

Abstract
Hsf1p, the heat-shock transcription factor from Saccharomyces cerevisiae, has a low level of constitutive transcriptional activity and is kept in this state through negative regulation. In an effort to understand this negative regulation, we developed a novel genetic selection that detects altered expression from the HSP26 promoter. Using this reporter strain, we identified mutations and dosage compensators in the Ras/cAMP signaling pathway that decrease cAMP levels and increase expression from the HSP26 promoter. In yeast, low cAMP levels reduce the catalytic activity of the cAMP-dependent kinase PKA. Previous studies had proposed that the stress response transcription factors Msn2p/4p, but not Hsf1p, are repressed by PKA. However, we found that reduction or elimination of PKA activity strongly derepresses transcription of the small heat-shock genes HSP26 and HSP12, even in the absence of MSN2/4. In a strain deleted for MSN2/4 and the PKA catalytic subunits, expression of HSP12 and HSP26 depends on HSF1 expression. Our findings indicate that Hsf1p functions downstream of PKA and suggest that PKA might be involved in negative regulation of Hsf1p activity. These results represent a major change in our understanding of how PKA signaling influences the heat-shock response and heat-shock protein expression.
THE heat-shock response is a highly conserved physiological response to severe changes in environmental conditions. It is characterized by an increase in the expression of heat-shock proteins (HSPs), which maintain protein homeostasis by alleviating protein misfolding defects and aggregation, thus protecting the cell from damage. In the yeast Saccharomyces cerevisiae, two response elements regulate heat-shock gene expression: the heat-shock element (HSE), which is bound by the heat-shock transcription factor Hsf1p (Sorger and Pelham 1988; Wiederrecht et al. 1988), and the stress response element (STRE), which is bound by the partially redundant transcription factors Msn2p and Msn4p (Martinez-Pastor et al. 1996; Schmitt and McEntee 1996). These two binding sites are distributed differentially among different heat-shock genes, causing Hsf1p and Msn2p/4p to have distinct contributions to the heat-induced expression of these genes (Treger et al. 1998; Boy-Marcotte et al. 1999; Amoros and Estruch 2001; Grably et al. 2002).
The heat-shock transcription factor is the primary transcriptional regulator for the eukaryotic heat-shock response (Jolly and Morimoto 2000; Pirkkala et al. 2001). In all eukaryotes, HSF binds HSEs in the promoters of most HSPs and strongly activates heat-shock gene expression in response to heat and other environmental stresses. In metazoans, multiple isoforms of HSF regulate tolerance to stresses, and they are also involved in developmental programs, including eye lens development and gametogenesis (Christians et al. 2003). The isoforms have unique and synergistic functions, with HSF1 as the predominant isoform for heat-induced HSP expression. In yeast, a single essential gene, HSF1, responds to various stresses and must therefore integrate diverse stimuli and elicit an appropriate transcriptional response (Sorger and Pelham 1988; Wiederrecht et al. 1988).
HSF activity must be tightly controlled to avoid inappropriate expression of heat-shock proteins (Nollen and Morimoto 2002). Thus, HSF is negatively regulated, kept in an inactive or low activity conformation prior to stress. In S. cerevisiae, a low level of Hsf1p activity is essential to maintain constitutive expression of HSPs necessary for normal cellular processes. Under normal growth conditions, Hsf1p is bound to strong HSEs (Jakobsen and Pelham 1988). Following heat shock, the occupancy of Hsf1p increases at all types of HSEs (Giardina and Lis 1995; Erkine et al. 1999; Hahn et al. 2004), and its transcriptional activity increases dramatically (Sorger and Pelham 1988). The phosphorylation state of Hsf1p also changes in response to stress, with some sites responsible for activation and others for attenuation of activity (Sorger and Pelham 1988; Høj and Jakobsen 1994; Hashikawa and Sakurai 2004). The constitutive and heat-induced phosphorylation of Hsf1p has been studied, but the signaling pathways that are responsible for the negative regulation and heat-induced activation have yet to be elucidated.
The Msn2p/4p transcription factors are not conserved from yeast to metazoans and are not required for viability. Under nonstress conditions, Msn2p/4p are localized to the cytoplasm; however, following stress they move into the nucleus where they drive the expression of their target genes (Schmitt and McEntee 1996; Gorner et al. 1998; Beck and Hall 1999). These genes include some of the same heat-shock genes regulated by Hsf1p, as well as genes involved in antioxidant and carbohydrate metabolism (Gasch et al. 2000; Causton et al. 2001). The control of Msn2p/4p seems to be primarily through negative regulation, with the Ras/cAMP pathway playing a major role (Gorner et al. 1998; Smith et al. 1998). As with Hsf1p, phosphorylation plays a critical role in the activity of Msn2p (Chi et al. 2001).
In this work, we have undertaken a novel genetic approach in an attempt to understand the signaling pathways that negatively regulate the expression of heat-shock genes in the absence of stress. For our first genetic selection, we have chosen the HSP26 promoter, which has been well characterized (Susek and Lindquist 1989; Susek and Lindquist 1990; Chen and Pederson 1993) and whose heat-induced expression is known to be regulated by Hsf1p and Msn2p/4p (Boy-Marcotte et al. 1999; Amoros and Estruch 2001). To identify regulators of HSP expression, we used a Tn7-insertional mutagenesis approach (Bachman et al. 2002; Uhl et al. 2003), which allowed us to efficiently identify loss-of-function alleles in two genes, CDC25 and RAM1. In a complementary approach, we used an overexpression library to identify two dosage compensators, PDE2 and MSI1.
These four genes, which are known components of the RAS/cAMP signaling cascade, dramatically increase the constitutive expression of HSP26 when they are either mutated or overexpressed. This increase of HSP26 expression is preserved even in an msn2/4Δ strain. The Ras/cAMP pathway regulates the activity of protein kinase A (PKA), a serine/threonine kinase that plays a role in stress resistance and nutrient signaling and specifically represses the activity of Msn2p (Broach 1991; Thevelein and de Winde 1999). Of the heat-shock genes we tested, reduction in the level of PKA activity strongly induces the Msn2p/4p-independent expression of the genes encoding two small heat-shock proteins, HSP26 and HSP12, and modestly induces HSP104. In contrast, the expression of the larger heat-shock proteins SSA3, SSA4, and HSP82 was unaffected by loss of PKA activity. By using a conditional allele of HSF1, we have shown that the increased expression of the small heat-shock genes observed in the absence of PKA is dependent on Hsf1p. Our results reveal that PKA plays a role in the negative regulation of Hsf1p-dependent transcription in addition to its already established role in repressing Msn2p/4p. In contrast to the role of PKA in the regulation of Msn2p/4p activity, which affects all STRE-containing genes, the role of PKA in the regulation of Hsf1p activity affects only a subset of HSE-containing genes. In addition, we have shown that Hsf1p is unlikely to be a direct target of PKA, as deletion of PKA causes an increase in Hsf1p phosphorylation.
MATERIALS AND METHODS
Plasmid construction:
The integrative plasmid pHN3102 contains 2888 bp upstream of the HSP26 start codon fused to the HIS3 selectable marker open reading frame. The HSP26 promoter sequences were generated by PCR amplification from the S. cerevisiae strain W303-1A using primers SF135 (GGGGTACCTAAGCATCAAAGAAGGTGCG) and SF136 (ATTCCTCGAGTTGTTTAGTTTGTTTGTTTGCTTTTTTGGATACC), which add KpnI and XhoI restriction sites to the 5′ and 3′ ends of the fragment, respectively. The cloning process introduces a 4-bp change in the HSP26 promoter just upstream of the start from CAAATTAAC ATG to CAAcTcgAg ATG. The HIS3 open reading frame, as well as 221 bp 3′ of the stop codon, were amplified from the pRS403 plasmid (Sikorski and Hieter 1989) using primers SF137 (ATTCCTCGAGATGACAGAGCAGAAAGCC) and SF138 (ATTCGCGGCCGCTTTCACACCGCATAGATCCG), which add XhoI and NotI sites to the 5′ and 3′ ends of the fragment, respectively. The HSP26 promoter and HIS3 ORF PCR fragments were cloned into the URA3-marked integrative plasmid pRS406 (Sikorski and Hieter 1989). The λTRP cDNA library was obtained from ATCC. Standard protocols were used to convert the phage library to the pTRP plasmid library, which expresses cDNAs from a GAL1 promoter (Elledge et al. 1991; Ramer et al. 1992).
