

Abstract
Individual contributions made by different calcium release and sequestration mechanisms to various aspects of excitable cell physiology are incompletely understood. SERCA, a sarco-endoplasmic reticulum calcium ATPase, being the main agent for calcium uptake into the ER, plays a central role in this process. By isolation and extensive characterization of conditional mutations in the Drosophila SERCA gene, we describe novel roles of this key protein in neuromuscular physiology and enable a genetic analysis of SERCA function. At motor nerve terminals, SERCA inhibition retards calcium sequestration and reduces the amplitude of evoked excitatory junctional currents. This suggests a direct contribution of store-derived calcium in determining the quantal content of evoked release. Conditional paralysis of SERCA mutants is also marked by prolonged neural activity-driven muscle contraction, thus reflecting the phylogenetically conserved role of SERCA in terminating contraction. Further analysis of ionic currents from mutants uncovers SERCA-dependent mechanisms regulating voltage-gated calcium channels and calcium-activated potassium channels that together control muscle excitability. Finally, our identification of dominant loss-of-function mutations in SERCA indicates novel intra- and intermolecular interactions for SERCA in vivo, overlooked by current structural models.
IN neurons and muscles, local calcium transients control activities of proteins that directly mediate physiological processes such as transmitter release and contraction. Different pools of calcium, acting over longer timescales, may also modulate a variety of properties of excitable cells including action potential generation and transmitter responsiveness. Thus, the function of calcium release or sequestration mechanisms that regulate rapid or longer-lasting calcium transients is of great importance (Berridge et al. 2000).
In neurons, calcium entry through voltage-gated channels tightly couples an action potential with transmitter release (Katz and Miledi 1969; Sheng et al. 1998; Catterall 1999). In muscles, calcium entry, through ligand-gated channels, is the key to excitation-contraction coupling (Melzer et al. 1995). Once calcium has entered the cell, it promotes the release of calcium from intracellular stores in the endo- or sarcoplasmic reticulum (ER or SR). This calcium-induced calcium release (CICR) synchronously augments levels of cytosolic calcium and depends on the gating of ryanodine receptors located in the SR membrane (Fabiato 1983; Gyorke et al. 2002). In neurons and muscles, removal of calcium by either pumping it out of the cell or sequestering it into intracellular stores restores cells to their resting state.
In addition to cytosolic calcium-buffering proteins (Roberts 1993) a significant contribution to intracellular calcium sequestration is made by membrane-bound calcium pumps that hydrolyze ATP to pump calcium either across the plasma membrane or into the ER/SR, the predominant store of intracellular calcium (Berridge et al. 2000). The importance of sarco-endoplasmic reticulum calcium ATPases (SERCA) in regulating excitability is most clearly demonstrated by the unexpected observation that genetic knockout for phospholamban, a SERCA inhibitor peptide, cures several mouse models of cardiac dysfunction (Minamisawa et al. 1999). Despite its importance, the contribution of SERCA to major aspects of neuromuscular physiology remains poorly understood. Most significantly, the relative importance of SERCA-dependent mechanisms in calcium sequestration and/or signaling has not been assessed critically. Even in cardiac disease where SERCA dysfunction has been implicated (Odermatt et al. 1996; Hasselbach 1998; Loke and MacLennan 1998; Gommans et al. 2002) the precise physiological underpinnings of these effects remain to be elucidated.
Here, we report the isolation of conditional mutations in the Drosophila SERCA gene (dSERCA, Ca-P60A) and the first detailed genetic analysis of the effects of SERCA inhibition on neuromuscular physiology. Since Drosophila neurons and muscles function by mechanisms largely similar to those in vertebrates (Suzuki and Kano 1977), these findings are likely to be broadly applicable. Our observations indicate the existence of (a) hitherto uncovered molecular interactions important for SERCA function in vivo, (b) SERCA-dependent mechanisms that contribute substantially to control of calcium pools at the presynaptic motor terminal, and (c) SERCA-dependent modulation of ionic currents that regulate muscle contraction and excitability. Although pharmacological perturbation of SERCA function has been extensively used for various studies (Toyoshima et al. 2000, 2003), our results compliment and significantly extend previous findings. They also offer avenues for the genetic analysis of store calcium function and provide fundamental new insight into SERCA functions in vivo.
MATERIALS AND METHODS
Drosophila stocks and culture:
The Canton-Special (CS) strain was used as the wild type in this study. The EP lines were a part of the Pernille Rorth collection (Rorth 1996); deletions were obtained from the Drosophila Stock Center, Bloomington, Indiana. Other dSERCA alleles and the genomic rescue construct for dSERCA were from G. Periz (Periz and Fortini 1999). The rescue construct contains the entire genomic region for dSERCA with all the introns and 4–5 kb of DNA on either side of the gene. All other Drosophila strains are a part of the Tata Institute of Fundamental Research or Ramaswami laboratory stock collection. Flies were reared on standard cornmeal-dextrose-yeast medium at 22°–25°.
EMS mutagenesis and screening:
For the mutagenesis, ethyl methanesulfonate (EMS), a chemical mutagen that induces mostly point mutations, was used. Two- to 3-day-old CS males were starved for 6 hr and introduced into bottles containing filter paper discs soaked in 0.75% EMS in 2% sucrose. The males were allowed to feed for 12 hr and mated to virgin CS females. A total of 200,000 F1 progeny were screened for paralysis at 40°. Any fly that paralyzed within 3 min was picked as a putative mutant and used to set up a mutant line by crossing to suitable males or virgins.
Behavioral assays:
The apparatus used for measurement of adult paralysis, the Sushi Cooker, has been described before (Ramaswami et al. 1993). Paralysis was empirically defined as the condition in which the animal lies on its back with little effective movement of the legs and wings. All flies tested were 1–2 days old. We define the restrictive temperature for all the behavioral analyses as the temperature at which 100% of flies paralyze in 3 min. Paralysis profiles were obtained by introducing 10 flies at a time in the cooker and recording the number of flies immobile at fixed intervals of time. Each data point represents the mean from three such runs.
Lethality staging:
Sibling crosses between heterozygous Ca-P60AKum170/+ and Ca-P60AKum295/+ were set up for lethality staging. In this cross, 25% of the progeny is homozygous for the Ca-P60A mutation and is expected to be lethal. The initial number of eggs laid was counted and the bottles were incubated at 25° for 24 hr at the end of which the number of unhatched eggs was determined. After a further incubation period of 30 hr, the number of second instar larvae was determined. The stage of lethality was defined as the point where the expected 25% lethality was observed. For each genotype, the data were averaged over five experiments, each consisting of at least 200 embryos. For determining the embryonic stage of death, freshly laid eggs were covered with halocarbon oil and observed under an inverted microscope throughout embryogenesis till hatching.
Recombination, deletion mapping, sequencing, and homology modeling:
A set of EP lines from the Pernille Rorth collection was used as second chromosome markers. w; CaP60AKum170/CyO females were crossed to w; EP/CyO males from various lines. The w; CaP60AKum170/EP progeny females were further crossed to w; CyO/Tft males. All Cy progeny was screened at 40° for 3 min, to assay for the presence of the CaP60AKum170 mutation, and scored for the w+ marker carried by the P element. Recombination frequency was calculated as number of recombinants/total number of flies × 100. At least 100 flies were screened for each EP line. About 500 flies were screened for the three lines closest to CaP60AKum170, EP938, EP348, and EP540.
For deletion analysis, various chromosomal deletions were tested for their ability to uncover the recessive lethal phenotype of CaP60AKum170. CaP60AKum170/CyO flies were crossed to flies carrying a specific deletion balanced over the CyO balancer chromosome. The progeny was screened for the presence of non-Cy flies. A total absence of non-Cy flies indicates that the mutation lies in the region that is deleted in a particular deficiency line.
The fly SERCA was homology modeled using Swiss-Model (www.expasy.org/swissmod/SWISS-MODEL.html) on the reported rabbit SERCA crystal structure (Toyoshima et al. 2000, 2003), using standard parameters. Drosophila SERCA shares a high degree of homology with various vertebrate isoforms (∼70%).
Generation of the CaP60AKumP52 allele and excision analysis:
To generate a P-element-tagged allele of CaP60A, w/w; EP938/CyO females were crossed to males from a line containing a source of the transposase, w/ Y; (Δ2-3)Sb/ TM6(Tb), to generate the “jump-starter” flies. The jump-starter males were further crossed to w/w; CaP60AKum170/CyO virgin females. A total of 1400 lines were set from the Cy progeny by crossing individual males, with a mobilized EP938 balanced over CyO, to w; CaP60AKum170/CyO virgin females. The progeny of these lines were screened for the absence of non-Cy flies. For excising the P element from the CaP60A locus, w; CaP60AKumP52/CyO females were mated to w;/Y; +/CyO; (Δ2-3)Sb/+ males and the F1, w/Y; CaP60AKumP52/+; (Δ2-3)Sb/+ male progeny was collected. These males were further crossed to w; CaP60AKum170/CyO females. All CyO progeny was screened for the presence of non-Sb, white-eyed males, in which the P element would have excised out. Such males were individually crossed to w; CaP60AKumP52/CyO females and the lines were screened for reversion of the lethal phenotype.
Plasmid rescue:
The EP transposable element in these flies allows plasmid rescue of flanking fly genomic DNA using the single EcoRI restriction site in the vector. Genomic DNA was isolated from CaP60AKumP52 flies and restricted with EcoRI using standard procedures. The DNA was ligated overnight and used for transformation of competent Escherichia coli. Colonies transformed with the rescued DNA fragment were analyzed by RFLP analysis. Flanking genomic DNA from an EcoRI-KpnI fragment was sequenced using automated sequencing. A BLAST search of the sequence was carried out using the BLASTn program at NCBI.
