

Abstract
Baeyer-Villiger monooxygenases (BVMOs) are biocatalysts that offer the prospect of high chemo-, regio-, and enantioselectivity in the organic synthesis of lactones or esters from a variety of ketones. In this study, we have cloned, sequenced, and overexpressed in Escherichia coli a new BVMO, cyclopentadecanone monooxygenase (CpdB or CPDMO), originally derived from Pseudomonas sp. strain HI-70. The 601-residue primary structure of CpdB revealed only 29% to 50% sequence identity to those of known BVMOs. A new sequence motif, characterized by a cluster of charged residues, was identified in a subset of BVMO sequences that contain an N-terminal extension of ∼60 to 147 amino acids. The 64-kDa CPDMO enzyme was purified to apparent homogeneity, providing a specific activity of 3.94 μmol/min/mg protein and a 20% yield. CPDMO is monomeric and NADPH dependent and contains ∼1 mol flavin adenine dinucleotide per mole of protein. A deletion mutant suggested the importance of the N-terminal 54 amino acids to CPDMO activity. In addition, a Ser261Ala substitution in a Rossmann fold motif resulted in an improved stability and increased affinity of the enzyme towards NADPH compared to the wild-type enzyme (Km = 8 μM versus Km = 24 μM). Substrate profiling indicated that CPDMO is unusual among known BVMOs in being able to accommodate and oxidize both large and small ring substrates that include C11 to C15 ketones, methyl-substituted C5 and C6 ketones, and bicyclic ketones, such as decalone and β-tetralone. CPDMO has the highest affinity (Km = 5.8 μM) and the highest catalytic efficiency (kcat/Km ratio of 7.2 × 105 M−1 s−1) toward cyclopentadecanone, hence the Cpd designation. A number of whole-cell biotransformations were carried out, and as a result, CPDMO was found to have an excellent enantioselectivity (E > 200) as well as 99% S-selectivity toward 2-methylcyclohexanone for the production of 7-methyl-2-oxepanone, a potentially valuable chiral building block. Although showing a modest selectivity (E = 5.8), macrolactone formation of 15-hexadecanolide from the kinetic resolution of 2-methylcyclopentadecanone using CPDMO was also demonstrated.
Bacteria of the genus Pseudomonas occupy a visible position in the clinical world and the environment at large. The present taxonomy of Pseudomonas consists of at least 85 validated species (44). Members of this diverse group of aerobic microorganisms have been studied for their medical, environmental, and industrial importance or their relevance in aspects of catabolic plasmids, virulence factors, biofilm formation, solvent tolerance, aromatic metabolism, biodegradation, biotransformations, etc. The many facets of Pseudomonas, including the more recent advancement in comparative genomics, have been captured in an impressive three-volume Pseudomonas series (47).
Few Pseudomonas species or strains have been isolated and shown to be capable of metabolizing aliphatic compounds, e.g., linear alkanes (for a review, see reference 63; http://umbbd.ahc.umn.edu/). Even fewer microorganisms have been found to grow on branched or cyclic alkanes (20, 31, 48, 51, 68). Wackett and Hershberger (65) noted that cycloalkanes or alicyclic hydrocarbons represent an “underappreciated” group of molecules, although they are prevalent in nature. Ranging in ring size from 3 to 17 carbons, many cyclic structures are found in steroids, plant oils, fragrances, and a variety of plant secondary metabolites (60). In petroleum mixtures, alicyclic hydrocarbons are estimated to represent up to 12% (wt/wt) of the total hydrocarbons (48). The structure of cyclododecane, a C12 compound, for example, has been compared to the aliphatic bridge components in coal (51). It is possible that activation or functionalization of these cyclic compounds can give rise to value-added products; lactones and dicarboxylic acids useful for polymer synthesis are two prime examples (3, 9, 59).
In this study, we have isolated a Pseudomonas strain that is able to grow on C11 to C15 cyclic ring compounds that include cyclopentadecanol and cyclopentadecanone. In particular, a new flavoprotein Baeyer-Villiger monooxygenase (BVMO), designated cyclopentadecanone monooxygenase (CpdB or CPDMO), was purified and characterized. Substrate profiling indicated it to be an accommodating and versatile biocatalyst in lactone production. In general, the BVMO-mediated ring expansion reaction of cyclic ketone to lactone constitutes a green chemistry alternative to chemical reagents by using molecular oxygen as the oxidant and producing more products than waste, besides offering the prospect of high chemo-, regio-, and enantioselectivity in the various oxidative reactions (1, 26, 28, 41, 56, 58).
MATERIALS AND METHODS
Bacterial strains, culture conditions, and plasmids.
All bacterial strains and plasmids used in this study are listed in Table 1. Pseudomonas sp. strain HI-70 was isolated from a soil sample from Osaka, Japan, by selective enrichment using a minimal salts medium (22), pH 7.0, containing cyclododecanol (1.0 g/liter) as the sole carbon source. To the best of available knowledge, this is the first gram-negative organism capable of growth on this large cyclic compound, two previous isolates being gram-positive rhodococci (32, 51). The HI-70 culture was maintained in the same medium containing 50% glycerol at −80°C. The growth temperature was 30°C for Pseudomonas and 37°C for the various Escherichia coli strains that were routinely cultured in standard LB medium. When necessary, the medium was supplemented with ampicillin (Ap; 100 μg/ml) or kanamycin (Km; 50 μg/ml for E. coli and 100 μg/ml for HI-70).
TABLE 1.
Bacterial strains and plasmids used in this study
| Bacterial strain or plasmid | Relevant characteristics | Source or reference |
|---|---|---|
| Bacterial strains | ||
| Pseudomonas sp. | ||
| HI-70 | Wild type, grows on cyclododecanol or cyclododecanone | This study |
| HI-70MB | cpdB::lacZ, Kmr; no growth on cyclododecanol or cyclododecanone | This study |
| E. coli | ||
| BL21(DE3) | F−ompT hsdSB(rB− mB−) gal dcm | Novagen |
| DH5α | supE44 thi-1 recA1 hsdR17 endA1 gyrA (Nalr) Δ(lacIZYA-argF)U169 deoR [φ80dlacΔ(lacZ)M15] | 49 |
| S17-1 | recA pro thi hsdR RP4-2-Tc::Mu-Km::Tn7 Tra+ Tpr Smr | 54 |
| XL1-Blue | recA1 endA1 gyrA96 thi hsdR17 supE44 relA1 [F′ lacIqZM15 Tn10(Tetr)] | 49 |
| Plasmids | ||
| pARO190 | Mobilizable vector; Apr | 45 |
| pARO-cpdB | pARO190 with a 2.9-kb EcoRV fragment containing cpdB from pCD200 | This study |
| pARO-cpdB::lacZ | cpdB::lacZ-Kmr; SalI-excised lacZ-Kmr cassette from pKOK6.1 in XhoI site of pARO-cpdB | This study |
| pKOK6.1 | lacZ-Kmr cassette | 30 |
| pSD80 | Expression vector with tac promoter; Apr | 55 |
| pUC19 | Cloning vector with lac promoter; Apr | 71 |
| pCD200 | pUC19 with a 4.2-kb BclI fragment from Pseudomonas sp. strain HI-70; Apr | This study |
| pCD201 | 1.8-kb EcoRIa fragment containing cpdB in pSD80; Apr | This study |
| pCpdBdelN | Deletion of CpdB N-terminal 54 amino acids in pCD201 | This study |
| pCpdBdel7 | Deletion of internal 7 amino acids in CpdB in pCD201 | This study |
| pCpdBS261A | Replacement of Ser261 by Ala in CpdB in pCD201 | This study |
| pCpdBG242A | Replacement of Gly242 by Ala in CpdB in pCD201 | This study |
EcoRI restriction endonuclease site introduced by PCR design.
Sequencing of the 16S rRNA.
The near-full-length 16S rRNA gene of strain HI-70 was amplified by PCR as previously described (23). Genomic DNA was obtained by the method of Wilson (70). The DNA sequencing reaction was carried out using the ABI PRISM BigDye Terminator (v. 3.1) cycle-sequencing kit (Applied Biosystems) in a GeneAmp PCR system 9700 (Perkin-Elmer). The thermal profile included 25 cycles of 30 s at 96°C (denaturation), 5 s at 50°C (reannealing), and 4 min at 60°C (DNA extension). The sequencing products were purified using a CENTRISEP column (Princeton Separations) and analyzed in an automated fluorescence sequencer (model 373A; Applied Biosystems). 16S rRNAs for comparison were obtained from the GenBank sequence database of the National Center for Biotechnology Information (NCBI; Bethesda, MD).
Cloning of the CPDMO-encoding (cpdB) gene.
Two conserved amino acid regions of the cyclohexanone monooxygenase (CHMO) (22), cyclopentanone monooxygenase (CPMO) (23) and steroid monooxygenase (42), were chosen to design two degenerate primers (5′-TGGYAYTGGAAYHGITAYCC-3′ and 5′-GCRTCRAANCCNGTIGC-3′) for PCR amplification (I = inosine; N = T, C, A, or G; R = A or G; and Y = C or T). The nucleotides correspond to amino acid positions 46 to 52 and 377 to 382, respectively, of the corrected Acinetobacter sp. strain NCIMB CHMO sequence (22, 29). The amplified DNA fragment (∼1 kb) was labeled by the digoxigenin-11-UTP system according to the manufacturer's instructions (Roche Molecular Biochemicals) and used to probe a Southern hybridization of strain HI-70 genomic DNA digested with a number of restriction enzymes. As a result, a 4.2-kb BclI fragment that was probed positive was chosen for cloning. This DNA was subsequently isolated from a 0.8% agarose gel and ligated to the E. coli plasmid pUC19, which had been linearized with BclI and dephosphorylated (49). Colony hybridization using the PCR product as a probe was carried out to screen for a positive clone. The resulting recombinant plasmid transformed in E. coli XL1-Blue was designated pCD200. DNA sequencing of the 4.2-kb insert was determined using first the universal and reverse primers of the pUC vector and then internal primers derived from the new sequences. The sequence was analyzed by GENETYX-Mac (Software Development Co., Ltd., Chiba, Japan) and the BLAST program of NCBI (2).
Inactivation of cpdB in strain HI-70.
The cpdB disruption mutant was constructed by inserting a lacZ-Kmr cassette from pKOK6.1 (30). The 2.9-kb EcoRV fragment containing cpdB from pCD200 was inserted into pARO190, yielding pARO-cpdB. The lacZ-Kmr cassette was excised as a SalI fragment and inserted into the XhoI site within the cpdB gene in pARO-cpdB, yielding pARO-cpdB::lacZ. The plasmid was inserted into strain HI-70 from E. coli S17-1 (pARO-cpdB::lacZ) by transconjugation. Kmr colonies were selected on minimal salts medium plates containing 0.3% succinate as the sole carbon source and 100 μg/ml kanamycin. Aps colonies were selected from Kmr colonies as a double crossover mutant and used for further experiments.
