

Abstract
Cryptosporidium parvum is an apicomplexan parasite that infects humans and ruminants. C. parvum isolated from cattle in northeastern Turkey and in Israel was genotyped using multiple polymorphic genetic markers, and the two populations were compared to assess the effect of cattle husbandry on the parasite's population structure. Dairy herds in Israel are permanently confined with essentially no opportunity for direct herd-to-herd transmission, whereas in Turkey there are more opportunities for transmission as animals range over wider areas and are frequently traded. A total of 76 C. parvum isolates from 16 locations in Israel and seven farms in the Kars region in northeastern Turkey were genotyped using 16 mini- and microsatellite markers. Significantly, in both countries distinct multilocus genotypes confined to individual farms were detected. The number of genotypes per farm was higher and mixed isolates were more frequent in Turkey than in Israel. As expected from the presence of distinct multilocus genotypes in individual herds, linkage disequilibrium among loci was detected in Israel. Together, these observations show that genetically distinct populations of C. parvum can emerge within a group of hosts in a relatively short time. This may explain the frequent detection of host-specific genotypes with unknown taxonomic status in surface water and the existence of geographically restricted C. hominis genotypes in humans.
Cryptosporidium parvum is considered a zoonotic pathogen, and it commonly infects ruminants worldwide. Severe infections are typical in children, immuncompromised individuals, and neonatal calves. In the course such infections calves can excrete large numbers (>109) of oocysts, which may find their way into public water supplies and cause waterborne outbreaks. Other routes of transmission, such as food-borne routes, contact with infected persons or animals (8), and recreational water, have also been documented (18, 19).
Like the life cycles of other organisms of the phylum Apicomplexa, the life cycle of Cryptosporidium alternates between asexual multiplication and a sexual phase characterized by the differentiation of gametes, fertilization, and meiosis (23). Sexual reproduction is expected to affect the population structure of C. parvum through the generation of recombinant genotypes. Genetic recombination has been demonstrated in experimentally infected mice (3).
In recent studies in Scotland (11, 12) the populations of C. parvum and of the closely related human-infecting species C. hominis were studied by comparing multilocus genotypes from calves and humans. The identification in these studies of genotypes of C. parvum restricted to humans was unexpected because animal-to-human transmission of C. parvum is thought to be common. Except for these studies, the population biology of Cryptosporidium species has not been investigated.
Although population studies of certain Cryptosporidium species may be relevant to understanding bovine cryptosporidiosis, the primary goal of this study was to assess whether the host's population structure can affect the parasite population and whether existing genotypic markers are sufficiently polymorphic to perform such an analysis. The system of dairy herd management in Israel, together with the high prevalence of cryptosporidiosis in newborn calves in that country, provided an ideal setting in which to test this possibility. We compared the C. parvum population structure in Israel with that in northeastern Turkey, where more-traditional livestock husbandry is expected to favor transmission of pathogens between herds. Using several indices, we quantified genetic diversity and linkage disequilibirium (LD) and described differences and similarities between C. parvum populations in these study areas.
MATERIALS AND METHODS
Geographic origin and collection of Cryptosporidium isolates.
Isolates were collected between September 2001 and May 2002 and in May 2005 on 14 farms located in the Kars region within 35 km of the city of Kars in northeastern Turkey (Fig. 1). Fecal samples from 149 calves were analyzed during the first collection period, and 23 samples were analyzed during the second collection period. The ages of the animals ranged from 2 to 30 days. Fecal smears were examined by modified acid-fast staining (10) for the presence of Cryptosporidium oocysts. For each animal, age, owner, and the presence or absence of diarrhea was recorded. For 17 isolates enough DNA was obtained for genotyping. DNA was extracted locally and transferred to Tufts University for genotyping. Two of 17 isolates were ultimately removed due multiple amplification failures, which left a total of 15 isolates.
FIG. 1.

Collection sites in the Kars region of northeastern Turkey. The locations of the farms are indicated by two-letter codes, as shown in Fig. 3.
