

Abstract
The mapping of quantitative trait loci (QTL) is an important research question in animal and human studies. Missing data are common in such study settings, and ignoring such missing data may result in biased estimates of the genotypic effect and thus may eventually lead to errant results and incorrect inferences. In this article, we developed an expectation-maximization (EM)–likelihood-ratio test (LRT) in QTL mapping. Simulation studies based on two different types of phylogenetic models revealed that the EM-LRT, a statistical technique that uses EM-based parameter estimates in the presence of missing data, offers a greater statistical power compared with the ordinary analysis-of-variance (ANOVA)-based test, which discards incomplete data. We applied both the EM-LRT and the ANOVA-based test in a real data set collected from F2 intercross studies of inbred mouse strains. It was found that the EM-LRT makes an optimal use of the observed data and its advantages over the ANOVA F-test are more pronounced when more missing data are present. The EM-LRT method may have important implications in QTL mapping in experimental crosses.
ANIMAL models and their corresponding genomes are highly useful for mapping traits that may apply to human diseases (Knoblauch and Lindpaintner 1999). Since genes are conserved throughout evolution, the identification of “evolutionary homologs” in animals is well appreciated in helping to find their counterparts in humans.
There are two primary methods for quantitative trait locus (QTL) mapping: (a) the single-marker method and (b) the interval-mapping method. The single-marker method is a traditional method for detecting the association between individual genetic markers and the quantitative trait of interest (Luo et al. 2000). The analysis-of-variance (ANOVA) represents the typical method applied in this kind of analysis. The interval-mapping method uses information provided by multiple linked markers to probabilistically assess potential QTL at chromosomal locations between such markers. In the interval-mapping approach developed by Lander and Botstein (1989), evidence for a putative QTL is summarized by a LOD (log of odds) score that exceeds a predefined threshold at a given chromosomal position.
The presence of missing data in studies usually lowers both the power of QTL mapping and the precision of parameter estimation, because the sample size for the incomplete data is less than it would be if the data were complete. In previous literature, the treatment of such a missing data problem is not adequate. Two simple methods have been most widely applied. One is simply to use the incomplete data by deleting all data records with any values missing, and it is called “listwise deletion.” A second approach is called “pairwise deletion,” which deletes those data records if either the phenotypic data or the genotypic data at the marker of interest are missing. In this article, we propose an expectation-maximization (EM)–likelihood-ratio test (LRT) to incorporate the flanking markers' information in the presence of missing marker data in the single-marker analysis. The LRT is derived from the maximum likelihood calculated using the EM algorithm based on all the observed data.
In the following section, we first introduce the mathematical model and notations, and then we derive the EM algorithm for maximum-likelihood estimation. Afterward, we describe the EM-LRT (or the EM-based Student's t-test) and the standard ANOVA-based tests (F-test and pairwise t-test). Then, we assess the validity of the EM-LRT at various sample sizes and various proportions of missing data, compare the performances of the proposed EM-based tests over the ANOVA-based tests through simulations, and evaluate whether or not it represents a more effective test for real data sets. Finally, we provide a summarization and some further discussions.
We have implemented the algorithm described in this article in the freely available statistical software R (Ihaka and Gentleman 1996). The code is available from the authors upon request.
MATERIALS AND METHODS
Model settings and notations:
Let us denote the genotypes at the trait marker locus A (the hypothesized QTL for the trait) as AA, Aa, and aa, the genotypes at its left-side flanking marker locus B as BB, Bb, bb, and the genotypes at its right-side flanking marker locus C as CC, Cc, and cc (note that we consider here only the biallelic markers, such as the simple sequence length polymorphisms). Let Y denote the phenotype value; let X1, X2, and X3 denote the respective genotype values at the loci A, B, and C, where X1 = 1, 2, and 3 denotes the three respective genotypes, AA, Aa, and aa, X2 = 1, 2, and 3 denotes the three respective genotypes, BB, Bb, and bb, and X3 = 1, 2, and 3 denotes the three respective genotypes, CC, Cc, and cc. Let μi denote E(Y|X1 = i), where i = 1, 2, and 3. Then, what we test here is H0: μ1 = μ2 = μ3 (locus A is not a QTL for Y), vs. Ha: μ1, μ2, and μ3 are not all equal (locus A is a QTL for Y).
This hypothesis test includes the test for both dominant and additive effects of the hypothesized QTL—locus A.
In practice, the genotype measure X1 at locus A may be missing for some animals. The usual approaches for missing data such as listwise deletion and pairwise deletion would simply exclude such animals from the ANOVA-based tests, resulting in a lower power to detect the QTL. Here, we propose an EM-based approach utilizing information of incomplete data, rather than discarding it. When there are missing data at locus A, the approach makes use of genotype data not only at locus A, but also at its two most closely linked markers, loci B and C. For the three linked markers, A, B, and C, there are a total of 27 possible genotype combinations {X1 = j, X2 = k, X3 = l}, where j, k, l = 1, 2, or 3. We denote the probabilities for the occurrence of each combination as pj,k,l = Pr(X1 = j, X2 = k, X3 = l).
By assuming a standard ANOVA model relating the phenotype Y to the genotype X1, we have
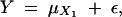 |
1 |
where ε ∼ N(0, σ2) and X1 can take one of the three possible genotype values of 1, 2, or 3 defined above. The complete data set in this case is {(Yi, X1,i, X2,i, X3,i), i = 1, … , n} for a sample size of n.
The log-likelihood of the complete data is 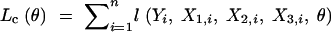 , where
, where
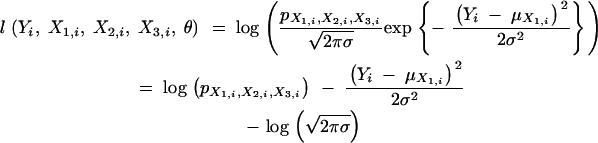 |
and θ = (μ1, μ2, μ3, σ2, p1,1,1, p1,1,2, p1,1,3, p1,2,1, p1,2,2, p1,2,3, p1,3,1, p1,3,2, p1,3,3, p2,1,1, p2,1,2, p2,1,3, p2,2,1, p2,2,2, p2,2,3, p2,3,1, p2,3,2, p2,3,3, p3,1,1, p3,1,2, p3,1,3, p3,2,1, p3,2,2, p3,2,3, p3,3,1, p3,3,2, p3,3,3). When there are missing data,
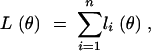 |
2 |
where liθ is defined as follows. First, if the phenotype Yi and the three genetic markers X1,i, X2,i, X3,i are all observed for the ith animal, obviously,
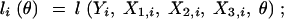 |
3 |
second, if the phenotypeYi is observed but some genetic markers are missing for the ith animal, then
 |
4 |
and third, if the phenotypeYi is missing for the ith animal,
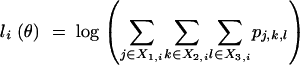 |
5 |
Here and in the following, the notation of summation ∑j∈X1,i denotes the summation over all possible values of X1,i. For example, if X1,i is observed to be 2, then the summation contains only one case (i.e., j = 2); on the other hand, if X1,i is missing, then the summation is taken over all three possible values j = 1, 2, and 3.