Subcloning of the cpdB gene in E. coli BL21.
Two primers containing an EcoRI restriction site (underlined) were synthesized (5′-CGGAATTCATGAGTCAGCTAATTCAAGAGC-3′ and 5′-CGGAATTCAATCAACGCTTGCGCTGCTG-3′) and used to amplify the cpdB gene contained in a 1.8-kb DNA fragment from pCD200. Pfu polymerase was used for the PCR amplification, and the thermal profile included 30 cycles of 1 min at 94°C, 1 min at 50°C, and 3 min at 72°C. The target DNA fragment was separated by gel electrophoresis on 1% agarose (1× Tris-acetate-EDTA), excised, and purified by the QIAEX II gel extraction kit (QIAGEN). The purified DNA and the pSD80 vector were each digested with EcoRI, ligated, and transformed in E. coli BL21 cells. The recombinant plasmid is designated pCD201. DNA sequencing was performed to exclude possible mutations of the amplified cpdB gene.
Growth conditions and cell disruption of E. coli BL21(pCD201).
E. coli BL21 harboring the pCD201 plasmid was maintained on LB medium containing glycerol (50%, vol/vol) at −80°C. For protein expression experiments, a fresh LB agar plate (1.5% agar) containing ampicillin (50 μg/ml) was prepared from the stock culture, and one colony was transferred to a 10-ml preculture (LB medium containing ampicillin, 50 μg/ml) and grown overnight. Main cultivation was carried out in 2-liter Erlenmeyer flasks containing 1 liter LB medium and ampicillin (50 mg), using 3 ml of the preculture. Cells were grown at 30°C, and CpdB production was induced by the addition of IPTG (isopropyl-β-d-thiogalactopyranoside) (1 mM). About 3 h after induction, the cells were harvested by centrifugation (5,000 × g, 20 min, 4°C) and washed twice with 20 mM Tris-HCl (pH 7.2). The pellet was resuspended in the same buffer, and crude extract was obtained by passing the cell suspension twice through a French pressure cell (SLM Instruments, Urbana, Ill.) at 20,000 lb/in2 followed by centrifugation (20,000 × g, 30 min, 4°C) to remove unbroken cells and debris.
Enzyme assay and kinetics.
The CPDMO enzyme activity was assayed in a reaction mixture (1 ml) containing Tris-HCl buffer (50 mM, pH 9.0), 0.1 mM NADPH, and an appropriate amount of enzyme (10 to 20 mU), and the reaction was started by adding 10 μl of substrate (e.g., 0.1 M cyclopentadecanone in n-propanol). The decrease in the absorbance of NADPH was measured spectrophotometrically at 340 nm. The specific activity was defined as μmol of NADPH (ɛ = 6.3 liter mmol−1 cm−1) oxidized per minute (U) per milligram of protein (U/mg). Protein concentration was determined by the method of Bradford (4). Kinetic parameters of CpdB and some of their variants (Km and kcat) were determined by using the double-reciprocal transformation (Lineweaver-Burk plot) of the Michaelis-Menten equation under steady-state conditions. Initial reaction rates were measured at 25°C in Tris-HCl buffer (50 mM, pH 9.0) using a total substrate concentration between 5 μM and 5 mM. The Km for NADPH was estimated with cyclopentadecanone as a substrate (0.1 mM) in a standard 1-ml assay using various NADPH concentrations from 5 μM to 160 μM.
Protein purification.
All purification procedures were performed at 4°C on an LKB II fast protein liquid chromatography system (Pharmacia, Uppsala, Sweden) (Table 2). The crude extract (one 10-ml aliquot per run, of a total of 45 ml from a 4-liter cell culture) was loaded on a Mono Q column (10/100) previously equilibrated with 20 mM Tris-HCl buffer (pH 7.2) (buffer A) by using a 10-ml superloop. The flow rate was 0.75 ml/min. The column was washed with buffer A until no protein could be detected in the flowthrough, and the enzyme was subsequently eluted with a linear gradient of 0 to 0.2 M NaCl in buffer A. Active fractions were collected, pooled, and concentrated by ultrafiltration (membrane exclusion size, 30 kDa) (in a 50-ml stirring cell; Amicon, United States) and applied to a Superose 6 HR (10/300) gel filtration column previously equilibrated with 20 mM Tris-HCl buffer (pH 7.2) containing 150 mM NaCl (buffer B). Protein was eluted with the same buffer (flow rate of 1 ml/min) and collected in 0.5-ml fractions. Active fractions were pooled and concentrated by ultrafiltration and applied to a HiPrep Sephacryl S-200 column (26/60) which had been previously equilibrated with buffer B. Protein was eluted using buffer B (flow rate of 2 ml/min), and active fractions were collected, pooled, and concentrated by ultrafiltration. The protein profile was monitored by its absorbance at 280 nm.
TABLE 2.
Purification of cyclopentadecanone monooxygenase
| Purification step | Total protein (mg) | Total activity (U) | Sp act (U/mg) | Yield (%) | Purification (fold) |
|---|---|---|---|---|---|
| Crude extract | 1,332 | 373 | 0.28 | 100 | 1 |
| Mono Q | 117 | 214 | 1.83 | 57 | 6.5 |
| Superose 6 | 62 | 156 | 2.52 | 42 | 9 |
| Sephacryl S200 | 19 | 75 | 3.94 | 20 | 14 |
SDS-PAGE and molecular mass determination.
Ten or 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) containing 0.1% SDS (35) was carried out using the discontinuous buffer system originally described by Davis (12) to determine the molecular mass of the denatured enzyme compared to the marker proteins (Bio-Rad, United States) and for purification control. The SDS-PAGE gels were silver stained (38). The molecular mass of the native enzyme was determined by gel filtration on a Superose 6 column and in reference to standard proteins (Pharmacia, Sweden).
Determination of amino-terminal sequence.
Purified enzyme, separated by SDS-PAGE, was blotted to a polyvinylidene difluoride membrane (Bio-Rad, United States). N-terminal sequence determination was performed with a sequencer (model 473A; Applied Biosystems) by the Edman and Henschen method (14). Phenylthiohydantoin amino acids were analyzed by high-pressure liquid chromatography (HPLC) with a reversed-phase column.
Determination and quantification of the prosthetic group of CPDMO.
Purified CPDMO (2 mg in 200 μl sodium phosphate buffer, 50 mM, pH 7.5) was boiled (52) and subsequently chilled on ice for 5 min. Precipitated protein was removed by centrifugation (20,000 × g, at 4°C), and the supernatant was analyzed by HPLC. Analysis was performed using a Waters Millenium32 liquid chromatography system (Waters 600E solvent delivery system, autosampler model 717, PDA detector model 996) on a reversed-phase column (CSC-Inertsil, 150A/ODS2, 5 μm, 25 cm by 0.46 cm; CSC, Montreal, Canada). The mobile phase consisted of 25% methanol in sodium phosphate buffer (10 mM, pH 6.0), and the flow rate was 1 ml/min (isocratic) at a constant temperature of 45°C. Different retention times of flavin adenine dinucleotide (FAD) (5.8 min) and flavin mononucleotide (FMN) (7.8 min) using pure standards (Sigma) were recorded (at 450 nm) and compared with the retention times of the sample.
Truncation and mutagenesis of cpdB sequence.
Four mutations in the CpdB sequence were constructed. Plasmid pCpdB-delN, lacking the first 54 amino acids of the N terminus of CpdB, was created as follows: primer CPDB-del (CGGAATTCATGAAAGACCCCTATGCCGA), containing an EcoRI restriction site (underlined) and ATG start codon (double underlined), was designed to amplify the truncated cpdB gene together with a C-terminal primer that was used for the subcloning of the cpdB gene. Pfu polymerase was used for the PCR amplification, and the thermal profile included 30 cycles of 1 min at 94°C, 1 min at 50°C, and 3 min at 72°C. The target DNA fragment was separated by gel electrophoresis on 1% agarose (1× Tris-acetate-EDTA), excised, and purified by the QIAEX II gel extraction kit (QIAGEN). The purified DNA and the pSD80 vector were each digested with EcoRI, ligated, and transformed in E. coli BL21 cells. The recombinant plasmid was designated pCpdB-del. DNA sequencing was performed to exclude possible mutations of the amplified cpdB gene.
A second mutant (pCpdB-del7) with a deletion of seven amino acids at positions 240-QTGNLEG-246 was carried out using the site-directed, ligase-independent mutagenesis procedure of Chiu et al. (10) as follows: four primers (two tailed and two nontailed), per Chiu et al. terminology, were synthesized to facilitate the desired deletion. In the primer design, the position of the deleted DNA sequence defines the placement of the other regions. The 5′-adaptor tail (18 bases, underlined), the region that will make up the overhang of the tailed primers [CDNB Rt, 5′-(ATCGCCGCCGGTATAGGAGTAGTCCCAGCGACTGGTTGT); CDNB Ft, 5′-(TCCTATACCGGCGGCGATTTGAAAGACAGCGCGTGGCC)], must be adjacent and immediately 5′ to the region deleted. CDNB Fs, 5′-(TTGAAAGACAAGCGCGTGGCC), is the complementary oligonucleotide region that is recognized by the gene-specific portion of the primers (normal capital letters) immediately 5′ to the 5′ adaptor tail. CDNB Rs, 5′-(GTAGTCCCAGCGACTGGTGTG), is the 3′ gene-specific region (in bold) immediately 3′ to the deleted sequence. Sequence deletion from pCpdB is accomplished by excluding the desired deletion region from the single PCR amplification described as follows.
A single PCR was performed containing the following components: 2.5 μl of 10× Pfx buffer, 200 μM each deoxynucleoside triphosphate, 1 mM MgSO4, 100 mM betaine, 10 pmol of each of the four primers, 100 pg of the 7.676-kb plasmid template pCD201, 0.5 U Taq DNA polymerase (New England Biolabs, Beverley, MA), 0.25 U platinum Pfx DNA polymerase (Invitrogen, Carlsbad, CA), and molecular biology-grade water to a final volume of 25 μl. The reaction was started by heating to 98°C for 2 min and then brought to 85°C, at which point the DNA polymerase mix was added. The PCR was subjected to a further 25 cycles of 95°C for 15 s, 58°C for 20 s, and 68°C for 3.5 min, adopting the conditions of the site-directed, ligase-independent mutagenesis technique. The PCR mixture was migrated on a 0.8% agarose gel, and the 7.676-kb fragment was isolated and purified with QIAEX II. An aliquot (100 to 200 ng) of linearized pCdpB-del7 was diluted 1:1 in H buffer (300 mM NaCl, 50 mM Tris [pH 9.0], 20 mM EDTA [pH 8.0]) and used for heteroduplex formation by denaturation at 99°C for 3 min followed by two hybridization cycles: 65°C for 5 min and 30°C for 15 min. One-third of the heteroduplex reaction mixture was used to transform CaCl2-competent E. coli BL21(DE3) cells.
