

Abstract
Loline alkaloids are produced by mutualistic fungi symbiotic with grasses, and they protect the host plants from insects. Here we identify in the fungal symbiont, Neotyphodium uncinatum, two homologous gene clusters (LOL-1 and LOL-2) associated with loline-alkaloid production. Nine genes were identified in a 25-kb region of LOL-1 and designated (in order) lolF-1, lolC-1, lolD-1, lolO-1, lolA-1, lolU-1, lolP-1, lolT-1, and lolE-1. LOL-2 contained the homologs lolC-2 through lolE-2 in the same order and orientation. Also identified was lolF-2, but its possible linkage with either cluster was undetermined. Most lol genes were regulated in N. uncinatum and N. coenophialum, and all were expressed concomitantly with loline-alkaloid biosynthesis. A lolC-2 RNA-interference (RNAi) construct was introduced into N. uncinatum, and in two independent transformants, RNAi significantly decreased lolC expression (P < 0.01) and loline-alkaloid accumulation in culture (P < 0.001) compared to vector-only controls, indicating involvement of lolC in biosynthesis of lolines. The predicted LolU protein has a DNA-binding site signature, and the relationships of other lol-gene products indicate that the pathway has evolved from various different primary and secondary biosynthesis pathways.
SEED-BORNE endophytic fungi—specifically the Epichloë species (asexual states, Neotyphodium species)—in symbiosis with cool-season grasses (Poaceae subfam. Poöideae) can impart to those grasses a variety of fitness enhancements including resistance to vertebrate and invertebrate herbivores, resistance to pathogens and parasites, enhanced phosphate uptake and nitrogen utilization, and increased tolerance of drought and heat (Bush et al. 1997; Malinowski and Belesky 2000; Schardl et al. 2004). Loline alkaloids produced by several Neotyphodium and Epichloë species are potent, broad-spectrum insecticides (Riedell et al. 1991; Dougherty et al. 1998; Wilkinson et al. 2000), have little or no antimammalian activities (Jackson et al. 1996), and sometimes accumulate in the plant to levels up to 20 mg g−1 plant dry weight (DW; Craven et al. 2001). These alkaloids have an unusual structure, comprising a saturated 1-aminopyrrolizidine-ring system, with a highly strained ether bridge between C-2 and C-7 (Petroski et al. 1989). Lolines are almost exclusively found in these grass-endophyte symbioses (Hartmann and Witte 1995; Bush et al. 1997); outside of the grasses they have been identified in only a few plant species in the families Fabaceae and Convolvulaceae (Hartmann and Witte 1995; Tofern et al. 1999). Genetic tests have confirmed linkage between capability of endophytes to produce lolines in planta and their bioprotective effects against aphids (Wilkinson et al. 2000). Improved survival under drought and/or competition of grass-endophyte symbiota possessing lolines has raised the possibility of additional roles of these alkaloids in host plant fitness and persistence (Malinowski and Belesky 2000; Schardl et al. 2004).
The biochemical pathway for the lolines is so far unknown. On the basis of certain structural similarities of lolines with plant pyrrolizidines (Hartmann and Witte 1995), Bush et al. (1993) proposed that lolines are produced by a pathway involving polyamines such as spermidine. However, results of precursor feeding studies have now ruled out this possibility and suggest that lolines are formed by a novel biosynthetic pathway from the amino acids l-proline and l-homoserine (Blankenship et al. 2005).
We have previously identified two genes, lolA and lolC, showing strong upregulation in loline-alkaloid-producing Neotyphodium uncinatum cultures and strict association with in symbio loline-alkaloid-production phenotypes among isolates of different endophyte species (Spiering et al. 2002). In this study we show that two gene clusters in the genome of N. uncinatum contain homologs of lolA and lolC, along with several additional genes whose predicted products showed relationships to enzymes typical of primary or secondary metabolic pathways. A functional test performed with RNA interference (RNAi) in N. uncinatum confirmed involvement of lolC in loline-alkaloid production. These findings provide first insight into the molecular genetics of loline-alkaloid production and constitute an important basis for detailed studies of the biosynthetic pathway and ecological roles of the lolines.
MATERIALS AND METHODS
Fungal strains and growth conditions:
N. uncinatum CBS 102646 (deposited in Centraalbureau voor Schimmelculture) and N. coenophialum ATCC 62374 (deposited in the American Type Culture Collection) were grown in shake-cultures as described by Blankenship et al. (2001). Loline-alkaloid production by N. uncinatum was induced in minimal medium (MM; Blankenship et al. 2001) with 15 mm urea and 20 mm sucrose as nitrogen and carbon sources, respectively. To suppress loline-alkaloid production, the fungus was grown in complex medium (CM) consisting of potato dextrose broth (PDB; Difco, Detroit) diluted 1:1 with MM (Spiering et al. 2002). In all experiments, three replicate culture plates of each treatment were used for loline-alkaloid analysis. For in symbio experiments, Lolium pratense (Festuca pratensis) and L. arundinaceum (F. arundinacea) plants symbiotic with N. uncinatum CBS 102646 and N. coenophialum ATCC 90664, respectively, were grown in the greenhouse.