Generation of antibodies against dSERCA peptides and immunohistochemistry:
Two 20-amino-acid stretches were selected from the dSERCA protein. The first, SS1 (ERGLTLDQIKANQKKYGPNE), is from aa 21 to aa 40 and the second peptide SS2 (TLKFVARKIADVPDVVVDRM) is from aa 983 to aa 1002. Generation of antibodies including peptide synthesis, conjugation to keyhole limpet hemocyanin, and affinity purification was carried out by Alpha Diagnostics (San Antonio, TX; http://www.4adi.com). Antibody titer was monitored after each booster immunization. The final bleeds were affinity purified and used at a concentration of 1:1000 for immunohistochemistry and 1:10,000 for Western blotting. For immunohistochemistry, larvae were dissected in calcium-free HL3 saline, fixed in 4% paraformaldehyde, and stained overnight in blocking solution containing 0.1% Triton X-100. Mouse anti-BiP (anti-KDEL, StressGen SPA-827) was used at 1:100. All secondary antibodies were Alexa dye-conjugated anti-rabbit and anti-mouse antibodies from Molecular Probes (Eugene, OR). Protein gels and Western blotting were according to standard procedures (Laemmli and Quittner 1974) and as per manufacturer's instructions (Amersham, Arlington Heights, IL).
Electrophysiology:
Adult dorsal longitudinal flight muscle recordings:
Intracellular recordings were made from the dorsal longitudinal flight muscle (DLM) e, f muscle fibers (King and Wyman 1980; Costello and Wyman 1986; Ikeda and Koenig 1988). Intact adults were immobilized on wax and a small hole was made in the dorsal part of the thorax where DLM e, f muscle fibers are positioned. Glass microelectrodes of 10–20 MΩ resistance were used to record the muscle potentials. Muscle stimulation was achieved by activating the DLM e, f motoneuron (MN5) through the giant fiber system (GFS; Thomas and Wyman 1982). The GFS was stimulated by a tungsten electrode placed in the eye of the fly.
Larval current recordings:
Calcium current recordings were made as described previously (Gielow et al. 1995). Recording saline to block all potassium currents contained: NaCl (77.5 mm), KCl (5 mm), MgCl2 (4 mm), NaHCO3 (2.5 mm), trehalose (50 mm), sucrose (115 mm), Hepes (5 mm), tetra-ethylammonium (TEA; 20 mm), 4-aminopyridine (4-AP; 1 mm), quinidine (0.1 mm), and BaCl2 (10 mm). All currents were Barium currents through voltage-gated calcium channels. For measurement of calcium-activated potassium currents (slowpoke currents), recordings were done in normal HL3 medium with 100 mm Ca2+ and 2 mm 4-AP to block the fast Shaker potassium current (I[A]). Voltage steps were in increments of 10 mV from a holding potential of −70 mV. The slowpoke current could be most clearly discerned at a voltage step to 0 mV, where there is maximal calcium current and the slower potassium currents are minimal (Elkins et al. 1986). This current could be completely suppressed by removing calcium from the recording medium. The currents were not normalized with capacitance measurements since all recordings were from muscle 6 in abdominal segment 2 of separate animals. Muscle surface areas were independently confirmed to be similar. The resting membrane potential was determined at the end of each voltage clamp experiment and only traces from animals with resting potentials more negative than −50 mV were included in the analysis. Traces were digitally leak subtracted and baseline adjusted for analysis. Representative traces are plotted using the electrophysiology module in SigmaPlot (Jandel Scientific, San Rafael, CA).
Larval EJC recordings:
Excitatory junction current (EJC) recordings were made in HL3 saline (Stewart et al. 1994) with 1 mm Ca2+ as described previously (Sanyal et al. 2003). All recordings were made from muscle 6 in segment A2 of wandering third instar larvae. Muscles were voltage clamped at −70 mV, and excitatory junctional currents were evoked, by stimulating the segmental nerves such that both neurons innervating muscle 6 were recruited. The nerves were stimulated at 0.5 Hz and a train of 25 stimuli was averaged for each recording. For spontaneous events, a 1-min continuous recording was used to determine mEJC amplitude and frequency. The traces were analyzed using the minianalysis software (Synaptosoft).
Calcium imaging at the larval neuromuscular junction:
The preparations were incubated for 40 min in nominally zero Ca2+ saline (CaCl2 was replaced with 2 mm MgCl2 in normal saline) containing 10 μm rhod-2/AM, 0.03% pluronic F-127 in the dark at 22°. The preparation was rinsed with HL3 medium three times. The fluorescence was excited at 545 nm and monitored at wavelengths >580 nm. To excite nerve terminals, pulses (1 msec and 2× threshold voltage) were delivered to the appropriate segmental nerve via a conventional glass suction electrode (20–30 μm inside diameter).
RESULTS
Isolation of conditional Kum mutants and their behavioral phenotypes:
Two independent mutant lines isolated in our screen for temperature-sensitive paralytic mutants showed identical dominant paralytic behavior at 40° (materials and methods). Both mutations were localized to the second chromosome and were recessively lethal. Genetic recombination analysis showed that the dominant paralytic phenotype cosegregated with the recessive lethal phenotype. The two mutations 170 and 295 were tentatively classified as allelic, because they not only mapped to the same chromosomal region, but also did not complement for the recessive lethal phenotype. Thus, the mutants were named Kumbhakarna170 (Kum170) and Kumbhakarna295 (Kum295). The conditional paralytic behavior of Kum170/+ and Kum295/+ heterozygous flies is documented in Figure 1A. The mutants, but not wild-type controls, paralyze at 40° within 3–5 min. When restored to permissive temperatures (20°), paralysis persists for 6–48 hr depending on the duration of prior exposure to restrictive temperature. (This prolonged inactive phase, unique among Drosophila temperature-sensitive (ts) paralytics, led to the gene being named after an eponymous mythological hero who slept for 6 months of the year). The prolonged recovery phase might derive from irreversible heat-sensitive alteration of the mutant protein or from chronic effects on membrane excitability (see later sections).
Figure 1.—

Phenotypes associated with conditional mutations in dSERCA. (A) Both Kum170 and Kum295 are dominant temperature-sensitive paralytics and paralyze between 39° and 40° in 3 min. (B) Heterozygous Kum larvae also paralyze on heating and exhibit prolonged contraction as compared to wild type. (C) Kum mutations are recessively lethal. Lethal embryos appear contracted inside the egg case and fail to hatch. (D) Paralysis in Kum animals is dependent on neural activity. para;Kum animals at temperatures restrictive for both mutations do not show the contracted phenotype. At temperatures permissive for para, the temperature-sensitive phenotype of Kum manifests itself following prior heating to restrictive temperatures.
Like adults, Kum larvae are also paralyzed at 40° within 5 min; paralyzed Kum larvae appear shortened due to severe contraction of the body wall musculature (Figure 1B). This observation suggested a role for the Kum gene product in suppressing muscle contraction. Consistent with this, many homozygous Kum295/Kum295 mutant embryos also appeared abnormally contracted inside the egg case (Figure 1C), although most Kum170/Kum170 animals developed normally and hatched into very sluggish first-instar larvae.
To distinguish between two possibilities, whether Kum mutations prolonged nerve-evoked muscle contraction or induced spontaneous muscle contraction, we examined Kum phenotypes in a parats background that allows conditional inactivation of neuronal voltage-gated Na channels encoded by the para locus (Loughney et al. 1989). In parats1; Kum double mutants, the characteristic contraction of Kum mutants was completely abolished so long as para-dependent nerve transmission was blocked (Figure 1D). Conversely, at temperatures permissive for para, previously heated Kum mutants contracted as expected. Together, these data indicate a need for the Kum gene product in neuromuscular function, specifically in limiting the duration of neurally evoked muscle contraction.
Kum mutants have mutations at the dSERCA locus:
Recombination mapping, using a set of independent, white-marked P-element inserts from the Pernille Rorth EP collection, placed Kum in the cytological interval 59A–60E, at the tip the right arm of the second chromosome. An analysis using overlapping deletions in this region (Figure 2A) further narrowed the location to 60A1–B1. To clone the Kumbhakarna gene, the EP938 transposon, which is located at 59F6–8 on the second chromosome, was mobilized to disrupt the Kum locus at 60A1–B1 (materials and methods). A lethal line generated from this mobilization, P52, failed to complement the lethality of Kum170, Kum295, or a chromosomal deficiency in the region, Df(2R)bw-S46. The phenotypes of this mutant line mapped to Kum and were caused by a P-element insertion as evidenced by successful and frequent reversion of all mutant phenotypes by remobilization of the P-transposon (materials and methods). This P-element-induced lethal allele has been named KumP52. This allele does not display temperature-sensitive paralysis like the EMS-induced alleles and is probably a hypomorphic allele. Genomic DNA flanking the P52 insertion was recovered and, when sequenced, found to be identical to the upstream regulatory region of the organellar type Ca2+-ATPase gene in Drosophila (dSERCA, CaP60A; Magyar et al. 1995). The location of the P element in the genome of the KumP52 flies is shown in Figure 2A. The P element separates the upstream regulatory elements of the gene from the TATA box and the coding region.