Yeast strains and media:
Strains were grown on either YPDA rich media (YPD supplemented with 40 mg/ml adenine) or synthetic complete media (SC) using as carbon sources either 2% dextrose (SC Dex) or a 2% galactose/2% raffinose mixture (SC Gal/Raf; Ausubel et al. 2004). When necessary, aminoglycosides were used at the following concentrations: 200 μg/ml Geneticin G418 (Invitrogen, Carlsbad, CA) and 100 μg/ml ClonNAT (Werner BioAgents, Jena, Germany). For growth comparisons of a given strain on SC medium containing 3-amino-1,2,4-triazole (3-AT), equal numbers of cells were plated on both nonselective and selective media. The minimal inhibitory concentration of 3-AT for the wild-type PHSP26-HIS3 reporter strain is ∼7 mm, as seen in Figure 1. This concentration was used in Figure 7 to demonstrate the weak phenotype of MSI1, while 30 mm was used as a more stringent selection for other experiments.
Figure 1.—

Creation of PHSP26-HIS3 reporter strain. (A) The HIS3 auxotrophic marker was fused to the HSP26 promoter and integrated in tandem with the native HSP26 gene. The targeted HSP26 locus on chromosome II is diagrammed. (B) Total RNA was isolated from the wild-type strain (W303-1A) and the PHSP26-HIS3 reporter strain (YHN802) that were either maintained at 30° or heat-shocked at 37° for 15 min. Northern blots were probed with HIS3 and HSP26 probes, with ACT1 as a loading control. (C) The wild-type PHSP26-HIS3 reporter strain (YHN802) was struck on nonselective SC Dex −Ura medium or selective SC Dex −Ura −His medium containing 7 mm 3-AT.
Figure 7.—

HSF1 is required for small HSP expression in strains lacking PKA activity. A tetracycline-regulatable dual expression system was used to create a conditional allele of HSF1 in the msn2/4Δ and the msn2/4Δ tpk1/2/3Δ strain backgrounds, creating strains YHN1172 and YHN1173, respectively. (A) Total RNA was isolated from the tetracycline-repressible HSF1 strain (YHN1172) that had been grown for 12 hr in minimal medium or minimal medium supplemented with 20 μg/ml of doxycycline (a tetracycline analog) and then either maintained at 30° or heat-shocked at 37° for 15 min. Northern blots were probed with HSP104, SSA4, and HSP26 probes, with ACT1 as a loading control. (B) Total RNA was isolated from an msn2/4Δ tpk1/2/3Δ strain with the wild-type HSF1 promoter (YHN1114) that had been grown for 12 hr at 30° in YPD or YPD supplemented with 20 μg/ml of doxycycline. The Northern blots were probed with HSP12- and HSP26-specific probes, with ACT1 as a loading control. (C) Total RNA was isolated from the msn2/4Δ tpk1/2/3Δ strain carrying the tetracycline-repressible allele of HSF1 (YHN1173) that had been grown for 12 hr at 30° in minimal media or minimal media supplemented with 20 μg/ml of doxycycline. The Northern blots were probed with HSP12- and HSP26-specific probes, with ACT1 as a loading control.
Yeast strains, listed in Table 1, are all derived from W303-1A (MATa ade2- 1 trp1-1 can1-100 leu2,3-112 his3-11,15 ura3-1). The HIS3 conditional reporter strains were generated by integration of BamHI-linearized pHN3102 into a diploid W303-1 strain, followed by subsequent sporulation to obtain strains YHN801 and YHN802, and into the msn2Δ msn4Δ strain YHN963 to obtain strain YHN966. The proper integration of the reporter was confirmed by PCR. Gene knockouts were performed as previously described (Guldener et al. 1996; Goldstein and McCusker 1999). For strains where serial disruptions using the kanMX4 cassette was necessary, the marker was excised using cre-loxP recombination (Guldener et al. 1996). All gene knockouts were confirmed by PCR analysis. In addition, we confirmed the msn2Δ msn4Δ alleles by showing a complete absence for the heat-induced expression of CTT1, a gene whose heat-induced expression is completely dependent on MSN2 and MSN4 (data not shown; Martinez-Pastor et al. 1996; Schmitt and McEntee 1996).
TABLE 1.
Strains used in this study
| Strain | Relevant genotypea |
|---|---|
| YHN801 | MATα HSP26::PHSP26-HIS3,URA3 |
| YHN802 | MATaHSP26::PHSP26-HIS3,URA3 |
| YHN917 | MATa/α ρ0 cir0 |
| YHN932 | MATaHSP26::PHSP26-HIS3,URA3 ram1-doh1::Tn7 |
| YHN934 | MATaHSP26::PHSP26-HIS3,URA3 ram1-doh3::Tn7 |
| YHN941 | MATaHSP26::PHSP26-HIS3,URA3 ram1-doh8::Tn7 |
| YHN946 | MATaHSP26::PHSP26-HIS3,URA3 cdc25-doh11::Tn7 |
| YHN947 | MATaHSP26::PHSP26-HIS3,URA3 cdc25-doh12::Tn7 |
| YHN963 | MATamsn2Δ::loxP msn4Δ::kanMX |
| YHN966 | MATaHSP26::PHSP26-HIS3,URA3 msn2Δ::loxP msn4Δ::kanMX |
| YHN1005 | MATα ram1Δ::kanMX |
| YHN1007 | MATα cdc25Δ::kanMX |
| YHN1077 | MATaHSP26::PHSP26-HIS3,URA3 msn2Δ::loxP msn4Δ::kanMX ram1Δ::kanMX |
| YHN1078 | MATaHSP26::PHSP26-HIS3,URA3 msn2Δ::loxP msn4Δ::kanMX cdc25Δ::kanMX |
| YHN1082 | MATaHSP26::PHSP26-HIS3,URA3 msn2Δ::loxP msn4Δ::kanMX tpk1Δ::loxP |
| YHN1083 | MATaHSP26::PHSP26-HIS3,URA3 msn2Δ::loxP msn4Δ::kanMX tpk2Δ::loxP |
| YHN1084 | MATaHSP26::PHSP26-HIS3,URA3 msn2Δ::loxP msn4Δ::kanMX tpk3Δ::loxP |
| YHN1085 | MATaHSP26::PHSP26-HIS3,URA3 msn2Δ::loxP msn4Δ::kanMX tpk1Δ::loxP tpk2Δ::loxP |
| YHN1086 | MATaHSP26::PHSP26-HIS3,URA3 msn2Δ::loxP msn4Δ::kanMX tpk1Δ::loxP tpk3Δ::loxP |
| YHN1087 | MATaHSP26::PHSP26-HIS3,URA3 msn2Δ::loxP msn4Δ::kanMX tpk2Δ::loxP tpk3Δ::loxP |
| YHN1090 | MATaHSP26::PHSP26-HIS3,URA3 msn2Δ::loxP msn4Δ::kanMX + pRS424GAL1b |
| YHN1091 | MATaHSP26::PHSP26-HIS3,URA3 msn2Δ::loxP msn4Δ::kanMX + PGAL1-PDE2-TRP1 |
| YHN1092 | MATaHSP26::PHSP26-HIS3,URA3 msn2Δ::loxP msn4Δ::kanMX + PGAL1-MSI1-TRP1 |
| YHN1114 | MATamsn2Δ::loxP msn4Δ::kanMX tpk1Δ::loxP tpk2Δ::loxP tpk3Δ::loxP |
| YHN1116 | MATaHSP26::PHSP26-HIS3,URA3 msn2Δ::loxP msn4Δ::kanMX tpk1Δ::loxP tpk2Δ::loxP tpk3Δ::loxP |
| YHN1126 | MATaHSP26::PHSP26-HIS3,URA3 ram1Δ::kanMX |
| YHN1128 | MATaHSP26::PHSP26-HIS3,URA3 cdc25Δ::kanMX |
| YHN1172 | MATamsn2Δ::loxP msn4Δ::kanMX PtetO2-HSF1, adhP-tetR-VP16, natMX4 adhP-tetR′-SSN6::LEU2 |
| YHN1173 | MATamsn2Δ::loxP msn4Δ::kanMX tpk1Δ::loxP tpk2Δ::loxP tpk3Δ::loxP PtetO2-HSF1, adhP-tetR-VP16, natMX4 adhP-tetR′-SSN6::LEU2 |
| YHN1189 | MATamsn2Δ::loxP msn4Δ::kanMX HSF1-13Myc::TRP1 |
| YHN1201 | MATamsn2Δ::loxP msn4Δ::kanMX tpk1Δ::loxP tpk2Δ::loxP tpk3Δ::loxP HSF1-13Myc::TRP1 |
All strains are derivatives of W303-1 and were constructed for this study.
The diploid ρ0 cir0 strain YHN917 was created from Y842 by standard protocols (Fox et al. 1991; Tsalik and Gartenberg 1998). Loss of the 2μ plasmid was confirmed by the absence of a PCR product for the 2μ-encoded REP1 gene. Loss of mitochondrial DNA was confirmed by an inability to utilize glycerol as the sole carbon source and a lack of mitochondrial staining with Hoechst 33258.