We propose estimating the parameters by maximizing the log-likelihood Lθ as defined in Equations 2–5 above and using the corresponding LRT in hypothesis tests.
Direct maximization of Lθ is difficult, as we can see in the complicated equations [(2)–(5)] shown above. The EM algorithm (Dempster et al. 1977; Little and Rubin 1987) is an appropriate method for computing the maximum-likelihood estimator θ̂ when missing data are present. In the following, we first derive formulas for the EM algorithm to maximize the log-likelihood Lθ. Then, we deduce the LRT using the EM estimations and compare its performance with the ordinary ANOVA-based tests.
EM algorithm:
We now derive the formulas of the EM algorithm for this problem following standard notations (McLachlan and Krishnan 1997).
We start with an initial estimate θ0 (which can be either the ANOVA estimate or any other reasonable estimate). At the (m + 1)th iteration, we update the current estimate θm by completing the E-step and the M-step as follows.
E-step:
Compute 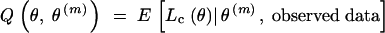 . The computation is simplified to
. The computation is simplified to
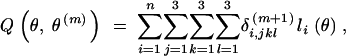 |
6 |
where 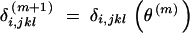 denotes the Pr(X1,i = j, X2,i = k, X3,i = l|observed data and θm). It can be computed according to the following formula: If Yi is observed,
denotes the Pr(X1,i = j, X2,i = k, X3,i = l|observed data and θm). It can be computed according to the following formula: If Yi is observed,
 |
7 |
if Yi is missing,
 |
8 |
Here φ{j ∈ X1,i, k ∈ X2,i, l ∈ X3,i} is the indicator function whether (j, k, l) is a possible value for (X1,i, X2,i, X3,i).
M-step:
Update the parameter estimate to the value 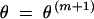 that maximizes Qθ, θm. The maximization over θ becomes rather simple if we further write out the expression
that maximizes Qθ, θm. The maximization over θ becomes rather simple if we further write out the expression
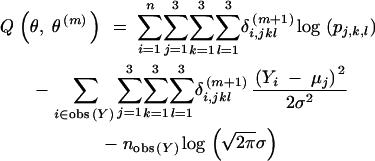 |
Here, obs(Y) denotes the set of i's where Yi is observed, and nobs(Y) = |obs(Y)|.
The maximization of the above expression is very similar to a linear model and we find explicitly the following updating formula:
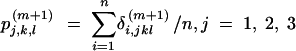 |
9 |
 |
10 |
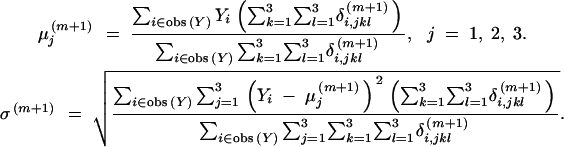 |
11 |
The E-step and M-step are then iterated until the estimate θm converges to an estimated value, θ̂.
Hypothesis testing:
To check whether locus A is a QTL for the trait of interest, Y, statistically we test the hypothesis H0: μ1 = μ2 = μ3 (locus A is not a QTL for Y) vs. Ha: μ1, μ2, and μ3 are not all equal (locus A is a QTL for Y). Here we first describe the ordinary ANOVA for single-marker analysis, which is the standard approach in the present literature (Rubattu et al. 1996; Vallejo et al. 1998; Poyan Mehr et al. 2003; Zhao and Meng 2003). When missing data are present, the ordinary ANOVA excludes all the data records with missing information on X1 or Y, and a subset of observations is left {(Yi, X1,i), i = 1, … , n*}, (n* ≤ n). The ordinary ANOVA then estimates the mean phenotype given the genotype data,
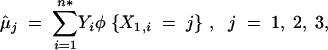 |
where φ is an indicator function. The variance is estimated by
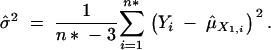 |
Then, an F-test is constructed by comparing σ̂2 with the between-group variance, σ̂2b,
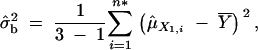 |
where 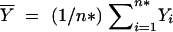 . Therefore, the F-test statistic is constructed as
. Therefore, the F-test statistic is constructed as
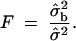 |
12 |
The F-test would reject H0 if F > Fα;2,n*−3, where Fα;2,n*−3 is the (1 − α)100th percentile of an F-distribution with d.f. = 2 and (n* − 3).
The ANOVA can also use the pairwise t-tests to examine the phenotypic difference between two particular genotypes. This pairwise t-test is used to evaluate H0: μj = μm vs. Ha: μj ≠ μm for pairs of genotypes j and m (e.g., j = 1 and m = 2, or j = 1 and m = 3, or j = 2 and m = 3). The T-statistic is calculated as
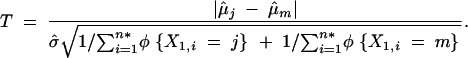 |
13 |
The t-test would reject H0 (therefore declare a phenotypic difference between genotypes j and m) when T > tα/2;n*−3, where tα/2;n*−3 is the (1 − α/2)100th percentile of a t-distribution with d.f. = (n* − 3).
As pointed out above, the power of the ordinary ANOVA is not optimal because it does not use information for those data records with either phenotype or genotype marker data missing. In the previous section, we proposed using the EM algorithm to incorporate information from the flanking loci (i.e., B and C) in the parameter estimation. Here we describe how to use these EM-based parameter estimates to develop a statistical test that replaces the corresponding F-test (or the pairwise t-test when applicable) in the ordinary ANOVA.
