

Abstract
The general stress response of yeast involves the induction of ∼200 genes in response to any one of several stresses. These genes are activated by Msn2 and repressed by the Srb10 kinase, a member of the mediator complex. Normally, Msn2 is exported from the nucleus, and Srb10 represses STRE gene expression. Under stress, Msn2 relocalizes to the nucleus and, with the relief of Srb10 repression, activates transcription. The stress response is rapid, but quickly attenuated. We show here that this attenuation is due to a nuclear-dependent degradation of Msn2. Msn2 rapidly disappeared from cells after heat or osmotic shock. This disappearance was not due to a change in MSN2 RNA levels, which remain constant during stress. Pulse-chase experiments confirmed the stress-dependent Msn2 degradation. The levels of Msn2 were significantly reduced in msn5 deletion cells that have been shown to constitutively retain Msn2 in the nucleus. The degradation was Srb10-dependent; Msn2 was not degraded in an srb10 deletion mutant. An Msn2 internal deletion mutant was insensitive to Srb10 repression, but was degraded by the Srb10-dependent mechanism. Thus, this mutation uncoupled Srb10 repression from degradation.
THE yeast Saccharomyces cerevisiae has a variety of regulatory systems that respond to and protect the cell from environmental stresses (Mager and De Kruijff 1995; Ruis and Schuller 1995; Estruch 2000; Hohmann 2002; Moye-Rowley 2002; O'Rourke et al. 2002). Many of these are specific to a given stress, such as the Hsf1-mediated heat-shock response and the Hog1-mediated osmotic-shock response. However, the general stress, or STRE, response involves the induction of ∼200 genes (Gasch et al. 2000; Causton et al. 2001) in response to any one of several stresses, including heat shock, osmotic shock, oxidative stress, low pH, glucose starvation, and others (Ruis and Schuller 1995; Hohmann 2002). STRE gene transcription is activated through the STRE element, 5′ CCCCT, located in several copies in their upstream regulatory regions (Schmitt and McEntee 1996). The response is characterized by a transitory increase in gene expression upon stress, but for any given STRE gene, the response may vary due to other, stress-specific regulatory elements (Gasch et al. 2000; Rep et al. 2000).
The STRE genes are activated upon stress by two functionally similar proteins, the major activators Msn2 and Msn4 (Marchler et al. 1993; Martinez-Pastor et al. 1996; Schmitt and McEntee 1996). Both proteins contain a zinc-finger binding domain at their C terminus that binds to the STRE element. Under nonstress conditions, Msn2 is localized in the cytoplasm, but upon any one of the general stresses, the protein relocalizes to the nucleus (Boy-Marcotte et al. 1998; Gorner et al. 1998, 2002; Garreau et al. 2000). Msn2 nuclear exclusion in unstressed cells is regulated by cAMP-dependent protein kinase A (cAPK) phosphorylation. Gorner et al. (2002) used fusion proteins to demonstrate that cAPK acted through at least two domains. Direct phosphorylation of the nuclear import signal of Msn2 prevented nuclear import and was responsive to glucose starvation but not to other general stresses. A second region, also sensitive to cAPK activity, controlled nuclear export. Nuclear exclusion through this region is dependent on the other general stresses and requires the exportin Msn5. Msn5 has been shown to control the nuclear localization of a variety of transcription factors, often recognizing differential phosphorylation (Kaffman et al. 1998; Blondel et al. 1999; DeVit and Johnston 1999; Komeili et al. 2000; Boustany and Cyert 2002; Kaplun et al. 2003; Queralt and Igual 2003). The nonstress exclusion of Msn2 from the nucleus may also be augmented by anchoring Msn2 in the cytoplasm. Beck and Hall (1999) found that Msn2 and Msn4 interacted with the cytoplasmic 14-3-3 protein Bmh2 and that this interaction was enhanced by TOR kinase activity. Rapamycin inhibition of TOR kinase activity resulted in rapid nuclear localization of Msn2 in unstressed cells (Gorner et al. 2002).
The Srb10 protein kinase and its cyclin-like regulatory subunit Srb11 also play a role in preventing STRE gene expression in unstressed cells (Cooper et al. 1997, 1999; Holstege et al. 1998; Cohen et al. 2003). Srb10 is active in unstressed cells, but under stress, the Srb11 regulatory subunit is rapidly degraded (Cooper et al. 1997, 1999). At least two different activities are reported for Srb10 that could be responsible for the regulation of STRE gene expression. Srb10/11 are members of the RNA polymerase II mediator complex that binds to the CTD tail of the largest polII subunit and plays a role in regulating transcriptional activation and repression (Liao et al. 1995; Myers and Kornberg 2000). Transcriptional initiation by polII requires phosphorylation of the CTD tail by Kin28, but Hengartner et al. (1998) demonstrated that Srb10 could block the formation of stable polII preinitiation complexes in vitro by prematurely phosphorylating the CTD tail. Thus, one mechanism for Srb10 inhibition of STRE gene expression might be the prevention of preinitiation complex formation at STRE genes. A second possible mechanism for Srb10 inhibition of STRE gene expression might involve Msn2 phosphorylation (Chi et al. 2001). However, it is unclear what the effect of this phosphorylation might be; Srb10-dependent phosphorylation of Msn2 was observed only during the initial period of stress. This stress-dependent phosphorylation cannot account for STRE derepression under nonstress conditions in an srb10Δ strain. In addition, Msn2 was observed to be in the nucleus in less than one-third of unstressed cells in the srb10Δ strain. Again, it is unclear whether this limited mislocalization can account for STRE gene induction in the srb10Δ strain.
Clearly a number of issues need to be resolved concerning the regulation of the general stress response. First, how is the initial burst of STRE gene expression attenuated to give the strong, transient response? It is possible that the activity, levels, or cellular compartmentalization of the activators Msn2 and Msn4 are regulated. Alternatively, a repression mechanism could be activated. Second, how is Srb10 repression achieved? Two different activities of Srb10 are outlined above. One or both of these may be responsible for STRE gene repression. In this study, we begin to answer these questions by demonstrating that Msn2 levels are reduced upon stress via a nuclear-specific, Srb10-dependent degradation. Also, we show that Srb10 repression can be eliminated by an internal deletion of Msn2, strongly suggesting that Srb10 represses through affecting Msn2 activity.
MATERIALS AND METHODS
Strains and cell growth:
The S. cerevisiae strains used in this study are presented in Table 1. The srb10Δ, msn2Δ, and msn4Δ deletion alleles constructed for this study were introduced into cells through one-step gene replacement (Rothstein 1983). The msn5Δ strain was purchased from EUROSCARF. The srb10Δ, msn2Δ, msn4Δ, and msn5Δ deletion alleles and the CYC7-lacZ integrated allele were combined in the various combinations presented in Table 1 through standard yeast crosses and dissections (Kaiser et al. 1994).
TABLE 1.
Yeast strains
| Strain | Genotype |
|---|---|
| RZ53-6 | MATα trp1 leu2 ura3 ade1 |
| MZ161-8A | MATα trp1 leu2 ura3 msn2::LEU2 |
| MZ161-8D | MATα trp1 leu2 ura3 his3 ade1 met15 msn2::LEU2 srb10::TRP1 msn5::kanMX |
| MZ161-39B | MATatrp1 leu2 ura3 his3 ade1 met15 msn2::LEU2 msn5::kanMX |
| MZ161-41 | MATα trp1 leu2 ura3 ade1 met15 msn2::LEU2 srb10::TRP1 |
| MZ171-60 | MATα trp1 leu2 ura3 ade1 msn2::LEU2 msn4::kanMX anb1::CYC7-lacZ |
| MZ171-48 | MATα trp1 leu2 ura3 his3 lys2 msn2::LEU2 msn4::kanMX srb10::TRP1 anb1::CYC7-lacZ |
| MZ171-15 | MATα trp1 leu2 ura3 his3 msn2::LEU2 msn4::kanMX msn5::kanMX anb1::CYC7-lacZ |
| MZ171-75 | MATα trp1 leu2 ura3 ade1his3 lys2 msn2::LEU2 msn4::kanMX srb10::TRP1 msn5::kanMX anb1::CYC7-lacZ |
Yeast cells were grown in synthetic SC medium (Kaiser et al. 1994). For plasmid maintenance, the appropriate nutritional marker was omitted, and for the [35S]methionine labeling experiments, methionine was omitted. For selection of the kanMX marker, geneticin was added to a final concentration of 200 μg/ml. Cells were grown at 30° for routine use, but grown in liquid at 25° with vigorous shaking to maintain nonstress conditions. Cells were transformed as described by Chen et al. (1992).