In Israel, fecal samples from calves that were 7 to 13 days old were obtained from 14 large farms with 250 to 750 milking cows each (Fig. 2). Two isolates, one from a horse from Neve Yarak and one from goat kid from Kseifa, did not originate from such farms. A total of 145 isolates were collected from calves. All calves sampled tested positive for C. parvum by acid-fast staining of fecal smears. A total of 61 isolates were randomly selected for genotyping; 17 of these were isolated between March and May 2004, 24 were isolated between September and November 2004, and 20 were isolated in April 2005. Oocysts from fecal samples estimated to contain at least 5 × 105 oocysts/ml were purified by sucrose flotation (13) and were transferred to Tufts University. Regulatory requirements of the U.S. Department of Agriculture and the Centers for Disease Control and Prevention for the transfer of pathogens to the United States were strictly followed, and the necessary permits were obtained from both agencies.
FIG. 2.

Collection sites in Israel.
DNA extraction and genotyping.
Oocyst DNA was extracted from gradient-purified oocysts using a High Pure template preparation kit (Roche Diagnostics, Indianapolis, Ind.) as previously described (22). DNA was eluted in 20 to 50 μl at a concentration equivalent to 104 oocysts/μl.
A total of 16 genetic markers (11 minisatellites and 5 microsatellites) were PCR amplified exactly as previously described (20), using the primers listed in Table 1. Briefly, initial denaturation was performed at 95°C for 10 min, and the denaturation step was followed by 45 cycles of 94 to 95°C for 1 s, 55 to 62°C (Table 1) for 2 to 5 s, and 72°C for 7 to 15 s. The presence of amplicons was initially assessed by melting curve analysis (22), and amplification products were fractionated on 15% polyacrylamide gels in Tris-borate-EDTA buffer. Sizes of alleles were determined by visual comparison with DNA molecular weight markers (Marker VIII; Roche Diagnostics). In addition to DNA markers, reference amplicons from C. parvum isolate MD (15) and C. hominis isolate TU502 (27) were loaded on all gels to facilitate unambiguous scoring of alleles. Micro- and minisatellite alleles were numbered according to amplicon length. Fluorescently labeled PCR products from seven of these markers (MSA, MSB, MSC, MSG, MS5, MS9, and TP14) were also obtained using 5′-labeled (CEQ WellRED D4; Beckman Coulter, Fullerton, CA) forward primer and the PCR conditions indicated above. Fluorescently labeled amplicons were fractionated with a CEQ 8000 genetic analysis system together with standard-600 size markers labeled with CEQ WellRED D1 to confirm the genotypes under denaturing electrophoretic conditions.
TABLE 1.
PCR primers and PCR annealing conditions
| Locus | Primer (5′-3′)
|
Annealing condition
|
||
|---|---|---|---|---|
| Forward | Reverse | Temp (°C) | Time (s) | |
| MSA | TAGGCTCGGGTTCAGA | GACTGTCACAAAAGTTAATCC | 60 | 2 |
| MSB | CTTTTGATCGCTTCTTTTCCA | GGGAGGCATAGGGATGA | 60 | 2 |
| MSC | AAATGGGTGTGGAGAAAAG | TTAGATAAAGATTGGTCTTGTC | 60 | 3 |
| MSD | CATCTCAAGAAATTCAGTCTC | CTCCTTTTGCTCCAGC | 60 | 2 |
| MSF | TCGGCCTCCTCTACAG | AGAAGAAAGCCAAGAAGGGT | 61 | 3 |
| MSG | TGGAATGATAATTGGACC | GGAGTTTCTGAGACAC | 61 | 3 |
| MSI | TCCTTGGATAAACCTGG | AGTGACGCATCTCAAAC | 55 | 3 |
| MS9 | ACCTGGAGTGTGATTTGG | GTTCTTGTTCAAAGTCA | 63 | 3 |
| Cp492 | TCATCTACCAGCACTAC | ACCAATAGTGTATCTTACATC | 58 | 2 |
| TP14 | GTTCACAGCCAACAGT | CATTTTGATTTTGGGAGT | 61 | 3 |
| 5B12 | AGGAGGAGGAGAAAAATAG | AATTCCCCATATTACTCTATTTGT | 56 | 2 |
Data analysis.