Clones were screened by PCR for the desired deletion using detection primers DEL R, 5′-(CAGGCCCTTTTCGGTGATGCGCTC), corresponding to the residues 414-ERITEKGL-421 of the CpdB sequence, and DEL F, 5′-(GGCGGCGATTTGAAAGACA), corresponding to the new sequence created by the seven-amino-acid deletion. Colonies were picked and grown, and plasmid DNA template was prepared using the QIAprep spin miniprep kit (QIAGEN, Hilden, Germany). An aliquot of 1 μl of plasmid DNA was used as template in a PCR (screening) mixture that contained 2.5 μl of 10× Taq buffer, 200 μM of each deoxynucleoside triphosphate, 10 pmol each of the DEL F and DEL R primers, 1 U of Taq DNA polymerase, and molecular biology-grade water to a final volume of 25 μl. Positive clones were identified by electrophoresis on a 0.8% agarose gel for the presence of a 547-bp fragment that would confirm the desired deletion. Further confirmation of the desired deletion was carried out by DNA sequencing.
Similarly, substitution mutagenesis in the cpdB gene (mutants S261A and G242A) was performed by the design of tailed primers carrying the mutation(s) (nucleotides in lowercase) on the target sequence in the 5′ adaptor tail and used in conjunction with the gene-specific primers (normal capital letters for the downstream primer and boldface letters for the upstream primer) to generate the desired overhangs. S261A Rt, 5′-(CTGGATTGcGGTGGCGCCGGTACCGATGATGGCCACGCG), S261A Rs, 5′-(GGTACCGATGATGGCCACGCG), S261A Ft, 5′-(GGCGCCACCgCAATCCAGGCTGTGCCACACCTGGCGGCC), and S261A Fs, 5′-(GCTGTGCCACACCTGGCGGCC) were used in the single PCR amplification to create the point mutation S261A. For creating the G242A mutant, the following four primers were used: G242A Ft, 5′-(GATCAGACCGcCAACCTGGAAGGTTTGAAAGACAAGCGC); G242A Fs, 5′-(GAAGGTTTGAAAGACAAGCGC); G242A Rt, 5′-(CAGGTTGGcGGTCTGATCGCCGCCGGTATAGGAGTAGTC); and G242A Rs, 5′(GCCGCCGGTATAGGAGTAGTC).
Fourier transform infrared spectroscopy.
A ReactIR 4000 (Mettler Toledo) was used to monitor the time course of bioconversion of cyclododecanone to lauryl lactone. In a total volume of 10 ml (Tris-HCl, 50 mM, pH 9.0) containing 0.5 mM NADPH and 200 mU of enzyme, the reaction was started by adding 50 μl of substrate (0.1 M in n-propanol), giving a final substrate concentration of 0.5 mM. The reaction was stopped at a desired reaction time (5, 10, 20, 30, or 60 min) by adding 200 μl of hexadecane, and samples were read.
Chemicals.
The Mono Q, Sephacryl S200, and Superose 6 were purchased from Pharmacia (Uppsala, Sweden). Marker proteins for gel filtration and SDS gel electrophoresis were purchased from Pharmacia (Uppsala, Sweden) and Bio-Rad, respectively. All ketone substrates were purchased from Sigma-Aldrich, except for 2-methylcyclopentadecanone, which was prepared as described below. Standards of racemic lactones were prepared by oxidation of the cyclic ketones with m-chloroperbenzoic acid as previously described (39). All other chemicals purchased from various manufacturers were of analytical grade quality or higher.
Synthesis of 2-methylcyclopentadecanone.
This was carried out according to the method of Meyer et al. (40). Since the kinetic methylation of cyclopentadecanone gave a mixture of mono- and dialkylated products which could not be easily separated, Corey's method (19) was used to first convert cyclopentadecanone into its N,N-dimethylhydrazone, which was then monoalkylated with methyl iodide. Hydrolysis of the methylated hydrazone then gave 2-methylcyclopentadecanone in a total 47% yield.
Whole-cell BVMO oxidations.
Biotransformation experiments using whole cells of E. coli BL21(DE3)(pCD201) were carried out essentially as previously described for CPMO or CHMO (23, 67). As a negative control, BL21(DE3) cells harboring pSD80 instead of pCD201 were used.
Analytical methods and techniques.
The sample was extracted with hexane or ethyl acetate, dried with Na2SO4, and analyzed by gas chromotography (GC)-mass spectrometry (MS), using a Hewlett-Packard HP6890 series gas chromatograph system connected to an HP5971 mass-selective detector (Hewlett-Packard) and a DB-1 capillary column (J & W Scientific, Folsom, CA). The temperature program consisted of a 3-min hold at 75°C, an increase to 220°C at 10°C/min, an increase to 300°C at 20°C/min, and a 3.5-min hold at 300°C. Peaks were identified by comparison of their retention times and of their mass spectra with those of authentic standards. Infrared (IR) spectra were measured on a ReactIR 4000 (Mettler Toledo). 1H and 13C nuclear magnetic resonance (NMR) analyses were recorded in CDCl3 solution at room temperature, unless otherwise stated, on a Varian XL-200, INOVA-400, or INOVA-500 (Varian, Inc.) spectrometer. The rotation of chiral compounds was determined on a Jasco DIP-140 digital polarimeter (courtesy of the Chemistry Department, McGill University) at room temperature.
Chiral-phase GC analyses.
Chiral capillary-column GC was performed on a Perkin-Elmer Sigma 2000 gas chromatograph employing a 0.25-mm by 30-m (0.25-μm film) β-Dex 225 column (Supelco Inc.) or another chiral-phase column (Chrompak Chiralsil-Dex CB; 25-m by 0.25-mm column). The GC instrument used a flame ionization detector and helium as the carrier gas. The injector and detector temperatures were maintained at 225°C and 300°C, respectively.
E (enantioselectivity) value was calculated by a computer program available at website http://www.orgc.tu-graz.ac.at, with nonlinear data fitting according to the equations of Chen et al. or Rakels et al. (7, 46).
Chiral-GC and spectroscopic data.
2-Methylcyclopentadecanone (compound no. 1): 1H-NMR (400 MHz, CDCl3) δ: 0.95 (d, J = 7 Hz, 3H), 1.20 to 1.40 (m, 22H), 1.56 (m, 1H), 1.69 (m, 1H), 2.37 (m, 1H), 2.51 (m, 1H), 2.60 (m, 1H) ppm. Fab-MS: 239.3 (M + 1)+, 256 (M + NH4)+, 261.3 (M + Na)+.
15-Hexadecanolide (compound no. 2): 1H-NMR (500 MHz, CDCl3) δ: 1.25 (d, J = 6 Hz, 3H), 1.30 to 1.45 (m, 20H), 1.60 (m, 3H), 1.75 (m, 1H), 2.30 (m, 2H), 4.99 (m, 1H) ppm. Fab-MS: 254 (M+). It was identical to the reported IR υmax cm−1: 1735.
7-Methyl-2-oxepanone (compound no. 4): 1H-NMR (250 MHz, CDCl3) δ: 1.36 (d, J = 6.5 Hz), 1.62 (m, 4H), 1.93 (m, 2H), 2.65 (m, 2H), 4.28 (m, 1H) ppm. 13C-NMR (63 MHz, CDCl3) δ: 22.5, 22.9, 28.2, 35.0, 36.2, 76.8, 175.6 ppm. EI-MS (m/z): 128 (1%, M+), 113 (2%), 84 (95%), 55 (100%).
4-Methyl-2-oxepanone (compound no. 6a) (75%) and 6-methyl-2-oxepanone (compound no. 6b) (25%) on chiral-GC: 4-methyl isomer (compound no. 6a) (only one enantiomer) with retention time (r.t.) 28.62 min, EI-MS (m/z): 128 (10%, M+), 69 (100%); and 2-methyl isomer (compound no. 6b) with two enantiomers (major enantiomer [96% ee] with r.t. 30.23 min and minor enantiomer with r.t. 30.92 min), EI-MS (m/z): 128 (10%, M+), 98 (100%).
5-Methyl-2-oxepanone (compound no. 8) on chiral-GC: the r.t. was 30.52 min, EI-MS (m/z): 128 (18%, M+), 98 (25%), 69 (85%), 56 (100%).
5-Ethyl-2-oxepanone (compound no. 10) on chiral-GC: the r.t. was 33.13 min; EI-MS (m/z): 142 (5%, M+), 112 (34%), 97 (70%), 83 (100%).
5-t-Butyl-2-oxepanone (compound no. 12) on chiral-phase GC: only one enantiomer with r.t. 27.26 min (99% ee); EI-MS (m/z): 171 (1%, M++1), 155 (3%), 114 (100%), 86 (90%).
cis-3,7-Dimethyl-2-oxepanone (compound no. 14) on chiral-phase GC: only one enantiomer (99% ee) with r.t. 23.60 min; EI-MS (m/z): 142 (3%, M+), 98 (82%), 56 (100%).
6-Methyl tetrahydropyran-2-one (compound no. 16) on chiral-phase GC: two enantiomers with r.t.'s of 27.25 min and 27.52 min (major enantiomer, 87% ee); EI-MS (m/z): 114 (18%, M+), 99 (20%), 70 (100%).
4,4,6-Trimethyl tetrahydropyran-2-one (compound no. 20) on chiral-phase GC: two enantiomers with r.t.'s of 15.76 min and 16.49 min (major enantiomer, 41% ee) at 82% conversion; EI-MS (m/z): 142 (8%, M+), 127 (12%), 98 (25%), 83 (30%), 70 (60%), 56 (100%).
Octahydro-benzo[b]oxepin-2-one (compound no. 22a) (78%) and octahydro-benzo[c]oxepin-1-one (compound no. 22b) (22%) on chiral-phase GC: the r.t.'s of major isomer (compound no. 22a) with two enantiomers were 36.47 min (major enantiomer, 33% ee) and 37.14 min, EI-MS (m/z): 168 (7%, M+), 125 (9%), 97 (20%), 84 (100%); and the r.t. of minor isomer (compound no. 22b) was 35.23 min (only one enantiomer, 99% ee), EI-MS (m/z): 168 (45%, M+), 139 (15%), 126 (17%), 108 (40%), 81 (72%), 67 (100%).
cis/trans-Octahydro-benzo[c]oxepin-3-one (compound no. 24a) (70%) cis-octahydro-7-oxa-benzo cyclohepten-6-one (compound no. 24b) (30%) on chiral-phase GC showed three isomers, the same as on normal-phase GC; the r.t.'s of cis/trans forms of major isomer (compound no. 24a) were 39.50 and 40.02 min; EI-MS (m/z): 168 (M+), 138 (100% for trans-, and 50% for cis-), 96 (95% for trans- and 100% for cis-). The r.t. of the minor isomer (compound no. 24b) was 38.66 min, EI-MS (m/z): 168 (6%, M+), 140 (30%), 109 (30%), 96 (100%).