To induce heat shock, cells were grown to midexponential phase at 25° and then diluted with an equal volume of medium equilibrated at 37°. Growth was continued at 37° and time points were taken for RNA blots, immunoblots, or β-galactosidase assays as indicated. Osmotic shock was induced in a similar fashion, except cells were diluted with an equal volume of medium containing 2 m sorbitol and growth continued at 25°. For the preparation of samples for RNA and protein blots, the cells were rapidly chilled by addition of a sample of the cell culture to liquid nitrogen or prechilled medium. The cells were then pelleted.
Plasmids:
The plasmid shuttle vectors YCplac22, YCplac111, YCplac33, YEplac195 (Gietz and Sugino 1988), and pFA6akanMX4 (Wach et al. 1997) have been described. The HA epitope-tagged TUP1 plasmid YCp(23)TUP1HA (Mennella et al. 2003) and the CYC7-lacZ fusion plasmid YCp7Z (Wright and Zitomer 1984) have been described. Epitope sequences are presented in Ausubel et al. (2000).
The following plasmids were constructed using standard techniques (Ausubel et al. 2000). Restriction enzymes were purchased from New England Biolabs (Beverly, MA) or MBI Fermentas. Taq polymerase was purchased from Fermentas, and T4 DNA ligase was purchased from Roche (Indianapolis). Enzymes were used as recommended by the vendor. The sequences of PCR primers are available upon request. Coordinates for genes are presented with the A of the ATG initiation codon as +1 and base pairs 5′-wards numbered in negative integers and 3′-wards in positive integers.
pBSMSN2BP:
A PCR fragment of MSN2 from −875 to +2997 with a BamHI site at the 5′ end and a PstI site at the 3′ end was generated from RZ53-6 genomic DNA. It was cloned into pBS+ (Stratagene, La Jolla, CA).
YCp(33)MSN2:
The BamHI-PstI MSN2 insert from pBSMSN2BP was subcloned into YCplac33.
YCp(33)MSN2HA:
A PCR fragment of MNS2 from −875 to +2115 (the end of the coding sequence) was generated with a BamHI site at the 5′ end and a PstI site at the 3′ end from pBSMSN2BP. This fragment was cloned into the BamHI-PstI sites of a YCp(23)TUP1HA derivative. The resulting plasmid YCp(23)MSN2HA contained the MSN2 upstream and coding sequences fused to four HA epitope tag coding sequences followed by a stop codon and 240 bp of 3′ sequences of TUP1. This fusion was subcloned into YCplac33 to create YCp(33)MSN2HA.
YEp(195)MSN2HA:
The BamHI-HindIII fragment containing the MSN2HA insert in YCp(33)MSN2HA was subcloned into the BamHI-HindIII sites of YEplac195.
YCp(33)MSN2ΔE:
The EcoRI fragment containing codons 304–478 of MSN2 was deleted from YCp(33)MSN2HA.
YCp(33)c-MSN2:
A sequence encoding four copies of the c-myc 9E10 epitope was inserted into the SalI site (codons 3 and 4) of YCp(33)MSN2.
pmsn2::LEU2:
A PCR fragment of LEU2 from −337 to +1207 with an XbaI site at the 5′ end and an XhoI site at the 3′ end was generated from YCplac111. This fragment was inserted into the XbaI (+561) and XhoI (+1925) sites of the MNS2 coding sequence of pBSMSN2BP. It was digested with BamHI and PstI for transformation of yeast to generate the msn2 null allele.
pmsn4::kanMX:
YCp(33)MSN4 was constructed by cloning into YCplac33 a PCR-amplified fragment of MSN4 from −655 to +3593 with a 5′ BamHI site and a 3′ EcoRI site generated from RZ53-6 genomic DNA. The kanMX insert in pFA6akanMX4 was PCR amplified with an MfeI at one end and an AflII site at the other and cloned into the AflII (−158) and MfeI (+2420) sites of MSN4 in YCp(33)MSN4. pmsn4::kanMX was digested with BamHI and EcoRI for integration into yeast.
psrb10::TRP1:
The PstI (−992)-BglII (+3244) fragment containing the SRB10 gene was subcloned from a YCp50 library clone into the PstI-BamHI sites of pUC9 (Yanisch-Perron et al. 1985). A 1.77-kb KpnI-SphI TRP1 PCR fragment generated from YCplac22 was inserted into the KpnI (−501)-SphI (+1307) sites in the above construct. The null allele was transformed into yeast as a PstI-SmaI fragment.
panb1::7Z:
The CYC7-lacZ fusion in YCp7Z was subcloned into the SalI-SmaI sites of pCYC1 containing a 2.5-kb fragment from the CYC1-ANB1 locus (Zitomer et al. 1979). The insert disrupted the ANB1 gene. The fusion was integrated into the yeast genome by transformation with a BglII-HindIII fragment.
RNA blots, β-galactosidase assays, and immunoblots:
RNA was prepared using the hot acid phenol extraction as described by Ausubel et al. (2000). Blots were carried out as described by Ausubel et al. (2000). The radiolabeled probes were prepared by the random primer method (Ausubel et al. 2000) from the following fragments: +500 to +1086 of ACT1, −141 to +264 of CYC7, +8 to +2115 of MSN2, −320 to +1456 of ALD3, and −686 to +696 of DDR2.
β-Galactosidase assays were performed as described by Kaiser et al. (1994). The errors indicate the standard deviations derived from multiple assays performed with several independent transformants.
Protein samples were prepared for immunoblotting by the addition of 2× SDS gel sample buffer (Ausubel et al. 2000) to the harvested cell pellet. The samples were immediately boiled for 5 min. The cell debris was pelleted, and the samples were either immediately loaded on gels or frozen at −20°. Frozen samples were boiled for 2 min, and the debris was repelleted before loading on gels. Immunoblots were performed as described by Ausubel et al. (2000). Monoclonal antibody against the c-myc 9E10 epitope and the HA epitope, goat anti-mouse IgG horseradish peroxidase conjugate, and the Western blotting luminol reagent were purchased from Santa Cruz Biotechnology. Rabbit polyclonal antibody against bacterial expressed and purified eIF5A encoded by the yeast TIF51A gene was prepared by Alexander Kastaniotis. Goat anti-rabbit IgG horseradish peroxidase conjugate was purchased from Bio-Rad Laboratories (Richmond, CA).