To determine if multilocus genotypes were present on different farms, the genetic distances between multilocus genotypes were calculated using Populations, version 1.2.28, downloaded from http://www.cnrs-gif.fr. Distances were based on the average square distance parameter (4) and were graphically displayed using TreeView (16). Linkage analysis between pairs of 10 polymorphic loci was performed using the web interface of Genepop at http://wbiomed.curtin.edu.au/genepop/. Using contingency tables, this program tests the association of alleles at either of two loci against the null hypothesis that genotypes at one locus are independent from genotypes at the other locus (17). Six monomorphic markers were excluded from this analysis. Since Cryptosporidium is haploid, a dummy allele was added to each allele number using Microsoft Excel. The associations between the number of pairs of loci in linkage disequilibirum and country, between multilocus genotypes present on more than one farm and country, and between the number of mixed genotypes and country were tested using the Fisher exact test with SigmaStat, version 2.0 (Systat Software, Point Richmond, Calif.). The goodness of fit between the observed distribution of nonamplifying markers and a theoretical Poisson distribution was tested using a G-test. In addition, the standardized index of association (IAS), a global measure of LD for multilocus genotypic data, was calculated with LIAN 3.1 using the web interface at http://adenine.biz.fh-weihenstephan.de/lian/ (6). The genetic diversity for each locus of the nine loci having at least two alleles in both study regions was also calculated with LIAN 3.1 using the following definition (7):
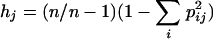 |
where hj is the genetic diversity at the jth locus, n is the number of isolates, and pij is the frequency of the ith allele at the jth locus. The mean genetic diversity was defined as the arithmetic mean for the nine polymorphic loci tested for both countries. Since LIAN does not tolerate missing alleles, replicate IAS and hj calculations were performed using alleles observed in other isolates to replace missing alleles in five isolates from Turkey. When mixed genotypes were present, two possibilities were considered, where either the larger or the smaller allele was used. These alternative calculations minimally affected IAS and hj and did not change the conclusions.
RESULTS
Taxonomic classification of isolates.
All isolates were initially identified as C. parvum based on the host, the host age, and the oocyst morphology. This identification was subsequently confirmed genotypically using the Lib13 PCR assay (GenBank accession number AF190627) as described previously (21). The polymorphism discriminates between C. parvum and C. hominis on the basis of a 4-bp insertion/deletion (indel) located on chromosome I (24) and does not amplify DNA from C. meleagridis, C. muris, and C. andersoni. The polymorphic markers used in this study also do not amplify C. muris, and markers MSA, MSC, MSE, MSF, MSG, and 1887 do not amplify C. meleagridis. Based on these observations, together with the host age and oocyst morphology, the isolates included in this study were classified as C. parvum.
Genotyping.
Initially, 11 minisatellite and 5 microsatellite markers were used to genotype 61 C. parvum isolates from Israel and 17 isolates from Turkey. Minisatellites MSE, MSK, and MS5 and microsatellites 1887 and 1962 were monomorphic and excluded from subsequent analyses. The remaining 11 markers are located on chromosome I (MSA and MSB), chromosome II (MSC, MSI, and 5B12), chromosome III (MSD), chromosome V (MSF and MS9), chromosome VI (MSG and Cp492), and chromosome VIII (TP14). No markers from chromosomes IV and VII were available. The Turkish isolates were not typed with MSD, and MSC was monomorphic in Israel. The remaining 10 polymorphic markers were successfully amplified from 61 isolates from Israel, whereas 13 single-locus genotypes could not be determined in Turkish isolates due to nonamplification (Fig. 3). There was no amplification at five loci (1887 included) for two isolates from Turkey (farm VK), and these isolates were excluded from further analyses and from Fig. 3. We tested whether the distribution of nonamplifying loci was clustered in certain isolates by tabulating their occurrence in all 78 isolates from both countries and comparing this distribution to that expected from a theoretical Poisson distribution. A significant deviation from the expected distribution was found (G = 18.09 and P < 0.001, as determined by a G test for goodness of fit [3 df]), indicating that there was clustering of nonamplifying loci among these isolates. This result could have indicated the presence of additional alleles with sequence polymorphisms in a priming site (null alleles) or that the quality of the DNA from certain Turkish isolates was inferior and negatively affected the PCR. The latter possibility was supported by microscopic analysis of acid-fast-stained fecal smears and oocyst preparations containing few oocysts in several fecal samples originating from Turkey.
FIG. 3.