The strain YHN1172 was created to allow tetracyline-repressible expression of HSF1. First, the native HSF1 promoter from YHN963 was replaced with a tetracycline operator cassette containing the repressor binding site (tetO2) and the TetR-VP16 tTA transactivator (Belli et al. 1998a,b). Then, the TetR′-SSN6 repressor from pCM245 was integrated at the leu2 locus by linearization with ClaI (Belli et al. 1998b), creating a wild-type LEU2 allele. This dual repression system allows HSF1 transcription to be strongly repressed by the addition of doxycycline to the growth media at a concentration of 20 μg/ml.
HSF1 was tagged with 13 tandem copies of the Myc epitope to enable efficient immunoprecipitation and detection of the Hsf1 protein. The 13Myc tagging cassette was amplified from the pFA6a-13Myc-TRP1 plasmid (Longtine et al. 1998) and integrated at the 3′ end of the HSF1 open reading frame in strains YHN963 and YHN1114 to generate strains YHN1189 and YHN1201, respectively.
In vitro Tn7 transposition mutagenesis:
The Tn7 donor plasmid pNB3 was a generous gift from Nurjana Bachman and Jef Boeke. This plasmid contains a mini-Tn7 transposon marked with LEU2, as well as the Escherichia coli πAN7 origin and kanamycin resistance marker (Bachman et al. 2002). Target DNA was prepared from the diploid ρ0 cir0 strain YHN917. The absence of mitochondrial and 2μ plasmid DNA prevents the transposon library from being heavily biased by these abundant sequences, which can frequently yield false positives. The genomic DNA was isolated by a CsCl banding protocol and then sonicated to yield fragments averaging 3–5 kb. The in vitro transposition reaction was performed as previously described (Bachman et al. 2002).
The reporter strain YHN802 was transformed with the mutagenized genomic DNA using the TRAFO protocol (Gietz and Woods 2002) and plated onto SC Dex lacking leucine and uracil. After 3 days of incubation at 30°, the mutagenized strains were replica plated onto 2% agar (to reduce background) and then onto SC Dex media lacking uracil, leucine, and histidine and supplemented with 30 mm 3-AT. These plates were incubated at 30° for an additional 3–5 days.
Candidate strains were crossed with the α-derivative of the reporter strain, YHN801, and sporulated. Dissected tetrads were tested for cosegregation of the Leu+ and His+ phenotypes. To excise the transposons as plasmids, genomic DNA from the candidate strains was digested with BamHI (to rescue only the right arm of the transposon) or NdeI or SphI (to rescue both arms of the transposon), self-ligated, and transformed into E. coli. Rescued plasmids were sequenced and the identity of the insertion site was determined using the BLAST tool on the Saccharomyces Genome Database website (Cherry et al. 1998; Ball et al. 2000).
RNA preparation and Northern blot analysis:
Total RNA was isolated from yeast cells using the hot acidic phenol method (Ausubel et al. 2004). RNA samples were fractionated on a 1% agarose denaturing gel containing formaldehyde and transferred by vacuum blotting onto ZetaProbe nylon membrane (Bio-Rad, Richmond, CA). Biotin-labeled DNA probes were generated by incorporation of Biotin-16-dUTP nucleotide (Roche, Indianapolis) into PCR products from cloned fragments of the HSP12, HSP26, SSA3, SSA4, HSP82, and HSP104 ORFs. For hybridization and detection, we used the North2South chemiluminescent detection kit (Pierce, Rockford, IL) and a Fluorchem 8800 cooled CCD detection system (Alpha Innotech).
Phosphorylation analysis:
Myc-tagged strains were grown to midlog phase and harvested by centrifugation, and the pellets were frozen in liquid N2 and stored at −70° for subsequent analysis. Pellets were resuspended in FA lysis buffer (50 mm HEPES, pH 7.5, 150 mm NaCl, 1 mm EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS) along with a yeast-specific protease inhibitor cocktail and a serine/threonine phosphatase inhibitor cocktail. The resuspended pellets were boiled for 10 min, and then the cells were disrupted with zirconia-silica beads in a Mini-Bead-Beater-8 (Biospec Products). The extracts were clarified by centrifugation and then incubated with agitation with 25 μl of anti-Myc(9E10) antibody conjugated agarose beads (Santa Cruz Biotechnology) for 90 min at room temperature. The beads were washed three times with FA lysis buffer plus protease inhibitors. Half of the beads were then incubated for 30 min at 37° with 25 units of calf intestinal alkaline phosphatase (New England Biolabs, Beverly, MA). The beads were then boiled in sample buffer and resolved on a 7.5% SDS-PAGE. Hsf1p was detected by Western blotting of the immunoprecipitates using an anti-Myc(9E10) antibody-HRP conjugate (Santa Cruz Biotechnology).
RESULTS
Isolation of mutations that derepress expression from the HSP26 promoter:
The Hsf1 protein is negatively regulated and is thereby kept in a low activity state prior to stress (Sorger and Pelham 1988; Wiederrecht et al. 1988). In an effort to understand the mechanism underlying this negative regulation, we have designed a genetic selection based on the promoter of the HSP26 gene fused to the HIS3 reporter gene. The HSP26 promoter was selected for the screen because of its low level of expression in cells grown at 30°, as well as its robust expression following heat shock (Chen and Pederson 1993; Amoros and Estruch 2001). The HIS3 gene encodes imidazolegylcerol-phosphate dehydratase (IGP dehydratase). The HIS3 gene was selected as a reporter because one can compensate for any background expression with the IGP dehydratase competitive inhibitor 3-AT (Horecka and Sprague 2000). Approximately 3 kb of the HSP26 promoter was cloned upstream of the HIS3 open reading frame and integrated into W303-1A derivatives in tandem with the native HSP26 gene at the HSP26 genomic locus (Figure 1A).
To determine whether expression from the PHSP26-HIS3 fusion mirrors that of the native HSP26 gene, we compared the levels of constitutive and heat-induced mRNAs in the parent strain (W303-1A) and a version of the parent strain that contains the PHSP26-HIS3 reporter (YHN802). The constitutive levels of HSP26 are undetectable in both strains, but are rapidly induced to comparable levels following a 15-min heat shock at 37°. The constitutive levels of HIS3 are undetectable in both strains, but the HIS3 level increases in the reporter strain (YHN802) following a 15-min heat shock at 37°, indicating that the PHSP26-HIS3 gene fusion is being regulated in a manner similar to that of HSP26 (Figure 1B). We also determined the ability of the reporter strain to grow on selective media lacking histidine in the presence of varying concentrations of 3-AT. The reporter strain (YHN802) is unable to grow on media in the presence of 7 mm 3-AT (Figure 1C), confirming the low level of constitutive expression from the PHSP26-HIS3 promoter fusion. We used this reporter strain to identify mutants involved in the stress response that are able to grow on selective media in the absence of stress, thereby generating derepression of HSP expression (doh) strains that exhibit increased constitutive HSP26 expression.
Mutations were generated using a Tn7-mediated mutagenesis protocol (Bachman et al. 2002). This approach uses a Tn7 transposase that lacks site selection specificity and therefore randomly integrates a mini-Tn7 transposon into sheared genomic S. cerevisiae DNA (Stellwagen and Craig 1997; Biery et al. 2000). In addition to a LEU2 selectable marker, the mini-Tn7 transposon contains E. coli sequences that facilitate direct rescue and identification of the integration site. Sheared yeast genomic DNA was combined in vitro with the mutant Tn7 transposase and the mini-Tn7 transposon to create a library of mutagenized genomic DNA that could be transformed into yeast.
Approximately 80,000 transformants of the PHSP26-HIS3 reporter strain (YHN802) were screened. After replica plating, we found 13 strains that grew on stringent selective media containing 30 mm 3-AT, greater than four times the minimal inhibitory concentration. As described below, five of these candidates (eventually) proved to have a doh phenotype. Two of the five strains demonstrated 2:2 meiotic cosegregation of the Leu+ transposon and His+ growth phenotypes following a backcross to the isogenic wild-type reporter strain. This suggests that a single causative chromosomal lesion was responsible for upregulated expression from the HSP26 promoter. The mini-Tn7 was isolated from strains containing the doh11 and doh12 alleles (YHN946 and YHN947). Sequencing revealed independent integrations in the Ras guanine nucleotide exchange factor CDC25 (Figure 2A). The reporter growth phenotype of the cdc25 insertions could be suppressed by the presence of a plasmid-borne copy of CDC25 (data not shown). Cdc25p catalyzes the exchange of guanine nucleotides on the two small GTPases, Ras1p and Ras2p, resulting in increased Ras signaling (Broek et al. 1987; Robinson et al. 1987). Inactivation of Cdc25p would reduce the downstream signaling functions of the Ras proteins.