Figure 2.—
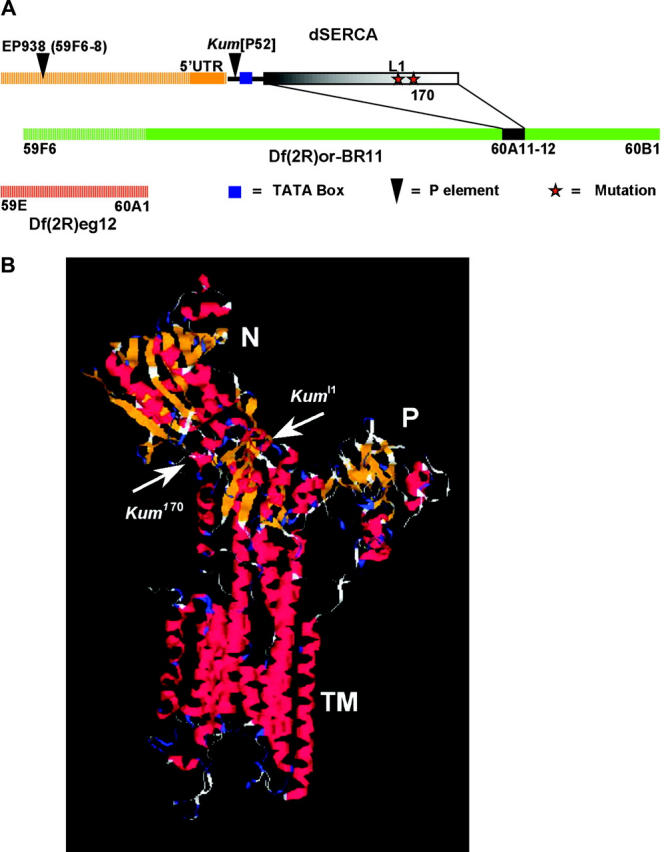
Kum mutations map to the dSERCA locus at 60A11–12. (A) Df(2R)or-BR11 uncovers it while Df(2R)eg12 does not. The EP938 P line was used to generate a P-element transposon-tagged allele of dSERCA, KumP52. Plasmid rescue of this line showed the downstream gene to be dSERCA and the insertion site to be just upstream of the TATA box. Sequencing one allele, CaP60AKum170, from this study and an allele isolated previously, CaP60Al1, identified the mutations in the coding region of dSERCA. (B) Fly SERCA shows a high degree of homology to vertebrate SERCA and can be homology modeled with a high degree of confidence. Shown are the sites of the mutations in CaP60AKum170 and CaP60Al1. Both mutations are in the “hinge” region of the molecule.
To confirm that the mutant phenotypes resulted from altered SERCA function we ensured first that the Kum alleles did not complement recently described EMS-induced dSERCA lethals (Periz and Fortini 1999) and second that the mutants carried lesions in the Ca2+-ATPase gene. In addition to noncomplementation observed between our Kum alleles and bona fide Ca2+-ATPase mutations, we discovered that one of the previously reported recessive lethal alleles, CaP60Al1, displays dominant paralytic phenotypes identical to Kum170 and Kum295 (henceforth called CaP60AKum170 and CaP60AKum295, respectively). Further sequence analysis revealed that CaP60AKum170 and CaP60Al1 carry single-base substitutions in Drosophila SERCA coding sequences, predicted to cause a Glu442-to-Lys mutation (CaP60AKum170) or a Cys318-to-Ser (CaP60Al1) substitution in the sequence of SERCA (Figure 2, A and B). The location of the two mutations is indicated on a homology-modeled 3D dSERCA structure (see materials and methods). Both mutations are located in the hinge domain and could potentially influence either ATP binding or conformational state of the molecule (Toyoshima et al. 2000, 2003). Together with the observation that the CaP60AKum170 and CaP60AKum295 recessive lethal phenotypes were completely rescued by a genomic transgene of dSERCA, these data establish that mutations at the dSERCA locus cause the phenotypes we observe in Kum mutants. We selected CaP60AKum170 for further extensive analysis because of the robustness of the phenotypes; however, all phenotypes are exhibited by CaP60AKum295 as well.
dSERCA is abundantly expressed in both neurons and muscle:
To visualize the distribution of the SERCA protein in flies, we raised antibodies against two carefully selected 20-amino-acid peptides derived from the fly SERCA sequence (see materials and methods). After immunization the antibodies were affinity purified using immobilized peptide columns and were used in all experiments. The antibody recognizes the predicted SERCA band at ∼100 kD in Western blots of total fly protein (Figure 3A, 1003 amino acids). The preimmune sera as well as antibody preadsorbed on free peptide failed to detect this band.
Figure 3.—
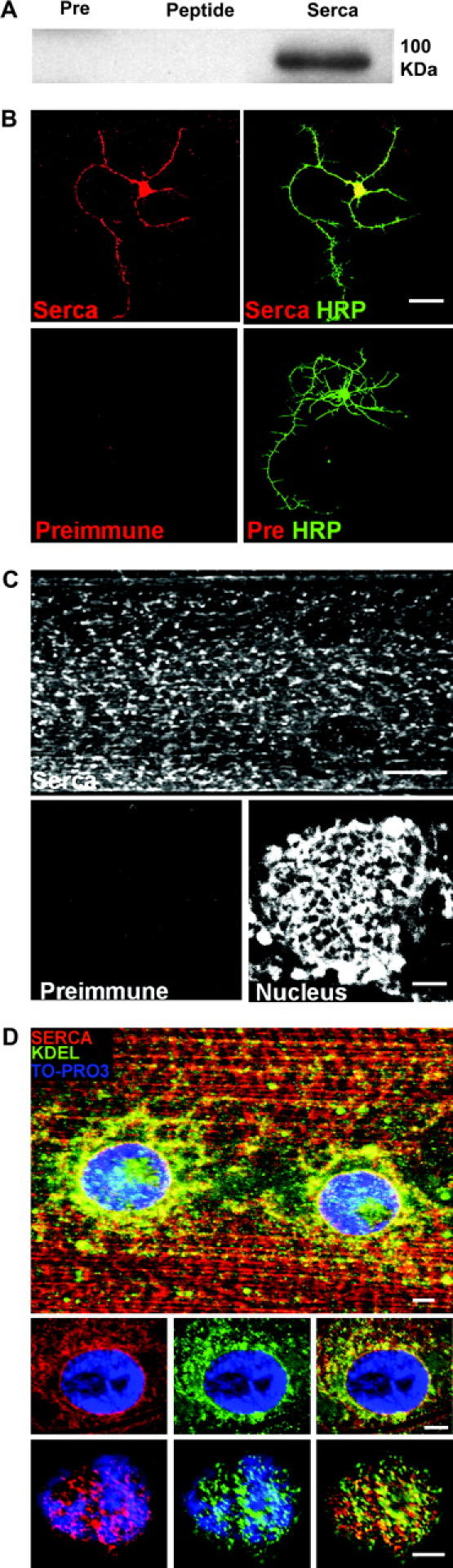
SERCA is highly expressed in both neurons and muscles. (A) A Western blot using protein purified from adult Drosophila and probed with the anti-SERCA antibody. A distinct band of ∼100 kD (the predicted size from a 1003-aa protein) is recognized. The preimmune serum and immune serum cross-adsorbed against the immunizing peptides fail to detect this band. (B) Primary neurons in culture stained with either preimmune serum or anti-SERCA antibodies with anti-HRP as a neuronal membrane marker. The anti-SERCA antibody stains the entire neuron as compared to the preimmune serum (bar, 10 μm). (C) Staining of larval body wall with the anti-SERCA antibody shows distinct cellular compartments (possibly SER). Also seen are membranous compartments continuous with the nuclear envelope (bottom right). (D) Double staining with anti-SERCA and anti-KDEL (that labels proteins retained in the ER) shows extensive colocalization, especially in membrane continuous with the nuclear membrane. Top: a maximum projection view of a shallow stack through a body wall muscle fiber. Bottom two rows: single planes at different depths through a muscle nucleus, demonstrating a high degree of overlap. The nucleus is shown in blue for reference (bar for muscle, 100 μm; for nucleus, 25 μm).
SERCA antibodies stain the body wall muscles strongly (Figure 3C). This is expected given that SERCA is known to form a large fraction of the total protein content of sarcoplasmic reticulum. The observed staining clearly highlights a membrane network (consistent with the ER) that extends to the nuclear envelope (Figure 3, C and D). To further show that our antibodies to SERCA label the ER, we double stained larval body wall preparations with anti-SERCA and anti-BiP (StressGen, SPA-827), an antibody that labels KDEL-containing proteins that are retained in the ER. Figure 3D clearly shows extensive colocalization between SERCA and KDEL-containing proteins, thus indicating that our antibody does indeed label SERCA in ER and nuclear membrane. The two bottom panels in Figure 3D are single planes at different depths through a muscle nucleus (shown using TO-PRO3 staining in blue) and further demonstrate the marked colocalization of an ER marker and SERCA. Neurons cultured from the ventral ganglia of wandering third instar larvae (Kraft et al. 1998) also stained positive for SERCA throughout the cell. (Figure 3B). These staining patterns, not seen with control preimmune serum, indicate that SERCA is highly expressed in intracellular membranes widely distributed through the cytoplasm of both neurons and muscles.
CaP60AKum170 mutations are loss-of-function mutations in dSERCA:
Mutations in dSERCA are likely to cause impaired calcium sequestration. However, the selection criteria we used in isolation of the mutants could have led to specific recovery of unusual alleles that might, for instance, sequester calcium at a faster rate. To firmly interpret our mutant phenotypes in terms of SERCA function, we tested whether the mutant effects on presynaptic calcium were similar to those observed in response to treatment with thapsigargin, an established SERCA inhibitor in vertebrates. The effect of thapsigargin has also been most simply and conclusively shown in Drosophila at the presynaptic nerve terminal (Kuromi and Kidokoro 2002). Hence, we used this assay to test the effect of our mutations in altering calcium dynamics in this paradigm.
At wild-type larval motor terminals, presynaptic calcium levels, assessed using Rhod-2-based calcium detection, are increased during a 4-min burst of 30-Hz nerve stimulation (Kuromi and Kidokoro 2002). Within 2 min after cessation of stimulation, calcium levels fall in wild-type animals to a value that remains significantly elevated compared to prestimulus levels. In the presence of thapsigargin (Tg), the initial peak of calcium during stimulation is higher, but after stimulation levels return to baseline rather than remain elevated. Together these data indicate that SERCA-dependent calcium sequestration limits the initial calcium peak and that release from internal stores contributes significantly to the sustained elevation of calcium observed following 30-Hz stimulation. The return to baseline and lack of sustained high levels of calcium are possibly due to the subsequent depletion of internal stores (Kuromi and Kidokoro 2002).