Pulse-chase experiments:
The degradation of radiolabeled Msn2-HA was determined as follows. MZ171-60 cells transformed with YEp(195)MSN2HA were grown to midexponential phase at 25°. A total of 20–30 μCi of [35S]methionine (>1000 Ci/mmol, Amersham Biosciences) was added to 6 ml of cells, and incubation at 25° with shaking was continued for 5 min. The labeling was terminated by the addition of unlabeled methionine to a final concentration of 1 mg/ml. Samples (1 ml) were taken immediately for the time zero and the nonspecific control samples, 2 ml was incubated at 25° with shaking for continued nonstress growth, and 2 ml was shifted to 37° with shaking for stress induction. Time points were taken from each culture at 20 and 60 min. All time points were added to prechilled tubes containing a cell pellet from 2 ml of the same strain without the HA-tagged Msn2. All subsequent steps were carried out between 0° and 4°. Cells were pelleted and resuspended in 0.5 ml lysis buffer (50 mm HEPES-KOH, pH 7.5, 140 mm NaCl, 1 mm EDTA, 1% TritonX-100, 0.1% sodium deoxycholate) plus a protease inhibitor cocktail (1 mm phenylmethylsulfonyl fluoride, 1 μg/ml pepstastin, 1 μg/ml leupeptin, 10 μg/ml aprotinin, 5 μg/ml antipain, 1 mm benzamidine) and broken by vigorous mixing with glass beads. The lysate was transferred to a clean tube, and the glass beads were washed with 0.5 ml TBS (10 mm Tris HCl, pH 7.5, 150 mm NaCl), which was combined with the lysate. The lysate was clarified by centrifugation for 20 min at 7700 × g. A 5-μl aliquot was removed from each sample for trichloroacetic acid precipitation to determine the radioactivity in total soluble protein, and a 20-μl sample was added to 20 μl of SDS gel sample buffer (Ausubel et al. 2000). A total of 2 μl of anti-HA epitope antibody (200 μg/ml) was added to the remainder of the cleared lysate for each time point, while 2 μl of anti-c-myc 9E10 antibody (200 μg/ml) was added to the control cleared lysate. The mixture was incubated for 1 hr and then 30 μl of pretreated protein A-Sepharose beads (Amersham Bioscience) was added to each sample and incubated on a rotating wheel for 1 hr. (Pretreated beads were prepared by incubating the beads for 1.5 hr at 4° with a cleared cell lysate prepared from a culture of cells lacking the Msn2-HA protein. A lysate from a 7-ml midexponential phase culture was used per 100 μl of beads.) The beads were washed four times with 1 ml of 0.1 m sodium phosphate, pH 8.0, and then the protein was eluted in 30 μl of SDS gel sample buffer at 90° for 5 min. The samples, normalized to the total counts in the cleared lysate, were fractionated by SDS gel electrophoresis (Ausubel et al. 2000) and the dried gel was exposed to a PhosphoImager screen.
RESULTS
The attenuation of the STRE response correlates with reduced Msn2 levels:
The STRE genes are induced in response to any one of a number of stresses. As shown in the RNA blot in Figure 1A and the graph in Figure 1B, the RNA levels for the STRE genes CYC7, ALD3, and DDR2 were induced within minutes after shifting cells from 25° to 37° and increased to peak levels after 10–15 min. However, RNA levels then fell to steady-state levels slightly higher than the basal, uninduced level. This attenuation of the stress response may reflect the needs of the cell for a very rapid build-up of stress-protective proteins, but then a lower steady-state maintenance level.
Figure 1.—

The induction and attenuation of the STRE gene RNAs in response to heat shock and osmotic shock. Cell cultures of RZ53-6 were grown to midexponential phase in SC medium and then subjected to either heat (A) or osmotic (C) shock as described in materials and methods. Samples were taken at the times designated above the lanes, RNA was prepared, and RNA blots were carried out. The hybridization probes used are listed in materials and methods. The RNA for each gene is indicated at the right of the blot. Because ALD3 and ACT1 and CYC7 and DDR2 RNAs are similar in size, blots were first hybridized with the MSN2, ACT1, and CYC7 probes and then stripped and hybridized with the ALD3 and DDR2 probes. The hybridization bands were visualized and quantitated using a Molecular Dynamics (Sunnyvale, CA) Storm 850, and the analyses are shown in B for heat-shock induction and in D for osmotic-shock induction. The levels of RNA for each STRE gene were normalized to the ACT1 RNA level for each time point and then divided by the peak expression level for that RNA. This allowed the best visualization of the induction and attenuation patterns and placed less emphasis on the barely detectable levels of RNA present at time zero.
To explore the basis for this attenuation of the STRE response, we investigated the levels of the major activator of the STRE genes, Msn2. Msn2 is found in the cytoplasm under nonstress conditions, but, upon any one of a number of general stresses, it localizes to the nucleus to activate transcription. An HA epitope-tagged version of MSN2 was constructed and transformed into a wild-type yeast strain. The levels of Msn2-HA were determined during heat shock by immunoblotting, and the results are presented in Figure 2A. The control (lane C) contained samples prepared from cells lacking the epitope-tagged protein and served to unambiguously identify Msn2-HA. As a control for protein loading, the blots were stripped and reprobed using polyclonal antisera against the non-STRE protein eIF5A. After shifting cells from 25° to 37°, the levels of Msn2-HA fell rapidly, fourfold within 10 min. To ascertain that this decay was not a function of the epitope tag or its position, we constructed c-Msn2, where four copies of the c-myc epitope were fused to the N terminus of Msn2. Both the sequence of the epitope and its position (N terminal rather than C terminal) varied in this new fusion. Nonetheless, cellular levels of c-Msn2 showed a similar rapid decrease upon heat shock (eightfold in 10 min; Figure 2B) as was observed for Msn2-HA. Both the MSN2HA (see Figure 5A) and the c-MSN2 alleles (data not shown) complemented an msn2Δmsn4Δ double deletion to give wild-type MSN2 induction of the STRE genes.
Figure 2.—
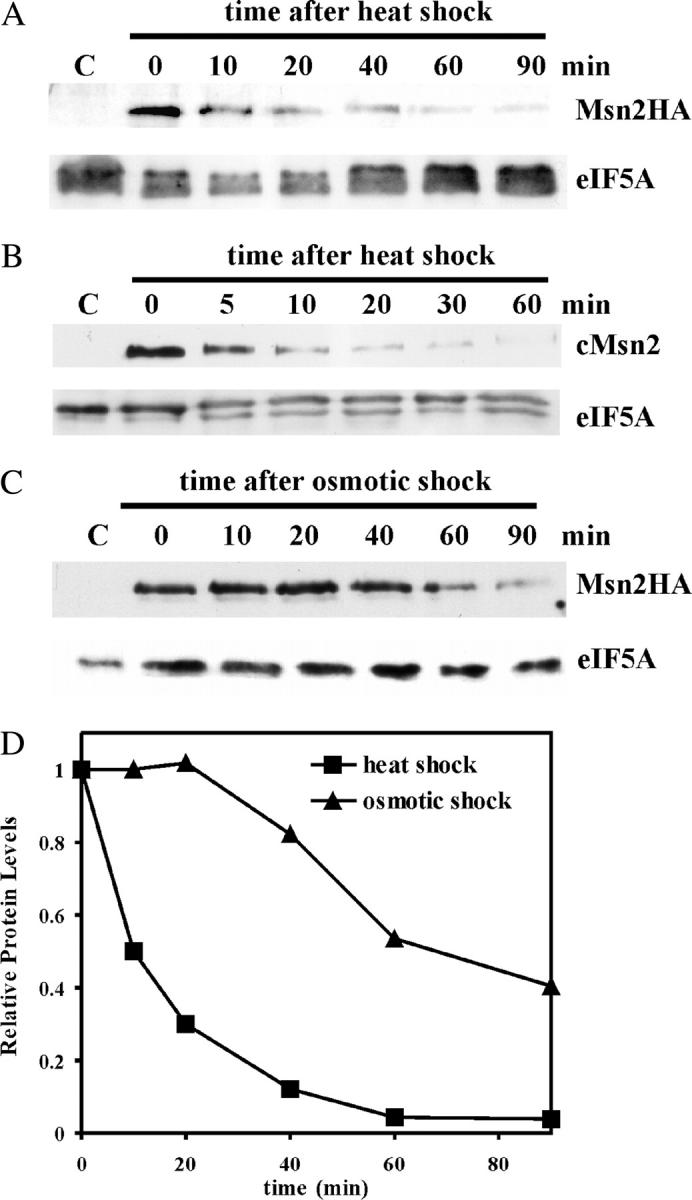
The disappearance of Msn2 upon heat shock and osmotic shock. Cell cultures of RZ53-6 transformed with either YCp(33)MSN2HA (A and C) or YCp(33)c-MSN2 (B) were grown to midexponential phase in SC minus uracil medium and then subjected to either heat (A and B) or osmotic (C) shock as described in materials and methods. Samples were taken at the times designated above the lanes and prepared for immunoblots as described in materials and methods. The lane designated C represents a sample taken from cells transformed with YCplac33, a plasmid lacking the epitope-tagged MSN2 allele. The immunoblots were first probed with anti-HA (A and C) or anti-c-myc (B) antiserum and then stripped and reprobed with eIF5A antiserum. The visualized bands are identified to the right of each blot. For the graph in D, the blots were quantitated by scanning the X-ray films and analyzing the band densities using the Molecular Dynamics ImageQuant 5.0 program. The Msn2 protein levels were normalized to eIF5A for each time point and then divided by the time zero value.