Figure 2.—

Isolation of doh alleles. (A) Location of the transposition insertion alleles that disrupt the ORFs of CDC25 and RAM1 and exhibit a doh phenotype. (B) The wild-type reporter strain (YHN802), the reporter strain harboring the doh8 ram1::Tn7 transposition allele (YHN941), and the reporter strain harboring the doh12 cdc25::Tn7 transposition allele (YHN947) were struck on nonselective SC Dex −Ura medium or selective SC Dex −Ura −His medium containing 30 mm 3-AT. (C) The wild-type reporter strain (YHN802), the reporter strain with a ram1Δ allele (YHN1126), and the reporter strain with a cdc25Δ allele (YHN1128) were struck on nonselective SC Dex −Ura medium or selective SC Dex −Ura −His medium containing 30 mm 3-AT.
Three of the 13 potential doh strains were sterile. Because two of the doh candidates affected the Ras/cAMP pathway, we thought it might be possible that the sterile doh candidates were at the locus of the farnesyltransferase β-subunit RAM1. The Ram1 protein is responsible for conferring substrate specificity to the Ram1/2 farnesyltransferase holoenzyme. The Ram1/2 holoenzyme adds a 15-carbon farnesyl lipid moiety to the C-terminal CaaX box motif of substrate proteins, including Ras and a-factor mating pheromone, enabling them to localize to the membrane and elicit signaling functions (He et al. 1991). The inability to synthesize mature a-factor could explain the sterility of these potential doh candidates, as they were in a MATa strain. Isolation and sequencing of the mini-Tn7 transposons from strains carrying the doh1, doh3, and doh8 alleles (YHN932, YHN934, and YHN941) showed that they contained independent insertions in RAM1 (Figure 2A). Reintegration of the rescued mini-Tn7 plasmid into a naive reporter strain at the RAM1 locus recapitulated the His+ growth phenotype (data not shown), supporting the idea that these disruptions in the RAM1 ORF are responsible for upregulation of the HSP26 promoter.
The five independent mini-Tn7 insertions in CDC25 and RAM1 upregulate expression from the HSP26 promoter, but it is unclear whether these insertions are hypomorphic or complete loss-of-function alleles. To test this, we deleted the complete open reading frames of CDC25 or RAM1 by PCR-generated allele replacement. Previous studies had indicated that RAM1 is not an essential gene, but that CDC25 appeared to be essential, at least in the SP1 and BY4730 strain backgrounds (Broek et al. 1987; He et al. 1991; Giaever et al. 2002). In our W303-1A strain background, we were able to obtain viable haploid strains of both null mutants (YHN1005 and YHN1007). Recent studies have also found that cdc25Δ is viable in the W303-1A strain background, presumably because of the presence of a functional copy of SDC25, which has homology to CDC25 (Folch-Mallol et al. 2004). We crossed the ram1Δ and cdc25Δ alleles into the wild-type reporter strain (YHN802) and found that the cdc25Δ and ram1Δ alleles behaved similarly to the cdc25::Tn7 and ram1::Tn7 alleles with respect to activation of the PHSP26-HIS3 reporter (Figure 2C). These results strongly suggest that the doh phenotype observed in the transposon insertion alleles is due to partial or complete loss of Cdc25p or Ram1p function.
Upregulation of the HSP26 promoter is maintained in the absence of MSN2/4:
It is well established that the Ras/cAMP pathway regulates the HSP26 promoter by modulating the nuclear localization of the partially redundant Msn2 and Msn4 transcription factors (Gorner et al. 1998; Smith et al. 1998). To determine whether the effect we observe is due to enhanced Msn2p/4p activity at the HSP26 promoter, we generated an msn2Δ msn4Δ version of the reporter strain called YHN966. Similar to the wild-type reporter strain (YHN802), this new reporter strain had undetectable levels of HIS3 or HSP26 mRNA at 30° and showed the same inability to grow on 3-AT plates (data not shown). The cdc25Δ and ram1Δ alleles were crossed into the msn2/4Δ reporter strain and resulted in increased expression from the HSP26 promoter, as determined by growth of the reporter strain on selective medium (Figure 3A). In addition, we found that the cdc25Δ and ram1Δ alleles upregulated the expression of HSP26 (Figure 3B). These results suggest that the doh phenotype is independent of Msn2p/4p.
Figure 3.—

MSN2 and MSN4 are not required for doh phenotypes. (A) The msn2/4Δ reporter strain (YHN966), the msn2/4Δ reporter strain with the ram1Δ allele (YHN1077), and the msn2/4Δ reporter strain with the cdc25Δ allele (YHN1078) were struck on nonselective SC Dex −Ura medium or selective SC Dex −Ura −His medium containing 30 mm 3-AT. (B) A Northern blot of total RNA isolated from the indicated strains grown at 30° was probed with a HSP26-specific probe, with ACT1 as a loading control.
High-level expression of PDE2 or MSI1 activates expression from the HSP26 promoter in an Msn2p/4p-independent manner:
In a complementary approach to the mini-Tn7 disruption, we searched for genes whose overexpression led to an increase in activity from the PHSP26-HIS3 reporter in the absence of Msn2p/4p. We used a cDNA library whose inserts are driven by the GAL1 promoter (Elledge et al. 1991; Ramer et al. 1992) and transformed the library into the msn2/4Δ reporter strain (YHN966). We isolated four clones whose doh phenotype was dependent on galactose as the carbon source. Retransformation of the plasmids into the YHN966 reporter strain confirmed the doh growth phenotype. Three of the clones were independent isolates of PDE2 and gave a robust phenotype, while the sole clone of MSI1 gave a weaker phenotype (Figure 4). PDE2 encodes the high-affinity cAMP phosphodiesterase, which antagonizes the Ras/cAMP signaling pathway by directly degrading cAMP (Sass et al. 1986). MSI1 was originally identified as a multicopy suppressor of IRA1, the GTPase-activating protein that negatively regulates Ras signaling. Overexpression of MSI1 can suppress an activated Ras/cAMP pathway and decrease levels of cAMP through a complex mechanism that is not yet fully understood (Ruggieri et al. 1989; Johnston et al. 2001). These results support the idea that the doh phenotype is Msn2p/4p independent and is due to alterations in the Ras/cAMP pathway.
Figure 4.—
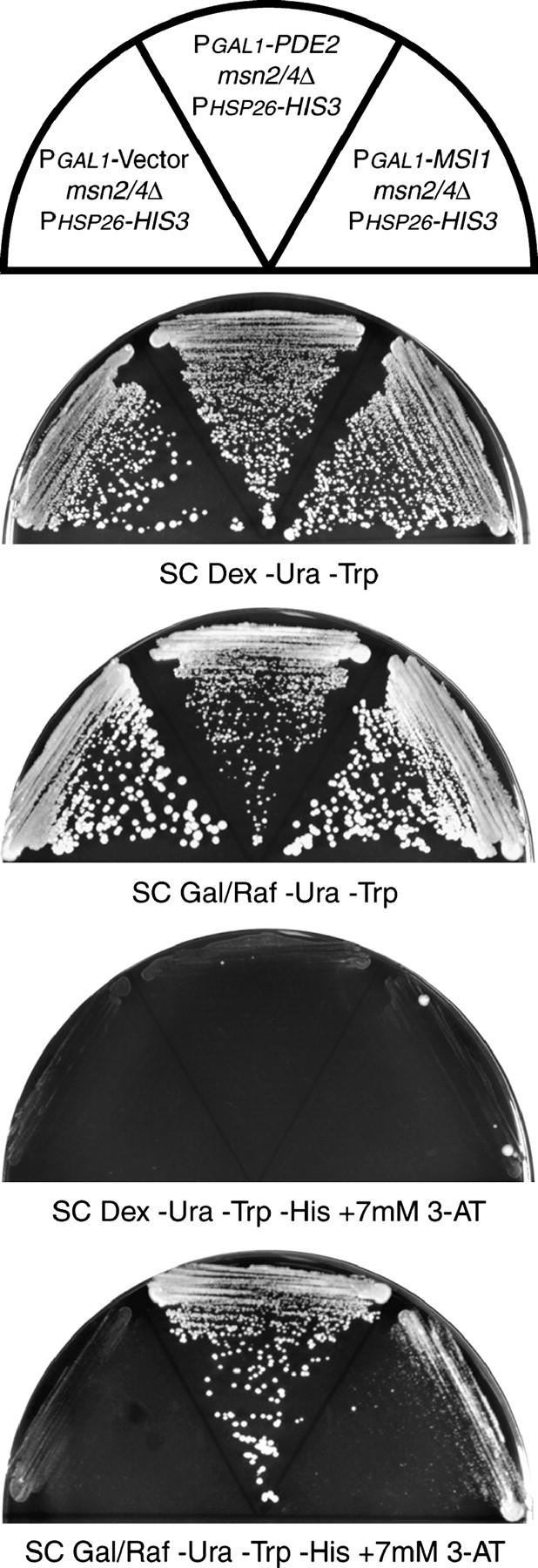
Overexpression of PDE2 or MSI1 activates the PHSP26-HIS3 reporter. The msn2/4Δ reporter strain with an empty vector (YHN1090), the msn2/4Δ reporter strain with the PGAL1-PDE2 plasmid (YHN1091), and the msn2/4Δ reporter strain with the PGAL1-MSI1 plasmid (YHN1092) were struck on nonselective SC Dex −Ura −Trp medium, nonselective SC Gal/Raf −Ura −Trp medium, selective SC Dex −Ura −Trp −His medium containing 7 mm 3-AT, and selective SC Gal/Raf −Ura −Trp −His medium containing 7 mm 3-AT.