At 22° in both Canton-S controls and CaP60AKum170, the rhod-2 fluorescence intensity in boutons increased during stimulation at 30 Hz and declined gradually after tetanus but stayed at a slightly elevated level for a prolonged time [Figure 4A, Kum170(unheated); and Figure 4B, Kum170(−heat), open circles with solid line, and CS (−heat), dotted line with open circles]. In CaP60AKum170 preparations treated with thapsigargin (20 um for 20 min), the rhod-2 fluorescence intensity increased to a higher peak during early phase and declined during tetanus. After tetanus, the fluorescence rapidly declined to the pretetanus level [Figure 4A, Kum170 +Tg (unheated); and Figure 4B, Kum170 +Tg, solid circles with solid line]. These patterns are similar to those observed in wild-type animals treated with Tg (Kuromi and Kidokoro 2002). When CaP60AKum170 was exposed to 40° for 15 min, the changes in rhod-2 fluorescence in boutons during and after tetanus showed the same patterns as those observed in CaP60AKum170 treated with thapsigargin at 22° [Figure 4A, Kum170(heated); and Figure 4B, Kum170 (+heat), solid line with triangles]. CS animals, on the other hand, behaved similarly to unheated CS controls and did not show either an initial peak or a return to pretetanus levels [Figure 4B, CS(+heat), dotted line with triangles]. Thus, mutant CaP60AKum170 animals showed conditional alterations in calcium handling similar to those observed in thapsigargin-treated animals. In addition to establishing CaP60AKum170 as a dominant loss-of-function allele of SERCA, these experiments serve to further establish that a SERCA-containing compartment and SERCA-dependent functions operate to control calcium dynamics in the presynaptic terminal.
Figure 4.—
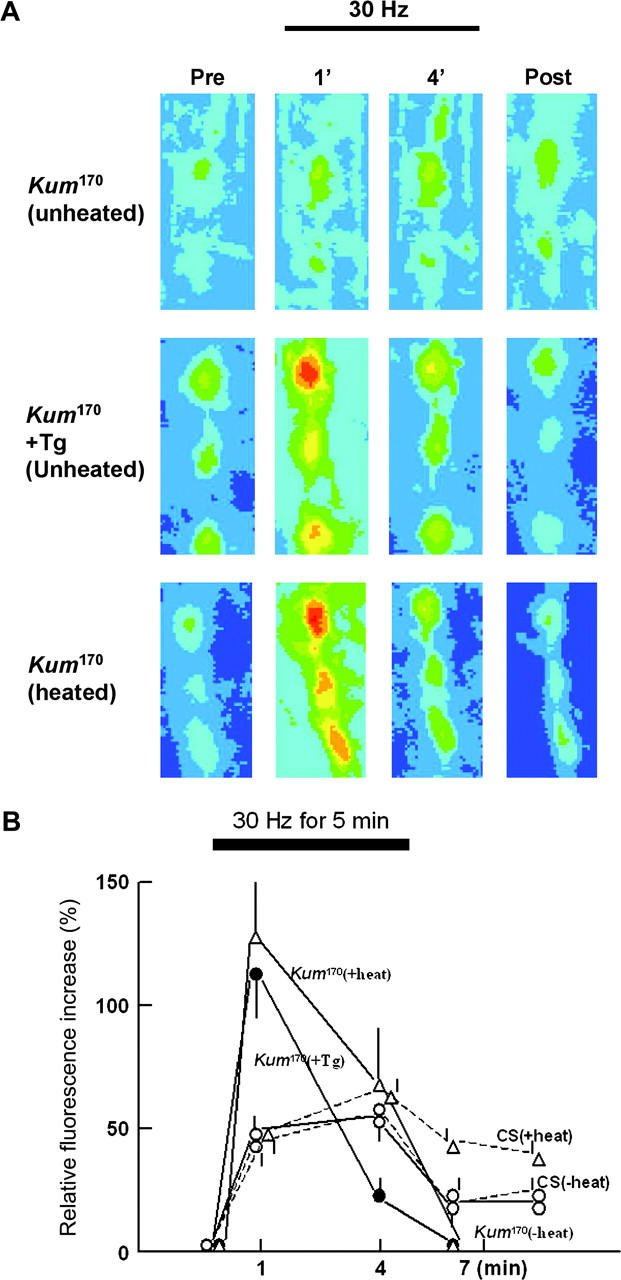
CaP60AKum170 mutants result in a loss of normal SERCA function and show elevated stimulus-dependent cytosolic calcium. (A) Rhod-2-loaded boutons are electrically stimulated at 30 Hz and pseudo-colored images of boutons are shown before stimulation (Pre), at ∼1 min (1') and 4 min (4') after the start of tetanic stimulation, and at 2 min after the end of stimulation (Post). Warmer colors correspond to higher [Ca2+]i. Untreated CaP60AKum170 at 22° (unheated), thapsigargin (20 um for 20 min)-treated CaP60AKum170 at 22° (+Tg), and CaP60AKum170 at 40° for 15 min (heated) are shown. (B) The ordinate shows relative fluorescence increase in boutons. The resting fluorescence (Fr) was subtracted from the stimulation-induced value (Ft), and the result (ΔF = Ft − Fr) was normalized to the resting fluorescence intensity (Ft) to give ΔF/Fr × 100. The abscissa indicates the time after the start of tetanic stimulation. Fluorescence in five or three boutons was measured in one preparation. Bars above or below each value are SEM. The numbers of samples tested for each genotype and treatment are as follows; CS (−heat), four; CS (+heat), five; CaP60AKum170 (−heat), five; CaP60AKum170 (+heat), seven; and CaP60AKum170 (+Tg), five.
Since the allele P52 does not have a paralytic phenotype, it is possible that the dominance of the loss-of-function alleles derives from a potentially oligomeric state of SERCA in the membrane. Thus, in these alleles, on heating, a majority of the oligomers are rendered nonfunctional, leading to paralysis. It is also possible that since in the dominant alleles, half the SERCA molecules on the ER/SR membrane are mutant, heat inactivation would alter the overall efficacy of calcium sequestration in these animals. In the P52 allele, on the other hand, SERCA molecules derived from the wild-type copy of the gene on the other chromosome might achieve “normal” density of functional SERCA molecules on the ER/SR membranes leading to normal function in the heterozygotes. Only in a homozygous condition is this allele lethal, probably owing to a critical reduction of SERCA molecules. A further consideration of this issue is provided in the discussion.
SERCA-dependent mechanisms control quantal content of evoked presynaptic release:
Since our experiments showed that presynaptic calcium levels are acutely controlled through SERCA function, we tested the effect of SERCA inhibition on evoked release at the presynapse. Two-electrode voltage clamp recordings done at the neuromuscular junction of untreated and treated wild-type and CaP60AKum170 third instar larvae show a significant reduction (∼50%) in evoked but not spontaneous release (Figure 5, A and B). (Under voltage-clamp conditions, only the postsynaptic response to presynaptically released neurotransmitter is measured, without any contribution from the voltage-gated channels.) This change, not seen at wild-type nerve terminals treated similarly (40° for 5 min, followed by recordings made at room temperature; wild type at room temperature, mean EJC amplitude is 40 ± 3 nA, n = 10, and after heating is 42 ± 2 nA, n = 8), is probably due to progressive depletion of calcium in the presynaptic compartment or to chronic inhibition of presynaptic calcium channels (see next section). Thus, SERCA-dependent calcium sequestration may contribute directly or indirectly in determining the number of vesicles that are released following depolarization of the nerve terminal. That there is no change in the mEJC size indicates normal vesicle filling and postsynaptic receptivity.
Figure 5.—

Store-derived calcium regulates quantal content of presynaptic release. (A) Representative traces showing reduction of mean EJC amplitude in CaP60AKum170 following heating at 40° for 5 min. The characteristics of spontaneous events remain relatively unchanged. Vertical bar is 10 nA for EJC and 1 nA for mEJC. Horizontal bar is 30 msec for EJC and 200 msec for mEJC. (B) Histograms depicting significantly reduced EJP amplitude in CaP60AKum170 on heating (P < 0.04) but unaltered mEJP amplitude and frequency. All error bars are SEM. − indicates untreated animals and + indicates animals heated at 40° for 5 min, prior to dissection and recordings at room temperature.
SERCA permits repetitive muscle action potential generation by preventing attenuation of voltage-gated calcium currents:
Following our observation of complete wing immobility in paralyzed CaP60AKum170 adults, we initially examined both spontaneous firing of the DLMs and their response to stimulation of the giant fiber pathway (King and Wyman 1980; Costello and Wyman 1986; Ikeda and Koenig 1988). In wild-type animals, recordings of spontaneous activity in flight muscles reveal intermittent bursts of action potential firing in DLMs (Figure 6A, top trace). These groups of action potential spikes are thought to be representative of the flight pattern (Ikeda and Koenig 1988). A single “spike” is composed of a slow rising phase, the excitatory postsynaptic potential (EPSP) representing response to released transmitter (glutamate) and a rapid action potential generated by the active responses of voltage-gated ion channels on muscle membrane. These characteristics are largely unchanged in unheated CaP60AKum170 animals. In contrast, in recordings made at room temperature after a 5-min exposure to 40°, CaP60AKum170 DLMs showed characteristic broad multipeaked action potentials progressing eventually to spike failure (Figure 6A, bottom traces).