4,5-Dihydro-1H-benzo[c]oxepin-3-one (compound no. 26a) (70%) and 8,9-dihydro-5H-7-oxabenzo-cyclohepten-6-one (compound no. 26b) (30%): the r.t.'s on chiral-phase GC were 41.16 (major isomer [compound no. 26a]) and 42.09 min (minor isomer [compound no. 26b]); EI-MS (m/z): 162 (55%, M+), 117 (100%).
3-Methyl-isochroman-1-one (compound no. 28): it was a racemic mixture that did not resolve on the chiral-phase GC, but gave one peak at 29.67 min; EI-MS (m/z): 162 (50%, M+), 117 (100%). However, the substrate 2-methyl-1-indone was resolved on this chiral-GC and showed poor enantioselectivity (E, ∼1).
Nucleotide sequence accession numbers.
The nucleotide sequences determined in this study have been deposited in the GenBank database under accession numbers AB232538 (4.2-kb BclI fragment) and AB234289 (16S rRNA).
RESULTS
16S rRNA identification and growth substrates of strain HI-70.
The 1,475-base-long 16S sequence of strain HI-70 was found to be 99% identical to that of Pseudomonas pseudoalcaligenes strain KF707, a well-known biphenyl/polychlorinated biphenyl degrader (18); high percentages of identity were also found with Pseudomonas resinovorans strain ATCC 14235T (98%), Pseudomonas stutzeri strains SP1402 and LS401 (97%), and several other pseudomonads not identified to the species level (BLAST search results not shown). The HI-70 strain is tentatively referred to as a Pseudomonas sp. Besides its growth on cyclododecanol or cyclododecanone as a sole carbon and energy source, strain HI-70 is also able to grow on cycloundecanone (C11), cyclotridecanone (C13), cyclopentadecanol (C15), and cyclopentadecanone (C15). It did not grow on smaller cyclic C5 to C10 compounds, such as cyclopentanol, cyclopentanone, cyclohexanol, cyclohexanone, cycloheptanol, cycloheptanone, cyclooctanol, cyclooctanone, cyclononanone, and cyclodecanone. Methyl-substituted compounds, e.g., cis/trans 2-methyl cyclohexanol and 2-methyl cyclohexanone, did not serve as growth substrates either.
Cloning and sequencing of a 4.2-kb BclI fragment containing cpdB.
The primers that were designed from two conserved regions of several known BVMO sequences amplified the expected ∼1-kb DNA fragment. A partial sequence was determined prior to using it as a probe to clone a 4.2-kb BclI genomic fragment of strain HI-70 in an E. coli pUC19 vector to produce the recombinant plasmid, pCD200. DNA sequencing of the insert (4.216 kb) and computer analysis revealed the presence of a 601-codon open reading frame that we designated cpdB (described in more detail below). This is flanked by genes for a LysR-type transcriptional regulator (designated cpdR) upstream in the opposite orientation and a possible lactone hydrolase (cpdC) downstream in the same orientation as cpdB (Fig. 1). The respective intergenic spaces are 409 bp and 94 bp, each containing the appropriate Shine-Dalgarno sequences; a possible promoter element (TTGACA---GAGAAT) is readily identified in the 409-bp intergenic sequence immediately before the cpdB gene but not in the cpdB-cpdC intergenic space. The 304-amino-acid CpdR is most related to a “probable transcriptional regulator” (GenBank no. AE004683.1) in the Pseudomonas aeruginosa PAO1 genome showing 34% sequence identity. CpdC was found to be a new member of the large family of lipase/esterases; the closest homologous sequence of 42% identity (GenBank no. AAS77233.1) was derived from an uncultured organism.
FIG. 1.

Localization of cpdB encoding cyclopentadecanone monooxygenase in between cpdR, which encodes a potential transcriptional regulator, and cpdC, which encodes a lactone hydrolase, within a 4.216-kb BclI chromosomal fragment of Pseudomonas sp. strain HI-70. The XhoI restriction site within the cpdB gene is the point of insertion of a lacZ-Kmr cassette resulting in a mutant strain designated HI-70MB. The numbers in parentheses refer to the nucleotide positions of the respective endonuclease cleavage sites.
Inactivation of the cpdB gene.
The cpdB gene was chromosomally inactivated by transcriptional fusion of cpdB::lacZ, using a lacZ-Kmr cassette from the pKOK6.1 vector (30). The resulting mutant, Pseudomonas sp. strain HI-70MB, was unable to grow on cyclododecanol, cyclododecanone, cycloundecanone, cyclotridecanone, cyclopentadecanol, and cyclopentadecanone, which are otherwise carbon and energy sources for strain HI-70.
In LB medium, a low level (<20 Miller units) of β-galactosidase expression was observed in HI-70MB cells. The presence of cyclododecanol or cyclododecanone increased the activity to 2,128 or 1,089 Miller units, respectively, after 15 h of cultivation. This observation indicated a possible regulation of transcription of cpdB gene by cpdR that is located upstream. However, regulation is outside the scope of this study.
Sequence characteristics of CpdB.
The conserved stretches of amino acids in CpdB, typical of BVMO sequences, are those of two possible Rossmann folds (79-GGGFGG-84 and 256-GTGATS-261), a so-called BVMO fingerprint (221-FKGHSFHTSRWD-232), and two other motifs (109-GG-110 and 431-DCLIYATG-438) (15, 62, 69). The underlined sequences correspond to the invariant or most-conserved residues in the various aligned BVMO sequences (15). The three highest scores in a BLAST search showing 49% to 54% sequence identity were provided by putative monooxygenases derived from the genome sequences of Bradyrhizobium japonicum strain USDA 110 (blr3857; gi 47118316), Streptomyces avermitilis strain MA-4680 (gi 57546753), and Mycobacterium avium subsp. paratuberculosis (gi 41398205), followed by a known BVMO (cyclododecanone monooxygenase, CddA; CDMO) that originated from Rhodococcus ruber strain SC1 (32). In a phylogenetic analysis (result not shown), CpdB is in a distinct cluster from the majority of the cloned BVMOs that function with lower cyclic ring compounds, steroids, or benzene ring-based substrates. Examples of the latter are the classical CHMO of Acinetobacter sp. (8, 22, 29), CPMO of Comamonas sp. strain NCIMB 9872 (23), steroid monooxygenase (42), hydroxyacetophenone monooxygenase (HAPMO) (25), and phenylacetone monooxygenase (PAMO) (17, 37).
N-terminal amino acid sequencing confirmed the first 10 residues (SQLIQEPAEA) of the CpdB protein predicted by its DNA sequence, while also establishing the fact that the CpdB protein is characterized by an N-terminal extension of 66 amino acids compared to the prototype CHMO sequence (Fig. 2A). This is like the CDMO or CddA of R. ruber SC1 and the HAPMO of Pseudomonas fluorescens strain ACB (25). Interestingly, taken together with four other uncharacterized BVMO sequences in the GenBank microbial database, it appears that a subset of BVMO sequences arises that is characterized by having an N-terminal extension ranging from 60 to 147 amino acids (numbered from the first G of the first consensus Rossmann fold sequence). A particularly interesting feature within this extension is a stretch of about 20 amino acids that is predominantly charged (consensus DLxALRxKYRxERDKRLRxD).
FIG. 2.

(A) Identification of a conserved cluster of charged amino acids in the N-terminal sequences of a subset of BVMO sequences. The highly conserved sequences are shaded. CHMO, CPMO, and PAMO are representative BVMOs that do not have the extraneous N-terminal extension. The FAD-binding motif (GxGxxG, in dark shade) serves as a point of reference. In HAPMO, the first 19 amino acids are not shown; an alternative alignment provided a better FAD-binding motif (25). The origins of the various sequences, besides CpdB (this study), are shown as superscripts in the far left column as follows: 1, B. japonicum (Q89NI1); 2, S. avermitilis (Q82IY8); 3, Streptomyces coelicolor (Q9RL17); 4, M. paratuberculosis (Q73U59); 5, R. ruber (Q938F6); 6, P. fluorescens ACB (AAK54073); 7, T. fusca (1W4X_A); 8, Comamonas sp. strain NCIMB 9872 (Q8GAW0); and 9, Acinetobacter sp. strain NCIMB 9871 (BAA86293). The characters in parentheses are GenBank accession or reference numbers. Put., putative; Hyp. Prot., hypothetical protein. (B) Sequence alignment showing an apparent insertional sequence in CpdB and related sequences in between the BVMO fingerprint and the second Rossmann fold motif, as indicated. Amino acids 240 to 246 in CpdB are the deleted region, and the underlined G and S residues indicate the G242A and S261A substitutions (this study). The references indicated by superscripts are as described in the legend to panel A.
Another notable feature of the CpdB sequence is an apparent insertion of 10 or 11 amino acids (amino acids 240 to 249) in between the “BVMO fingerprint” (amino acids 221 to 232) and the second Rossmann fold sequence (amino acids 256 to 261; Fig. 2B). This is also found in CDMO and the four uncharacterized BVMO sequences (Fig. 2B).
Biochemical properties and purification of CpdB.
Knowledge of the cpdB nucleotide sequence was used to construct an overexpression clone in the IPTG-inducible plasmid pSD80 in order to verify its protein production and enzyme activity and to facilitate protein purification. In pCD201, the CpdB protein was found to be expressed readily, with an observed molecular mass of 63.5 kDa by SDS-PAGE analysis which confirms the predicted molecular mass of 68,313. No appropriate protein band of this size was detectable in the control cells containing the pSD80 vector only. By gel filtration, the molecular mass of the native CdnB was found to be 64 kDa, indicating a monomeric enzyme.
The enzyme was purified to homogeneity using a three-step procedure, based on anion exchange chromatography on Mono Q and two steps of molecular size exclusion chromatography on Superose 6 and Sephacryl S200, respectively. The specific activity of CpdB was enriched about 14 times, rendering 3.94 U/mg and a yield of 20% (Table 2). Several other purification trials using ammonium sulfate precipitation followed by hydrophobic interaction chromatography (phenyl, butyl, and octyl Sepharose) failed to produce an active enzyme preparation.
Cofactor and stability of CpdB.
The free FAD concentration was determined by HPLC using the supernatant of boiled purified enzyme. This was estimated to be 0.65 mol/mol protein. However, in the same fraction, 0.11 mol/mol FMN was detected (data not shown). CpdB requires NADPH for its activity; replacement by NADH resulted in greatly decreased catalytic activity (<5%). The Km for NADPH was established to be 24 μM.