Deletion of PKA induces expression from the HSP26 and HSP12 promoters:
We have isolated several factors that repress Ras/cAMP activity and result in upregulation of expression from the HSP26 promoter in an Msn2p/4p-independent manner. In yeast, the primary effector of the Ras/cAMP pathway is PKA (Broach 1991; Thevelein and de Winde 1999). In S. cerevisiae, PKA consists of the inhibitory regulatory subunit Bcy1p, which responds to cAMP levels, and three partially redundant catalytic subunits, Tpk1p, Tpk2p, and Tpk3p. To test the role of PKA kinase activity in the regulation of HSP26, we deleted each of the three redundant catalytic subunits of yeast PKA in an msn2/4Δ reporter strain. Deletion of individual or pairs of TPK subunits (strains YHN1082–1087; Table 1) had no effect on the ability of the reporter strain to grow on selective media (data not shown). Deletion of all three TPK subunits is lethal in most strains, including ours, but a concomitant deletion of MSN2/4 can suppress this lethality (Smith et al. 1998). We deleted all three TPK subunits in the msn2/4Δ reporter strain, creating strain YHN1116. Deletion of the PKA catalytic subunits caused an increase in expression of the reporter, as seen by the ability of the strain to grow on selective medium (Figure 5). These results suggest that each of the Tpk catalytic subunits is redundant for the doh phenotype. PKA is known to repress the activity of Msn2p/4p; however, our results suggest that the doh phenotype must represent a different mechanism of PKA regulatory activity.
Figure 5.—

Loss of PKA activates the PHSP26-HIS3 reporter. The msn2/4Δ reporter strain (YHN966) and the msn2/4Δ reporter strain with the tpk1/2/3Δ alleles (YHN1116) were struck on nonselective SC Dex −Ura medium or selective SC Dex −Ura −His medium containing 30 mm 3-AT.
Our results show that loss of PKA activity increases the expression of the PHSP26-HIS3 reporter. To confirm that HSP26 expression was increased in the msn2/4Δ tpk1/2/3Δ strain, we examined levels of HSP26 by Northern blot analysis. In comparison to the TPK1/2/3 strain, the tpk1/2/3Δ strain had dramatically increased expression for HSP26 (Figure 6). To determine whether this phenotype was specific to the HSP26 promoter, we also examined the effect of deletion of PKA on the expression of several different heat-shock promoters in an msn2/4Δ background. HSP12 showed the same dramatic increase as HSP26. However, expression of HSP104 was increased only very slightly, while expression of SSA3, SSA4, and HSP82 was unaffected (Figure 6). In contrast to PKA regulation of Msn2p/4p, which involves subcellular localization and therefore does not differentiate between different STRE-containing promoters (Gorner et al. 1998; Smith et al. 1998), PKA regulation of this Msn2p/4p-independent mechanism shows promoter selectivity.
Figure 6.—
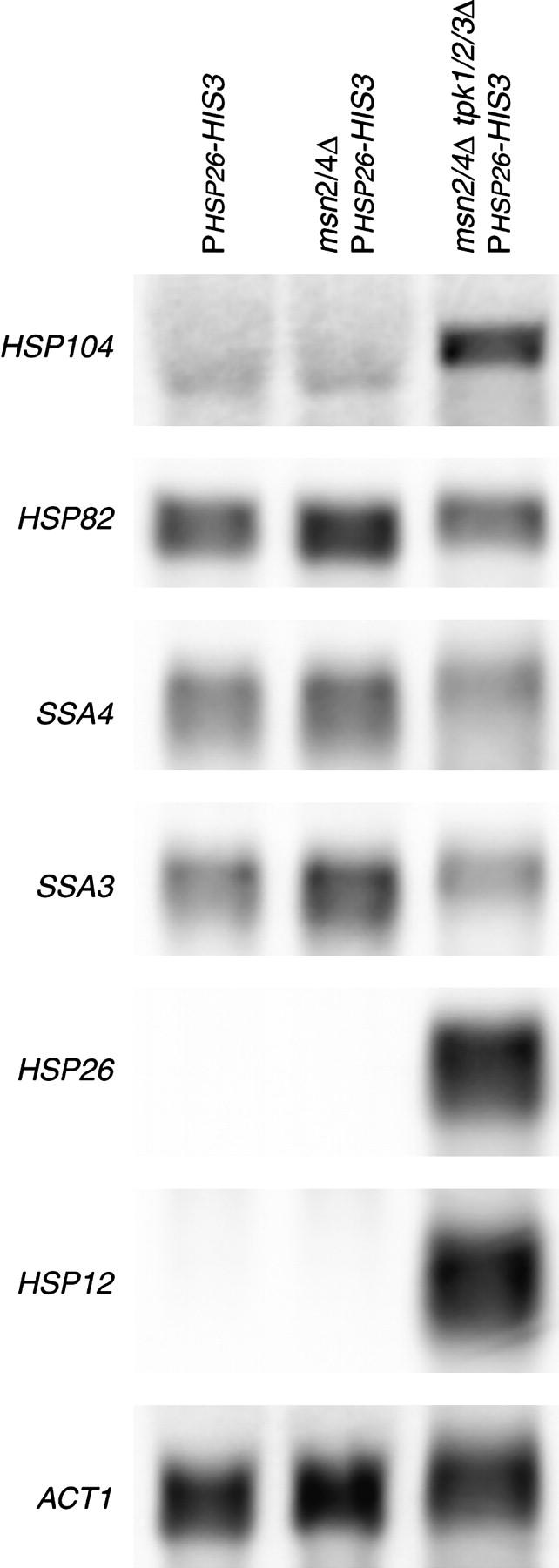
Loss of PKA specifically derepresses small heat-shock gene expression. (A) Total RNA was isolated from the wild-type reporter strain (YHN802), the msn2/4Δ reporter strain (YHN966), and the msn2/4Δ reporter strain with the tpk1/2/3Δ alleles (YHN1116). The Northern blot was probed with HSP12-, HSP26-, SSA3-, SSA4-, HSP82-, or HSP104-specific probes, with ACT1 as a loading control.
Induction of HSP26 and HSP12 in strains with compromised PKA function requires HSF1 expression:
The heat-inducible expression of HSP12 and HSP26 is known to be dependent on both Msn2p/4p and Hsf1p (Boy-Marcotte et al. 1999; Amoros and Estruch 2001; Hahn et al. 2004). We have shown that deletion of PKA increases expression of HSP12 and HSP26 independent of Msn2p/4p. To determine whether the PKA regulation of these genes is dependent on Hsf1p activity, we needed to construct a conditional allele of HSF1. Because HSF1 is required for viability under all conditions, we used a regulatable expression system developed to study essential genes (Belli et al. 1998a,b). We constructed a conditional promoter-replacement allele of HSF1 using a tetracycline-regulatable dual expression system that represses HSF1 transcription in the presence of doxycycline. To test the conditional allele of HSF1, it was integrated into an msn2/4Δ strain background to generate strain YHN1172. The growth of this strain is strongly inhibited on plates containing doxycycline, consistent with the essential role of HSF1 (data not shown). In addition, the presence of doxycycline inhibited the heat-shock induction of HSE-containing genes, including HSP104, SSA4, and HSP26 (Figure 7A) as well as HSP82, SSA3, and HSP12 (data not shown). This supports the idea that Hsf1p levels had been depleted in the presence of doxycycline. Incubation of a tpk1/2/3Δ msn2/4Δ (YHN1114) strain with the wild-type HSF1 promoter did not inhibit HSP12 or HSP26 expression (Figure 7B), indicating that doxycycline alone does not affect HSP expression.