Genotype analysis of C. parvum from Israel (a) and Turkey (b). The column on the left shows the isolate number. For the host species C indicates bovine and H indicates equine. Two isolates from Turkey with more than one amplification failure are not shown. Israeli locations are grouped according to geographical region, as indicated. Zero indicates no amplification. The numbers indicate amplicon lengths (in base pairs).
Mixed genotypes.
Visual inspection of the genotypic data suggested that there was a higher proportion of mixed genotypes (loci with two alleles) in Turkey than in Israel (Fig. 3). Because Cryptosporidium is haploid and multicopy genes are absent (except for the ribosomal genes [9]), the presence of more than one electrophoretic band indicates that there is a mixed population. We favor this interpretation over the alternative view that there are nonspecific amplification products, because each putative allelic band detected in mixed profiles was also identified as a single allele in other isolates from the same farm or from the same region (Fig. 3) or in geographically unrelated isolates (20). In 610 loci (61 isolates and 10 loci) for the Israeli isolates, five mixed profiles (0.8%) were identified. In contrast, for the 157 loci typed in the Turkish isolates, 23 mixed profiles (14% of the total) were found. The proportion of mixed profiles was significantly higher in Turkey (P < 0.001, as determined by Fisher’s exact test).
Geographic distribution of multilocus genotypes.
A total of 14 multilocus genotypes were identified on 14 farms and at two additional locations (Neve Yarak and Kseifa) in Israel, which was equivalent to 0.88 isolate/location. The number of multilocus genotypes per farm was higher in Turkey. However, because of the presence of multiple mixed genotypes in individual isolates, the exact number of multilocus genotypes from farms OK and VK could not be determined. For instance, in isolate TK41 (farm OK), in which seven loci with two alleles each were detected, the possible number of unique multilocus genotypes ranged from a minimum of 2 to a maximum of 128 (27). Similarly, between 2 and 8 (23) distinct multilocus genotypes could be present in isolate TK23 from farm VK. There was no ambiguity in the number of Israeli isolates as no isolate had more than one locus with a biallelic profile and such profiles were scored as two multilocus genotypes. The number of isolates per farm in Turkey calculated from these data ranged from 3 to 24, clearly exceeding what was found in Israel (Table 2), even without adjustment for the larger number of isolates per farm in Israel.
TABLE 2.
C. parvum populations in Turkey and Israel
| Result for C. parvum population in:
|
|||
|---|---|---|---|
| Parameter | Turkey | Israel | Significance (P) |
| Mean genetic diversity | 0.525 | 0.244 | 0.003 |
| Pair of loci in LD (%)a | 42 | ||
| IASb | 0.130 | 0.01c | |
| No. of multilocus genotypes/farm | ≥3 | 0.88 | |
| Proportion of multifarm genotypesd | 0.125 | 0.357 | NSe |
| % of biallelic loci (% of all typed loci) | 14 | 0.8 | <0.001 |
Results obtained with Genepop.
Calculated by LIAN.
Probability that IAS is different from zero.
Multilocus genotypes present on more than one farm.
NS, no significant difference between countries.
Probably the most interesting finding is that a majority of genotypes were limited to single farms. When all possible allele combinations were included, only farms OK and VK in Turkey shared multilocus genotypes. In Israel, Naan and Sheller, Sheller and Neve Yarak, Hefer, HofHaSharon, BeitHalevy, and Azayra, and HofHaSharon, BeitHalvey, and Beit Dagan shared multilocus genotypes (Fig. 4). Whereas in Turkey 1/8 (12%) of the theoretically possible genotypes occurred on more than one farm, in Israel 5/14 (36%) were present on multiple farms. The proportions of shared genotypes in the two countries are not significantly different (P = 0.35, as determined by Fisher’s exact test).
FIG. 4.

Multilocus C. parvum genotypes in Israel. Genotypes are indicated in columns, and farms are indicated in rows. The numbers are numbers of isolates.
Linkage analysis.
To further analyze the population structure of C. parvum in Israel, we tested for LD between pairs of loci using Genepop. This analysis was not performed for the Turkish isolates due to the relatively small number of isolates and the presence in some isolates of several mixed loci. For the isolates from Israel, 45 pairwise tests of association between loci were performed using 10 polymorphic loci, and the corresponding P values were determined for each pair of loci. A total of 19/45 (42%) tests showed a significant (P < 0.05) association. LD was also detected by calculating the IAS (6). Consistent with the confinement of most multilocus genotypes to single farms, the IAS in Israel was significantly (P = 0.001) different from 0 (linkage equilibrium) (Table 2) and the values for variance of pairwise differences were greater than the 95% critical value.