Basically, the F-test in the ordinary ANOVA is replaced by the LRT in the EM approach as follows: (a) use the EM algorithm of (6)–(11) to find the parameter estimate θ̂, and then compute the log-likelihood Lθ̂ according to (1); (b) fit the parameters again under H0 (by the EM algorithm with formulas described in the next paragraph) to yield an estimate θ̂0, and compute the log-likelihood Lθ̂0; and (c) compute the likelihood-ratio statistic (LRS),
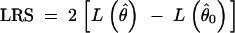 |
14 |
The LRT will reject H0 if LRS > χ2α, where χ2α is the (1 − α)100th percentile of the χ2-distribution with d.f. = 1.
The calculation of the LRS according to Equation 14 requires the calculations of both the maximum log-likelihood Lθ̂ under Ha and the maximum log-likelihood Lθ̂0 under H0. We have provided in the previous section EM formulas for fitting θ̂ in Equations 7–11. Here we describe EM formulas for fitting the parameters θ̂0 under H0. The EM algorithm under H0 is simpler because μ1 = μ2 = μ3 = μ. Therefore, we would estimate μ by the overall sample mean under H0. Correspondingly, the variance is estimated by the sample variance. That is, we can get the estimates without going through any iterations:
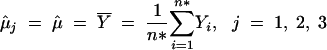 |
10′ |
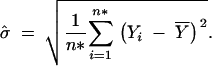 |
11′ |
Thus, for estimating pj,k,l's, we need to iterate only between the E-step,
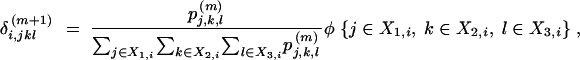 |
8′ |
and the M-step,
 |
9′ |
The estimate θ̂0 consists of μ̂j in (10′), σ̂ in (11′), and p̂j,k,l's that are the values of (9′) at convergence. Then θ̂0 is plugged into Equation 1 to calculate Lθ̂0, which is then used to compute the LRS in (14).
The pairwise t-test in the ordinary ANOVA is replaced by a corresponding adjusted t-test in the EM approach. Since μ̂j − μ̂m = ∑i∈obs(Y)Yi∑k,l(δi,jkl/∑i∈obs(Y)∑k,lδi,jkl − δi,mkl/∑i∈obs(Y)∑k,lδi,mkl), the variance of μ̂j − μ̂m is approximately ∑i∈obs(Y)[∑k,l(δi,jkl/∑i∈obs(Y)∑k,lδi,jkl − δi,mkl/∑i∈obs(Y)∑k,lδi,mkl)]2σ̂2. The adjusted t-test statistic, T, for testing the pair of genotypes j and m is
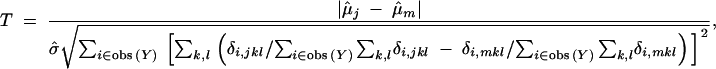 |
15 |
where μ̂j, μ̂m, and σ̂ are from the EM estimate, θ̂. The t-test would reject H0 when T > tα/2;n−30, where tα/2;n−30 is the (1 − α/2)100th percentile of a t-distribution with d.f. = (n − 30).
As the proportion of missing data increases, but is kept below the upper limit such that the type I error is not inflated, we would expect the EM-LRT to perform better than the ANOVA-based test in the single-marker analysis.
Comparison with the interval-mapping method:
The proposed EM-LRT above uses the genotype information at flanking marker loci to allow more efficient QTL detection at the trait locus when there are missing genotype or phenotype data. The idea of using genotype information at flanking marker loci for capturing information of incomplete data is similar to the idea adopted by the interval-mapping method (Lander and Botstein 1989). The interval-mapping method also uses the EM algorithm to incorporate flanking markers' genotype information for inferring the association (expressed as a LOD score) of the phenotypic trait with genetic variation at any given point between the two flanking markers, but there is a significant difference between our method and the interval-mapping method. First, the main strategy is different. Our method is exactly a single-marker test when no data are missing, and it uses information of the flanking markers only when data are missing at the marker of interest; in contrast, the interval-mapping method intends to “screen” any given point, locus X, in the interval bracketed by two linked markers, assuming (a) genotypic variation at such theoretical point exists and (b) its recombination rates from the two flanking markers are correctly specified. Therefore, the trait locus X is a putative locus and is totally unobserved, and the interval-mapping method uses recombination rates, rB and rC, to compute the conditional probabilities 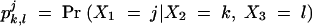 , thus reducing the number of parameters to 2. However, such reduction of the number of parameters is valid only if the underlying assumptions regarding the recombination rates (i.e., rB and rC in Figure 1) hold. Our proposed EM-LRT, on the other hand, makes no assumptions on the recombination rates (i.e., rB and rC), but instead it computes pjk,l through pj,k,l = Pr(X1 = j, X2 = k, X3 = l), only if there are some incomplete phenotype data or genotype data at locus A (Figure 1). For convenience of mathematical derivation, we have written our formula in terms of pj,k,l. Hence our EM-LRT involves 27 pj,k,l's and we did not reduce them to two parameters, rB and rC, which are used in interval-mapping methods. However, the trade-off is that our EM-LRT is more generic with no model assumptions on the specification of recombination rates: for example, for very tightly linked markers, it has been shown that the rate of recombination is no longer a monotone function of the physical distance (Thompson et al. 1988), and the assumption of the interval-mapping method would appear to be overly strong. Under such circumstances, when there are missing data, our EM-LRT is still valid. We therefore consider our EM-LRT as a complimentary method for the interval-mapping method, particularly when markers are very densely spaced (<1 cM).
, thus reducing the number of parameters to 2. However, such reduction of the number of parameters is valid only if the underlying assumptions regarding the recombination rates (i.e., rB and rC in Figure 1) hold. Our proposed EM-LRT, on the other hand, makes no assumptions on the recombination rates (i.e., rB and rC), but instead it computes pjk,l through pj,k,l = Pr(X1 = j, X2 = k, X3 = l), only if there are some incomplete phenotype data or genotype data at locus A (Figure 1). For convenience of mathematical derivation, we have written our formula in terms of pj,k,l. Hence our EM-LRT involves 27 pj,k,l's and we did not reduce them to two parameters, rB and rC, which are used in interval-mapping methods. However, the trade-off is that our EM-LRT is more generic with no model assumptions on the specification of recombination rates: for example, for very tightly linked markers, it has been shown that the rate of recombination is no longer a monotone function of the physical distance (Thompson et al. 1988), and the assumption of the interval-mapping method would appear to be overly strong. Under such circumstances, when there are missing data, our EM-LRT is still valid. We therefore consider our EM-LRT as a complimentary method for the interval-mapping method, particularly when markers are very densely spaced (<1 cM).