To test the Hsf1p dependence of the doh phenotype, the HSF1 conditional allele was created in an msn2/4Δ tpk1/2/3Δ strain background to generate strain YHN1173. The growth of this strain was also strongly inhibited by doxycycline (data not shown). Because of the tpk1/2/3Δ, YHN1173 has the expected high levels of HSP26 and HSP12 in the absence of doxycycline (Figure 7C). Incubation of YHN1173 with doxycycline resulted in a dramatic reduction of the HSP26 and HSP12 transcripts (Figure 7C). These results indicate that activation of small HSP expression by the absence of PKA activity requires HSF1 expression.
Deletion of PKA induces hyperphosphorylation of Hsf1p:
Increases in Hsf1p transcriptional activity from either heat shock or glucose starvation are associated with hyperphosphorylation of the protein (Sorger and Pelham 1988; Sorger 1990; Jakobsen and Pelham 1991; Høj and Jakobsen 1994; Hahn and Thiele 2004; Hashikawa and Sakurai 2004). Hyperphosphorylation can be detected as a decrease in the electrophoretic mobility of Hsf1p and confirmed by loss of that altered mobility upon treatment with phosphatase. To determine whether deletion of PKA alters Hsf1p phosphorylation, we analyzed the effect of the tpk1/2/3Δ on the electrophoretic mobility of Hsf1p with and without phosphatase treatment. To facilitate this, we tagged the Hsf1p allele at the C terminus with 13 copies of the Myc epitope and generated strains with the genotypes msn2/4Δ HSF1-13Myc (YHN1189) and msn2/4Δ tpk1/2/3Δ HSF1-13Myc (YHN1201). The 13Myc tag does not disrupt Hsf1p function, as crossing the HSF1-13Myc allele into the PHSP26-HIS3 reporter background in the presence and absence of the TPK catalytic subunits gives the same phenotype as the wild-type HSF1 allele with respect to growth on selective media (data not shown).
Myc-tagged Hsf1p was immunoprecipitated from strains YHN1189 and YHN1201 grown at 30° and compared to Myc-tagged Hsf1p immunoprecipitated from YHN1189 that had been heat-shocked at 40° for 30 min. As seen before, heat shock decreases the electrophoretic mobility of Hsf1p (Sorger and Pelham 1988; Sorger 1990; Hahn and Thiele 2004; Hashikawa and Sakurai 2004). However, deletion of the Tpk catalytic subunits also decreased the electrophoretic mobility of Hsf1p (Figure 8). To confirm that this altered mobility was due to an increase in phosphorylation, we treated the immunoprecipitated samples with phosphatase. The increase in electrophoretic mobility upon phosphatase treatment is consistent with the idea that PKA deletion, like heat shock, increases the phosphorylation state of Hsf1p. Further experiments will be needed to determine whether the increase in phosphorylation is responsible for the increase in Hsf1p's transcriptional activity and, if so, what kinase is responsible for the phosphorylation.
Figure 8.—

Deletion of PKA increases Hsf1p phosphorylation. The phosphorylation state of Myc-tagged Hsf1 was analyzed in strains YHN1172 (HSF1-13Myc::TRP1 msn2/4Δ) and YHN1173 (HSF1-13Myc::TRP1 msn2/4Δ tpk1/2/3Δ). Yeast expressing Myc-tagged Hsf1 were grown at 30° (heat shock −) or heat-shocked at 40° for 30 min (heat shock +). Hsf1-13Myc was enriched by immunoprecipitation from cell extracts and split into two pools. These samples were incubated with and without calf intestinal alkaline phosphatase (CIP +/−) and resolved on a 7.5% SDS-PAGE, and Hsf1-13Myc was detected by Western blotting with Myc antibodies.
DISCUSSION
All cells require the ability to carefully coordinate their gene expression to compensate for environmental perturbations. It is just as important to be able to repress the expression of HSPs in the absence of stress as it is to upregulate their expression in the presence of heat or other stresses. We have developed a genetic selection to identify genes that when mutated or overexpressed can increase constitutive (i.e., nonstressed) expression from a heat-shock promoter. In this study, we used the HSP26 promoter and found that the Ras signaling pathway, specifically the cAMP-dependent protein kinase PKA, represses heat-shock gene expression through a new mechanism that is dependent on Hsf1p.
Previous studies of regulation of HSP expression had led to the suggestion that Ras signaling via PKA did not affect Hsf1p activity (Engelberg et al. 1994). This apparent misconception occurred because activated Ras alleles do not appear to further repress Hsf1p's already low level of constitutive activity. Additional studies had shown that the cAMP/PKA signaling pathway could regulate the activity of two other heat-inducible transcription factors, Msn2p and Msn4p. Msn2p and Msn4p bind to STRE sequences that are found in some, but not all, heat-shock promoters. PKA can regulate the cellular localization and thus activity of Msn2p (Gorner et al. 1998; Smith et al. 1998). This solidified the opinion that PKA regulation of heat-shock expression was not occurring through Hsf1p, but rather through Msn2p/4p. However, recent studies suggest that the regulation of heat-shock genes through the Ras/cAMP signaling pathway cannot be solely attributed to the Msn2/4 proteins (Boy-Marcotte et al. 1999; Grably et al. 2002; Aranda and del Olmo 2003; Versele et al. 2004). In support of these studies, we have now shown specifically that inactivation of PKA in an msn2/4Δ background can still increase heat-shock gene expression. Furthermore, we have used conditional expression of HSF1 through a PtetO2-HSF1 allele to show that the increased heat-shock gene expression in the absence of PKA is dependent on Hsf1p. These results represent a major change in our understanding of how PKA signaling influences the heat-shock response and heat-shock protein expression.
Hsf1p is required for the expression of only a subset of heat-shock genes in the absence of PKA. Deletion of PKA dramatically increases Hsf1-dependent expression at HSP12 and HSP26, but has only a limited effect on HSP104 and no detectable effect on SSA3, SSA4, or HSP82. Comparison of the promoter sequences of these six heat-shock genes does not immediately suggest a reason why HSP12 and HSP26 are regulated by PKA in a manner different from that of the other four genes. In addition, comparison of the constitutive HSE occupancies of all six genes (Hahn et al. 2004) does not show a correlation with PKA sensitivity. Because of the promoter selectivity, the influence of PKA on Hsf1p activity is unlikely to affect a global aspect of Hsf1p, such as its ability to bind HSEs or increase its transcriptional activity, but instead must affect a particular promoter-specific aspect of Hsf1p activity.
One possible explanation for why regulation of HSP12 and HSP26 is different from that of the other heat-shock genes is based on their functions as heat-shock proteins. The Hsp12 and Hsp26 proteins are part of a group called the small heat-shock proteins (sHsp's). Within the protein chaperone network, the sHsp's form a reservoir for nonnative refoldable proteins. In addition, they have specific roles in apoptosis, cytoskeletal organization, and formation of the eye lens (Haslbeck 2002). In contrast, the large heat-shock proteins, including the SSA family, HSP82, and HSP104, assist in the refolding of nonnative proteins and the disruption of aggregated proteins (Ellis 1990; Buchner 1996). Given the unique role of the small heat-shock proteins compared to their larger colleagues, it is not surprising that their expression is coregulated to some extent.
In addition to increasing the Hsf1p-dependent expression of some heat-shock genes, deletion of PKA also causes an increase in Hsf1p phosphorylation (Figure 8). Further analyses are needed to determine what sites are phosphorylated. From the crude SDS-PAGE analysis, it appears that the hyperphosphorylation pattern of Hsf1p differs whether it is activated by PKA deletion or heat shock. Given that Hsf1p phosphorylation increases upon deletion of PKA, it is unlikely that Hsf1p is a direct target of PKA phosphorylation. Instead, these data support a model where PKA represses an activating kinase under constitutive conditions.
It is also possible that the hyperphosphorylation of Hsf1p seen in the absence of PKA is not directly related to the increase in Hsf1p-dependent transcription. If this were the case, then a PKA deletion could affect Hsf1p transcriptional activity by activating an ancillary transcription factor that is normally repressed in the presence of PKA. During the preparation of this manuscript, a new publication reported that Srb9p, a component of the general RNA polymerase II transcription apparatus, is a direct target of PKA. Inactivation of PKA or deletion of SRB9 caused an increase in expression at several promoters, including HSP12 and HSP26 (Chang et al. 2004). Expression at other heat-shock promoters was not studied, nor was the contribution of Hsf1p to this phenotype. We have deleted the SRB9 gene in YHN966, the msn2/4Δ reporter strain, and found that it did not exhibit a strong growth phenotype on selective media (data not shown). Although this suggests that Srb9p is independent of the doh phenotype, further studies will be needed to confirm this hypothesis.