Figure 5.—

The effect of deletions of MSN2MSN4, MSN5, and SRB10 on heat-shock induction of CYC7-lacZ expression. The MZ171 series of yeast strains all contained the msn2Δmsn4Δ double deletions, an integrated CYC7-lacZ fusion, and the additional relevant markers: MZ171-60 (none), MZ171-48 (srb10Δ), MZ171-15 (msn5Δ), and MZ171-75 (msn5Δsrb10Δ). Each strain was transformed with YCp(33)MSN2HA (A) or YCpMSN2ΔE (B). MZ171-60 was also transformed with the vector YCplac33. Cell cultures were grown to midexponential phase in SC minus uracil medium and then subjected to heat shock as described inmaterials and methods. Samples were taken at various times after stress, and cellular levels of β-galactosidase were determined. The strain designations are as above with MZ171-60 YCplac33 transformants labeled msn2 msn4 and YCp(33)MSN2HA (A) and YCp(33)MSN2ΔE (B) transformants labeled WT. (○) WT; (⋄) msn5; (□) srb10; (♦) srb10 msn5; (•) msn2 msn4.
A similar attenuation of the STRE response and the stress-induced decay of Msn2 were observed with osmotic shock. To induce osmotic shock, cells were diluted with an equal volume of 2 m sorbitol, and samples were taken at various times for RNA and immunoblots. CYC7, ALD3, and DDR2 RNA levels showed a delayed induction compared to heat shock, but again the response was attenuated (Figure 1, C and D). Msn2-HA levels decreased after the addition of sorbitol with a delay that correlated with the delayed increase in RNA levels (Figure 2C). Thus, attenuation of the transcription and the drop in Msn2 levels appears to be a general property of the STRE response.
The stress-induced loss of Msn2 is due to stress-dependent Msn2 degradation:
The reduction of Msn2 protein levels could result from a stress-induced repression of MSN2 transcription combined with an intrinsically unstable Msn2 protein that rapidly disappears when it is no longer synthesized. However, as can be seen in Figure 1, A and C, MSN2 RNA levels were unaffected by heat shock or osmotic shock.
To demonstrate that Msn2 is degraded more rapidly under stress directly, pulse-chase experiments were carried out. For these experiments, cells carrying the MSN2HA allele on a multicopy plasmid were used to increase sensitivity. The increased copy number had little effect on the heat-shock-dependent disappearance of Msn2; the protein still disappeared from the cell within 60 min after cells were shifted to 37° as seen in the immunoblot in Figure 3A. For the pulse-chase experiment, cells were grown at 25°, and total protein was labeled with [35S]methionine for 5 min. An excess of cold methionine was added to terminate labeling, and after removing samples for the time zero and nonspecific control, the culture was split in two. In one, cell growth was continued at 25° (nonstress), and in the other, cells were shifted to 37° (heat shock). The label incorporated into total protein was monitored by trichloroacetic acid precipitation. No further incorporation occurred after the addition of unlabeled methionine, and no decrease in radioactivity in total protein occurred during the chase at either 25° or 37°. This lack of global protein decay was also evident in the image of total radioactive protein fractionated by SDS gel electrophoresis (Figure 3B). Thus, heat shock had no effect on total protein turnover. However, the amount of labeled Msn2 did disappear from the cells much more rapidly at 37° compared to 25° as determined after Msn2-HA immunoprecipitation and gel-fractionation (Figure 3C). The radioactivity in Msn2 was quantitated normalizing to the radioactivity in total soluble protein for each sample over three experiments. The results clearly show that Msn2 degraded at a faster rate under heat shock compared to nonstress conditions (Figure 3D).
Figure 3.—

The differential degradation of 35S-Msn2-HA under nonstress and heat shock. (A) MZ171-60 cells transformed with YEp(195)MSN2HA were subjected to heat shock for the designated times, and the protein was subjected to immunoblot as described in materials and methods. The immunoblots were first probed with anti-HA antibody and then stripped and reprobed with eIF5A antiserum. (B) MZ171-60 cells transformed with YEp(195)MSN2HA were labeled with [35S]methionine for 5 min, the label was chased, and growth was continued under nonstress (25°) and heat-shock (37°) conditions as described in materials and methods. Total soluble protein from each time point and the control (C) were size fractionated by SDS polyacrylamide gel electrophoresis. (C) The Msn2-HA in the cleared lysate samples from B was subjected to immunoprecipitation with anti-HA antibody (time points) or anti-c-myc 9E10 antibody (C, control) as described in materials and methods. The proteins in the immunoprecipitates were size fractionated by SDS polyacrylamide gel electrophoresis. The volumes of each sample loaded were normalized to the total trichloroacetic acid insoluble radioactivity in each cleared lysate. (D) The radioactivity in Msn2 in C was quantitated using a PhosphoImager. The values were normalized to radioactivity in total protein and then normalized to time zero.
Msn2 degradation is dependent on nuclear localization:
Msn2 cellular localization is regulated by the nuclear exportin Msn5. Under nonstress conditions, Msn2 is phosphorylated, and this modification is the signal for export by Msn5. Under stress, Msn2 is dephosphorylated and, consequently, is retained in the nucleus. We reasoned that the degradation of Msn2 might be dependent on nuclear localization and, therefore, investigated the stability of Msn2 in the absence of Msn5. Initially, we determined the levels of Msn2-HA by immunoblot in cells carrying a deletion of MSN5 and an MSN2HA construct (Figure 4A). For samples prepared from equivalent numbers of unstressed cells, the level of Msn2-HA in the msn5Δ strain was one-fifth that in wild type. This low level Msn2-HA remained unchanged during a heat-shock time course (Figure 4B). Thus, Msn2 cellular levels are maintained at a low, steady state when Msn2 is constitutively retained into the nucleus.
Figure 4.—

The effect of msn5Δ and srb10Δ mutations on Msn2 degradation. The yeast strains MZ161-8A (msn2Δ), MZ161-39B (msn2Δ msn5Δ), MZ161-41 (msn2Δsrb10Δ), and MZ161-8D (msn2Δ msn5Δsrb10Δ) were each transformed with YCp(33)MSN2HA. Cell cultures were grown on SC minus uracil medium to midexponential phase, and cell extracts were prepared for immunoblots as described in materials and methods. For each blot, the C lane contained a sample prepared from MZ161-8A cells transformed with the YCplac33 vector. Each immunoblot was probed sequentially with anti-HA and anti-eIF5A antisera. (A) Extracts were prepared from nonstressed cultures of the above cells. The numbers below the lanes represent the fraction of mutant to wild type of Msn2-HA protein after normalization to eIF5A for four experiments. (B) MZ161-39B transformants were subjected to heat shock, and samples were taken at the indicated times. (C) MZ161-41 transformants were subjected to heat shock, and samples were taken at the indicated times.
As might be expected, these low levels of Msn2 protein gave lower induction levels of the STRE genes. The induction of the CYC7-lacZ fusion in msn5Δ cells upon heat shock is shown in Figure 5A. The induction of the CYC7-lacZ fusion was significantly reduced in the deletion strain compared to the MSN5 wild-type cells.
The strains used in the experiments presented in Figure 5A carried a deletion of the wild-type MSN2 and MSN4 genes and were transformed with an MSN2HA construct. The wild-type induction profile compared to that in the cells transformed with the empty vector (msn2 msn4) indicated that the epitope-tagged protein complemented the mutant alleles.
The Srb10 repressor is required for the stress-dependent Msn2 degradation:
Expression of the STRE genes is repressed under nonstress conditions by the kinase Srb10, a member of the mediator complex. Srb10 prematurely phosphorylates the CTD tail of the large subunit of RNA polymerase II blocking preinitiation complex formation and also phosphorylates Msn2. Under stress, the cyclin-like Srb10 regulatory subunit Srb11 is degraded, allowing induction. The effect of the srb10Δ allele on the expression of the CYC7-lacZ fusion is shown in Figure 5A. Under nonstress conditions, loss of the repressor caused increased, but not fully induced, STRE gene expression. This derepression was dependent on Msn2 and Msn4; the triple srb10Δmsn2Δmsn4Δ deletion resulted in complete loss of induction (3.2 ± 0.4 units of β-galactosidase after 90 min of heat shock and 6.4 ± 1.1 units after 120 min of osmotic shock).