Bacterial strains and plasmids:
The cosmid pMOCosX (Orbach 1994) and plasmid pCB1004 (Carroll et al. 1994) were used in genomic library construction and DNA cloning, respectively, in Escherichia coli strain XL1-Blue (Bullock et al. 1987). These plasmids contain the hygromycin B-phosphotransferase (hph) gene under control of constitutive promoters of the cpc-1 gene in Neurospora crassa (pMOCosX) and the trpC gene in Aspergillus nidulans (pCB1004; hph cassette). Bacterial cultures were grown on LB plates or in LB medium with shaking (200 rpm) at 37° for 16 hr. Plasmid DNA was isolated from bacterial cultures by the method of Ahn et al. (2000).
Fungal genomic DNA isolation and genome walking:
Fungal genomic DNA was isolated by the method of Al-Samarrai and Schmid (2000). To amplify long (>1 kb) genomic DNA segments, 5 ng of genomic DNA was amplified in 50-μl reaction volumes, by using the LA-PCR version 2.1 kit per manufacturer's instructions (Takara Shuzo, Otsu, Shiga, Japan). Reactions were performed in a GeneAmp PCR System 2400 thermocycler (Perkin-Elmer, Boston). PCR conditions were 95° for 60 sec; 7 cycles of 95° for 20 sec and 70° for 8 min; 28 cycles of 95° for 20 sec and 67° for 8 min; and a final 16 min at 67°. To PCR walk into unknown genomic regions, the Universal Genome Walker kit (CLONTECH, Palo Alto, CA) and N. uncinatum or N. coenophialum genomic DNA were used and manufacturer's instructions were followed for generating genome-walker libraries. PCR primers for genome walking were designed to anneal to sites in known genomic DNA regions and synthesized by Integrated DNA Technologies (IDT; Coralville, IA). For gene-copy-specific genome-walking PCRs, a single nucleotide polymorphism (SNP) was incorporated into the terminal 3′-nucleotide position of each genome-walking primer. The amplified DNA fragments were purified with the QIAquick PCR purification kit (QIAGEN, Valencia, CA) following the manufacturer's instructions and sequenced as described below.
Genomic DNA library construction and screening and cosmid sequencing:
A partial N. uncinatum genomic DNA library was constructed in cosmid vector pMOCosX as previously described for construction of a genomic library from Claviceps purpurea (Wang et al. 2004). A total of 2613 primary clones were obtained and arrayed in 384-well microtiter plates. To identify cosmid clones containing lolA and lolC genes, plates were PCR screened by the method of Wang et al. (2004), with primers lolA-3′ and lolA-5′ (for detection of lolA) and primers lolC-3′a and lolC-5′a (for detection of lolC). To obtain random Tn7 insertions for sequencing, the TnsABC transposase/Tn7 transposon-based genome-priming system (GPS-1; New England Biolabs, Beverly, MA) was used following the manufacturer's instructions. GPS-tagged cosmids were electroporated into E. coli XL1-Blue, and colonies were selected on kanamycin and ampicillin. Approximately 200 independent GPS-tagged clones were obtained and partially sequenced. To obtain a contiguous sequence of the cosmid clone, sequences from all GPS-tagged clones were assembled using PhredPhrap and checked in Consed (University of Washington, http://www.phrap.org/). Gaps between contigs were closed by primer walking.
DNA sequencing:
Sequencing of DNA was performed with the BigDye Version 3 Terminator cycle sequence kit (Applied Biosystems, Foster City, CA) on an Applied Biosystems 310 or 3100 DNA analyzer. PCR fragments of genomic DNA were sequenced with the PCR primers and with primers designed on sequences thereby obtained. For high-throughput sequencing of genomic library clones, the CEQ2000XL DNA analysis system (Beckman Coulter, Fullerton, CA) with the CEQ DTCS-Quick Start kit (Beckman Coulter) was used.