Recently, Hahn and Thiele (2004) have shown that the yeast AMPK homolog, Snf1p, functions as an activator of Hsf1p activity under glucose-limiting conditions. These conditions also decrease the production of cAMP and hence reduce the activity of PKA, which we have proposed to be a repressor of Hsf1p activity under normal growth conditions. Despite the complementary roles of Snf1p and PKA as activator and repressor under opposing conditions, their influence on heat-shock gene expression does not totally overlap. For example, Snf1p and PKA both influence Hsf1-dependent expression of HSP26, but only Snf1p can influence SSA3 expression (Hahn and Thiele 2004). In addition, the Snf1 kinase appears to modify Hsf1p directly, while PKA is unlikely to directly phosphorylate Hsf1p in vivo. Therefore, these two glucose-regulated kinases can each affect heat-shock gene expression via Hsf1p in a unique way. Further studies on the identification of the sites of phosphorylation, as well as the kinase(s) responsible for the PKA-dependent modifications, will help us further resolve this new twist in the complex regulation of the activity of the heat-shock transcription factor.
Acknowledgments
We thank Nurjana Bachman, Jef Boeke, Jeffrey Field, Marc Gartenberg, and Jim Broach for providing reagents and technical advice; Shelley Berger for use of the Mini-Bead-Beater-8; John Wagner and Meera Sundaram for comments on the manuscript; and Amanda Bulman, Laura Conlin, and Dawn Eastmond for useful discussions. This work was supported in part by funds from the University of Pennsylvania School of Medicine (to H.C.M.N.), by National Institutes of Health training grants 5-T32-GM-08216-15 and 5-T32-HL-07027 (to S.B.F.), and by the Roy and Diana Vagelos Scholars Program in the Molecular Life Sciences (to E.S.A.). N.L.C. is an Investigator of the Howard Hughes Medical Institute.
References
- Amoros, M., and F. Estruch, 2001. Hsf1p and Msn2/4p cooperate in the expression of Saccharomyces cerevisiae genes HSP26 and HSP104 in a gene- and stress type-dependent manner. Mol. Microbiol. 39: 1523–1532. [DOI] [PubMed] [Google Scholar]
- Aranda, A., and M. del Olmo, 2003. Response to acetaldehyde stress in the yeast Saccharomyces cerevisiae involves a strain-dependent regulation of several ALD genes and is mediated by the general stress response pathway. Yeast 20: 747–759. [DOI] [PubMed] [Google Scholar]
- Ausubel, F. M., R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman et al. (Editors), 2004 Current Protocols in Molecular Biology. Greene Publishing Associates/John Wiley & Sons.
- Bachman, N., M. C. Biery, J. D. Boeke and N. L. Craig, 2002. Tn7-mediated mutagenesis of Saccharomyces cerevisiae genomic DNA in vitro. Methods Enzymol. 350: 230–247. [DOI] [PubMed] [Google Scholar]
- Ball, C. A., K. Dolinski, S. S. Dwight, M. A. Harris, L. Issel-Tarver et al., 2000. Integrating functional genomic information into the Saccharomyces Genome Database. Nucleic Acids Res. 28: 77–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck, T., and M. N. Hall, 1999. The TOR signalling pathway controls nuclear localization of nutrient-regulated transcription factors. Nature 402: 689–692. [DOI] [PubMed] [Google Scholar]
- Belli, G., E. Gari, M. Aldea and E. Herrero, 1998. a Functional analysis of yeast essential genes using a promoter-substitution cassette and the tetracycline-regulatable dual expression system. Yeast 14: 1127–1138. [DOI] [PubMed] [Google Scholar]
- Belli, G., E. Gari, L. Piedrafita, M. Aldea and E. Herrero, 1998. b An activator/repressor dual system allows tight tetracycline-regulated gene expression in budding yeast. Nucleic Acids Res. 26: 942–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biery, M. C., F. J. Stewart, A. E. Stellwagen, E. A. Raleigh and N. L. Craig, 2000. A simple in vitro Tn7-based transposition system with low target site selectivity for genome and gene analysis. Nucleic Acids Res. 28: 1067–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boy-Marcotte, E., G. Lagniel, M. Perrot, F. Bussereau, A. Boudsocq et al., 1999. The heat shock response in yeast: differential regulations and contributions of the Msn2p/Msn4p and Hsf1p regulons. Mol. Microbiol. 33: 274–283. [DOI] [PubMed] [Google Scholar]
- Broach, J. R., 1991. Ras-regulated signaling processes in Saccharomyces cerevisiae. Curr. Opin. Genet. Dev. 1: 370–377. [DOI] [PubMed] [Google Scholar]
- Broek, D., T. Toda, T. Michaeli, L. Levin, C. Birchmeier et al., 1987. The S. cerevisiae CDC25 gene product regulates the RAS/adenylate cyclase pathway. Cell 48: 789–799. [DOI] [PubMed] [Google Scholar]
- Buchner, J., 1996. Supervising the fold: functional principles of molecular chaperones. FASEB J. 10: 10–19. [PubMed] [Google Scholar]
- Causton, H. C., B. Ren, S. S. Koh, C. T. Harbison, E. Kanin et al., 2001. Remodeling of yeast genome expression in response to environmental changes. Mol. Biol. Cell 12: 323–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, Y. W., S. C. Howard and P. K. Herman, 2004. The Ras/PKA signaling pathway directly targets the Srb9 protein, a component of the general RNA polymerase II transcription apparatus. Mol. Cell 15: 107–116. [DOI] [PubMed] [Google Scholar]
- Chen, J., and D. S. Pederson, 1993. A distal heat shock element promotes the rapid response to heat shock of the HSP26 gene in the yeast Saccharomyces cerevisiae. J. Biol. Chem. 268: 7442–7448. [PubMed] [Google Scholar]
- Cherry, J. M., C. Adler, C. Ball, S. A. Chervitz, S. S. Dwight et al., 1998. SGD: Saccharomyces genome database. Nucleic Acids Res. 26: 73–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi, Y., M. J. Huddleston, X. Zhang, R. A. Young, R. S. Annan et al., 2001. Negative regulation of Gcn4 and Msn2 transcription factors by Srb10 cyclin-dependent kinase. Genes Dev. 15: 1078–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christians, E. S., Q. Zhou, J. Renard and I. J. Benjamin, 2003. Heat shock proteins in mammalian development. Semin. Cell Dev. Biol. 14: 283–290. [DOI] [PubMed] [Google Scholar]
- Elledge, S. J., J. T. Mulligan, S. W. Ramer, M. Spottswood and R. W. Davis, 1991. Lambda YES: a multifunctional cDNA expression vector for the isolation of genes by complementation of yeast and Escherichia coli mutations. Proc. Natl. Acad. Sci. USA 88: 1731–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis, R. J., 1990. The molecular chaperone concept. Semin. Cell Biol. 1: 1–9. [PubMed] [Google Scholar]
- Engelberg, D., E. Zandi, C. S. Parker and M. Karin, 1994. The yeast and mammalian Ras pathways control transcription of heat shock genes independently of heat shock transcription factor. Mol. Cell. Biol. 14: 4929–4937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erkine, A. M., S. F. Magrogan, E. A. Sekinger and D. S. Gross, 1999. Cooperative binding of heat shock factor to the yeast HSP82 promoter in vivo and in vitro. Mol. Cell. Biol. 19: 1627–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folch-Mallol, J. L., L. M. Martínez, S. J. Casas, R. Yang, C. Martínez-Anaya et al., 2004. New roles for CDC25 in growth control, galactose regulation and cellular differentiation in Saccharomyces cerevisiae. Microbiology 150: 2865–2879. [DOI] [PubMed] [Google Scholar]
- Fox, T. D., L. S. Folley, J. J. Mulero, T. W. McMullin, P. E. Thorsness et al., 1991. Analysis and manipulation of yeast mitochondrial genes. Methods Enzymol. 194: 149–165. [DOI] [PubMed] [Google Scholar]
- Funk, M., R. Niedenthal, D. Mumberg, K. Brinkmann, V. Ronicke et al., 2002. Vector systems for heterologous expression of proteins in Saccharomyces cerevisiae. Methods Enzymol. 350: 248–257. [DOI] [PubMed] [Google Scholar]
- Gasch, A. P., P. T. Spellman, C. M. Kao, O. Carmel-Harel, M. B. Eisen et al., 2000. Genomic expression programs in the response of yeast cells to environmental changes. Mol. Biol. Cell 11: 4241–4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaever, G., A. M. Chu, L. Ni, C. Connelly, L. Riles et al., 2002. Functional profiling of the Saccharomyces cerevisiae genome. Nature 418: 387–391. [DOI] [PubMed] [Google Scholar]
- Giardina, C., and J. T. Lis, 1995. Dynamic protein-DNA architecture of a yeast heat shock promoter. Mol. Cell. Biol. 15: 2737–2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz, R. D., and R. A. Woods, 2002. Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol. 350: 87–96. [DOI] [PubMed] [Google Scholar]