Figure 1.—

A schematic illustrating (a) EM-LRT and (b) the interval-mapping method. The shaded inverted triangles indicate observed markers, the open inverted triangle indicates the putative locus.
RESULTS
Assessment of the validity of EM-LRT in finite samples:
EM-LRT is a valid test asymptotically; however, its validity for finite sample sizes needs to be carefully checked. We used extensive simulations to assess the validity of the EM-LRT for various sample sizes under various proportions of missing data.
We simulated a data set of n animals with the phenotype measurement (Yi) and three genetic markers (X1,i, X2,i, X3,i) for each animal i: {(Yi, X1,i, X2,i, X3,i)}, where i = 1, … , n. The phenotype Y for each animal was generated according to the linear model: Equation 1, with parameters μ1 = μ2 = μ3 = 100 and σ = 10. We assigned pj,k,l to be proportional to (4 − j) + (4 − k) + (4 − l). [We initially intended to simulate pj,k,l proportional to j + k + l. However, as j = 1 denotes the homozygous wild-type genotype, it should have higher probability than j = 3. Hence, we used the transformations (4 − j) to flip the probabilities.] We then randomly dropped phenotype observations at the trait marker locus A according to a missing probability.
For each data set, we first fitted the EM estimates θ̂ through iterations of Equations 6–11. The iteration started with the initial estimates:
 |
The iteration would stop when a convergence criterion of 10−4 relative change was met. Next, we fitted the EM estimates θ̂0 again under H0 through Equations 8′–11′. Then θ̂ and θ̂0 were used in computing the LRS in (14).
We repeatedly ran the simulation 1000 times. For each simulated data set, we computed the EM-LRT (14) and recorded their values. The empirical type I error of EM-LRT was calculated as the proportions of the 1000 data sets where H0 was rejected at the significance level α = 0.05.
We simulated for n = 50, 100, 200, 500, and 1000, respectively. For each sample size of n, we increased the missing probability from 10% upward, until the type I error exceeds the nominal significance level α = 0.05 significantly (that is, it exceeds by two standard deviations, 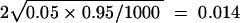 ). Table 1 shows the type I error for EM-LRT for various sample sizes.
). Table 1 shows the type I error for EM-LRT for various sample sizes.
TABLE 1.
The empirical type I error of EM-LRT over 1000 simulations
| Proportion of missing genotype
|
|||||||
|---|---|---|---|---|---|---|---|
| Sample size (n) | 10% | 20% | 30% | 40% | 50% | 60% | 70% |
| 50 | 0.059 | 0.068 | — | — | — | — | — |
| 100 | 0.043 | 0.055 | 0.072 | — | — | — | — |
| 200 | 0.046 | 0.055 | 0.061 | 0.060 | 0.060 | 0.086 | — |
| 500 | 0.054 | 0.055 | 0.049 | 0.048 | 0.052 | 0.058 | 0.088 |
| 1000 | 0.060 | 0.048 | 0.056 | 0.051 | 0.048 | 0.046 | 0.081 |
Note that the type I error calculations were made only at those proportions of missing data when the EM-LRT remains valid or when the type I error starts to be inflated.
As shown in Table 1, for a small sample size (n = 50), the EM-LRT is valid for up to 10% missing observations. When n = 100, the EM-LRT is valid when as much as 20% data were missing. When n = 200, the EM-LRT can tolerate up to 50% missing data. These simulations showed that we have to be careful in applying the EM-LRT. For a small sample (e.g., n = 40), which is often encountered in real-world experiments, the type I error rates were 0.060 and 0.077 for 10 and 20% missing, respectively. Thus, for n = 40 (see the real example in III shown below), we can still use EM-LRT if 10% or fewer observations are missing. When there are ≥200 animals, we can use the tests with up to half of all observations missing.
To evaluate the accuracy of parameter estimates, we calculated the coefficient of variability (CV) for each model parameter estimate. CV is conventionally defined as 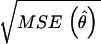 /θ, where MSE(θ̂) denotes the mean squared error of the estimate for parameter θ over 1000 simulation runs. Table 2 shows the average CV for estimates of pj,k,l's, μj's, and σ2. (It turned out the CVs for estimates of pj,k,l's were rather similar and thus we presented only their average values.)
/θ, where MSE(θ̂) denotes the mean squared error of the estimate for parameter θ over 1000 simulation runs. Table 2 shows the average CV for estimates of pj,k,l's, μj's, and σ2. (It turned out the CVs for estimates of pj,k,l's were rather similar and thus we presented only their average values.)
TABLE 2.
The average CVs for the parameter estimates of EM-LRT over 1000 simulations
| Proportion of missing genotype
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Sample size (n) |
Parameters of interest |
10% | 20% | 30% | 40% | 50% | 60% | 70% |
| 50 | pj,k,l | 0.767 | 0.812 | — | — | — | — | — |
| 50 | μj | 0.023 | 0.025 | — | — | — | — | — |
| 50 | σ2 | 0.106 | 0.110 | — | — | — | — | — |
| 100 | pj,k,l | 0.536 | 0.571 | 0.603 | — | — | — | — |
| 100 | μj | 0.018 | 0.019 | 0.020 | — | — | — | — |
| 100 | σ2 | 0.071 | 0.073 | 0.074 | — | — | — | — |
| 200 | pj,k,l | 0.386 | 0.402 | 0.423 | 0.453 | 0.487 | 0.541 | — |
| 200 | μj | 0.013 | 0.013 | 0.014 | 0.015 | 0.016 | 0.019 | — |
| 200 | σ2 | 0.051 | 0.051 | 0.052 | 0.053 | 0.053 | 0.055 | — |
| 500 | pj,k,l | 0.241 | 0.253 | 0.267 | 0.285 | 0.306 | 0.336 | 0.387 |
| 500 | μj | 0.008 | 0.008 | 0.009 | 0.009 | 0.010 | 0.011 | 0.014 |
| 500 | σ2 | 0.032 | 0.031 | 0.031 | 0.032 | 0.032 | 0.033 | 0.033 |
| 1000 | pj,k,l | 0.172 | 0.180 | 0.188 | 0.198 | 0.216 | 0.239 | 0.269 |
| 1000 | μj | 0.006 | 0.006 | 0.006 | 0.007 | 0.007 | 0.008 | 0.009 |
| 1000 | σ2 | 0.021 | 0.023 | 0.022 | 0.022 | 0.023 | 0.023 | 0.023 |
Note that the average CVs for the parameter estimates were calculated only at those proportions of missing data when the EM-LRT remains valid or when the type I error starts to be inflated.