In crude extracts, CpdB has a half-life of about 50 days at 4°C, whereas the purified enzyme retained its full activity when stored at −80°C for several months. Occasionally, protein precipitation was observed when CpdB was stored at 4°C for longer terms (>1 week), which could be prevented in part by adding FAD (protein:FAD ratio, 1:10). However, enzyme activity loss could not be recovered by the addition of FAD or FMN. Furthermore, heating of purified CpdB during the denaturing step in SDS sample buffer using reducing conditions (>5 min at 95°C) led to total degradation of the protein accompanied by the appearance of many additional bands in SDS-PAGE (data not shown).
Substrate specificity and kinetic properties of CpdB.
Optimum enzyme activity was obtained at 40°C and pH 9.0 using Tris-HCl buffer (50 mM). CpdB has the highest affinity towards cyclopentadecanone (5.8 μM) and the highest reaction speed (kcat) towards cyclotridecanone and methylcyclopentadecanone (6.5 and 6.6 s−1, respectively). However, the catalytic efficiency (kcat/Km) favors cyclopentadecanone (7.2 × 105 M−1 s−1) as a substrate (Fig. 3).
FIG. 3.

Comparison of the kinetic parameters of CpdB of Pseudomonas sp. strain HI-70 towards long-chain cyclic ketones. C11, C12, C13, C15, and C15+1 are cycloundecanone, cyclododecanone, cyclotridecanone, cyclopentadecanone, and 2-methycyclopentadecanone, respectively. The bars represent the Km values in μM. Solid line, kcat; dashed line, catalytic efficiency (kcat/Km).
CpdB is also able to oxidize a variety of alkyl-substituted cyclic ketones, particularly 2-methyl-, 2-methoxy-, and 2-ethoxycyclohexanone (Fig. 4). The activity of CpdB towards these substrates is 29 to 34% higher than that of cyclopentadecanone. However, the respective Km values are 790, 80, and 170 μM, compared to 6 μM for cyclopentadecanone. 4-Ethylcyclohexanone, in contrast to 4-methylcyclohexanone or 3-methylcyclohexanone, appeared to be a good substrate, having 80% of the cyclopentadecanone activity, but the affinity is low. Interestingly, the dimethyl-substituted 2,6-dimethylcyclohexanone is a poor substrate compared to the 2-methylated ketone.
FIG. 4.

The relative activities of purified CpdB toward a range of large and small cyclic ketones and bicyclic ketones. The specific activity (3.9 U/mg) toward cyclopentadecanone (substrate 4) is taken as 100%. n.d., not determined.
CpdB is also active towards saturated bicyclic ketones, such as 1- and 2-decalone. Both of these compounds displayed an equivalent high catalytic activity, but the affinity appears to favor 1-decalone with a Km value of 21 μM that approximates those of the large cyclic compounds. CpdB is also capable of using tetralones as substrates, but the enzyme appears to have a preference for substitutions of the keto group in the 2- or β- position, viz., 1-methyl-2-tetralone and β-tetralone. The results confirmed that conjugated ketones were generally poor substrates for this enzyme. Chloro-substituted cyclic ketones, such as 2-chloro-cycloheptanone, 2-chloro-cyclooctanone, and 2-chloro-cyclododecanone, are poor substrates for CpdB (results not shown).
ReactIR detection of product formation.
Using purified CpdB and cyclododecanone as the substrate, we monitored product formation (lauryl lactone) and substrate depletion with Fourier transform IR spectroscopy-based ReactIR 4000 spectroscopy. Figure 5 shows the overlay spectra obtained for the conversion of cyclododecanone (1,714 cm−1) to lauryl lactone (1,741 cm−1) as a function of time. The oxidations of cyclopentadecanone and 2-methylcyclopentadecanone were also followed using the React IR4000, and their lactone products were confirmed by extraction with ethyl acetate (EtOAc) and identified by comparison with authentic samples on GC-MS and capillary GC (data not shown).
FIG. 5.
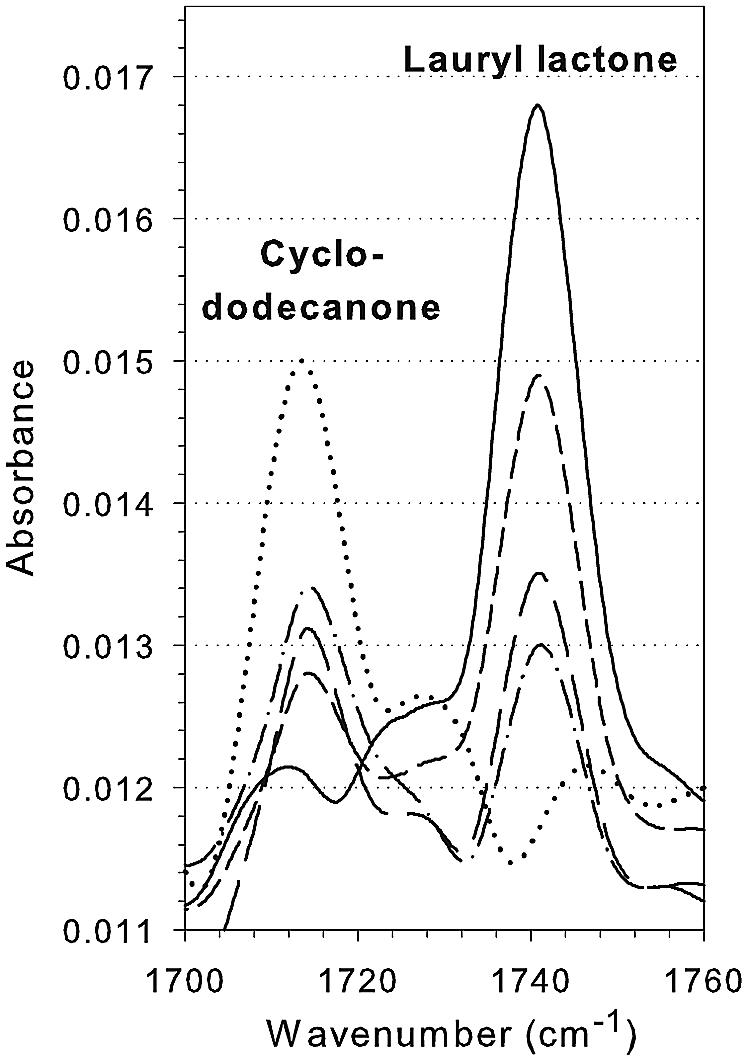
Overlaid infrared spectra of a time course monitoring of cyclododecanone conversion to lauryl lactone catalyzed by CpdB using the ReactIR 4000. A quantity of 0.2 U of the purified enzyme was used. · · · · · , 0 min; - · - · , 5 min; — —, 10 min; - - -, 20 min; —, 60 min.
Truncation and mutagenesis of CpdB. (i) pCpdB-delN.
To probe the possible function of the CpdB N-terminal extension, pCpdB-delN was created, which lacked the first 55 codons of cpdB. As a result, the mutant protein with the correct molecular size (62 kDa; calculated size, 61,876 Da) was found to be expressed as a soluble protein. N-terminal sequencing confirmed the first 10 amino acids of the truncated CdnB protein (MKDPYADPIV; methionine was introduced by the PCR primer). However, this truncated protein had less than 5% of the wild-type activity.
(ii) pCpdB-del7.
Deletion of amino acids 240-QTGNLEG-246 in pCpdB-del7 produced a correctly sized protein that was found in the particulate fraction and enzymatically inactive (data not shown). No attempt was made to solubilize the inclusion bodies.
(iii) G242A and S261A mutants.
In contrast to pCpdBdel7, a substitution within the seven-amino-acid region (G242A mutant) and a substitution within the second Rossmann fold motif (S261A mutant) produced a soluble and active protein of the equivalent quantity as the native protein. Kinetic parameters of the purified enzymes from these mutants were determined and compared to the wild-type enzyme (Table 3). In general, the catalytic efficiency (kcat/Km) of the G242A mutant did not change significantly for the three substrates and NADPH. In contrast, the S261A mutant showed an apparent increase in affinity towards NADPH as indicated by a threefold-lower Km than that of the wild-type protein (Table 3). The S261A mutant also showed an increased stability (75% activity after 50 days at 4°C) compared to the wild-type enzyme, whereas the G242A mutant showed a decreased activity and stability (half-life of about 30 days at 4°C; results not shown).
TABLE 3.
Summary of the kinetic parameters of CpdB and its mutants
| Substrate | Km (μM) | kcat (s−1) | kcat/Km (M−1 s−1) |
|---|---|---|---|
| Cyclododecanone (C12) | |||
| CpdB (wt)a | 34.0 ± 1.2 | 2.7 | 7.9 × 104 |
| CpdB (S261A) | 24.5 ± 2.5 | 3.1 | 1.3 × 105 |
| CpdB (G242A) | 38.3 ± 2.3 | 2.0 | 5.2 × 104 |
| Cyclotridecanone (C13) | |||
| CpdB (wt) | 12.5 ± 0.8 | 6.5 | 5.2 × 105 |
| CpdB (S261A) | 9.1 ± 1.5 | 5.3 | 5.8 × 105 |
| CpdB (G242A) | 14.4 ± 0.8 | 3.6 | 5.2 × 105 |
| Cyclopentadecanone (C15) | |||
| CpdB (wt) | 5.8 ± 0.9 | 4.2 | 7.2 × 105 |
| CpdB (S261A) | 7.0 ± 2.1 | 4.9 | 7.0 × 105 |
| CpdB (G242A) | 6.5 ± 2.1 | 2.7 | 4.2 × 105 |
| NADPH | |||
| CpdB (wt) | 24.0 ± 2.1 | NAb | NA |
| CpdB (S261A) | 7.8 ± 1.2 | NA | NA |
| CpdB (G242A) | 32.4 ± 3.0 | NA | NA |
wt, wild type.
NA, not applicable.
Kinetic resolution of 2-methylcyclopentadecanone (compound no. 1).
(The compound numbers refer to the bold and underlined numbers in Tables 4 and 5). Whole-cell biotransformation was carried out using E. coli BL21(DE3)(pCD201) cells induced by IPTG to test the possibility of production of enantiopure 15-hexadecanolide as a possible application of CPDMO in asymmetric organic synthesis (33). It turned out that only 10% conversion could be achieved with one-stage fermentation, and the lactone (compound no. 2) had 65% ee (S). A prolonged fermentation (three stage) yielded 30% conversion and a lactone ee of 59% (Table 4). The biotransformation mixture was extracted by EtOAc and monitored using two chiral-phase GC columns. The Supelco Beta DEX 225 chiral-phase column did not resolve the lactone enantiomers, but it could separate the ketone substrate and lactone product. This problem was resolved by using a Chrompak Chiralsil-Dex CB column as previously described (36). The absolute configuration of 15-hexadecanolide was also assigned by comparison with the value in the literature (36).
TABLE 4.