- Goldstein, A. L., and J. H. McCusker, 1999. Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae. Yeast 15: 1541–1553. [DOI] [PubMed] [Google Scholar]
- Gorner, W., E. Durchschlag, M. T. Martinez-Pastor, F. Estruch, G. Ammerer et al., 1998. Nuclear localization of the C2H2 zinc finger protein Msn2p is regulated by stress and protein kinase A activity. Genes Dev. 12: 586–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grably, M. R., A. Stanhill, O. Tell and D. Engelberg, 2002. Hsf1p and Msn2/4p can exclusively or cooperatively activate the yeast HSP104 gene. Mol. Microbiol. 44: 21–35. [DOI] [PubMed] [Google Scholar]
- Guldener, U., S. Heck, T. Fielder, J. Beinhauer and J. H. Hegemann, 1996. A new efficient gene disruption cassette for repeated use in budding yeast. Nucleic Acids Res. 24: 2519–2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn, J. S., and D. J. Thiele, 2004. Activation of the Saccharomyces cerevisiae heat shock transcription factor under glucose starvation conditions by Snf1p protein kinase. J. Biol. Chem. 279: 5169–5176. [DOI] [PubMed] [Google Scholar]
- Hahn, J. S., Z. Hu, D. J. Thiele and V. R. Iyer, 2004. Genome-wide analysis of the biology of stress responses through heat shock transcription factor. Mol. Cell. Biol. 24: 5249–5256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashikawa, N., and H. Sakurai, 2004. Phosphorylation of the yeast heat shock transcription factor is implicated in gene-specific activation dependent on the architecture of the heat shock element. Mol. Cell. Biol. 24: 3648–3659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haslbeck, M., 2002. sHsps and their role in the chaperone network. Cell. Mol. Life Sci. 59: 1649–1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, B., P. Chen, S. Y. Chen, K. L. Vancura, S. Michaelis et al., 1991. RAM2, an essential gene of yeast, and RAM1 encode the two polypeptide components of the farnesyltransferase that prenylates a-factor and Ras proteins. Proc. Natl. Acad. Sci. USA 88: 11373–11377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Høj, A., and B. K. Jakobsen, 1994. A short element required for turning off heat shock transcription factor: evidence that phosphorylation enhances deactivation. EMBO J. 13: 2617–2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horecka, J., and G. F. Sprague, Jr., 2000. Use of imidazoleglycerolphosphate dehydratase (His3p) as a biological reporter in yeast. Methods Enzymol. 326: 107–119. [DOI] [PubMed] [Google Scholar]
- Jakobsen, B. K., and H. R. Pelham, 1988. Constitutive binding of yeast heat shock factor to DNA in vivo. Mol. Cell. Biol. 8: 5040–5042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobsen, B. K., and H. R. Pelham, 1991. A conserved heptapeptide restrains the activity of the yeast heat shock transcription factor. EMBO J. 10: 369–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston, S. D., S. Enomoto, L. Schneper, M. C. McClellan, F. Twu et al., 2001. CAC3(MSI1) suppression of RAS2(G19V) is independent of chromatin assembly factor I and mediated by NPR1. Mol. Cell. Biol. 21: 1784–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolly, C., and R. I. Morimoto, 2000. Role of the heat shock response and molecular chaperones in oncogenesis and cell death. J. Natl. Cancer Inst. 92: 1564–1572. [DOI] [PubMed] [Google Scholar]
- Longtine, M. S., A. McKenzie, III, D. J. Demarini, N. G. Shah, A. Wach et al., 1998. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14: 953–961. [DOI] [PubMed] [Google Scholar]
- Martinez-Pastor, M. T., G. Marchler, C. Schuller, A. Marchler-Bauer, H. Ruis et al., 1996. The Saccharomyces cerevisiae zinc finger proteins Msn2p and Msn4p are required for transcriptional induction through the stress response element (STRE). EMBO J. 15: 2227–2235. [PMC free article] [PubMed] [Google Scholar]
- Nollen, E. A., and R. I. Morimoto, 2002. Chaperoning signaling pathways: molecular chaperones as stress-sensing ‘heat shock’ proteins. J. Cell Sci. 115: 2809–2816. [DOI] [PubMed] [Google Scholar]
- Pirkkala, L., P. Nykanen, L. Sistonen, Y. Kawazoe, J. Inazawa et al., 2001. Roles of the heat shock transcription factors in regulation of the heat shock response and beyond. FASEB J. 15: 1118–1131. [DOI] [PubMed] [Google Scholar]
- Ramer, S. W., S. J. Elledge and R. W. Davis, 1992. Dominant genetics using a yeast genomic library under the control of a strong inducible promoter. Proc. Natl. Acad. Sci. USA 89: 11589–11593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson, L. C., J. B. Gibbs, M. S. Marshall, I. S. Sigal and K. Tatchell, 1987. CDC25: a component of the RAS-adenylate cyclase pathway in Saccharomyces cerevisiae. Science 235: 1218–1221. [DOI] [PubMed] [Google Scholar]
- Ruggieri, R., K. Tanaka, M. Nakafuku, Y. Kaziro, A. Toh-e et al., 1989. MSI1, a negative regulator of the Ras-cAMP pathway in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 86: 8778–8782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sass, P., J. Field, J. Nikawa, T. Toda and M. Wigler, 1986. Cloning and characterization of the high-affinity cAMP phosphodiesterase of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 83: 9303–9307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt, A. P., and K. McEntee, 1996. Msn2p, a zinc finger DNA-binding protein, is the transcriptional activator of the multistress response in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 93: 5777–5782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski, R. S., and P. Hieter, 1989. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122: 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, A., M. P. Ward and S. Garrett, 1998. Yeast PKA represses Msn2p/Msn4p-dependent gene expression to regulate growth, stress response and glycogen accumulation. EMBO J. 17: 3556–3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorger, P. K., 1990. Yeast heat shock factor contains separable transient and sustained response transcriptional activators. Cell 62: 793–805. [DOI] [PubMed] [Google Scholar]
- Sorger, P. K., and H. R. Pelham, 1988. Yeast heat shock factor is an essential DNA-binding protein that exhibits temperature-dependent phosphorylation. Cell 54: 855–864. [DOI] [PubMed] [Google Scholar]
- Stellwagen, A. E., and N. L. Craig, 1997. Gain-of-function mutations in TnsC, an ATP-dependent transposition protein that activates the bacterial transposon Tn7. Genetics 145: 573–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susek, R. E., and S. Lindquist, 1990. Transcriptional derepression of the Saccharomyces cerevisiae HSP26 gene during heat shock. Mol. Cell. Biol. 10: 6362–6373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susek, R. E., and S. L. Lindquist, 1989. hsp26 of Saccharomyces cerevisiae is related to the superfamily of small heat shock proteins but is without a demonstrable function. Mol. Cell. Biol. 9: 5265–5271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thevelein, J. M., and J. H. de Winde, 1999. Novel sensing mechanisms and targets for the cAMP-protein kinase A pathway in the yeast Saccharomyces cerevisiae. Mol. Microbiol. 33: 904–918. [DOI] [PubMed] [Google Scholar]
- Treger, J. M., A. P. Schmitt, J. R. Simon and K. McEntee, 1998. Transcriptional factor mutations reveal regulatory complexities of heat shock and newly identified stress genes in Saccharomyces cerevisiae. J. Biol. Chem. 273: 26875–26879. [DOI] [PubMed] [Google Scholar]
- Tsalik, E. L., and M. R. Gartenberg, 1998. Curing Saccharomyces cerevisiae of the 2 micron plasmid by targeted DNA damage. Yeast 14: 847–852. [DOI] [PubMed] [Google Scholar]
- Uhl, M. A., M. Biery, N. Craig and A. D. Johnson, 2003. Haploinsufficiency-based large-scale forward genetic analysis of filamentous growth in the diploid human fungal pathogen C. albicans. EMBO J. 22: 2668–2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Versele, M., J. M. Thevelein and P. Van Dijck, 2004. The high general stress resistance of the Saccharomyces cerevisiae fil1 adenylate cyclase mutant (Cyr1Lys1682) is only partially dependent on trehalose, Hsp104p and overexpression of Msn2/4p-regulated genes. Yeast 21: 75–86. [DOI] [PubMed] [Google Scholar]
- Wiederrecht, G., D. Seto and C. S. Parker, 1988. Isolation of the gene encoding the S. cerevisiae heat shock transcription factor. Cell 54: 841–853. [DOI] [PubMed] [Google Scholar]