The phosphorylation of Msn2 by the Srb10 kinase was reported to be only partially responsible for the nuclear export of Msn2 in nonstressed cells, and, therefore, we investigated the effect of the loss of Srb10 on the stress-dependent degradation of Msn2. An srb10Δ strain carrying the MSN2HA construct was subjected to heat shock, and the levels of Msn2-HA were determined by immunoblot. The results shown in Figure 4A indicated that Msn2-HA levels were somewhat reduced in the deletion, but were not significantly reduced further upon stress (Figure 4C).
If Msn2 is not degraded in the srb10Δ mutant, then we would expect the attenuation of STRE gene transcription after stress to be lost. RNA blots were performed to determine the levels of the STRE mRNAs upon heat shock. As can be seen in the RNA blot in Figure 6A, the RNA levels for the STRE genes CYC7, ALD3, and DDR2 increased initially upon stress. This induction was expected due to the relocalization of Msn2 into the nucleus. It agrees with the increase observed for CYC7-lacZ expression in Figure 5A. The magnitude of the increase was not as great as that in wild-type cells (see Figure 1) due to the partially derepressed levels of RNA in unstressed srb10Δ cells. The attenuation of the STRE levels was greatly reduced; that is, RNA levels remained high during stress. Nonetheless, there was a modest, less than twofold, attenuation of transcription. The low level of this effect is more apparent in Figure 6C in which the data from Figures 1B and 6B are plotted together on a log scale. Although the attenuation in srb10Δ cells is clearly modest, it cannot be explained by the lack of reduction of Msn2 levels upon stress in the srb10Δ cells. The small effect could be due to changes in Msn2 activity or perhaps to Msn4 activity. Overall, however, the loss of most attenuation in this mutant correlated with the loss of Msn2 degradation.
Figure 6.—
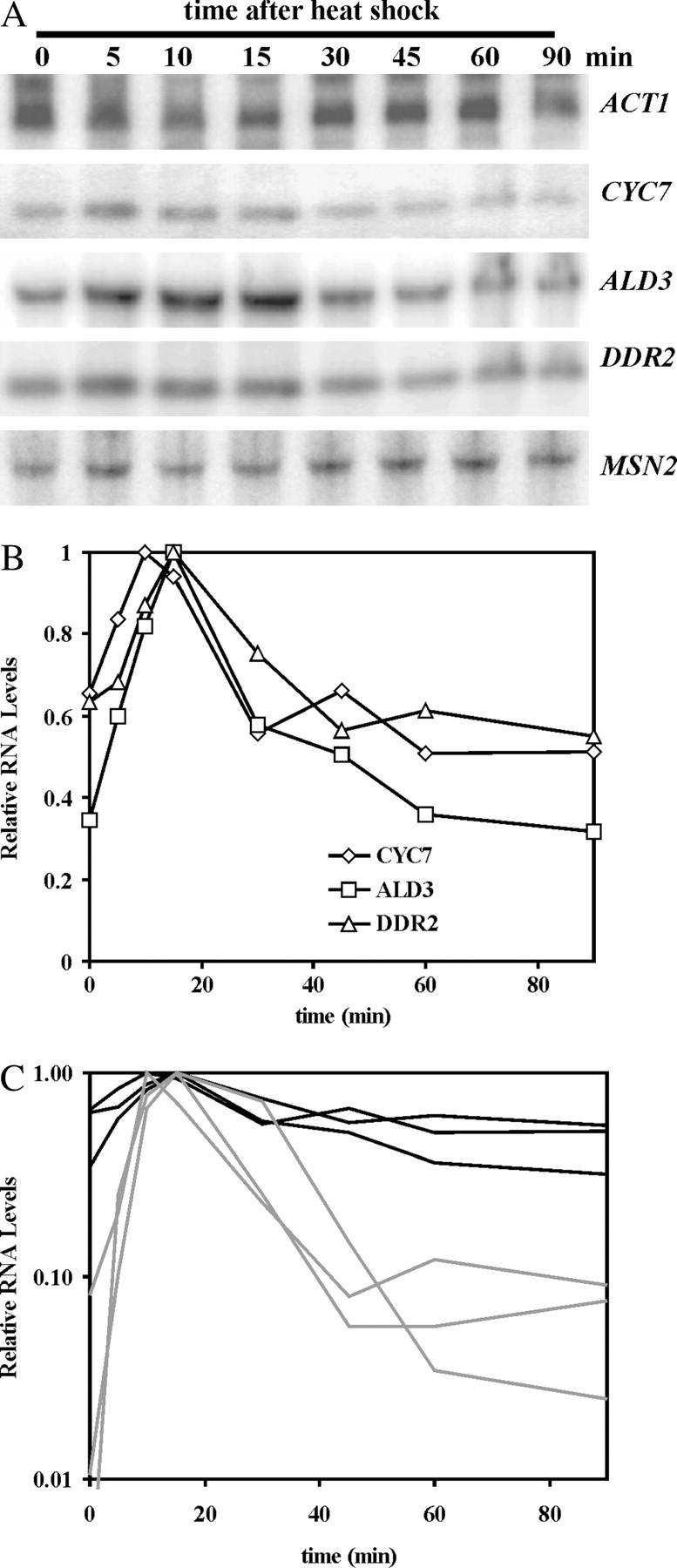
The induction and attenuation of STRE gene RNA in an srb10Δ mutant. Cell cultures of MZ161-41 transformed with YCp(33)MSN2HA were grown to midexponential phase in SC minus uracil medium and then subjected to heat shock as described in materials and methods. Samples were taken at the times designated above the lanes, RNA was prepared, and RNA blots were carried out. The hybridization probes used are listed in materials and methods. The RNA for each gene is indicated at the right of the blot. The hybridization bands were visualized (A) and quantitated (B) as described for Figure 1. The analyses shown in B represent the averages for two independent blots. The plot in C represents a replotting of the data in B (black lines) with that in Figure 1B (gray lines) on a log scale for the y-axis.
The interactions between the msn5Δ and the srb10Δ alleles were explored in terms of the effects on Msn2-HA stability and CYC7-lacZ expression. Msn2 is severely reduced in cells carrying both deletions as evidenced in Figure 4A, while CYC7-lacZ expression remained uninducible (Figure 5A). Thus, the effect of msn5Δ was epistatic to srb10Δ. Again the results are somewhat surprising. If Srb10 is required for Msn2 degradation, then why should the constitutive localization of Msn2 in the nucleus caused by the msn5 deletion be epistatic to the absence of Srb10? It appears that while Srb10 promotes the degradation of Msn2, it may not be the sole determinant. It appears likely that, even in the absence of Srb10, Msn2 is more susceptible to proteolysis in the nucleus than in the cytoplasm.
Separable Msn2 domains are targets for degradation and Srb10 repression:
In the initial experiments using the MNS2HA allele, we constructed an internal deletion to confirm the Msn2 identification in the immunoblots. This deletion took advantage of two in-frame EcoRI sites, which, when ligated together, resulted in the msn2ΔE allele lacking codons 304 to 478 of MSN2's 705 codons and containing the HA epitope tag. The protein produced retained the DNA binding domain at the C terminus and was capable of transcriptional activation of the CYC7-lacZ fusion (Figure 5B). However, β-galactosidase levels were high in unstressed, wild-type cells, showing the same uninduced level and heat-shock induction profile as that for wild-type Msn2-HA in srb10Δ cells. These results suggested that Msn2ΔE activity was not responsive to Srb10 repression. To determine if this was the case, the induction profile during heat shock was determined for the CYC7-lacZ fusion in srb10Δ cells. As seen in Figure 5B, there was no difference between the nonstress levels of β-galactosidase in the wild-type and deletion strains, indicating that the Msn2ΔE protein indeed lacked sequences responsive to Srb10 repression. The srb10Δ strain did show a somewhat altered induction profile, the significance of which is not clear at this time.