Baeyer-Villiger oxidation of monocyclic ketones by CPDMO
 |
 |
Conversion yield was based on the EtOAc-extracted sample determined by chiral-phase GC analysis on a β-Dex 225 column or Chrompak Chiral-Dex CB column and referred to an internal standard.
ee, enantiomeric excess equal to [R] − [S]/[R] + [S] (%). Values of ee were based on chiral-phase GC analysis. conv. yield, conversion yield.
E(ee), E(enantiomeric ratio) value calculated based on product ee(P) and remaining substrate ee(S), E = ln[(1 − ee(S)/(1 + ee(S)/ee/(P))]/ln[(1 + ee(S)/(1 + ee(S)/ee(P))] (46) or based on product ee(P) and conversion yield referred to in equation in footnote e.
(S) or (R), absolute configuration of products.
E[c, ee(P)], E(enantiomeric ratio) value calculated based on product ee(P) and conversion yield c, then E[c, ee(P)] = ln[1 − c(1 + ee(P))]/ln[1 − c(1 − ee(P))] (7).
Isomer ratio is the ratio of two regioisomeric lactones.
Enantioselectivity is the product ee for prochiral substrate and E (enantiomeric ratio) for racemic substrate.
Enantioselectivity (E) calculated from the plot at 4 h, 32% conversion; the regioisomer ratio was 1 (945% ee):5.2 (99% ee).
TABLE 5.
Baeyer-Villiger oxidations of bicyclic ketones by CPDMO
 |
Conversion yield was based on the EtOAc-extracted sample determined by normal-phase or chiral-phase GC analysis and referred to an internal standard.
Stereoselectivities refer to regioisomeric lactone ratio, and the given enantiomeric excess values are based on determinations of β-Dex 225 chiral-phase column GC analysis.
Product isomer contents were based on both normal-phase GC-MS and chiral-phase GC analyses.
Biotransformation products of small monocyclic ketones.
In whole-cell biotransformation, 2-, 3-, or 4-methyl-substituted cyclohexanones all gave good percent conversions. In particular, 2-methylcyclohexanone (compound no. 3) gave an excellent enantioselectivity, E > 200. CPDMO oxidized it with complete (S)-selectivity, giving a product (compound no. 4) with specific rotation [α]D of −16, c 10, in CH2Cl2 and >99% ee in chiral-phase GC determination. Oxidation of 3-methylcyclohexanone (compound no. 5) gave two regioisomers of lactone, compound no. 6b and compound no. 6a (the ratio of lactone was 1:3), although with high enantioselectivity. Oxidation of 4-methyl and 4-ethyl cyclohexanones showed that CPDMO behaved like the CHMO of Acinetobacter sp. strain NCIMB 9871 that gave the highly optical pure lactone by desymmetrization of prochiral substrate (57).
4-t-Butyl cyclohexanone (compound no. 11) was transformed to the corresponding lactone (compound no. 12) in good yield and excellent enantioselectivity of 99% ee (S). CPDMO also accepts cis-2,6-dimethylcyclohexanone (compound no. 13) as a substrate, with 74% conversion yield at 20 h with high enantioselectivity (99% ee). Since the 2,6-dimethylcyclohexanone sample was a cis/trans mixture (5% trans-isomer), we also found that trans-2,6-dimethylcyclohexanone was a relatively poor substrate of CPDMO.
The five-member ring ketone system generally gave a poor performance (Table 4). However, a multi-methyl-substituted cyclopentanone such as 2,4,4-trimethylcyclopentanone (compound no. 19) turned out to be a good substrate; it was oxidized to the corresponding lactone (compound no. 20) in a 78% conversion yield with little enantioselectivity (E = 15).
Biotransformation products of bicyclic ketones.
Table 5 summarizes the Baeyer-Villiger oxidations of bicyclic ketones mediated by E. coli BL21(DE3)(pCD201) whole cells. A racemic trans-1-decalone (compound no. 21) containing 10% cis-1-decalone was tested in the whole-cell Baeyer-Villiger oxidation. We found that both enantiomers of trans-1-decalone were good substrates, whereas both cis-1-decalone enantiomers were not. The conversion yield at 20 h reached 80%. This reaction gave two regioisomers of lactone product in a 3.6:1 ratio. The major one (compound no. 22a) gave 33% ee, whereas the minor one (compound no. 22b) gave 98% ee based on chiral-GC results. However, the conversion yield did not improve after prolonged incubation.
With 2-decalone (compound no. 23), which was a mixture of 20% of cis-2-decalone and 80% of trans-2-decalone, three regioisomer lactone products were obtained in a total conversion of 23% at 20 h. The cis isomer was completely consumed, and the two enantiomers were oxidized to two alternate lactone regioisomers. Such behavior is similar to the monocyclic ketone system described earlier. In this case, the cis isomer is a better substrate than the trans isomer. β-Tetralone (compound no. 25) and 2-methyl-1-indone (compound no. 27) were also shown to be good substrates for CPDMO, although the stereoselectivity was not high.
DISCUSSION
This study describes the cloning, purification, and characterization of CPDMO, a new type I BVMO that contains FAD as a cofactor and uses NADPH as an electron donor (26). CPDMO is a valuable addition to a short but growing list of cloned BVMOs as regio- and/or enantioselective biocatalysts (for a review, see reference 26; for recent reports, see references 5, 6, 16, 17, 25, and 64).
The sequence of CPDMO is characterized by an N-terminal extension of 66 amino acids compared to that of the prototypical CHMO of Acinetobacter sp. strain NCIMB 9871 (22, 29). HAPMO (640 amino acids), an aromatic monooxygenase of P. fluorescens ACB, is by far the largest BVMO, with an N-terminal extension of about 135 residues (25). This N-terminal portion was deemed important for the structural integrity of the dimeric protein, since deletion of the first 115 amino acids rendered the protein inactive and insoluble (25). In the case of CPDMO, truncation of the 55 N-terminal amino acids in pCpdB-delN resulted in the expression of a soluble but almost inactive protein. Presumably, this resulted in an apoprotein which had lost its ability to bind FAD efficiently (demonstrated by an increased 280/440 nm ratio). Nonetheless, this N-terminal extension in CPDMO, as well as in HAPMO, is expected to constitute an additional domain besides the two-domain architecture, consisting of a FAD-binding and an NADPH-binding domain, found in the three-dimensional structure of a PAMO that originated from the genome of a thermophilic actinomycete, Thermobifida fusca (37). It will be interesting to find the possible contribution of the newly identified cluster of charged amino acids to the function of CPDMO or in a related protein.
A second sequence characteristic of CPDMO and its few related proteins is the presence of a 9- or 10-amino-acid insertion between the BVMO fingerprint and the second Rossmann fold (Fig. 2B). According to the three-dimensional structure of PAMO, the fingerprint amino acids constitute a linker region that connects the FAD-binding domain and the NADPH domain (37), and they are not part of the active site. We reckoned that this region may specify some substrate determinants towards the large cyclic ketones. Unfortunately, deletion of seven amino acids in pCpdB-del7 rendered the protein insoluble and inactive. However, a single amino acid substitution in the G242A mutant produced a variant that retained 70% of the wild-type enzyme activity. In the absence of structural information regarding a BVMO that is complexed with its substrate or cofactor (37), we are not able to make any definite statement about the G242A variant at this time.
With reference to the PAMO structure, seven of the eight residues involved in FAD binding are conserved in CPDMO. These amino acids are W112, Y117, D123, Y129, R387, F439, and M500; the corresponding residues in the PAMO sequence are W55, Y60, D66, Y72, R337, F389, and M446, respectively (37). The exception is A442 of PAMO which appears to be replaced by T494 in CPDMO in the present sequence alignment. CPDMO R387 is the equivalent of R337 of PAMO, which is part of the active site and strictly conserved among BVMOs (37). Also, extrapolating from the studies on HAPMO (27) and PAMO, the R280 and K386 of CPDMO are predicted to represent NADPH specificity determinants. In the PAMO structure these residues are R217 and K336 (37).
The most interesting mutant derived from this study is S261A, which led to a significantly higher affinity of the variant enzyme towards NADPH, with a Km of 8 μM compared to the 24 μM value of the native CPDMO. The former value fits well with the few determined NADPH Km values of CPMO, CHMO, and HAPMO that are <3 μM, 7 to 10 μM, and 64 μM, respectively (25, 53, 61). It appeared that in the native CPDMO sequence, a serine substitution at position 261 that is a deviation from the consensus dinucleotide binding motif (GXGXXG or A) in the second Rossmann fold motif led to a decreased affinity towards the cofactor. In addition, the S261A mutant led to an increased stability by almost doubling its half-life (90 days versus the 50 days of the wild-type enzyme).
CPDMO activity was initially assayed for its conversion of cyclododecanone to lauryl lactone using the conventional NADPH oxidation. As a proof of concept, we have used the state-of-the-art ReactIR 4000 technology that employs mid-infrared spectroscopy and a diamond probe to monitor the progress of this BVMO reaction as a function of time. Previously, Dadd et al. (11) had used a silicon probe of ReactIR to follow the enzyme kinetics involved in nitrile biocatalysis using whole cells of Rhodococcus rhodochrous LL100-21. This technique provided excellent quantitative and qualitative real-time data of nitrile biocatalytic reactions that are largely water soluble. As shown in Fig. 5, conversion of the ketone group of cyclododecanone to a lactone by the purified CPDMO is readily monitored at the appropriate wavelength as a function of time. In a separate study, we have followed the bioconversion of cyclododecanone to lauryl lactone in a two-phase bioreactor and whole-cell system for the preparation of the water-insoluble lactone (J. Yang, M.-J. Lorraine, D. Rho, and P. C. K. Lau, unpublished data). Lauryl lactone is a compound that is not readily available by chemical production (59).
CPDMO is also capable of producing oxacyclohexadecan-2-one, a musk compound responsible for the pleasant odor of angelica root oil used as a fixative in perfumery (trade name Exaltolide [the Merck Index]). However, we are particularly interested in knowing whether CPDMO is capable of producing chiral products, as has been shown for several BVMOs that demonstrated good enantio- or regioselectivity (41, 56, 58). To prove the concept, CPDMO was used to produce a 15-hexadecanolide that represents a rare example of macrolactones known in the literature. (R)-15-hexadecanolide is one of the three active components of a stink bug Piezodorus hybneri pheromone that elicits aggregation behavior in males. This stink bug commonly infests leguminous crops, such as soybeans and kidney beans (36). The two stereoisomers of hexadecanolides are otherwise synthesized by a solvent-intensive chemical route involving at least six steps using the Yamaguchi or Mitsunobu macrolactonization reaction of a chiral material, (R)-15-hydroxyhexadecanoic acid (33). As it turned out, although a one-step biotransformation is possible, CPDMO was able to make only the nonactive S-stereoisomer, besides giving low enantioselectivity (59% ee; E = 5.8).
