., and B. Burchell. 1988. Expression of human liver epoxide hydrolase in Saccharomyces pombe. Biochem. J. 251:931-933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Janssen, D. B., F. Pries, J. van der Ploeg, B. Kazemier, P. Terpstra, and B. Witholt. 1989. Cloning of 1,2-dichloroethane degradation genes of Xanthobacter autotrophicus GJ10 and expression and sequencing of the dhlA gene. J. Bacteriol. 171:6791-6799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kawasaki, H., K. Tsuda, I. Matsushita, and K. Tonomura. 1992. Lack of homology between two haloacetate dehalogenase genes encoded on a plasmid from Moraxella sp. strain B. J. Gen. Microbiol. 138:1317-1323. [DOI] [PubMed] [Google Scholar]
- 25.Liu, J. Q., T. Kurihara, S. Ichiyama, M. Miyagi, S. Tsunasawa, H. Kawasaki, K. Soda, and N. Esaki. 1998. Reaction mechanism of fluoroacetate dehalogenase from Moraxella sp. B. J. Biol. Chem. 273:30897-30902. [DOI] [PubMed] [Google Scholar]
- 26.Lutje Spelberg, J. H., R. Rink, R. M. Kellogg, and D. B. Janssen. 1998. Enantioselectivity of a recombinant epoxide hydrolase from Agrobacterium radiobacter. Tetrahedron 9:459-466. [Google Scholar]
- 27.Manoj, K. M., A. Archelas, J. Baratti, and R. Furstoss. 2001. Microbiological transformations. Part 45. A green chemistry preparative scale synthesis of enantiopure building blocks of eliprodil: elaboration of a high substrate concentration epoxide hydrolase-catalyzed hydrolytic kinetic resolution process. Tetrahedron 57:695-701. [Google Scholar]
- 28.Mira, A., H. Ochman, and N. A. Moran. 2001. Deletional bias and the evolution of bacterial genomes. Trends Genet. 17:589-596. [DOI] [PubMed] [Google Scholar]
- 29.Misawa, E., C. K. Chion, I. V. Archer, M. P. Woodland, N. Y. Zhou, S. F. Carter, D. A. Widdowson, and D. J. Leak. 1998. Characterisation of a catabolic epoxide hydrolase from a Corynebacterium sp. Eur. J. Biochem. 253:173-183. [DOI] [PubMed] [Google Scholar]
- 30.Morisseau, C., J. K. Beetham, F. Pinot, S. Debernard, J. W. Newman, and B. D. Hammock. 2000. Cress and potato soluble epoxide hydrolases: purification, biochemical characterization, and comparison to mammalian enzymes. Arch. Biochem. Biophys. 378:321-332. [DOI] [PubMed] [Google Scholar]
- 31.Nardini, M., I. S. Ridder, H. J. Rozeboom, K. H. Kalk, R. Rink, D. B. Janssen, and B. W. Dijkstra. 1999. The X-ray structure of epoxide hydrolase from Agrobacterium radiobacter AD1. An enzyme to detoxify harmful epoxides. J. Biol. Chem. 274:14579-14586. [PubMed] [Google Scholar]
- 32.Nellaiah, H., C. Morisseau, A. Archelas, R. Furstoss, and C. Baratti Jacques. 1996. Enantioselective hydrolysis of p-nitrostyrene oxide by an epoxide hydrolase preparation from Aspergillus niger. Biotechnol. Bioeng. 49:70-77. [DOI] [PubMed] [Google Scholar]
- 33.Ollis, D. L., E. Cheah, M. Cygler, B. Dijkstra, F. Frolow, S. M. Franken, M. Harel, S. J. Remington, I. Silman, J. Schrag, et al. 1992. The α/β hydrolase fold. Protein Eng. 5:197-211. [DOI] [PubMed] [Google Scholar]
- 34.Reetz, M. T., C. Torre, A. Eipper, R. Lohmer, M. Hermes, B. Brunner, A. Maichele, M. Bocola, M. Arand, A. Cronin, Y. Genzel, A. Archelas, and R. Furstoss. 2004. Enhancing enantioselectivity of an epoxide hydrolase by directed evolution. Org. Lett. 6:177-180. [DOI] [PubMed] [Google Scholar]
- 35.Rink, R., M. Fennema, M. Smids, U. Dehmel, and D. B. Janssen. 1997. Primary structure and catalytic mechanism of the epoxide hydrolase from Agrobacterium radiobacter AD1. J. Biol. Chem. 272:14650-14657. [DOI] [PubMed] [Google Scholar]
- 36.Rink, R., J. H. Lutje Spelberg, R. J. Pieters, J. Kingma, M. Nardini, R. M. Kellogg, B. W. Dijkstra, and D. B. Janssen. 1999. Mutation of tyrosine residues involved in the alkylation half reaction of epoxide hydrolase from Agrobacterium radiobacter AD1 results in improved enantioselectivity. J. Am. Chem. Soc. 121:7417-7418. [Google Scholar]