Since the rapid stress-dependent Msn2 degradation is also dependent on Srb10, it was of interest to determine whether the Msn2ΔE also lacked this Srb10 response. We investigated the heat-shock dependence of Msn2ΔE degradation (Figure 7A). As expected, the protein migrated faster than the wild-type protein in the denaturing gel (compare the second and third lanes). The levels of the deletion protein fell rapidly upon shifting the temperature from 25° to 37°. The rate of degradation was similar to that observed for the Msn2-HA protein, indicating that the proteolytic target sequence(s) remained in the deletion protein. Furthermore, the degradation of the Msn2ΔE protein was dependent upon Srb10 as seen in Figure 7B, indicating that the Srb10-dependent degradation sequences remained. Thus, it appears that there are distinct sequences in Msn2 that respond to Srb10 repression vs. Srb10 signaled degradation.
Figure 7.—
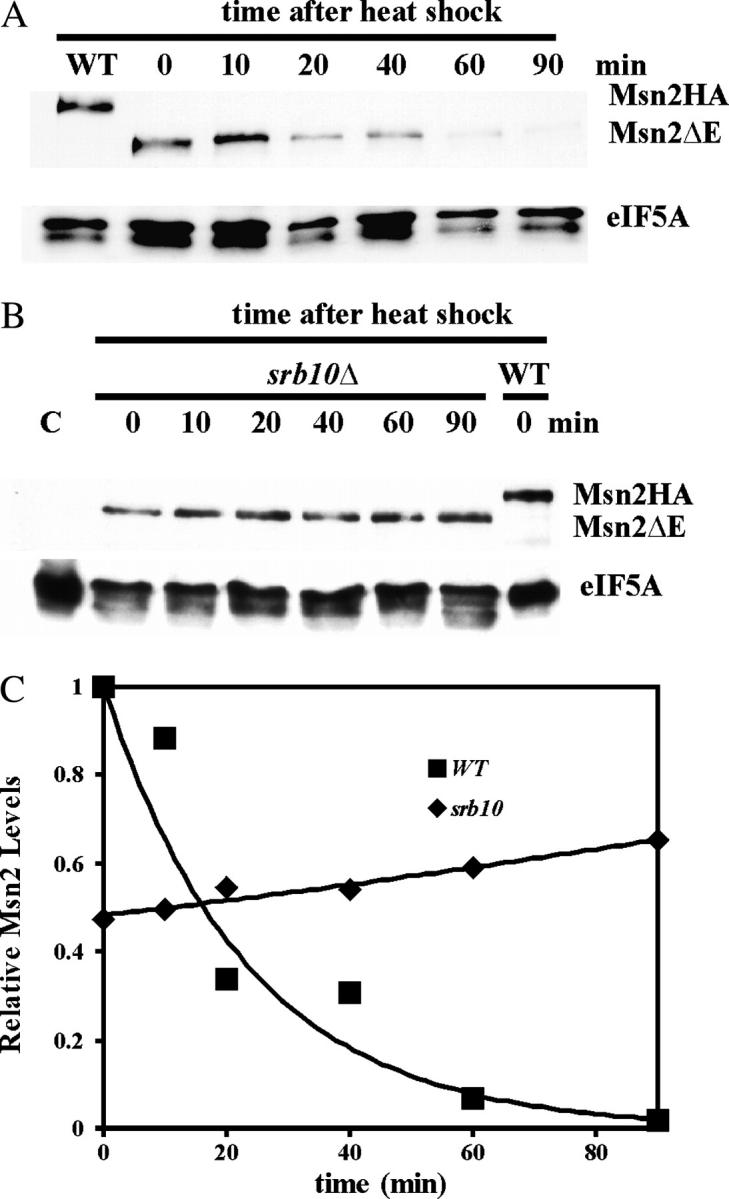
The effect of the internal ΔE deletion on Msn2 degradation. The yeast strains MZ161-8A (msn2Δ; A) and MZ161-41 (msn2Δsrb10Δ; B) were each transformed with YCp(33)MSN2ΔE. In addition, MZ161-8A was transformed with YCplac33 (for lanes marked C) or YCp(33)MSN2HA (for lanes marked WT). Cell cultures were grown on SC minus uracil medium to midexponential phase, and cell extracts were prepared for immunoblots as described in materials and methods. Each immunoblot was probed sequentially with anti-HA and anti-eIF5A antisera. The positions of the full-length Msn2HA and the ΔE deletion mutant are shown to the right of the blot.
Finally, we determined that Msn2ΔE, like the wild-type Msn2-HA protein, failed to induce CYC7-lacZ expression during heat shock in msn5Δ and srb10Δmsn5Δ cells (Figure 5B). This lack of STRE gene induction in the msn5Δ mutant suggests that the Msn2ΔE protein is still subject to nuclear-dependent degradation.
DISCUSSION
The induction of ∼200 general stress response genes represents the cell's response to an immediate environmental crisis. The protective proteins must accumulate very rapidly, which requires a burst of mRNA synthesis. However, as cells adapt to the stress, a lower steady-state level of these proteins may be required. Gasch et al. (2000) have proposed that this initial burst of synthesis represents a “loading dose.” They also note from their expression array data that the stress response can vary from gene to gene. These differences, at least in some cases, are due to combinations of transcription factors that control the response of specific genes to specific stresses and severity of stresses. For example, some Msn2-regulated genes are also induced by Hot1 while others are also regulated by Yap1 (Gasch et al. 2000; Rep et al. 2000). The CYC7 gene used in this study is regulated weakly by Hap1, an oxygen-dependent transcriptional activator, and Rox1, an oxygen-dependent repressor (Zitomer et al. 1987; Cerdan and Zitomer 1988; Lowry and Zitomer 1988), in addition to Msn2 and Msn4. Hence, each STRE gene may have a different induction/attenuation profile. The major question addressed here is: How is the Msn2-dependent transitory induction of the STRE genes achieved? The answer appears to be a result of the combination of cellular localization and nuclear-dependent degradation of the Msn2 activator and transcriptional repression by the Srb10 kinase.
The results reported here demonstrate that Msn2 is degraded by a nuclear-dependent proteolytic system. Chi et al. (2001) reported a contradictory finding. Using a similarly epitope-tagged version of MSN2, they stated that there was no change in the stability of Msn2 upon heat shock. They did not present the data, but judging from their general experimental protocol, pulse-chase experiments were carried out similar to those presented here. In our experience, Msn2 is highly labile in cell extracts, and it is possible that the rapid degradation after cell breakage masked the differential protein levels in stressed and unstressed cells. We found that accurate measurements of differential Msn2 cellular levels required chilling cells rapidly and using a battery of protease inhibitors. By this approach, we have demonstrated that prelabeled HA epitope-tagged Msn2 disappeared more rapidly from heat-shocked vs. nonstress cells. In addition, we showed that two different epitope-tagged constructs of Msn2 rapidly disappeared from the cell upon heat shock or osmotic shock. The disappearance of the protein occurred despite the persistence of the MSN2 mRNA. We propose a nuclear-dependent proteolytic system because Msn2 degradation occurred constitutively in an msn5Δ strain. Msn5 is an exportin that is responsible for transporting Msn2 out of the nucleus in unstressed cells (Gorner et al. 2002). Deletion of MSN5 causes constitutive nuclear localization of an overexpressed Msn2-GFP fusion, but in our constructs, Msn2 levels were substantially reduced, reflecting the constitutive degradation. Chi et al. (2001) also reported that Msn2 was partially excluded from the nucleus in a fraction of unstressed srb10Δ cells, and we found that the Msn2 level was somewhat reduced under nonstress conditions in this mutant. Thus, there is an excellent correlation between the retention of Msn2 in the nucleus and reduced protein levels.