Nonetheless, CPDMO displays a wide spectrum of Baeyer-Villiger oxidations of cyclic ketones that includes methyl-substituted short-chain cyclic compounds (C5 and C6) and bicyclic ketones, such as decalone and β-tetralone. These are poor substrates of the well-characterized Acinetobacter CHMO and CPMO from Comamonas sp. strain NCIMB 9872 (56, 66). CPDMO is particularly useful for the synthesis of both enantiomers of the 7-methyl-2-oxepanone lactone (compound no. 4). CPDMO was able to synthesize 7-methyl-2-oxepanone from 2-methylcyclohexanone (compound no. 3) with an impressive E value of >200 and almost complete (S)-selectivity (99% ee). However, such a good enantioselective result was previously found to be achieved by a recombinant CDMO derived from Rhodococcus SC1 but not seven other BVMOs (34). The enantiomer of 7-methyl-2-oxepanone (compound no. 4) is a desirable chiral building block and substitute for chiral ω-2-hydroxyl heptanoic acid that was recently used for the synthesis of a daumone, a pheromone that has an antiaging effect in worms (24). However, ω-2-hydroxy-heptanoic acid moiety was synthesized from a four-step chemical transformation that is quite expensive and dangerous, requiring regiospecific Grignard-type alkylation of (R)-(+)-propylene oxide with 5-bromo-1-pentene and tetrahydrofuran solvent and later oxidation of the double bond to acid (24). Alternatively, racemic 7-methyl-2-oxepanone (compound no. 4) can be prepared by reaction of the 2-methycyclohexanone (compound no. 3) with m-chloroperbenzoic acid in dichloromethane (39).
In summary, the discovery and purification of CPDMO pave the way for its possible structural determination, knowledge of which will provide great value in further biotechnology development or application, besides serving as a guide for possible targeted changes in the primary structure of CPDMO to increase its biocatalytic potential. Substrate profiling of CPDMO constitutes a quintessential first step towards the possibility of a desirable bioproduction of commercially useful compounds. Recently, Hilker et al. provided the first example of a Baeyer-Villiger oxidation that operated at near-kilogram scale of a (±)-bicyclo[3.2.0] hept-2-en-6-one substrate for the production of optically pure lactones (21).
Finally, this study reiterates the need to isolate and characterize new microorganisms in the environment, not only because we want to enrich the little that we know about microbial diversity or community structure, but also because new microorganisms may have interesting new metabolic or biocatalytic properties (13, 43, 50). No strain, no gain.
Acknowledgments
The first three authors made equal contributions to this work.
We thank K. Morley and R. Kazlauskas for help with the chiral-GC column and analysis and C. Beaulieu and the J. Hawari laboratory for analytical help with GC-MS.
This research was financially supported in part by a Kansai University Grant-in-Aid for the Promotion of Advanced Research in a Graduate Course, 2005, and by a High-Tech Research Center Project for Private Universities matching fund subsidy from MEXT (Ministry of Education, Culture, Sports, Science, and Technology), 2002 to 2006.
This paper is dedicated to D. T. Gibson, who is retiring and whose influence on biodegradation and biotransformation goes beyond aromatic metabolism.
Footnotes
This publication is issued as NRCC number 47265.
REFERENCES
- 1.Alphand, V., G. Carrea, R. Wohlgemuth, R. Furstoss, and J. M. Woodley. 2003. Towards large-scale synthetic applications of Baeyer-Villiger monooxygenases. Trends Biotechnol. 21:318-323. [DOI] [PubMed] [Google Scholar]
- 2.Altschul, S. F., T. L. Madden, A. A. Schaffer, J. Zhang, Z. Zhang, W. Miller, and D. J. Lipman. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ayala, M., and E. Torres. 2004. Enzymatic activation of alkanes: constraints and prospective. Appl. Catal. A 272:1-13. [Google Scholar]
- 4.Bradford, M. M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248-254. [DOI] [PubMed] [Google Scholar]
- 5.Brzostowicz, P. C., M. S. Blasko, and P. E. Rouviere. 2002. Identification of two gene clusters involved in cyclohexanone oxidation in Brevibacterium epidermidis strain HCU. Appl. Microbiol. Biotechnol. 58:781-789. [DOI] [PubMed] [Google Scholar]
- 6.Brzostowicz, P. C., D. M. Walters, S. M. Thomas, V. Nagarajan, and P. E. Rouviere. 2003. mRNA differential display in a microbial enrichment culture: simultaneous identification of three cyclohexanone monooxygenases from three species. Appl. Environ. Microbiol. 69:334-342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen, C.-S., Y. Fujimoto, G. Girdaukas, and C. J. Sih. 1982. Quantitative analyses of biochemical kinetic resolutions of enantiomers. J. Am. Chem. Soc. 104:7294-7299. [Google Scholar]
- 8.Chen, Y.-C. J., O. P. Peoples, and C. T. Walsh. 1988. Acinetobacter cyclohexanone monooxygenase: gene cloning and sequence determination. J. Bacteriol. 170:781-789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheng, Q., S. M. Thomas, and P. Rouviere. 2002. Biological conversion of cyclic alkanes and cyclic alcohols into dicarboxylic acids: biochemical and molecular basis. Appl. Microbiol. Biotechnol. 58:704-711. [DOI] [PubMed] [Google Scholar]
- 10.Chiu, J., P. E. March, R. Lee, and D. Tillett. 2004. Site-directed, ligase-independent mutagenesis (SLIM): a single-tube methodology approaching 100% efficiency in 4 h. Nucleic Acids Res. 32:e174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dadd, M. R., D. C. Sharpe, A. J. Pettman, and C. J. Knowles. 2000. Real-time monitoring of nitrile biotransformations by mid-infrared spectroscopy. J. Microbiol. Methods 41:69-75. [DOI] [PubMed] [Google Scholar]
- 12.Davis, B. J. 1964. Disk electrophoresis. II. Method and application to human serum proteins. Ann. N. Y. Acad. Sci. 121:404-427. [DOI] [PubMed] [Google Scholar]
- 13.Demain, A. L. 2000. Small bugs, big business: the economic power of the microbe. Biotechnol. Adv. 18:499-514. [DOI] [PubMed] [Google Scholar]
- 14.Edman, P., and A. Henschen. 1975. Sequence determination, p. 232-279. In S. B. Needleman (ed.), Protein sequence determination. A source book for methods and techniques, 2nd ed. Springer-Verlag KG, Berlin, Germany.
- 15.Fraaije, M. W., N. M. Kamerbeek, W. J. van Berkel, and D. B. Janssen. 2002. Identification of a Baeyer-Villiger monooxygenase sequence motif. FEBS Lett. 518:43-47. [DOI] [PubMed] [Google Scholar]
- 16.Fraaije, M. W., N. M. Kamerbeek, A. J. Heudekamp, R. Fortin, and D. B. Janssen. 2004. The prodrug activator EtaA from Mycobacterium tuberculosis is a Baeyer-Villiger monooxygenase. J. Biol. Chem. 279:3354-3360. [DOI] [PubMed] [Google Scholar]
- 17.Fraaije, M. W., J. Wu, D. P. Heuts, E. W. van Hellemond, J. H. Spelberg, and D. B. Janssen. 2005. Discovery of a thermostable Baeyer-Villiger monooxygenase by genome mining. Appl. Microbiol. Biotechnol. 66:393-400. [DOI] [PubMed] [Google Scholar]
- 18.Furukawa, K., and T. Miyazaki. 1986. Cloning of a gene cluster encoding biphenyl and chlorobiphenyl degradation in Pseudomonas pseudoalcaligenes. J. Bacteriol. 166:392-398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Graham, R. J., and L. Weiler. 1991. The stereoselective alkylation and conformational analysis of 15-hexadecanolide. Tetrahedron Lett. 32:1027-1030. [Google Scholar]
- 20.Harayama, S., Y. Kasai, and A. Hara. 2004. Microbial communities in oil-contaminated seawater. Curr. Opin. Biotechnol. 15:205-214. [DOI] [PubMed] [Google Scholar]
- 21.Hilker, I., R. Wohlgemuth, V. Alphand, and R. Furstoss. 2005. Microbial transformations 59: first kilogram scale asymmetric microbial Baeyer-Villiger oxidation with optimized productivity using a resin-based in situ SFPR strategy. Biotechnol. Bioeng. 92:702-710. [DOI] [PubMed] [Google Scholar]
- 22.Iwaki, H., Y. Hasegawa, M. Teraoka, T. Tokuyama, H. Bergeron, and P. C. K. Lau. 1999. Identification of a transcriptional activator (ChnR) and a 6-oxohexanoate dehydrogenase (ChnE) in the cyclohexanol catabolic pathway in Acinetobacter sp. NCIMB 9871 and localization of the genes that encode them. Appl. Environ. Microbiol. 65:5158-5162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iwaki, H., Y. Hasegawa, S. Wang, M. M. Kayser, and P. C. K. Lau. 2002. Cloning and characterization of a gene cluster involved in cyclopentanol metabolism in Comamonas sp. strain NCIMB 9872 and biotransformations effected by Escherichia coli-expressed cyclopentanone 1,2-monooxygenase. Appl. Environ. Microbiol. 68:5671-5684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jeong, P. Y., M. Jung, Y. H. Yim, H. Kim, M. Park, E. Hong, W. Lee, Y. H. Kim, K. Kim, and Y. K. Paik. 2005. Chemical structure and biological activity of the Caenorhabditis elegans dauer-inducing pheromone. Nature 433:541-545. [DOI] [PubMed] [Google Scholar]
- 25.Kamerbeek, N. M., M. J. Moonen, J. G. Van Der Ven, W. J. Van Berkel, M. W. Fraaije, and D. B. Janssen. 2001. 4-Hydroxyacetophenone monooxygenase from Pseudomonas fluorescens ACB. A novel flavoprotein catalyzing Baeyer-Villiger oxidation of aromatic compounds. Eur. J. Biochem. 268:2547-2557. [DOI] [PubMed] [Google Scholar]
- 26.Kamerbeek, N. M., D. B. Janssen, W. J. H. van Berkel, and M. W. Fraaije. 2003. Baeyer-Villiger monooxygenases, an emerging family of flavin-dependent biocatalysts. Adv. Synth. Catal. 345:667-678. [Google Scholar]