DISCUSSION
Because of geographical differences in cattle husbandry, studying C. parvum populations in farm animals can provide information relevant for understanding the epidemiology of cryptosporidiosis, including cryptosporidiosis affecting humans. Although the effect of herd management on Cryptosporidium prevalence has been investigated (25), population studies have become feasible only since the identification of suitable genetic markers in the species (1, 2, 11, 20). The high prevalence of bovine cryptosporidiosis in Israel (5, 14), together with the confinement of herds, facilitated analysis of the population structure of C. parvum and a comparison with that in Turkey, where more-traditional husbandry is expected to provide more opportunities for herd-to-herd transmission. In the two study areas the distances between farms were similar (Fig. 1 and 2), which left the population structure of the host as one of the main variables relevant to parasite transmission. Considering the relative proximity of different farms in both study sites, the small number of isolates shared among farms was surprising. This is particularly the case for Turkey, where opportunities for transmission between herds seem to be common.
The most striking difference between the two countries was the higher proportion of animals infected with mixed parasite populations in Turkey. Consistently, the number of multilocus genotypes per farm was also higher in this country, even when the most conservative estimate was used, as was the genetic diversity. In agreement with the confinement of most multilocus genotypes to individual farms, LD was detected in Israel, and the IAS was significantly different from 0 (linkage equilibrium). We interpret the higher genetic diversity, higher number of multilocus genotypes per farm, and higher proportion of genotypically mixed isolates in Turkey as indications of a less stable population structure, probably as a consequence of herd-to-herd transmission.
Since IAS values from different studies are comparable, we noticed that values similar to those found here were observed when C. parvum isolates from human and bovine sources in Scotland were analyzed as a single population (11, 12). This suggests that human and bovine hosts inhabiting the same geographical area essentially behave like different “herds” harboring distinct parasite populations. Based on these observations, it appears that human C. parvum cryptosporidiosis is not always zoonotic, as typically assumed (8, 14), because frequent animal-to-human transmission would eliminate host-specific population substructuring and LD. Consequently, these observations raise the possibility that human-to-human transmission of C. parvum may be more important than has been assumed. The unique multilocus genotypes encountered in a horse and a goat from Israel may extend the model of host-specific C. parvum subpopulations to other livestock species.
Because of the presence of numerous genotypically mixed isolates in Turkey, we were unable to determine the number of multilocus genotypes. Where multiple mixed profiles were found within a multilocus genotype, a theoretically maximal number of genotypes could be calculated by assuming that all possible allele combinations were present in the population. Since this number greatly exceeded the number of isolates that were actually typed, the number remains hypothetical. To investigate how parasite populations within herds are structured, multiple genomes from the same herd need to be isolated and genotyped individually. Such an analysis is currently difficult, if not impossible, to perform, because it is not possible to isolate and propagate single sporozoites at this time. Alternatively, analysis of individual oocysts, which contain four genomes, and populations derived from such oocysts would still provide meaningful information. We hypothesize that such an analysis would show that parasites that infect individual herds are panmictic.
Probably the most obvious question raised by our analysis is why genetically distinct populations of C. parvum have emerged on different farms. Because many kibbutzim have existed for about 70 years, these populations have emerged in a relatively short time. The first possibility is that this situation resulted from the introduction into a herd of a small number of founders with an infected animal. The infection then spread to the entire herd, either because the herd was not infected or because the founder phenotype was more virulent. Alternatively, different conditions present on certain farms may have favored the outgrowth of certain genotypes. Positive selection implies the existence of different conditions on different farms, which makes this scenario less likely since all kibbutzim raise the same Israeli Holstein breed and use similar methods of husbandry and the climatic conditions are also similar.
In conclusion, in this study we examined the effect of the host population structure on the population structure of C. parvum and found that, regardless of the method of herd management, C. parvum populations were clearly structured according to farms. These observations are consistent with previous analyses of human and bovine C. parvum populations which revealed the emergence of genetically distinct genotypes in segregated host populations. This process may be the initial step leading to the differentiation of species-specific genotypes (26), which, given sufficient time, could evolve into reproductively separated populations or different species.