There is precedent for nuclear-dependent degradation of proteins, and there are at least two cases that bear some parallels to the Msn2 degradation reported here. Far1 is a cyclin-dependent kinase inhibitor that regulates the cell cycle in response to pheromone and plays a role in cell polarity. It is specifically degraded in the nucleus in a Cdc4 ubiquitin ligase-26S proteosome-dependent pathway (Blondel et al. 1999, 2000). Like Msn2, Far1 is stabilized by nuclear export via Msn5. The transcriptional activator Gcn4 is degraded in the nucleus, again via the Cdc4 pathway (Chi et al. 2001). Interestingly, like Msn2, Gcn4 is phosphorylated by Srb10, and it is this phosphorylation, at least in part, that targets it for degradation. This Srb10 targeting appears to be the case also for Msn2; Msn2 is not rapidly degraded in srb10Δ cells. Chi et al. (2001) reported that Srb10 phosphorylated Msn2 in vivo immediately after heat shock, which correlates with its nuclear import and degradation. This phosphorylation was only transitory, which might reflect the findings that Srb11, the Srb10 regulatory subunit, is degraded upon stress (Cooper et al. 1997, 1999). Hence, the immediate, rapid drop in Msn2 levels might be due to an Srb10/11-dependent rapid degradation, but the continuing lower basal levels of Msn2 that persist long into stress may be due to a higher degradation rate for Msn2 in the nucleus compared to that in the cytoplasm. Therefore, these three systems that control Far1, Gcn4, and Msn2 levels use shared components to different ends.
In addition to activation by Msn2 and Msn4, the STRE genes are repressed by Srb10 (Cooper et al. 1997, 1999; Holstege et al. 1998; Cohen et al. 2003). Deletion of the SRB10 gene resulted in a partial derepression of the CYC7-lacZ gene, and this derepression still required the function of Msn2 or Msn4. Srb10 repression is relieved upon stress by the degradation of the cyclin-like Srb11 regulatory subunit of Srb10 (Cooper et al. 1997). Srb10 has been proposed to function in at least two ways. First, as indicated above, it phosphorylates Msn2 (Chi et al. 2001). While this phosphorylation was observed only immediately after stress, it is quite possible that it occurs under nonstress conditions but was not detected by pulse-labeling experiments because so little of the cellular Msn2 is found in the nucleus; the pool that might be in the nucleus and phosphorylated by Srb10 at any instant would be either quickly exported by Msn5 (and dephosphorylated at the Srb10 site) or degraded. Consequently, only the large nuclear pool that accumulates immediately after stress would be detected. Thus, repression by direct phosphorylation of Msn2 by Srb10 cannot be ruled out. Second, Srb10 can prematurely phosphorylate the CTD tail of the large subunit of RNA polymerase II, resulting in the failure to form an active preinitiation complex (Hengartner et al. 1998). Such a mechanism for repression requires a recognition of STRE genes to prevent inhibition of all polII transcription. The only defining sequence of an STRE gene is the STRE element. We observed that an STRE-lacZ fusion contains five STRE elements as the sole regulatory elements fused to a non-STRE gene TATA box and lacZ coding sequence is both activated by Msn2 and repressed by Srb10 (our unpublished data). Thus, if the CTD phosphorylation activity of Srb10 is responsible for repression of the STRE genes, it must be regulated by a protein that recognizes the STRE element, possibly Msn2 and/or Msn4.
Repression of Msn2 activation of the STRE genes by Srb10 is independent of Msn2 degradation. This conclusion follows logically from the fact that the positive regulator of Srb10, Srb11, is not present in cells during stress when Msn2 is degraded and was experimentally confirmed by our finding that an internal deletion of MSN2, MSN2ΔE, generated a protein that was degraded upon stress but was not repressed by Srb10. This finding also suggests that Srb10 repression acts on Msn2 rather than another protein bound to the STRE elements.
Combining all these features of STRE gene regulation, we propose the following model for induction and attenuation of the STRE response. Under nonstress conditions, the cells are preloaded with Msn2. Preloading requires that Msn2 be phosphorylated and exported from the nucleus by Msn5 to protect it from degradation. This phosphorylation is cAPK dependent, but Srb10 may also play a minor role. Also under nonstress conditions, the Srb10 kinase prevents preinitiation complex formation at STRE genes by premature phosphorylation of polII. This repression may require a small amount of Msn2 in the nucleus to define the STRE genes for Srb10. This communication bears the Srb10 signature phosphorylation of Msn2, which aids in Msn2 export/degradation, thereby keeping the nonstress nuclear pool of Msn2 low. Upon stress, the Srb10/Srb11 repression is relieved, perhaps by its dissociation from the mediator. cPKA phosphorylation of Msn2 is turned off, resulting in a rapid accumulation of the protein in the nucleus through a loss of export. Msn2 can then activate transcription, giving a burst of STRE gene transcription. However, as Msn2 relocalizes to the nucleus, its levels decrease rapidly via an Srb10-dependent degradation. Consequently, activator levels fall and transcription of the STRE genes is downregulated. If the stress conditions persist, Srb10 becomes inactive through the degradation of its Srb11 regulatory subunit. Consequently, STRE gene expression persists at a low level as the balance between the synthesis and Srb10-independent, nuclear-dependent degradation of Msn2 leads to a new steady-state level of the activator.
The role of Msn4 in this response is not known. While Msn4 is often described as an iso-form of Msn2, the sequences of these proteins are quite divergent outside the C-terminal DNA binding domain. Therefore, it is quite possible that Msn4 plays a somewhat different role in the STRE response. This question is currently under investigation.
Acknowledgments
This study was supported by a grant from the National Institutes of Health. We thank Margye Zitomer for the construction of strains and Evelina Loghin for technical assistance.
References
- Ausubel, F. M., R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman et al., 2000 Current Protocols in Molecular Biology. John Wiley & Sons, New York.
- Beck, T., and M. N. Hall, 1999. The TOR signalling pathway controls nuclear localization of nutrient-regulated transcription factors. Nature 402: 689–692. [DOI] [PubMed] [Google Scholar]
- Blondel, M., P. M. Alepuz, L. S. Huang, S. Shaham, G. Ammerer et al., 1999. Nuclear export of Far1p in response to pheromones requires the export receptor Msn5p/Ste21p. Genes Dev. 13: 2284–2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blondel, M., J. M. Galan, Y. Chi, C. Lafourcade, C. Longaretti et al., 2000. Nuclear-specific degradation of Far1 is controlled by the localization of the F-box protein Cdc4. EMBO J. 19: 6085–6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boustany, L. M., and M. S. Cyert, 2002. Calcineurin-dependent regulation of Crz1p nuclear export requires Msn5p and a conserved calcineurin docking site. Genes Dev. 16: 608–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boy-Marcotte, E., M. Perrot, F. Bussereau, H. Boucherie and M. Jacquet, 1998. Msn2p and Msn4p control a large number of genes induced at the diauxic transition which are repressed by cyclic AMP in Saccharomyces cerevisiae. J. Bacteriol. 180: 1044–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Causton, H. C., B. Ren, S. S. Koh, C. T. Harbison, E. Kanin et al., 2001. Remodeling of yeast genome expression in response to environmental changes. Mol. Biol. Cell 12: 323–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerdan, M. E., and R. S. Zitomer, 1988. Oxygen-dependent upstream activation sites of Saccharomyces cerevisiae cytochrome c genes are related forms of the same sequence. Mol. Cell. Biol. 8: 2275–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, D. C., B. C. Yang and T. T. Kuo, 1992. One-step transformation of yeast in stationary phase. Curr. Genet. 21: 83–84. [DOI] [PubMed] [Google Scholar]