It can be seen that the ancillary parameters pj,k,l were estimated less accurately compared to the estimates of the main parameters μj and σ2 across the board. However, because pj,k,l's are parameters that are used only in the adjustment of the impacts of the missing data on the main parameters, the main parameters of interest (i.e., μj's and σ2) were not much affected by the accuracies of the estimates of pj,k,l's. All parameters were estimated more accurately when the sample size n became larger. As a result, the EM-LRT is a valid test for increasingly greater missing proportions as n becomes larger.
Power comparison of EM-LRT with ANOVA-based tests:
To compare the power of EM-LRT with that of the ANOVA-based test, we conducted simulation studies using two types of phylogenetic models.
Simulation models:
In the simulations performed, genetic markers were generated according to two phylogenetic models (Figure 2). Let A, B, and C denote the wild-type alleles and a, b, c their corresponding mutant alleles for the three loci, A, B, and C, respectively. We assume that the A → a event has arisen before either B → b or C → c occurred, and B → b or C → c events occurred only on the aBC haplotype. In model I, the B → b took place first on the ancestral haplotype aBC, followed by the mutation of locus C on the haplotype abC, resulting in four distinctive haplotypes: ABC, aBC, abC, and abc. In model II, the mutation at locus B took place first on the ancestral haplotype aBC, followed by the mutation of locus C on the haplotypes bearing either the wild-type allele (i.e., aBC) or the mutant allele (i.e., abC) at locus B, resulting in five distinctive haplotypes: ABC, aBC, abC, aBc, and abc.
Figure 2.—

Two phylogenetic models for three linked loci (i.e., A, B, and C) residing on the same chromosome, all starting from an ancestral haplotype aBC, which arose from its founder haplotype, ABC (i.e., A → a is the most ancestral event, and B → b or C → c took place only on the aBC haplotype). In model I, the B → b event took place first on the ancestral haplotype ABC, followed by the the C → c event occurring on the AbC haplotype only, resulting in four distinct haplotypes. In model II, the mutation at locus B took place first on the ancestral haplotype ABC, followed by the mutation of locus C on the haplotypes bearing either the wild-type allele (i.e., ABC) or the mutant allele (i.e., AbC) at locus B, resulting in five distinctive haplotypes. Open circles, aBC; shaded circles, AbC; hatched circles, aBc; solid circles, abC.
In model I, we assume that C → c occurred only on the abC haplotype, as shown in Figure 2. Let pa denote the proportion of the “a” allele in the population, pb denote the probability of the B → b event conditional on the A → a event, and pc denote the probability of the C → c event conditional on the B → b event. Two variants of model I were considered:
model IA: the genotype measures X1, X2, and X3 refer to loci A, B, and C, respectively (e.g., genotype “aaBbCC” corresponds to X1 = 3, X2 = 2, X3 = 1);
model IB: the genotype measures X1, X2, and X3 refer to loci B, A, and C, respectively (e.g., genotype aaBbCC now corresponds to X1 = 2, X2 = 3, X3 = 1).
In model II, we considered the case where B → b and C → c events were independent (see Figure 2); without loss of generality, we assume that B → b occurred before C → c. Under this model, pa and pb were defined similarly as we defined in model I, but pc is defined as the probability of the C → c event conditional on the A → a event.
In our simulations, we considered the following parameter settings for pa, pb, and pc: pb = pc = 0.8, and pa of values 0.1, 0.2, and 0.4. For example, pa = 0.2 would mean that the a allele is present in 20% of the population, and hence ∼32% of the animals have the genotype Aa and 4% have the genotype aa.
Simulation and fitting procedures:
For these models, we simulated for n = 200: {(Yi, X1,i, X2,i, X3,i)}, where i = 1, … , n. The phenotype Y for each animal is again generated according to the linear model—Equation 1, with parameters μ1 = 100 − Δ, μ2 = 100, μ3 = 100 + Δ, and σ = 10. Here we randomly dropped values from each variable with a probability, pm. We conducted simulations under two scenarios: (a) pm = 10% and (b) pm = 20%. Note that in our simulations used for assessing the validity of EM-LRT in finite samples, the missing proportion refers to the missing probability of X1. Here, pm refers to the missing probability of all variables, Y, X1, X2, and X3. The validity of the EM-LRT for the simulation used here was verified by checking the values of the empirical type I error rates (i.e., when Δ = 0).
For each model setting, we repeatedly ran the simulation 1000 times. For each simulated data set, we computed the EM-LRT (14) and ANOVA F-test (12) statistics and recorded their values. The empirical powers of the EM-LRT and F-test were calculated as the proportions of data sets where H0 was rejected at the significance level α = 0.05. Figures 3 and 4 display the empirical powers from the 1000 simulation runs for pa = 0.2 and 0.4, respectively. The statistical powers were calculated and compared for all three models (IA, IB, and II), for various values of Δ and for different missing probabilities (10 and 20% on the left-hand and right-hand sides, respectively for Figures 3–5). The simulated Δ values were defined as Kσ/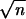 , where K = 0, 1, 2, … For the simulation runs with pa = 0.1 (Figure 5), we replaced the comparison between EM-LRT (14) and ANOVA F-test (12) with the comparison between the EM-adjusted t-test (15) and the ANOVA t-test (13) for the following reason: when the minor allele (a) frequency is low (pa = 0.1), it would be expected that only ∼1% of animals would carry the aa genotype. Since a total of 200 animals were in each simulation, there were on average <2 animals with the aa genotype in most simulated data sets. In many simulation runs, there was not a single observation in the aa genotype group. Therefore, in this case, the phenotypic comparison is needed only between the pair of genotypes AA and Aa, with respective mean values denoted as μ1 and μ2. It was thus more appropriate to compare the power of the EM-adjusted t-test (15) with that of the ordinary ANOVA t-test (13).
, where K = 0, 1, 2, … For the simulation runs with pa = 0.1 (Figure 5), we replaced the comparison between EM-LRT (14) and ANOVA F-test (12) with the comparison between the EM-adjusted t-test (15) and the ANOVA t-test (13) for the following reason: when the minor allele (a) frequency is low (pa = 0.1), it would be expected that only ∼1% of animals would carry the aa genotype. Since a total of 200 animals were in each simulation, there were on average <2 animals with the aa genotype in most simulated data sets. In many simulation runs, there was not a single observation in the aa genotype group. Therefore, in this case, the phenotypic comparison is needed only between the pair of genotypes AA and Aa, with respective mean values denoted as μ1 and μ2. It was thus more appropriate to compare the power of the EM-adjusted t-test (15) with that of the ordinary ANOVA t-test (13).