Figure 6.—
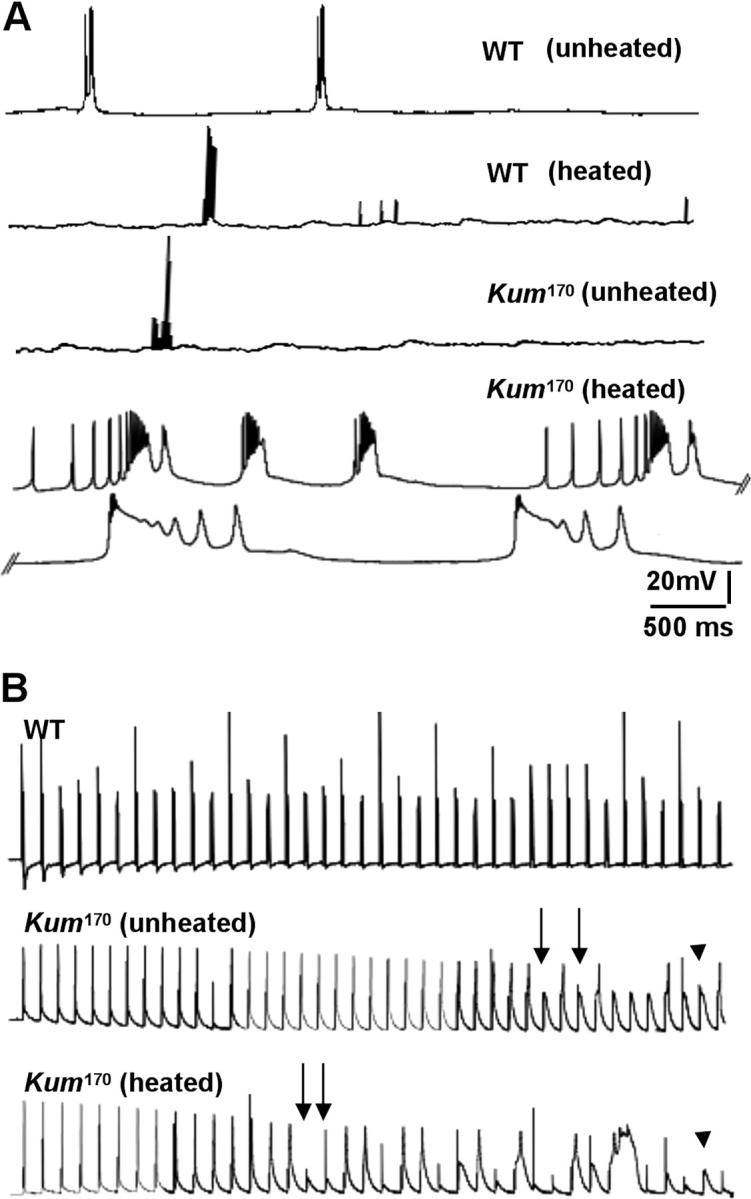
Normal SERCA function permits repetitive action potential generation: CaP60AKum170 mutations show action potential failures. (A) Sample traces showing spontaneous activity in DLMs of unheated and heated wild-type (top traces) and unheated and heated CaP60AKum170 animals (bottom traces). Wild-type DLMs are essentially inactive with occasional bursts of firing. This pattern is unchanged on heating and is similar in unheated CaP60AKum170 animals. However, on heating, CaP60AKum170 animals display obvious spike broadening leading to eventual spike failure. (B) Representative traces showing that at a stimulation frequency of 30 Hz, heated CaP60AKum170 animals exhibit spike failure before unheated CaP60AKum170 animals while wild-type animals show no failures at all. Further, spike broadening is regularly observed prior to failures in the mutants.
To more carefully define the origin of this phenotype, we analyzed DLM responses elicited by controlled stimulation of the motor nerve. Frequencies up to 50 Hz were used to evoke action potentials. Wild-type DLMs “follow” these stimulation frequencies with high fidelity. In contrast, untreated CaP60AKum170 DLM fibers show frequent action potential failures at 30 Hz (Figure 6B, arrows in middle trace). That this phenotype derives from mutations at the dSERCA locus is further proved by the fact that in heat-exposed CaP60AKum170 animals, failures are observed at even lower frequencies of ∼10 Hz. At a given frequency of stimulation, failures are observed earlier in heated CaP60AKum170 animals than in unheated CaP60AKum170 animals (Figure 6B, compare with arrows in bottom trace). The failures could derive from either defects in action potential generation in muscles or reduced presynaptic neurotransmitter release (leading to subthreshold EPSPs that do not lead to spiking; Figure 6B, arrowhead), or both.
Thus, we next tested if SERCA-dependent mechanisms in muscle serve to regulate excitability and action potential generation. Action potentials in Drosophila muscle are initiated by voltage-gated calcium currents. Since we observed a high incidence of spike failures in heated CaP60AKum170 animals, we tested whether voltage-gated calcium currents in muscles are altered in CaP60AKum170 mutants after heat exposure. Larval body wall muscles are particularly convenient and well established for voltage-clamp analysis of calcium currents (Gielow et al. 1995). We specifically measured the voltage-gated inward calcium current (by blocking all potassium channels—see materials and methods) in heated and unheated CaP60AKum170 animals. We find that voltage-gated calcium currents are dramatically and chronically reduced in heated CaP60AKum170 animals (Figure 7, A and B). In contrast, unheated CaP60AKum170 animals show calcium currents that are comparable to wild type. This reduced voltage-gated calcium current can account completely for spike failure, even in the presence of normal EPSPs. Thus, under normal conditions, SERCA-dependent calcium sequestration may serve to independently regulate the excitability of muscle by controlling signaling from intracellular stores of calcium that lead to inactivation of voltage-gated calcium channels.
Figure 7.—
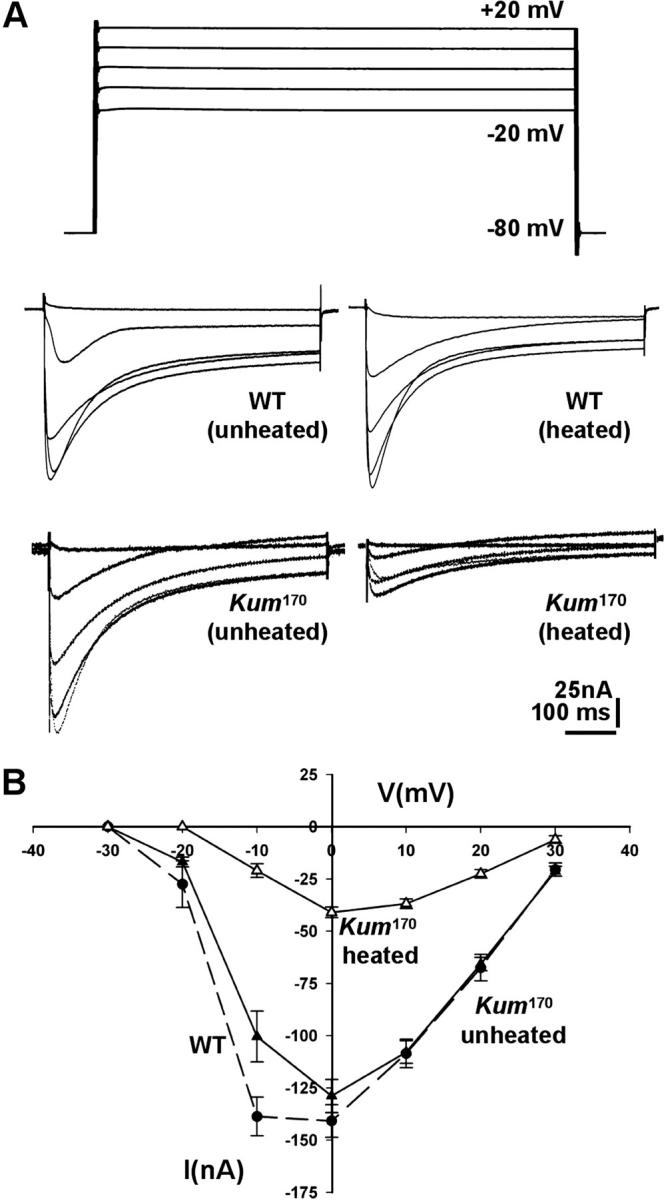
Voltage-gated calcium currents in heated CaP60AKum170 muscles are dramatically reduced. (A) Representative traces of voltage-gated calcium currents from unheated and heated wild-type (top) and CaP60AKum170 (bottom) larvae. Holding potential is −80 mV and traces corresponding to voltage steps to −20 mV through +20 mV in increments of 10 mV are shown. Voltage-gated calcium currents are severely reduced in heated CaP60AKum170 animals. (B) IV curve measuring peak barium currents through voltage-gated calcium channels in larval muscle 6 of segment A2 in unheated and heated CaP60AKum170 animals. An IV curve from wild-type animals is shown for reference (heating wild-type larvae does not cause any change in the IV curve).
SERCA-mediated calcium sequestration influences muscle action potential waveform by modulating calcium-activated potassium currents:
As mentioned before, spike failure in CaP60AKum170 muscles is preceded in several instances by spike broadening (Figure 8A, arrow). Such a multipeaked prolonged action potential has been described previously in slowpoke mutants (Atkinson et al. 1991, 2000). In these animals, a defect in the calcium-activated potassium channel encoded by the slo gene results in a slower falling phase of an action potential. This is because the calcium-activated outward potassium current is abolished or reduced. In CaP60AKum170 animals a potential mechanism by which this can happen is due to run-down internal calcium stores that likely play an important role in activating the slo channels.