- Chi, Y., M. J. Huddleston, X. Zhang, R. A. Young, R. S. Annan et al., 2001. Negative regulation of Gcn4 and Msn2 transcription factors by Srb10 cyclin-dependent kinase. Genes Dev. 15: 1078–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen, T. J., K. Lee, L. H. Rutkowski and R. Strich, 2003. Ask10p mediates the oxidative stress-induced destruction of the Saccharomyces cerevisiae C-type cyclin Ume3p/Srb11p. Eukaryot. Cell 2: 962–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper, K. F., M. J. Mallory, J. B. Smith and R. Strich, 1997. Stress and developmental regulation of the yeast C-type cyclin Ume3p (Srb11p/Ssn8p). EMBO J. 16: 4665–4675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper, K. F., M. J. Mallory and R. Strich, 1999. Oxidative stress-induced destruction of the yeast C-type cyclin Ume3p requires phosphatidylinositol-specific phospholipase C and the 26S proteasome. Mol. Cell. Biol. 19: 3338–3348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVit, M. J., and M. Johnston, 1999. The nuclear exportin Msn5 is required for nuclear export of the Mig1 glucose repressor of Saccharomyces cerevisiae. Curr. Biol. 9: 1231–1241. [DOI] [PubMed] [Google Scholar]
- Estruch, F., 2000. Stress-controlled transcription factors, stress-induced genes and stress tolerance in budding yeast. FEMS Microbiol. Rev. 24: 469–486. [DOI] [PubMed] [Google Scholar]
- Garreau, H., R. N. Hasan, G. Renault, F. Estruch, E. Boy-Marcotte et al., 2000. Hyperphosphorylation of Msn2p and Msn4p in response to heat shock and the diauxic shift is inhibited by cAMP in Saccharomyces cerevisiae. Microbiology 146(Pt. 9): 2113–2120. [DOI] [PubMed] [Google Scholar]
- Gasch, A. P., P. T. Spellman, C. M. Kao, O. Carmel-Harel, M. B. Eisen et al., 2000. Genomic expression programs in the response of yeast cells to environmental changes. Mol. Biol. Cell 11: 4241–4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz, R. D., and A. Sugino, 1988. New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene 74: 527–534. [DOI] [PubMed] [Google Scholar]
- Gorner, W., E. Durchschlag, M. T. Martinez-Pastor, F. Estruch, G. Ammerer et al., 1998. Nuclear localization of the C2H2 zinc finger protein Msn2p is regulated by stress and protein kinase A activity. Genes Dev. 12: 586–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorner, W., E. Durchschlag, J. Wolf, E. L. Brown, G. Ammerer et al., 2002. Acute glucose starvation activates the nuclear localization signal of a stress-specific yeast transcription factor. EMBO J. 21: 135–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengartner, C. J., V. E. Myer, S. M. Liao, C. J. Wilson, S. S. Koh et al., 1998. Temporal regulation of RNA polymerase II by Srb10 and Kin28 cyclin-dependent kinases. Mol. Cell 2: 43–53. [DOI] [PubMed] [Google Scholar]
- Hohmann, S., 2002. Osmotic stress signaling and osmoadaptation in yeasts. Microbiol. Mol. Biol. Rev. 66: 300–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holstege, F. C., E. G. Jennings, J. J. Wyrick, T. I. Lee, C. J. Hengartner et al., 1998. Dissecting the regulatory circuitry of a eukaryotic genome. Cell 95: 717–728. [DOI] [PubMed] [Google Scholar]
- Kaffman, A., N. M. Rank, E. M. O'Neill, L. S. Huang and E. K. O'Shea, 1998. The receptor Msn5 exports the phosphorylated transcription factor Pho4 out of the nucleus. Nature 396: 482–486. [DOI] [PubMed] [Google Scholar]
- Kaiser, C., S. Michaelis and A. Mitchell, 1994 Methods in Yeast Genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Kaplun, L., Y. Ivantsiv, A. Bakhrat and D. Raveh, 2003. DNA damage response-mediated degradation of Ho endonuclease via the ubiquitin system involves its nuclear export. J. Biol. Chem. 278: 48727–48734. [DOI] [PubMed] [Google Scholar]
- Komeili, A., K. P. Wedaman, E. K. O'Shea and T. Powers, 2000. Mechanism of metabolic control. Target of rapamycin signaling links nitrogen quality to the activity of the Rtg1 and Rtg3 transcription factors. J. Cell Biol. 151: 863–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao, S. M., J. Zhang, D. A. Jeffery, A. J. Koleske, C. M. Thompson et al., 1995. A kinase-cyclin pair in the RNA polymerase II holoenzyme. Nature 374: 193–196. [DOI] [PubMed] [Google Scholar]
- Lowry, C. V., and R. S. Zitomer, 1988. ROX1 encodes a heme-induced repression factor regulating ANB1 and CYC7 of Saccharomyces cerevisiae. Mol. Cell. Biol. 8: 4651–4658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mager, W. H., and A. J. De Kruijff, 1995. Stress-induced transcriptional activation. Microbiol. Rev. 59: 506–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler, G., C. Schuller, G. Adam and H. Ruis, 1993. A Saccharomyces cerevisiae UAS element controlled by protein kinase A activates transcription in response to a variety of stress conditions. EMBO J. 12: 1997–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Pastor, M. T., G. Marchler, C. Schuller, A. Marchler-Bauer, H. Ruis et al., 1996. The Saccharomyces cerevisiae zinc finger proteins Msn2p and Msn4p are required for transcriptional induction through the stress response element (STRE). EMBO J. 15: 2227–2235. [PMC free article] [PubMed] [Google Scholar]
- Mennella, T. A., L. G. Klinkenberg and R. S. Zitomer, 2003. Recruitment of Tup1-Ssn6 by yeast hypoxic genes and chromatin-independent exclusion of TATA binding protein. Eukaryot. Cell 2: 1288–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moye-Rowley, W. S., 2002. Transcription factors regulating the response to oxidative stress in yeast. Antioxid. Redox. Signal. 4: 123–140. [DOI] [PubMed] [Google Scholar]
- Myers, L. C., and R. D. Kornberg, 2000. Mediator of transcriptional regulation. Annu. Rev. Biochem. 69: 729–749. [DOI] [PubMed] [Google Scholar]
- O'Rourke, S. M., I. Herskowitz and E. K. O'Shea, 2002. Yeast go the whole HOG for the hyperosmotic response. Trends Genet. 18: 405–412. [DOI] [PubMed] [Google Scholar]
- Queralt, E., and J. C. Igual, 2003. Cell cycle activation of the Swi6p transcription factor is linked to nucleocytoplasmic shuttling. Mol. Cell. Biol. 23: 3126–3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rep, M., M. Krantz, J. M. Thevelein and S. Hohmann, 2000. The transcriptional response of Saccharomyces cerevisiae to osmotic shock. Hot1p and Msn2p/Msn4p are required for the induction of subsets of high osmolarity glycerol pathway-dependent genes. J. Biol. Chem. 275: 8290–8300. [DOI] [PubMed] [Google Scholar]
- Rothstein, R. J., 1983. One-step gene disruption in yeast. Methods Enzymol. 101: 202–211. [DOI] [PubMed] [Google Scholar]
- Ruis, H., and C. Schuller, 1995. Stress signaling in yeast. BioEssays 17: 959–965. [DOI] [PubMed] [Google Scholar]
- Schmitt, A. P., and K. McEntee, 1996. Msn2p, a zinc finger DNA-binding protein, is the transcriptional activator of the multistress response in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 93: 5777–5782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wach, A., A. Brachat, C. Alberti-Segui, C. Rebischung and P. Philippsen, 1997. Heterologous HIS3 marker and GFP reporter modules for PCR-targeting in Saccharomyces cerevisiae. Yeast 13: 1065–1075. [DOI] [PubMed] [Google Scholar]
- Wright, C. F., and R. S. Zitomer, 1984. A positive regulatory site and a negative regulatory site control the expression of the Saccharomyces cerevisiae CYC7 gene. Mol. Cell. Biol. 4: 2023–2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanisch-Perron, C., J. Vieira and J. Messing, 1985. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene 33: 103–119. [DOI] [PubMed] [Google Scholar]
- Zitomer, R. S., D. L. Montgomery, D. L. Nichols and B. D. Hall, 1979. Transcriptional regulation of the yeast cytochrome c gene. Proc. Natl. Acad. Sci. USA 76: 3627–3631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zitomer, R. S., J. W. Sellers, D. W. McCarter, G. A. Hastings, P. Wick et al., 1987. Elements involved in oxygen regulation of the Saccharomyces cerevisiae CYC7 gene. Mol. Cell. Biol. 7: 2212–2220. [DOI] [PMC free article] [PubMed] [Google Scholar]