Figure 3.—

Power estimation and comparison of the EM-LRT and ANOVA F-test when P(A → a) = 20%. The points plotted indicate the empirical proportion of tests (by use of a nominal level α = 0.05) that rejected the H0 among 1000 simulated data sets. K = Δ/σ/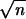 . Plots on the left correspond to cases with 10% missing data. Plots on the right correspond to cases with 20% missing data. * indicates cases where P < 0.05, and ** indicates cases where P < 0.005. Here “P” refers to the P-value of Wilcoxon rank-sum tests comparing the power difference between the EM-LRT and the F-test. Solid diamonds denote the power of the EM-LRT; solid squares denote the power of the ANOVA F-test.
. Plots on the left correspond to cases with 10% missing data. Plots on the right correspond to cases with 20% missing data. * indicates cases where P < 0.05, and ** indicates cases where P < 0.005. Here “P” refers to the P-value of Wilcoxon rank-sum tests comparing the power difference between the EM-LRT and the F-test. Solid diamonds denote the power of the EM-LRT; solid squares denote the power of the ANOVA F-test.
Figure 4.—

Power estimation and comparison of the EM-LRT and ANOVA F-test when P(A → a) = 40%. The points plotted indicate the empirical proportion of tests (by use of a nominal level α = 0.05) that rejected the H0 among 1000 simulated data sets. K = Δ/σ/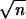 . Plots on the left correspond to cases with 10% missing data. Plots on the right correspond to cases with 20% missing data. * indicates those cases where P < 0.05, and ** indicates those cases where P < 0.005. Here “P” refers to the P-value of Wilcoxon rank-sum tests comparing the power difference between the EM-LRT and the F-test. Solid diamonds denote the power of the EM-LRT; solid squares denote the power of the ANOVA F-test.
. Plots on the left correspond to cases with 10% missing data. Plots on the right correspond to cases with 20% missing data. * indicates those cases where P < 0.05, and ** indicates those cases where P < 0.005. Here “P” refers to the P-value of Wilcoxon rank-sum tests comparing the power difference between the EM-LRT and the F-test. Solid diamonds denote the power of the EM-LRT; solid squares denote the power of the ANOVA F-test.
Figure 5.—

Power estimation and comparison of the EM-adjusted t-test and the ordinary t-test when P(A → a) = 10%. The points plotted indicate the empirical proportion of tests (by use of a nominal level α = 0.05) that rejected the H0 among 1000 simulated data sets. K = Δ/(σ/n). Plots on the left correspond to cases with 10% missing data. Plots on the right correspond to cases with 20% missing data. * indicates those cases where P < 0.05, and ** indicates those cases where P < 0.005. Here “P” refers to the P-value of Wilcoxon rank-sum tests comparing the power difference between the EM-adjusted t-test and the ordinary t-test. Solid diamonds denote the power of the EM-adjusted t-test; solid squares denote the power of the ordinary t-test.
Power comparisons:
For a hypothesis test, a type I error occurs if H0 is rejected when it is true. If H0 holds, a correct test should have a type I error rate ≤ α. The H0 in this case was represented by Δ = 0 (or equivalently, K = 0) or the left-most case in Figures 3–5. It can be seen that in those cases the empirical type I error rates for both the EM-LRT and the ANOVA F-test were close to α = 0.05, confirming that they were both valid tests.
The power of a test is defined as one minus the type II error. Among valid tests with correct type I error rates, it is clear that a test with a higher power is preferred. It can be seen from Figures 3 and 4 that the empirical powers of EM-LRT were higher than the empirical powers of the F-test. Due to simulation variations, however, a higher empirical power does not necessarily mean the real power is higher. To see whether the difference in power is statistically significant, we conducted a pairwise nonparametric test (the Wilcoxon rank-sum test) on the 1000 pairs of P-values for EM-LRTs and F-tests. The cases where the powers of EM-LRTs are statistically significantly higher are indicated by asterisks in the figures.
As illustrated in Figures 3 and 4, when pm = 10%, the power of the EM-LRT was significantly higher than that of F-test when K > 3 for models IA, IB, and II. And when pm = 20%, the EM-LRT started to outperform the F-test when K = 2. Not surprisingly, the power improvement of EM-LRT over the F-tests became more significant when more data were missing.
The comparison results shown in Figure 5 were similar to those of Figures 3 and 4: when 10% of data were missing, the EM-adjusted t-test started to significantly outperform the ordinary ANOVA t-test for K = 3 or 4; when 20% of data were missing, the better performance started when K = 2.
Application to a real data set in experimental crosses:
As an illustration, we applied the proposed method to a real data set based on an F2 intercross study. This data set, based on a previously published report (Rosen and Williams 2001), consisted of a total of 36 mice from an F2 intercross between a strain with low brain weight (A/J) and a strain with high brain weight (BXD5). Brain volume, striatal volume, striatal neuron number, striatal neuron number residual, striatal volume residual, and brain weight were measured using standard procedures. We studied a total of 13 microsatellite markers—9 markers on chromosome 10 (D10Mit106, D10Mit3, D10Mit194, D10Mit61, D10Mit186, D10Mit266, D10Mit233, D10Mit179, and D10Mit180), and 4 markers on chromosome 18 (D18Mit20, D18Mit120, D18Mit122, and D18Mit184). The map locations of the loci studied were obtained from Ensembl (http://www.ensembl.org/Mus_musculus/).
The P-values of both the ANOVA F-test and EM-LRT are displayed in Tables 2 and 3.
TABLE 3.