Figure 8.—
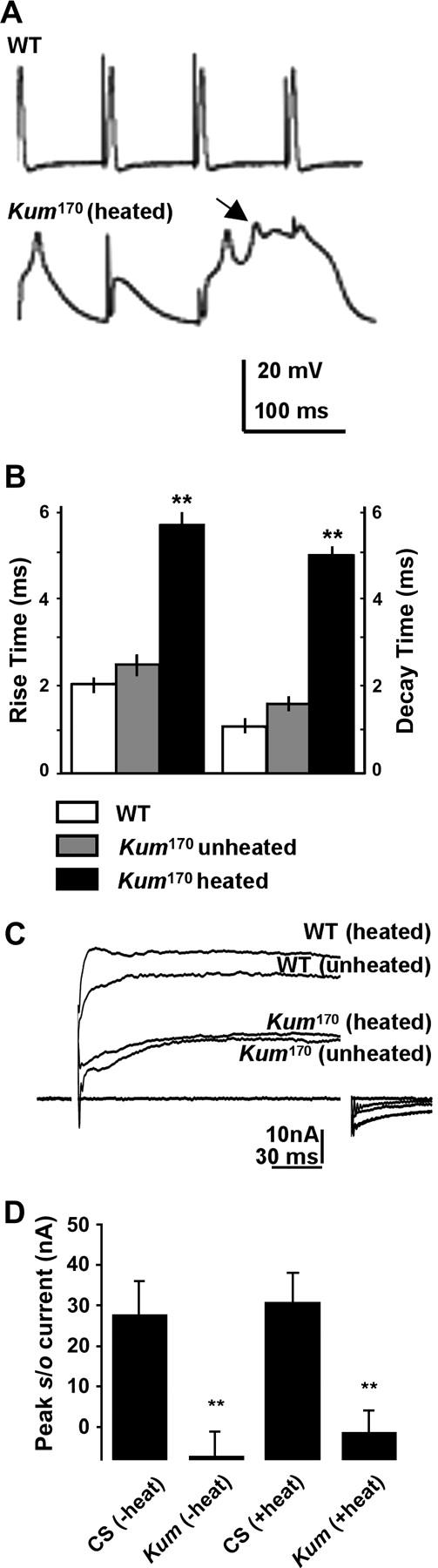
SERCA function controls action potential dynamics. (A) Representative traces from wild-type and heated CaP60AKum170 animals. Action potential failures in the CaP60AKum170 animals are preceded by spike broadening (arrows) and multipeaked action potentials. (B) Both the rise time and half-decay time of action potentials in heated CaP60AKum170 mutants are significantly increased as compared to wild-type and unheated mutants. Histogram represents mean values obtained from several spike trains from at least six separate animals in each case. (C) Representative traces to show the calcium-activated potassium current (slowpoke current) in heated and unheated wild-type and CaP60AKum170 larvae. Slo currents are reduced in both unheated and heated CaP60AKum170 animals as compared to wild type. (D) Quantification of the peak slo current 10 msec after the voltage step to 0 mV. Measurement at this voltage step and time point gives the best estimate of the state of the slo current. CaP60AKum170 animals show minimal slo currents under these conditions.
In addition to the broadening of spikes in CaP60AKum170 animals, we also noted that both the rising and falling phases of the action potential are slower. Figure 8B shows a comparison of the rise time and half decay time of consecutive action potential trains from wild-type, CaP60AKum170 unheated, and CaP60AKum170 heated animals. It is readily apparent that both the rise and decay are significantly slower in heated CaP60AKum170 animals. Likely explanations for these phenomena derive from effects on the two predominant channels that contribute to these components of an insect muscle action potential. Rise times are influenced most acutely by alterations in the inward calcium current, and thus a slow rise time could derive from the reduced voltage-gated calcium current. Similarly, the time for decay of the action potential is influenced by outward potassium currents. The observed increase in decay time could, therefore, derive from reduced calcium-activated potassium currents.
To directly test if the calcium-activated potassium currents are indeed reduced in CaP60AKum170 animals, we measured the outward slowpoke current from larval muscles. This was done by recording currents under voltage clamp in normal Ringer's solution containing 100 mm Ca2+ and 2 mm 4-AP (Elkins et al. 1986; materials and methods). Under these conditions a voltage step to 0 mV elicited a robust calcium-dependent outward current that represents the slo current. As seen in Figure 8, C and D, slo currents are highly reduced in both unheated and heated CaP60AKum170 animals. This correlates satisfactorily with the action potential broadening observed in both unheated and heated CaP60AKum170 animals and gives an accurate estimate of the state of cytosolic calcium in these larvae. Thus, SERCA-dependent maintenance of store calcium is critical for normal activation of slowpoke channels and recovery during repetitive action potentials.
Impairment of dSERCA function in muscles is sufficient to phenocopy the CaP60AKum170 mutations:
Since SERCA is found at high levels in both neurons and muscle and impairment of SERCA function results in both neural and muscle phenotypes, it is conceivable that the conditional paralytic phenotype in CaP60AKum170 mutations derives from defective SERCA function in both these tissues. To determine neural and muscular contributions to the paralytic phenotype of SERCA mutants, we made use of the GAL4-UAS system (Brand and Perrimon 1993) and cloned the mutated SERCA cDNA from CaP60AKum170 into the pUAST vector (materials and methods). We further generated flies carrying this transgene (UAS-CaP60AKum170) and crossed them to GAL4 lines that drove expression from UAS- CaP60AKum170 in neurons, muscles, or both tissues (Table 1). The effect of driving this dominant negative SERCA protein was assayed by a simple test for paralysis at restrictive temperatures.
TABLE 1.
Expression of mutantCaP60AKum170 in muscles is necessary and sufficient to phenocopy theCaP60AKum170 mutation
| Genotype | Larval paralysis |
Adult paralysis |
|---|---|---|
| CaP60AKum170 | +++ | +++ |
| Neural | ||
| C155 | − | − |
| Cha | − | − |
| C380 | − | − |
| Ubiquitous | ||
| Armadillo | − | − |
| Shibire | − | − |
| Tubulin | ++ | − |
| Muscle | ||
| MHC | + | − |
| 24B | ++ | − |
| Mef2 | +++ | ++ |
Overexpression of the mutant CaP60AKum170 protein in muscles mimics the CaP60AKum170 mutant phenotypes. Expression in neural tissue does not lead to any observable paralysis. Adult and larval paralysis is compared to that in CaP60AKum170 animals incubated at the restrictive temperature of 40° for 3 min.
Neural expression of CaP60AKum170 using elavC155 (pan-neural), cha (cholinergic neurons), or C380 (predominantly motor neurons) GAL4 drivers did not cause any observable paralysis at restrictive temperatures. Amplified expression in all neurons using elavC155; UAS-GAL4 (Hassan et al. 2000) also proved to be ineffective at causing behavioral paralysis. Thus, while SERCA function in neurons is likely to be important, a disruption of this function via CaP60AKum170 overexpression is not sufficient to lead to dominant paralysis.
In a different set of experiments, we used three ubiquitous GAL4 drivers (arm-GAL4, tubulin-GAL4, and shi-GAL4) and three muscle drivers (MHC-GAL4, 24B-GAL4, and mef2-GAL4). While tubulin-GAL4 and 24B-GAL4 animals showed larval paralysis, mef2 GAL4 animals showed both larval and adult paralysis completely undistinguishable from that in CaP60AKum170 (Table 1). Taken together, these observations clearly suggest that strong expression of mutant dSERCA protein in muscles, which probably alters the relative stoichiometry of wild-type vs. mutant SERCA molecules, is necessary and sufficient to duplicate the mutant paralytic phenotype and further tighten the causal link between mutations in the SERCA gene and observed phenotypes.
Myocyte enhancer factor-2 (Mef-2) is a transcription factor that is absolutely necessary for the proper development and differentiation of all classes of Drosophila musculature. A scan of the upstream region of the Ca-P60A gene for conserved transcription factor binding sites revealed two closely spaced, highly conserved binding sites for Mef-2. The first site occurs between 465 and 474 (ggttTAAAaa) and the second site occurs between 481 and 490 (tttcTAAAta) 5′ from the transcription start site. Both the sites occur on the plus strand and have a score of 1 for the conserved core sequence (PromoterInspector program at Genomatrix). It is conceivable that the spatio-temporal pattern of expression of dMef-2 also determines the physiologically relevant accuracy of SERCA expression and hence mef-2-GAL4 is the most potent in replicating the dSERCA mutant phenotypes.
To conclusively test if electrophysiological defects observed in muscles of CaP60AKum170 mutants are present in the mef2-GAL4; UAS-CaP60AKum170 animals, we recorded spontaneous and evoked responses from DLMs of both heated and unheated mef2-GAL4, UAS-CaP60AKum170 flight muscles. As shown in Figure 9, A and B, it is clear that DLM firing in mef2-GAL4, UAS-CaP60AKum170 animals is qualitatively similar to that in CaP60AKum170 mutants. Thus, genetically mosaic animals with wild-type nervous systems but SERCA-mutant muscle show all defects in flight muscle physiology characteristic of CaP60AKum170 mutants in both spontaneously firing (Figure 9A) and giant fiber stimulated (Figure 9B) recordings (arrow marks failures and arrowhead indicates presence of robust EPSPs). In control experiments, none of these effects are observed in either the mef2-GAL4 line or the UAS-CaP60AKum170 line alone (data not shown). These results suggest that the behavioral phenotype of temperature-sensitive paralysis in CaP60A mutants is predominantly, if not wholly, a consequence of SERCA inhibition and altered excitability in muscle.
Figure 9.—

Expression of dominant-negative SERCA protein in muscles is necessary and sufficient to phenocopy the CaP60AKum170 mutation. (A) Representative traces recorded from normal and heated Mef2-GAL4, UAS- CaP60AKum170 adults. The top trace shows spontaneous firing in the DLMs. Like the CaP60AKum170 mutants, action potentials show broadening with occasional failures. (B) Responses to controlled stimulation using the giant fiber pathway with and without heating. Here too, spike broadening is followed by frequent failures. This phenotype is exacerbated in response to heating as expected from the conditional nature of the mutation.