- 27.Kamerbeek, N. M., M. W. Fraaije, and D. B. Janssen. 2004. Identifying determinants of NADPH specificity in Baeyer-Villiger monooxygenases. Eur. J. Biochem. 271:2107-2116. [DOI] [PubMed] [Google Scholar]
- 28.Kelly, D. R., P. W. H. Wan, and J. Tang. 1998. Flavin monooxygenases—uses as catalysts for Baeyer-Villiger ring expansion and heteroatom oxidation, p. 536-587. In H.-J. Rehm and G. Reed (ed.), Biotechnology, vol. 8a. Biotransformations I. Wiley-VCH, Weinheim, Germany. [Google Scholar]
- 29.Kneller, M. B., M. J. Cheesman, and A. E. Rettie. 2001. ESI- and MALDI-MS analysis of cyclohexanone monooxygenase from Acinetobacter NCIB 9871. Biochem. Biophys. Res. Commun. 282:899-903. [DOI] [PubMed] [Google Scholar]
- 30.Kokotek, W., and W. Lotz. 1989. Construction of a lacZ-kanamycin resistance cassette, useful for site-directed mutagenesis and as a promoter probe. Gene 84:467-471. [DOI] [PubMed] [Google Scholar]
- 31.Koma, D., Y. Sakashita, K. Kubota, Y. Fujii, F. Hasumi, S.-Y. Chung, and M. Kubo. 2004. Degradation pathways of cyclic alkanes in Rhodococcus sp. NDKK48. Appl. Microbiol. Biotechnol. 66:92-99. [DOI] [PubMed] [Google Scholar]
- 32.Kostichka, K., S. M. Thomas, K. J. Gibson, V. Nagarajan, and Q. Cheng. 2001. Cloning and characterization of a gene cluster for cyclododecanone oxidation in Rhodococcus ruber SC1. J. Bacteriol. 183:6478-6486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kuwahara, S., T. Tsuruta, W. S. Leal, and O. Kodama. 1998. Synthesis of both enantiomers of 15-hexadecanolide, a sex pheromone component of the stink bug, Piezodorus hybneri. Biosci. Biotechnol. Biochem. 62:1261-1263. [DOI] [PubMed] [Google Scholar]
- 34.Kyte, B. G., P. Rouviere, Q. Cheng, and J. D. Stewart. 2004. Assessing the substrate selectivities and enantioselectivities of eight novel Baeyer-Villiger monooxygenases toward alkyl-substituted cyclohexanones. J. Org. Chem. 69:12-17. [DOI] [PubMed] [Google Scholar]
- 35.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 36.Leal, W. S., S. Kuwahara, X. Shi, H. Higuchi, C. E. B. Marino, M. Ono, and J. Meinwald. 1998. Male-released sex pheromone of the stink bug Piezodorus hybneri. J. Chem. Ecol. 24:1817-1829. [Google Scholar]
- 37.Malito, E., A. Alfieri, M. W. Fraaije, and A. Mattevi. 2004. Crystal structure of a Baeyer-Villiger monooxygenase. Proc. Natl. Acad. Sci. USA 101:13157-13162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marcinka, K., C. Roehring, and S. Kluge. 1992. Changes in protein patterns of pea plants systematically infected with red clover mottle virus. Biochem. Physiol. Pflanz. (BPP) 188:187-193. [Google Scholar]
- 39.Meinwald, J., J. J. Tufariello, and J. J. Hurst. 1964. Fused small-ring compounds. I. Synthesis of some trans-bicyclo[3.2.0]heptanes and trans-bicyclo[4.2.0]octanes. J. Org. Chem. 29:2914-2919. [Google Scholar]
- 40.Meyer, A. I., D. R. Williams, S. White, and G. W. Erickson. 1981. An asymmetric synthesis of acyclic and macrocyclic alpha-alkyl ketones. The role of (e)- and (z)-lithioenamines. J. Am. Chem. Soc. 103:3088-3093. [Google Scholar]
- 41.Mihovilovic, M. D., B. Müller, and P. Stanetty. 2002. Monooxygenase mediated Baeyer-Villiger oxidations. Eur. J. Org. Chem. 2002:3711-3730. [Google Scholar]
- 42.Morii, S., S. Sawamoto, Y. Yamauchi, M. Miyamoto, M. Iwami, and E. Itagaki. 1999. Steroid monooxygenase of Rhodococcus rhodochrous: sequencing of the genomic DNA, and hyperexpression, purification, and characterization of the recombinant enzyme. J. Biochem. (Tokyo) 126:624-631. [DOI] [PubMed] [Google Scholar]
- 43.Muraki, T., M. Taki, Y. Hasegawa, H. Iwaki, and P. C. K. Lau. 2003. Prokaryotic homologs of the eukaryotic 3-hydroxyanthranilate 3,4-dioxygenase and 2-amino-3-carboxymuconate-6-semialdehyde decarboxylase in the 2-nitrobenzoate degradation pathway of Pseudomonas fluorescens strain KU-7. Appl. Environ. Microbiol. 69:1564-1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Palleroni, N. J., and E. R. B. Moore. 2004. Taxonomy of pseudomonads: experimental approaches, p. 3-44. In J.-L. Ramos (ed.), Pseudomonas. I. Genomics, life style and molecular architecture. Kluwer Academic/Plenum Publishers, New York, N.Y.
- 45.Parke, D. 1990. Construction of mobilizable vectors derived from plasmids RP4, pUC18 and pUC19. Gene 93:135-137. [DOI] [PubMed] [Google Scholar]
- 46.Rakels, J. L. L., A. J. J. Straathof, and J. J. Heijnen. 1993. A simple method to determine the enantiomeric ratio in enantioselective biocatalysis. Enzyme Microb. Technol. 15:1051-1056. [DOI] [PubMed] [Google Scholar]
- 47.Ramos, J.-L. 2004. Pseudomonas, vol. I-III. Kluwer Academic/Plenum Publishers, New York, N.Y.
- 48.Rios-Hernandez, L. A., L. M. Gieg, and J. M. Suflita. 2003. Biodegradation of an alicyclic hydrocarbon by a sulfate-reducing enrichment from a gas condensate-contaminated aquifer. Appl. Environ. Microbiol. 69:434-443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 50.Schaechter, M., R. Kolter, and M. Buckley. 2004. Microbiology in the 21st century: where are we and where are we going? American Academy of Microbiology, Washington, D.C.
- 51.Schumacher, J. D., and R. M. Fakoussa. 1999. Degradation of alicyclic molecules by Rhodococcus ruber CD4. Appl. Microbiol. Biotechnol. 52:85-90. [DOI] [PubMed] [Google Scholar]
- 52.Shen, A. L., and C. B. Kasper. 2000. Differential contributions of NADPH-cytochrome P450 oxidoreductase FAD binding site residues to flavin binding and catalysis. J. Biol. Chem. 275:41087-41091. [DOI] [PubMed] [Google Scholar]
- 53.Sheng, D., D. P. Ballou, and V. Massey. 2001. Mechanistic studies of cyclohexanone monooxygenase: chemical properties of intermediates involved in catalysis. Biochemistry 40:11156-11167. [DOI] [PubMed] [Google Scholar]
- 54.Simon, R., U. Priefer, and A. Pühler. 1983. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram negative bacteria. Bio/Technology 1:784-789. [Google Scholar]
- 55.Smith, S. P., K. R. Barber, S. D. Dunn, and G. S. Shaw. 1996. Structural influence of cation binding to recombinant human brain S100b: evidence for calcium-induced exposure of a hydrophobic surface. Biochemistry 35:8805-8814. [DOI] [PubMed] [Google Scholar]
- 56.Stewart, J. D. 1998. Cyclohexanone monooxygenase: a useful reagent for asymmetric Baeyer-Villiger reactions. Curr. Org. Chem. 2:195-216. [Google Scholar]
- 57.Stewart, J. D., K. W. Reed, C. A. Martinez, J. Zhu, G. Chen, and M. M. Kayser. 1998. Recombinant baker's yeast as a whole-cell catalyst for asymmetric Baeyer-Villiger oxidations. J. Am. Chem. Soc. 120:3541-3548. [Google Scholar]
- 58.ten Brink, G. J., I. W. Arends, and R. A. Sheldon. 2004. The Baeyer-Villiger reaction: new developments toward greener procedures. Chem. Rev. 104:4105-4124. [DOI] [PubMed] [Google Scholar]
- 59.Thomas, S. M., R. DiCosimo, and V. Nagarajan. 2002. Biocatalysis: applications and potentials for the chemical industry. Trends Biotechnol. 20:238-242. [DOI] [PubMed] [Google Scholar]
- 60.Trudgill, P. 1986. Terpenoid metabolism by Pseudomonas, p. 483-525. In J. R. Sokatch and L. N. Ornston (ed.), The bacteria: a treatise on structure and function. The biology of Pseudomonas. Academic Press Inc., Orlando, Fla.
- 61.Trudgill, P. 1990. Cyclopentanone 1,2-monooxygenase from Pseudomonas NCIMB 9872. Methods Enzymol. 188:77-81. [DOI] [PubMed] [Google Scholar]
- 62.Vallon, O. 2000. New sequence motifs in flavoproteins: evidence for common ancestry and tools to predict structure. Proteins 38:95-114. [DOI] [PubMed] [Google Scholar]
- 63.Van Beilen, J. B., and B. Witholt. 2004. Alkane degradation by pseudomonads, p. 397-423. In J.-L. Ramos (ed.), Pseudomonas. III. Biosynthesis of macromolecules and molecular metabolism. Kluwer Academic/Plenum Publishers, New York, N.Y.
- 64.Van Beilen, J. B., F. Mourlane, M. A. Seeger, J. Kovac, Z. Li, T. H. Smits, U. Fritsche, and B. Witholt. 2003. Cloning of Baeyer-Villiger monooxygenases from Comamonas, Xanthobacter and Rhodococcus using polymerase chain reaction with highly degenerate primers. Environ. Microbiol. 5:174-182. [DOI] [PubMed] [Google Scholar]
- 65.Wackett, L. P., and C. D. Hershberger. 2001. Biocatalysis and biodegradation: microbial transformation of organic compounds. ASM Press, Washington, D.C.
- 66.Wang, S. 2003. Ph.D. thesis. University of New Brunswick, Canada.
- 67.Wang, S., G. Chen, M. M. Kayser, H. Iwaki, P. C. K. Lau, and Y. Hasegawa. 2002. Baeyer-Villiger oxidations catalyzed by engineered microorganisms: Enantioselective synthesis of δ-valerolactones with functionalized chains. Can. J. Chem. 80:613-621. [Google Scholar]
- 68.Watkinson, R. J., and P. Morgan. 1990. Physiology of aliphatic hydrocarbon-degrading microorganisms. Biodegradation 1:79-92. [DOI] [PubMed] [Google Scholar]
- 69.Wierenga, R. K., P. Terpstra, and W. G. Hol. 1986. Prediction of the occurrence of the ADP-binding beta alpha beta-fold in proteins, using an amino acid sequence fingerprint. J. Mol. Biol. 187:101-107. [DOI] [PubMed] [Google Scholar]
- 70.Wilson, K. 1994. Preparation of genomic DNA from bacteria, p. 2.4.1-2.4.5. In F. A. Ausubel, R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, J. A. Smith, and K. Struhl (ed.), Current protocols in molecular biology. John Wiley & Sons, Inc., New York, N.Y.
- 71.Yanisch-Perron, C., J. Vieira, and J. Messing. 1985. Improved M13 phage cloning vectors and host strain: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene 33:103-119. [DOI] [PubMed] [Google Scholar]