Identification of possible open reading frames and protein signature sites:
To search for possible open reading frames (ORFs) in both strands of genomic sequence, with the search parameters for the N. crassa genome, genomic DNA sequences were entered into the FGENESH gene prediction program (http://www.softberry.com/berry.phtml?topic=fgenesh&group=programs&subgroup=gfind; Salamov and Solovyev 2000). BLASTX similarity searches (Altschul et al. 1997) in the NCBI nr database (http://www.ncbi.nlm.nih.gov/BLAST/) were also performed to help identify coding regions. To search for highly significant matches, the BLASTX searches were first performed on long (>20 kb) contiguous sequences; to identify possible DNA regions having lower significant similarity (E > 10−7) to known sequences masked by matches with higher similarity, BLASTX searches were repeated with smaller (2–3 kb) segments from the large contiguous sequences. On the basis of the gene-prediction and BLASTX search results, primers were designed on the sequence just outside of the ORFs predicted by FGENESH (∼20–100 bp up- or downstream of the predicted 5′- and 3′-ends, respectively). To check expression of the predicted genes, the primers were used in reverse transcription PCR (RT-PCR) on RNA from loline-alkaloid-producing fungal cultures or endophyte-infected plant tissues. The cDNAs thus obtained were sequenced. cDNA 5′- and 3′-ends were amplified by rapid amplification of cDNA ends (RACE) methods (Schaefer 1995), using gene-specific primers (GSPs) oriented in the 5′- or 3′-direction and CLONTECH's universal PCR primer or 5′- or 3′-universal primer in the reverse transcription of RNA. Alternatively, cDNA ends were amplified by using the GSPs in combination with the vector primers tripleX 5′ amp or tripleX 3′ LD and cDNA from a previously constructed cDNA library (Spiering et al. 2002). Approximately 20 ng of total cDNA was used in pre-PCR with the GSPs in 20-μl reactions with thermal parameters of 95° for 60 sec and then 25 cycles of 95° for 20 sec, 64° for 20 sec, and 72° for 60 sec. The products were diluted 1:10 and 2 μl was used in 40-μl PCR reactions with a GSP and the corresponding RACE or vector primer (95° for 1 min; 5 cycles of 95° for 20 sec and 70° for 60 sec; and 27 cycles of 95° for 20 sec, 64° for 20 sec, and 72° for 60 sec). PCR products were purified and sequenced.
To identify conserved amino acid signature motifs, the protein sequences deduced from ORFs identified within the cDNA sequences were used in BLASTP and conserved domain (CD) similarity searches within the nr database and in searches of the Prosite database (http://us.expasy.org/prosite/).
Detection of lol-gene expression:
RNA was extracted from fungal cultures and plant tissues and total cDNA was synthesized from total RNA as described previously (Spiering et al. 2002). Primers suffixed “cDNA” in supplementary Table 1 (at http://www.genetics.org/supplemental/) were used in PCR with the total cDNA as template. Diagnostic PCRs with N. coenophialum cDNA were performed with “cDNA” primers specific to the LOL-2 genes. Approximately 10 ng of total cDNA was used in PCR with Takara's LA-PCR version 2.1 kit and the cDNA primers in 20-μl reactions with the following temperature regime: 95° for 60 sec; 7 cycles of 95° for 20 sec and 72° for 2 min; and 30 cycles of 95° for 20 sec, 64° for 20 sec, and 72° for 2 min. To test for possible contaminating genomic DNA, PCRs were performed on the RNA preparations used in the RT-PCR, using primers for detection of lolA-1 and seqX (see supplementary Table 1 at http://www.genetics.org/supplemental/) and the same PCR conditions used for detection of cDNAs (see above). In addition, PCRs on total cDNA were performed repeatedly with lolC-1 cDNA primers (see supplementary Table 1 at http://www.genetics.org/supplemental/), amplifying DNA regions spanning several introns. In none of the RNAs and cDNAs used in the gene expression profiling was any contaminating genomic DNA detected.