DISCUSSION
Cellular calcium dynamics are subject to stringent spatio-temporal control, especially in excitable cells. While the role for intracellular stores of calcium in this context is generally held to be important, details of how these stores may modulate the functioning of neurons and muscles are incompletely understood. SERCA is a key player in regulating intracellular calcium and its physiological effects as underscored by the observation that SERCA dysfunction underlies several different forms of cardiac disease (Gommans et al. 2002). In representative animal models of cardiac disease while impairment of SERCA activity has been shown to cause arrhythmic contraction, mechanisms by which SERCA affects electrical properties of cardiac cells remain incompletely described.
Although pharmacological perturbation of SERCA in other model systems using relatively specific drugs such as thapsigargin and cyclopiazonic acid has been used extensively, the elegant simplicity, precision, and clarity of a genetic analysis remains unparalleled. Moreover, unlike vertebrates, Drosophila has a single highly conserved SERCA gene, making analysis of the physiological contribution of SERCA particularly feasible. Our results not only reinforce and further develop previous findings, but also open up the possibility of detailed structure-function analysis in vivo and screens for genetic modifiers of SERCA function.
Thus, in this report, we isolate, characterize, and utilize conditional mutations in dSERCA to address specific questions about the role of SERCA-dependent calcium uptake and intracellular reserves of calcium in neuromuscular physiology. Using this genetic approach, we show that (1) SERCA regulates calcium sequestration and quantal content in neurons; (2) SERCA inhibition results in altered properties of voltage-gated ion channels on plasma membrane that contribute to normal muscle firing properties; and (3) we can provide evidence for novel intra- or intermolecular interactions that govern SERCA function in vivo. While some of these findings are admittedly anticipated (extensive distribution of SERCA in both neurons and muscles), others, such as the chronic regulation of ion channels by SERCA-dependent mechanisms, are surprisingly novel and further refine our understanding of the role of this key protein.
SERCA regulates presynaptic calcium sequestration and quantal content:
Conditional mutant alleles of dSERCA have allowed us to analyze immediate or early effects of SERCA perturbation on the physiology of excitable cells. In both heated CaP60AKum170 mutants and thapsigargin-treated wild-type larval motor terminals, stimulation at 30 Hz results in an unusually high initial increase in cytosolic calcium compared to a relatively modest increase in the wild type. This provides direct evidence for a SERCA-dependent mechanism in sequestering presynaptic calcium during high-frequency stimulation. Within 2–4 min of sustained 30-Hz stimulation, the early calcium peak, sustained in wild-type terminals, falls in SERCA-inhibited terminals. Our data currently do not distinguish between two explanations for this phenomenon: (a) that calcium entry into the nerve terminal is reduced under conditions of SERCA inhibition; or (b) that release from intracellular calcium stores, not replenished during SERCA inhibition, may contribute to the initial calcium peak. More instructively, after cessation of 30-Hz stimulations, intracellular calcium that remains significantly elevated in control preparations falls in SERCA-inhibited terminals to levels below those seen in controls (Figure 4). This phenomenon is most easily explained by postulating calcium release from intracellular stores to persist after cessation of stimulation in the wild type but not in SERCA-inhibited synapses in which calcium stores have been depleted. Together these data provide important genetic support for the conclusions based on pharmacological evidence that SERCA-mediated calcium sequestration is operative in the presynaptic terminal (Matias et al. 2002; Lauri et al. 2003).
Reduced cytosolic calcium as observed in our CaP60AKum170 mutants might have an effect on presynaptic release properties. Our results indicate that this is indeed the case. Evoked excitatory junction potentials are reduced in heated CaP60AKum170 animals as compared to unheated ones. Comparable heat treatment (40° for 5 min, followed by recordings at room temperature) in wild-type animals has negligible effects on evoked release. This suggests an effect of altered calcium sequestration (leading to the depletion of intracellular stores) on the probability of evoked release.
Altered synaptic transmission and membrane excitability in SERCA mutants:
The role of SERCA in calcium sequestration in muscles has been well established by several previous analyses (East 2000). However, these analyses make few predictions for SERCA function in regulating membrane excitability or synaptic transmission. The importance of SERCA in cardiac dysfunction is well documented. However, the remarkable finding that SERCA potentiation can suppress multiple unrelated cardiac myopathies has not been explained at a physiological or mechanistic level. Thus, it was unexpected for us to observe failure of synaptically driven action potentials, as well as a range of abnormalities in action-potential waveforms including slow rise time, slow decay time, and abnormal spike broadening in SERCA mutants. These defects were observed in the dorsal flight muscles of adult animals that normally follow stimulation frequencies of as high as 100 Hz with high fidelity. The presence of discernible EPSPs in SERCA-inhibited muscle indicates that these defects in muscle physiology might derive primarily from alterations in the active responses of muscles to initial neurotransmitter-induced depolarization. Furthermore, strong expression of the mutant SERCA protein in muscles using the GAL4-UAS system resulted in temperature-sensitive paralysis and electrophysiological defects similar to the CaP60AKum170 mutation. Thus, calcium sequestration by SERCA plays a critical role in maintaining the nature and fidelity of active muscle responses to synaptic stimulation.
More detailed, direct analysis of the major inward current carried by a voltage-gated calcium channel reveals that SERCA regulates the availability of voltage-gated calcium channels. CaP60AKum170 animals on heating show a dramatic reduction in voltage-gated calcium currents. Such a reduction is likely to cause failures of muscle action potentials even at low frequencies of stimulation. At this stage we can hypothesize only about the mechanism by which there is a reduction in calcium currents. Existing data suggest a feedback inactivation of calcium channels due to initial high levels of cytosolic calcium (Burgoyne and Weiss 2001). There might also be additional mechanisms, e.g., kinase signaling, by which alteration of SERCA-mediated calcium uptake can signal to voltage-gated channels on the cell surface and chronically attenuate calcium currents through these channels (Peterson et al. 1999; Wang et al. 2002, 2003; Hoeffer et al. 2003). In this scenario, SERCA may normally function as a clamp to control cytosolic calcium levels.
In addition to voltage-gated calcium channels, calcium-activated potassium channels also depend on SERCA function. Characteristically prolonged action potentials in CaP60AKum170, also observed in slowpoke mutants, can be explained by altered calcium-activated potassium current in these mutants (Atkinson et al. 1991, 2000). Direct measurement of the peak slo current shows a severely reduced calcium-activated potassium current in CaP60AKum170 animals (Figure 8). Although our data do not provide a conclusive mechanistic explanation for the phenomenon, it is possible that the effects are a direct consequence of reduced cytosolic calcium.
At present, very little is known about how signaling pathways regulate voltage-gated calcium channels or Slowpoke channels in Drosophila; thus, additional studies will be required to pinpoint the pathways that are involved in this process. However, our demonstration that ion-channel properties are sensitive to SERCA dysfunction provides at least one potential explanation for how a wide range of defects in cardiac physiology may be suppressed by stimulating SERCA function.
New insights into SERCA function in vivo:
Given that current structural models for SERCA generally consider a monomeric protein, the dominance of the conditional paralytic phenotype was surprising. It potentially indicated an unusual neomorphic (gain-of-function) effect on SERCA function. However, the remarkably similar effects of CaP60AKum mutations and the SERCA antagonist thapsigargin on presynaptic calcium fluxes strongly argue against this possibility. The dominant loss-of-function effect of three different dSERCA mutations we describe cannot be explained by haplo-insufficiency, the requirement of two wild-type alleles, because heterozygous deficiencies in this region are viable and normal. Rather the observed dominance suggests that existing structural models for SERCA should be further refined by incorporating intermolecular interactions. If SERCA exists as oligomers in the native state, then inhibiting half the SERCA molecules can have a dominant effect through the overall inhibition of all calcium transporting complexes. Given the importance of SERCA in regulation of various physiological processes, for instance, cardiac rhythm, and its relevance as a drug target (Minamisawa et al. 1999), this is an issue of some significance.
In conclusion, we describe the effects of mutating SERCA and altering intracellular calcium stores on neuromuscular physiology. We demonstrate that loss-of-function mutations in dSERCA lead to conditional dominant phenotypes. We also show that SERCA regulates function of ion channels on the plasma membrane, which contributes to generation and waveform of muscle action potentials. Further studies will be aimed at elucidating the link between SERCA dysfunction and ion channel modulation. Conserved signaling pathways other than direct calcium-mediated feedback may also be involved in this phenomenon. Investigation of structure-function relationships of SERCA and screens to isolate modifiers of CaP60AKum170 are also feasible.
Acknowledgments
We acknowledge members of the Ramaswami, Krishnan, and Levine laboratories and Konrad Zinsmaeir for helpful comments, criticisms, and suggestions. We also thank Carl Boswell for help with confocal imaging at the Molecular and Cellular Biology confocal facility, René Luedeman for help with neuronal cultures, Charles Hoeffer and Patricia Estes for help with Western analysis, and Patricia Estes for help with microinjection. C.C. acknowledges support from grant NS 28495 to R. Levine. Support for this work came in part from a Cancer Biology Training Grant [National Institutes of Health (NIH) T32 CA09213] to S.S.; grants from the Department of Science and Technology, Department of Biotechnology (India), and Tata Institute of Fundamental Research intramural funds to K.S.K.; and NIH grants to M.R. (NS 34889 and DA 15485).