TheP-values of the ANOVAF-test for associations between the phenotypes and genetic markers for the mouse data
| Phenotype
|
||||||
|---|---|---|---|---|---|---|
| Genetic marker | Brain volume |
Striatal volume |
Striatal neuron no. |
Striatal neuron no. residual |
Striatal volume residual |
Brain weight |
| D10Mit106 | 0.1506 | 0.0055 | 0.2186 | 0.8832 | 0.0441 | 0.0240 |
| D10Mit3 | 0.2361 | 0.0781 | 0.4253 | 0.4546 | 0.0568 | 0.0261 |
| D10Mit194 | 0.4302 | 0.0135 | 0.4712 | 0.3219 | 0.1759 | 0.0229 |
| D10Mit61 | 0.0555 | 0.0020 | 0.2007 | 0.1048 | 0.2857 | 0.0118 |
| D10Mit186 | 0.0062 | 0.0004 | 0.0225 | 0.0062 | 0.2073 | 0.0037 |
| D10Mit266 | 0.0640 | 0.0029 | 0.1382 | 0.2667 | 0.0754 | 0.0438 |
| D10Mit233 | 0.0749 | 0.0032 | 0.1314 | 0.2031 | 0.0553 | 0.0290 |
| D10Mit179 | 0.1523 | 0.0463 | 0.4410 | 0.5620 | 0.4758 | 0.1031 |
| D10Mit180 | 0.1185 | 0.0521 | 0.5966 | 0.6470 | 0.7244 | 0.0697 |
| D18Mit20 | 0.2476 | 0.0955 | 0.1081 | 0.0837 | 0.3491 | 0.0012 |
| D18Mit120 | 0.5902 | 0.3389 | 0.9843 | 0.4053 | 0.2581 | 0.0037 |
| D18Mit122 | 0.2092 | 0.2850 | 0.4006 | 0.2872 | 0.8266 | 0.0208 |
| D18Mit184 | 0.6908 | 0.4677 | 0.2811 | 0.1631 | 0.8803 | 0.0904 |
P-values <0.01 are in italics.
Since few missing observations were present in the data, the differences in P-values were very small between the ANOVA F-tests (Table 3) and the EM-LRT (Table 4). Both methods showed that D10Mit186 affects most phenotypes in the study. Also, two markers on chromosome 18, D18Mit20 and D18Mit120, significantly affect brain weight.
TABLE 4.
TheP-values of EM-LRT for associations between the phenotypes and genetic markers for the mouse data
| Phenotype
|
||||||
|---|---|---|---|---|---|---|
| Genetic marker |
Brain volume |
Striatal volume |
Striatal neuron no. |
Striatal neuron no. residual |
Striatal volume residual |
Brain weight |
| D10Mit106 | 0.1268 | 0.0035 | 0.1904 | 0.8733 | 0.0332 | 0.0171 |
| D10Mit3 | 0.2057 | 0.0568 | 0.3822 | 0.5158 | 0.077 | 0.0232 |
| D10Mit194 | 0.3488 | 0.0101 | 0.5458 | 0.3501 | 0.2025 | 0.0123 |
| D10Mit61 | 0.0426 | 0.0012 | 0.1735 | 0.0854 | 0.2549 | 0.0079 |
| D10Mit186 | 0.0039 | 0.0002 | 0.0160 | 0.0039 | 0.1796 | 0.0022 |
| D10Mit266 | 0.0391 | 0.0012 | 0.1067 | 0.2279 | 0.0492 | 0.0261 |
| D10Mit233 | 0.0536 | 0.0016 | 0.1003 | 0.1952 | 0.0339 | 0.0197 |
| D10Mit179 | 0.1284 | 0.0350 | 0.4093 | 0.5334 | 0.4448 | 0.0838 |
| D10Mit180 | 0.1284 | 0.0350 | 0.4093 | 0.5334 | 0.4448 | 0.0838 |
| D18Mit20 | 0.4458 | 0.1223 | 0.2379 | 0.1652 | 0.3077 | 0.0010 |
| D18Mit120 | 0.5360 | 0.3015 | 0.9819 | 0.3581 | 0.1829 | 0.0017 |
| D18Mit122 | 0.2882 | 0.2364 | 0.4726 | 0.3259 | 0.8005 | 0.0125 |
| D18Mit184 | 0.8812 | 0.2694 | 0.4598 | 0.1603 | 0.9425 | 0.0281 |
P-values <0.01 are in italics.
To illustrate the effects of missing genotype observations, we randomly dropped 10% of the genotype observations at the interested locus and recalculated the P-values of the ANOVA and EM-LRT. Table 5 presents the P-values of the ANOVA F-test and EM-LRT for all phenotypes of interest with and without the dropped D10Mit186 genotype data. Similarly, Table 6 presents the P-values of the ANOVA F-test and EM-LRT for brain weight with and without the dropped D18Mit20 and D18Mit120 genotype data.
TABLE 5.
TheP-values of the ANOVAF-test and the EM-LRT for associations between the phenotypes and genetic markerD10Mit186 for the mouse data with various proportions of missing genotype data
| Proportion of missing D10Mit186 genotype |
|||
|---|---|---|---|
| Phenotype | Estimation method |
0% | 10% |
| Brain volume | ANOVA F-test | 0.0062 | 0.0014 |
| EM-LRT | 0.0039 | 0.0032 | |
| Striatal volume | ANOVA F-test | 0.0003 | 0.0180 |
| EM-LRT | 0.0002 | 0.0001 | |
| Striatal neuron no. | ANOVA F-test | 0.0225 | 0.0231 |
| EM-LRT | 0.0159 | 0.0165 | |
| Striatal neuron no. | ANOVA F-test | 0.0062 | 0.0107 |
| Residual | EM-LRT | 0.0039 | 0.0039 |
| Striatal volume | ANOVA F-test | 0.2072 | 0.3731 |
| Residual | EM-LRT | 0.1796 | 0.1664 |
| Brain weight | ANOVA F-test | 0.0036 | 0.0004 |
| EM-LRT | 0.0022 | 0.0014 | |
P-values <0.01 are in italics.
TABLE 6.
TheP-values of the ANOVAF-test and the EM-LRT for association between brain weight and genetic markersD18Mit20 andD18Mit120 for the mouse data with various proportions of missing genotype data
| Proportion of missing genotype |
|||
|---|---|---|---|
| Genetic marker |
Estimation method |
0% | 10% |
| D18Mit20 | ANOVA F-test | 0.0011 | 0.0166 |
| EM-LRT | 0.0010 | 0.0030 | |
| D18Mit120 | ANOVA F-test | 0.0037 | 0.0261 |
| EM-LRT | 0.0017 | 0.0011 | |
P-values <0.01 are in italics.
As we can see from these tables, P-values for the ANOVA F-tests were more sensitive to the dropped phenotype data than were those for the EM-LRT. For example, in Table 6, the ANOVA tests are no longer able to detect the association at the α = 0.01 level with brain weight when 10% of genotype observations at the interested locus were dropped while the EM-LRT can still detect the association under the same condition. On the other hand, as shown in Table 5, the effect of dropping 10% D10Mit186 genotype data is less pronounced. The results produced by ANOVA tests led to the same conclusions on the associations of the D10Mit186 genotype with all the phenotypes except the striatal neuron number residual. The ANOVA test was not able to detect the association between the D10Mit186 genotype and the striatal neuron number residual when 10% of data were missing while the EM-LRT could still detect the association. By and large, we see that the EM-LRT improves the statistical power over the case when all missing data were excluded.