Acknowledgments
Financial support from the National Institute of Allergy and Infectious Diseases (grant AI52781) is gratefully acknowledged.
Lülüfer Tamer, Mersin University, Turkey, kindly provided logistical support. We thank Alex Grimberg, Massey University, New Zealand, for critical comments on the manuscript.
REFERENCES
- 1.Cacciò, S., F. Spano, and E. Pozio. 2001. Large sequence variation at two microsatellite loci among zoonotic (genotype C) isolates of Cryptosporidium parvum. Int. J. Parasitol. 31:1082-1086. [DOI] [PubMed] [Google Scholar]
- 2.Feng, X., S. M. Rich, D. Akiyoshi, J. K. Tumwine, A. Kekitiinwa, N. Nabukeera, S. Tzipori, and G. Widmer. 2000. Extensive polymorphism in Cryptosporidium parvum identified by multilocus microsatellite analysis. Appl. Environ. Microbiol. 66:3344-3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Feng, X., S. M. Rich, S. Tzipori, and G. Widmer. 2002. Experimental evidence for genetic recombination in the opportunistic pathogen Cryptosporidium parvum. Mol. Biochem. Parasitol. 119:55-62. [DOI] [PubMed] [Google Scholar]
- 4.Goldstein, D. B., L. A. Zhivotovsky, K. Nayar, A. R. Linares, L. L. Cavalli-Sforza, and M. W. Feldman. 1996. Statistical properties of the variation at linked microsatellite loci: implications for the history of human Y chromosomes. Mol. Biol. Evol. 13:1213-1218. [DOI] [PubMed] [Google Scholar]
- 5.Grinberg, A., A. Markovics, J. Galindez, N. Lopez-Villalobos, A. Kosak, and V. M. Tranquillo. 2002. Controlling the onset of natural cryptosporidiosis in calves with paromomycin sulphate. Vet. Rec. 151:606-608. [DOI] [PubMed] [Google Scholar]
- 6.Haubold, B., and R. R. Hudson. 2000. LIAN 3.0: detecting linkage disequilibrium in multilocus data. Linkage analysis. Bioinformatics 16:847-848. [DOI] [PubMed] [Google Scholar]
- 7.Haubold, B., M. Travisano, P. B. Rainey, and R. R. Hudson. 1998. Detecting linkage disequilibrium in bacterial populations. Genetics 150:1341-1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hunter, P. R., S. Hughes, S. Woodhouse, Q. Syed, N. Q. Verlander, R. M. Chalmers, K. Morgan, G. Nichols, N. Beeching, and K. Osborn. 2004. Sporadic cryptosporidiosis case-control study with genotyping. Emerg. Infect. Dis. 10:1241-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Le Blancq, S. M., N. V. Khramtsov, F. Zamani, S. J. Upton, and T. W. Wu. 1997. Ribosomal RNA gene organization in Cryptosporidium parvum. Mol. Biochem. Parasitol. 90:463-478. [DOI] [PubMed] [Google Scholar]
- 10.Ma, P., and R. Soave. 1983. Three-step stool examination for cryptosporidiosis in 10 homosexual men with protracted watery diarrhea. J. Infect. Dis. 147:824-828. [DOI] [PubMed] [Google Scholar]
- 11.Mallon, M., A. MacLeod, J. Wastling, H. Smith, B. Reilly, and A. Tait. 2003. Population structures and the role of genetic exchange in the zoonotic pathogen Cryptosporidium parvum. J. Mol. Evol. 56:407-417. [DOI] [PubMed] [Google Scholar]
- 12.Mallon, M. E., A. MacLeod, J. M. Wastling, H. Smith, and A. Tait. 2003. Multilocus genotyping of Cryptosporidium parvum type 2: population genetics and sub-structuring. Infect. Genet. Evol. 3:207-218. [DOI] [PubMed] [Google Scholar]
- 13.McNabb, S. J., D. M. Hensel, D. F. Welch, H. Heijbel, G. L. McKee, and G. R. Istre. 1985. Comparison of sedimentation and flotation techniques for identification of Cryptosporidium sp. oocysts in a large outbreak of human diarrhea. J. Clin. Microbiol. 22:587-589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miron, D., J. Kenes, and R. Dagan. 1991. Calves as a source of an outbreak of cryptosporidiosis among young children in an agricultural closed community. Pediatr. Infect. Dis. J. 10:438-441. [DOI] [PubMed] [Google Scholar]
- 15.Okhuysen, P. C., S. M. Rich, C. L. Chappell, K. A. Grimes, G. Widmer, X. Feng, and S. Tzipori. 2002. Infectivity of a Cryptosporidium parvum isolate of cervine origin for healthy adults and interferon-gamma knockout mice. J. Infect. Dis. 185:1320-1325. [DOI] [PubMed] [Google Scholar]
- 16.Page, R. D. 1996. TreeView: an application to display phylogenetic trees on personal computers. Comput. Appl. Biosci. 12:357-358. [DOI] [PubMed] [Google Scholar]
- 17.Raymond, F., and F. Rousset. 1992. GENEPOP on the web. Population Genetics software package, ed. 3.4. Laboratoire de Génétique et Environment, Montpellier, France.