References
- Atkinson, N. S., G. A. Robertson and B. Ganetzky, 1991. A component of calcium-activated potassium channels encoded by the Drosophila slo locus. Science 253: 551–555. [DOI] [PubMed] [Google Scholar]
- Atkinson, N. S., R. Brenner, W. Chang, J. Wilbur, J. L. Larimer et al., 2000. Molecular separation of two behavioral phenotypes by a mutation affecting the promoters of a Ca-activated K channel. J. Neurosci. 20: 2988–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge, M. J., P. Lipp and M. D. Bootman, 2000. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell. Biol. 1: 11–21. [DOI] [PubMed] [Google Scholar]
- Brand, A. H., and N. Perrimon, 1993. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118: 401–415. [DOI] [PubMed] [Google Scholar]
- Burgoyne, R. D., and J. L. Weiss, 2001. The neuronal calcium sensor family of Ca2+-binding proteins. Biochem. J. 353: 1–12. [PMC free article] [PubMed] [Google Scholar]
- Catterall, W. A., 1999. Interactions of presynaptic Ca2+ channels and snare proteins in neurotransmitter release. Ann. NY Acad. Sci. 868: 144–159. [DOI] [PubMed] [Google Scholar]
- Costello, W. J., and R. J. Wyman, 1986. Development of an indirect flight muscle in a muscle-specific mutant of Drosophila melanogaster. Dev. Biol. 118: 247–258. [DOI] [PubMed] [Google Scholar]
- East, J. M., 2000. Sarco(endo)plasmic reticulum calcium pumps: recent advances in our understanding of structure/function and biology (review). Mol. Membr. Biol. 17: 189–200. [DOI] [PubMed] [Google Scholar]
- Elkins, T., B. Ganetzky and C. F. Wu, 1986. A Drosophila mutation that eliminates a calcium-dependent potassium current. Proc. Natl. Acad. Sci. USA 83: 8415–8419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabiato, A., 1983. Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. Am. J. Physiol. 245: C1–14. [DOI] [PubMed] [Google Scholar]
- Gielow, M. L., G. G. Gu and S. Singh, 1995. Resolution and pharmacological analysis of the voltage-dependent calcium channels of Drosophila larval muscles. J. Neurosci. 15: 6085–6093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gommans, I. M., M. H. Vlak, A. de Haan and B. G. van Engelen, 2002. Calcium regulation and muscle disease. J. Muscle Res. Cell Motil. 23: 59–63. [DOI] [PubMed] [Google Scholar]
- Gyorke, S., I. Gyorke, V. Lukyanenko, D. Terentyev, S. Viatchenko-Karpinski et al., 2002. Regulation of sarcoplasmic reticulum calcium release by luminal calcium in cardiac muscle. Front. Biosci. 7: d1454–d1463. [DOI] [PubMed] [Google Scholar]
- Hassan, B. A., N. A. Bermingham, Y. He, Y. Sun, Y. N. Jan et al., 2000. atonal regulates neurite arborization but does not act as a proneural gene in the Drosophila brain. Neuron 25: 549–561. [DOI] [PubMed] [Google Scholar]
- Hasselbach, W., 1998. The Ca(2+)-ATPase of the sarcoplasmic reticulum in skeletal and cardiac muscle. An overview from the very beginning to more recent prospects. Ann. NY Acad. Sci. 853: 1–8. [DOI] [PubMed] [Google Scholar]
- Hoeffer, C. A., S. Sanyal and M. Ramaswami, 2003. Acute induction of conserved synaptic signaling pathways in Drosophila melanogaster. J. Neurosci. 23: 6362–6372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda, K., and J. H. Koenig, 1988. Morphological identification of the motor neurons innervating the dorsal longitudinal flight muscle of Drosophila melanogaster. J. Comp. Neurol. 273: 436–444. [DOI] [PubMed] [Google Scholar]
- Katz, B., and R. Miledi, 1969. Spontaneous and evoked activity of motor nerve endings in calcium Ringer. J. Physiol. 203: 689–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King, D. G., and R. J. Wyman, 1980. Anatomy of the giant fibre pathway in Drosophila. I. Three thoracic components of the pathway. J. Neurocytol. 9: 753–770. [DOI] [PubMed] [Google Scholar]
- Kraft, R., R. B. Levine and L. L. Restifo, 1998. The steroid hormone 20-hydroxyecdysone enhances neurite growth of Drosophila mushroom body neurons isolated during metamorphosis. J. Neurosci. 18: 8886–8899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuromi, H., and Y. Kidokoro, 2002. Selective replenishment of two vesicle pools depends on the source of Ca2+ at the Drosophila synapse. Neuron 35: 333–343. [DOI] [PubMed] [Google Scholar]
- Laemmli, U. K., and S. F. Quittner, 1974. Maturation of the head of bacteriophage T4. IV. The proteins of the core of the tubular polyheads and in vitro cleavage of the head proteins. Virology 62: 483–499. [DOI] [PubMed] [Google Scholar]
- Lauri, S. E., Z. A. Bortolotto, R. Nistico, D. Bleakman, P. L. Ornstein et al., 2003. A role for Ca2+ stores in kainate receptor-dependent synaptic facilitation and LTP at mossy fiber synapses in the hippocampus. Neuron 39: 327–341. [DOI] [PubMed] [Google Scholar]
- Loke, J., and D. H. MacLennan, 1998. Malignant hyperthermia and central core disease: disorders of Ca2+ release channels. Am. J. Med. 104: 470–486. [DOI] [PubMed] [Google Scholar]
- Loughney, K., R. Kreber and B. Ganetzky, 1989. Molecular analysis of the para locus, a sodium channel gene in Drosophila. Cell 58: 1143–1154. [DOI] [PubMed] [Google Scholar]
- Magyar, A., E. Bakos and A. Varadi, 1995. Structure and tissue-specific expression of the Drosophila melanogaster organellar-type Ca(2+)-ATPase gene. Biochem. J. 310(Pt. 3): 757–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matias, C., J. C. Dionisio and M. E. Quinta-Ferreira, 2002. Thapsigargin blocks STP and LTP related calcium enhancements in hippocampal CA1 area. Neuroreport 13: 2577–2580. [DOI] [PubMed] [Google Scholar]
- Melzer, W., A. Herrmann-Frank and H. C. Luttgau, 1995. The role of Ca2+ ions in excitation-contraction coupling of skeletal muscle fibres. Biochim. Biophys. Acta 1241: 59–116. [DOI] [PubMed] [Google Scholar]
- Minamisawa, S., M. Hoshijima, G. Chu, C. A. Ward, K. Frank et al., 1999. Chronic phospholamban-sarcoplasmic reticulum calcium ATPase interaction is the critical calcium cycling defect in dilated cardiomyopathy. Cell 99: 313–322. [DOI] [PubMed] [Google Scholar]
- Odermatt, A., P. E. Taschner, V. K. Khanna, H. F. Busch, G. Karpati et al., 1996. Mutations in the gene-encoding SERCA1, the fast-twitch skeletal muscle sarcoplasmic reticulum Ca2+ATPase, are associated with Brody disease. Nat. Genet. 14: 191–194. [DOI] [PubMed] [Google Scholar]
- Periz, G., and M. E. Fortini, 1999. Ca(2+)-ATPase function is required for intracellular trafficking of the Notch receptor in Drosophila. EMBO J. 18: 5983–5993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson, B. Z., C. D. DeMaria, J. P. Adelman and D. T. Yue, 1999. Calmodulin is the Ca2+ sensor for Ca2+ -dependent inactivation of L-type calcium channels. Neuron 22: 549–558. [DOI] [PubMed] [Google Scholar]
- Ramaswami, M., S. Rao, A. van der Bliek, R. B. Kelly and K. S. Krishnan, 1993. Genetic studies on dynamin function in Drosophila. J. Neurogenet. 9: 73–87. [DOI] [PubMed] [Google Scholar]
- Roberts, W. M., 1993. Spatial calcium buffering in saccular hair cells. Nature 363: 74–76. [DOI] [PubMed] [Google Scholar]
- Rorth, P., 1996. A modular misexpression screen in Drosophila detecting tissue-specific phenotypes. Proc. Natl. Acad. Sci. USA 93: 12418–12422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanyal, S., R. Narayanan, C. Consoulas and M. Ramaswami, 2003. Evidence for cell autonomous AP1 function in regulation of Drosophila motor-neuron plasticity. BMC Neurosci. 4: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng, Z. H., R. E. Westenbroek and W. A. Catterall, 1998. Physical link and functional coupling of presynaptic calcium channels and the synaptic vesicle docking/fusion machinery. J. Bioenerg. Biomembr. 30: 335–345. [DOI] [PubMed] [Google Scholar]
- Stewart, B. A., H. L. Atwood, J. J. Renger, J. Wang and C. F. Wu, 1994. Improved stability of Drosophila larval neuromuscular preparations in haemolymph-like physiological solutions. J. Comp. Physiol. A Sens. Neural Behav. Physiol. 175: 179–191. [DOI] [PubMed] [Google Scholar]
- Suzuki, N., and M. Kano, 1977. Development of action potential in larval muscle fibers in Drosophila melanogaster. J. Cell Physiol. 93: 383–388. [DOI] [PubMed] [Google Scholar]
- Thomas, J. B., and R. J. Wyman, 1982. A mutation in Drosophila alters normal connectivity between two identified neurones. Nature 298: 650–651. [DOI] [PubMed] [Google Scholar]
- Toyoshima, C., M. Nakasako, H. Nomura and H. Ogawa, 2000. Crystal structure of the calcium pump of sarcoplasmic reticulum at 2.6 A resolution. Nature 405: 647–655. [DOI] [PubMed] [Google Scholar]
- Toyoshima, C., H. Nomura and Y. Sugita, 2003. Structural basis of ion pumping by Ca(2+)-ATPase of sarcoplasmic reticulum. FEBS Lett. 555: 106–110. [DOI] [PubMed] [Google Scholar]
- Wang, B., C. Bolduc and K. Beckingham, 2002. Calmodulin UAS-constructs and the in vivo roles of calmodulin: analysis of a muscle-specific phenotype. Genesis 34: 86–90. [DOI] [PubMed] [Google Scholar]
- Wang, B., K. M. Sullivan and K. Beckingham, 2003. Drosophila calmodulin mutants with specific defects in the musculature or in the nervous system. Genetics 165: 1255–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]