- 37.Sachdev, D., and J. M. Chirgwin. 2000. Fusions to maltose-binding protein: control of folding and solubility in protein purification. Methods Enzymol. 326:312-323. [DOI] [PubMed] [Google Scholar]
- 38.Schanstra, J. P., R. Rink, F. Pries, and D. B. Janssen. 1993. Construction of an expression and site-directed mutagenesis system of haloalkane dehalogenase in Escherichia coli. Protein Expr. Purif. 4:479-489. [DOI] [PubMed] [Google Scholar]
- 39.Smit, M. S. 2004. Fungal epoxide hydrolases: new landmarks in sequence-activity space. Trends Biotechnol. 22:123-129. [DOI] [PubMed] [Google Scholar]
- 40.Stapleton, A., J. K. Beetham, F. Pinot, J. E. Garbarino, D. R. Rockhold, M. Friedman, B. D. Hammock, and W. R. Belknap. 1994. Cloning and expression of soluble epoxide hydrolase from potato. Plant J. 6:251-258. [DOI] [PubMed] [Google Scholar]
- 41.Steinreiber, A., and K. Faber. 2001. Microbial epoxide hydrolases for preparative biotransformations. Curr. Opin. Biotechnol. 12:552-558. [DOI] [PubMed] [Google Scholar]
- 42.Tang, Y.-F., J.-H. Xu, Q. Ye, and B. Schulze. 2001. Biocatalytic preparation of (S)-phenyl glycidyl ether using newly isolated Bacillus megaterium ECU1001. J. Mol. Catal. B 13:61-68. [Google Scholar]
- 43.van Loo, B., J. H. Lutje Spelberg, J. Kingma, T. Sonke, M. G. Wubbolts, and D. B. Janssen. 2004. Directed evolution of epoxide hydrolase from A. radiobacter toward higher enantioselectivity by error-prone PCR and DNA shuffling. Chem. Biol. 11:981-990. [DOI] [PubMed] [Google Scholar]
- 44.Venter, J. C., K. Remington, J. F. Heidelberg, A. L. Halpern, D. Rusch, J. A. Eisen, D. Wu, I. Paulsen, K. E. Nelson, W. Nelson, D. E. Fouts, S. Levy, A. H. Knap, M. W. Lomas, K. Nealson, O. White, J. Peterson, J. Hoffman, R. Parsons, H. Baden-Tillson, C. Pfannkoch, Y.-H. Rogers, and H. O. Smith. 2004. Environmental genome shotgun sequencing of the Sargasso Sea. Science 304:66-74. [DOI] [PubMed] [Google Scholar]
- 45.Visser, H., S. Vreugdenhil, J. A. de Bont, and J. C. Verdoes. 2000. Cloning and characterization of an epoxide hydrolase-encoding gene from Rhodotorula glutinis. Appl. Microbiol. Biotechnol. 53:415-419. [DOI] [PubMed] [Google Scholar]
- 46.Wahler, D., and J.-L. Reymond. 2002. The adrenaline test for enzymes. Angew. Chem. Int. 41:1229-1232. [DOI] [PubMed] [Google Scholar]
- 47.Weijers, C. A. G. M. 1997. Enantioselective hydrolysis of aryl, alicyclic and aliphatic epoxides by Rhodoturola glutinis. Tetrahedron 8:639-647. [Google Scholar]
- 48.Weijers, C. A. G. M., A. K. Botes, M. S. van Dyk, and J. A. M. de Bont. 1998. Enantioselective hydrolysis of unbranched aliphatic 1,2-epoxides by Rhodotorula glutinis. Tetrahedron 9:467-473. [Google Scholar]
- 49.Zhao, L., B. Han, Z. Huang, M. Miller, H. Huang, D. S. Malashock, Z. Zhu, A. Milan, D. E. Robertson, D. P. Weiner, and M. J. Burk. 2004. Epoxide hydrolase-catalyzed enantioselective synthesis of chiral 1,2-diols via desymmetrization of meso-epoxides. J. Am. Chem. Soc. 126:11158-11159. [DOI] [PubMed] [Google Scholar]
- 50.Zocher, F., M. M. Enzelberger, U. T. Bornscheuer, B. Hauer, W. Wohlleben, and R. D. Schmid. 2000. Epoxide hydrolase activity of Streptomyces strains. J. Biotechnol. 77:287-292. [DOI] [PubMed] [Google Scholar]
- 51.Zou, J., B. M. Hallberg, T. Bergfors, F. Oesch, M. Arand, S. L. Mowbray, and T. A. Jones. 2000. Structure of Aspergillus niger epoxide hydrolase at 1.8 Å resolution: implications for the structure and function of the mammalian microsomal class of epoxide hydrolases. Struct. Fold. Des. 8:111-122. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.