DISCUSSION
In this article, we presented an EM-LRT using flanking markers information in single-marker analysis to utilize information contained in incomplete data. By using both simulated and real data sets, we demonstrated that EM-LRT utilizing incomplete data is a valid test for finite samples with moderate proportions of missing values and is a more powerful test compared to ordinary ANOVA-based tests that discarded all missing data from the analysis.
Missing information on either genotype or phenotype can obscure the true genetic effect (Sen and Churchill 2001). To reduce the proportion of missing data, the best solution is to repeat the experiment, but it can be costly and time-consuming. The EM algorithm is a standard maximum-likelihood estimation method for handling missing data (Dempster et al. 1977). In the present context, the method fractionally assigns (E-step) the incomplete data to their theoretically possible values on the basis of the current estimates of the parameters and then revises the parameter estimates to maximize (the M-step) the likelihood on the basis of the pseudo-complete data. This two-step, alternating iteration procedure is repeated until convergence can be reached. Statistical theory guarantees that the observed data likelihood increases to a maximum via the algorithm, and thus the EM-LRT can be performed validly (Dempster et al. 1977). Likelihood methods with the EM algorithm allow the recovery of much of the lost information and make statistically efficient use of the data. In the simulated data sets, the EM-LRT outperforms the ANOVA-based tests at various marker allele frequencies, and the differences in statistical power became increasingly more pronounced with an increasing portion of missing data or an increasing value of Δ (Figures 3–5). In the real data set example on inbred mouse strains, we found that with 10% missing data the significant associations of D18Mit20 and D18Mit120 with brain weight could still be detected by EM-LRT, but not by ANOVA-based tests. Taken together, we argue that the EM-LRT is an attractive statistical method that can utilize information from incomplete data.
The EM-LRT is a valid test asymptotically (i.e., a large n). For finite samples, our simulations indicated that, for n = 100, the method can tolerate up to 20% missing genotype data; for n = 200, the method can tolerate up to 50% missing genotype data. Thus, there is another potential application of the proposed EM-LRT for a combined analysis of different studies. For example, suppose in study I (with a sample size of n1) that we already collected phenotype data and genotype data on D10Mit61 and D10Mit266, and later we decide to study other nearby genetic markers, say D10Mit186 as well as D10Mit61 and D10Mit266 in a new, independent study, study II (with a sample size of n2). We might combine study I with study II by treating the D10Mit186 genotype data as missing in study I, and then the EM-LRT can be used to detect the association between the phenotype of interest and D10Mit186 by merging studies I and II together (with a sample size of n1 + n2). When we use this approach to combine different studies, we have to pay particular attention to the assumption of “missing at random.” That is, the genotype missing probability is not related to the phenotype value. This can be ensured by checking that the animals in different studies come from exactly the same genetic backgrounds (e.g., common F0 parents) under the same experimental and breeding conditions. The tests developed in this article can be applied to the combined (studies I and II altogether) data provided that each of the new markers selected is independent from study I. In other words, the new genetic marker is not selected because the flanking markers already showed associations with the phenotype in study I. If the new genetic marker is selected because of an association observed in regard to the flanking markers in study I, then a sequential design is needed. How to adjust our tests for the sequential design is an interesting research topic that deserves further investigation.
Acknowledgments
We are grateful to the two anonymous reviewers for their comments and suggestions. We thank Glenn D. Rosen at the Beth Israel Deaconess Medical Center, Harvard Medical School for providing the mouse inbred strain data.
References
- Dempster, A. P., N. M. Laird and D. B. Rubin, 1977. Maximum-likelihood estimation from incomplete data via the EM algorithm. J. R. Stat. Soc. Ser. B 39: 1–38. [Google Scholar]
- Ihaka, R., and R. Gentleman, 1996. R: a language for data analysis and graphics. J. Comp. Graph. Stat. 5: 299–314. [Google Scholar]
- Knoblauch, M., and K. Lindpaintner, 1999. Use of animal models to search for candidate genes associated with essential hypertension. Curr. Hypertens. Rep. 1: 25–30. [DOI] [PubMed] [Google Scholar]
- Lander, E. S., and D. Botstein, 1989. Mapping Mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics 121: 185–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little, R. J. A., and D. B. Rubin, 1987 Statistical Analyses With Missing Data. Wiley, New York.
- Luo, Z. W., S. H. Tao and Z-B. Zeng, 2000. Inferring linkage disequilibrium between a polymorphic marker locus and a trait locus in natural populations. Genetics. 156: 457–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLachlan, G. J., and T. Krishnan, 1997 The EM Algorithm and Extensions. Wiley, New York.
- Poyan Mehr, A., A. K. Siegel, P. Kossmehl, A. Schulz, R. Plehm et al., 2003. Early onset albuminuria in Dahl rats is a polygenetic trait that is independent from salt loading. Physiol. Genomics 14: 209–216. [DOI] [PubMed] [Google Scholar]
- Rosen, G. D., and R. W. Williams, 2001. Complex trait analysis of the mouse striatum: independent QTLs modulate volume and neuron number. BMC Neurosci. 2: 5–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubattu, S., M. Volpe, R. Kreutz, U. Ganten, D. Ganten et al., 1996. Chromosomal mapping of quantitative trait loci contributing to stroke in a rat model of complex human disease. Nat. Genet. 13: 429–434. [DOI] [PubMed] [Google Scholar]
- Sen, S., and G. A. Churchill, 2001. A statistical framework for quantitative trait mapping. Genetics 159: 371–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, E. A., S. Deeb, D. Walker and A. G. Motulsky, 1988. The detection of linkage disequilibrium between closely linked markers: RFLPs at the AI-CIII apolipoprotein genes. Am. J. Hum. Genet. 42: 113–124. [PMC free article] [PubMed] [Google Scholar]
- Vallejo, R. L., L. D. Bacon, H. C. Liu, R. L. Witter, M. A. Groenen et al., 1998. Genetic mapping of quantitative trait loci affecting susceptibility to Marek's disease virus induced tumors in F2 intercross chickens. Genetics 148: 349–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, J., and J. Meng, 2003. Genetic analysis of loci associated with partial resistance to Sclerotinia sclerotiorum in rapeseed (Brassica napus L.). Theor. Appl. Genet. 106: 759–764. [DOI] [PubMed] [Google Scholar]