- 18.Sorvillo, F. J., K. Fujioka, B. Nahlen, M. P. Tormey, R. Kebabjian, and L. Mascola. 1992. Swimming-associated cryptosporidiosis. Am. J. Public Health 82:742-744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stafford, R., G. Neville, C. Towner, and B. McCall. 2000. A community outbreak of Cryptosporidium infection associated with a swimming pool complex. Commun. Dis. Intell. 24:236-239. [PubMed] [Google Scholar]
- 20.Tanriverdi, S., and G. Widmer. 2006. Differential evolution of repetitive sequences in Cryptosporidium parvum and Cryptosporidium hominis. Infect. Genet. Evol. 6:113-122. [DOI] [PubMed] [Google Scholar]
- 21.Tanriverdi, S., M. O. Arslan, D. E. Akiyoshi, S. Tzipori, and G. Widmer. 2003. Identification of genotypically mixed Cryptosporidium parvum populations in humans and calves. Mol. Biochem. Parasitol. 130:13-22. [DOI] [PubMed] [Google Scholar]
- 22.Tanriverdi, S., A. Tanyeli, F. Baslamisli, F. Koksal, Y. Kilinc, X. Feng, G. Batzer, S. Tzipori, and G. Widmer. 2002. Detection and genotyping of oocysts of Cryptosporidium parvum by real-time PCR and melting curve analysis. J. Clin. Microbiol. 40:3237-3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tzipori, S. 1988. Cryptosporidiosis in perspective. Adv. Parasitol. 27:63-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Widmer, G., D. Akiyoshi, M. A. Buckholt, X. Feng, S. M. Rich, K. M. Deary, C. A. Bowman, P. Xu, Y. Wang, X. Wang, G. A. Buck, and S. Tzipori. 2000. Animal propagation and genomic survey of a genotype 1 isolate of Cryptosporidium parvum. Mol. Biochem. Parasitol. 108:187-197. [DOI] [PubMed] [Google Scholar]
- 25.Xiao, L., R. P. Herd, and G. L. Bowman. 1994. Prevalence of Cryptosporidium and Giardia infections on two Ohio pig farms with different management systems. Vet. Parasitol. 52:331-336. [DOI] [PubMed] [Google Scholar]
- 26.Xiao, L., I. M. Sulaiman, U. M. Ryan, L. Zhou, E. R. Atwill, M. L. Tischler, X. Zhang, R. Fayer, and A. A. Lal. 2002. Host adaptation and host-parasite co-evolution in Cryptosporidium: implications for taxonomy and public health. Int. J. Parasitol. 32:1773-1785. [DOI] [PubMed] [Google Scholar]
- 27.Xu, P., G. Widmer, Y. Wang, L. S. Ozaki, J. M. Alves, M. G. Serrano, D. Puiu, P. Manque, D. Akiyoshi, A. J. Mackey, W. R. Pearson, P. H. Dear, A. T. Bankier, D. L. Peterson, M. S. Abrahamsen, V. Kapur, S. Tzipori, and G. A. Buck. 2004. The genome of Cryptosporidium hominis. Nature 431:1107-1112. [DOI] [PubMed] [Google Scholar]