lolC RNAi construct:
A lolC-RNAi construct was created by PCR (LA-PCR version 2.1 kit) using 10 ng genomic DNA from N. coenophialum strain ATCC 62374 in 100-μl reactions (N. coenophialum has only a single copy of the lolC gene, and the DNA regions targeted for amplification were 100% identical to those in LOL-2 in N. uncinatum). With primers RNAi 1 and RNAi 2 (see supplementary Table 1 at http://www.genetics.org/supplemental/), a DNA fragment was amplified (95° for 60 sec and then 35 cycles of 95° for 25 sec, 64° for 30 sec, and 72° for 90 sec) containing 769 bp promoter and 5′-untranslated regions (5′-UTRs) before the ATG of the lolC ORF, 98 bp of the first exon, and 54 bp of the first intron. In a second PCR (95° for 60 sec; 5 cycles of 95° for 25 sec, 62° for 30 sec, and 72° for 30 sec; and 30 cycles of 95° for 25 sec and 68° for 30 sec) with primers RNAi 3 and RNAi 4, a DNA fragment was generated containing 8 bp of lolC first intron forward sequence, followed by 98 bp of the first exon and 114 bp of the 5′-UTR of lolC in reverse-complement orientation. After PCR, both fragments were purified as described above, pooled, digested with XbaI (XbaI sites were built into primers RNAi 2 and RNAi 3), and ligated (Fast-Link ligation kit; Epicentre, Madison, WI) to give a 1.1-kb fragment. The fragment was digested with XhoI and KpnI (sites built into the 5′-ends of primers RNAi 1 and RNAi 4, respectively), ligated into XhoI and KpnI-cut pCB1004, and electroporated into E. coli XL1-Blue cells, and then transformants were selected on chloramphenicol. The construct, pKAES178 (Figure 1), was isolated with a QIAGEN Plasmid Midi kit. The integrity of the lolC-RNAi construct was checked by sequencing into the insert from flanking vector regions. Since sequencing of regions containing the first exon was hampered by secondary structures, diagnostic digests with different restriction enzymes and electrophoretic analysis of the restriction fragments was also performed to confirm that the desired construct had been obtained.
Figure 1.—

Construct for RNAi of lolC genes. (A) Map of the plasmid construct. (B) Map of the lolC sequences in the construct. Open and shaded boxes are, respectively, the 5′-UTR and coding sequences in exon 1. (C) Sequence of the lolC first intron (lowercase letters) between the complementary first exon sequences (uppercase letters). Underlined sequence indicates the putative intron branch point, and italicized sequence indicates the XbaI site introduced for cloning.
Fungal transformation:
Protoplasts were prepared from a 7-day-old half-strength PDB culture of N. uncinatum CBS 102646. The mycelium was treated for 4 hr at 30° with 7 mg ml−1 Novozyme 234 (Novo Industri AS, Bagsvaerd, Denmark) and 3 mg ml−1 Glucanex (Novo Industri AS) with bovine serum albumin (Sigma, St. Louis) added at 5 mg ml−1. The protoplasts were harvested and then electroporated with 5 μg of DraI-linearized pKAES178 or pCB1004 (vector-only control) as described by Tsai et al. (1992). After electroporation, the protoplasts were mixed with 4 ml of regeneration medium (Panaccione et al. 2001) and plated onto regeneration medium plates with 80 μg ml−1 hygromycin B (Calbiochem; San Diego). After ∼6 weeks of growth at 22°, viable fungal colonies were transferred onto potato dextrose agar (PDA) with 80 μg ml−1 hygromycin B for sporulation and then single-spore isolated three times on the same medium.
Loline-alkaloid analysis:
Loline-alkaloid extraction from freeze-dried culture filtrates and quantification by gas chromatography were performed as described by Blankenship et al. (2001). Reported is the sum of all lolines produced in culture, e.g., loline, N-acetylnorloline, and N-formylloline. The lower limit of detection in the assay was 10 μg (g DW)−1.
Quantification of lolC expression:
Primers and probes for TaqMan real-time PCR were designed with Primer Express software (Applied Biosystems) following the manufacturer's specifications. Primers and probe searches were performed on lolC-1 and lolC-2 sequences. Sequences contained within pKAES178 were excluded from this search. Primers lolC forw, lolC-1 rev, and lolC-2 rev (see supplementary Table 1 at http://www.genetics.org/supplemental/) were designed for amplification of cDNA from the lolC orthologs. To specifically discriminate between expression of lolC-1 and lolC-2 in the real-time PCR assay, TaqMan probes specific to lolC-1 (lolC-1 RT-PCR probe) or lolC-2 (lolC-2 RT-PCR probe) were designed to span two SNPs for 3′-labeling with a minor groove binding (MGB) nonfluorescent quencher (dihydrocyclopyrroloindole tripeptide; Kutyavin et al. 2000). As a quantification standard, the coding sequence of the β-tubulin (tub2) gene in N. uncinatum (L06946) was used to design the PCR primers, tub2 forw and tub2 rev, and the fluorescently labeled tub2 RT-PCR probe (see supplementary Table 1 at http://www.genetics.org/supplemental/). TaqMan probes 5′-labeled with 6-fluorescein (6-FAM) reporter and 3′-labeled with MGB were synthesized by Applied Biosystems, and forward and reverse primers were synthesized by IDT. Real-time PCR was performed with the TaqMan One-Step RT-PCR Master Mix reagents kit (Applied Biosystems). Reactions (25 μl) were set up in duplicate in 96-optical-well plates, and PCR was performed in an Applied Biosystems PRISM 7700 (PCR cycling: 48° for 30 min, 95° for 10 min, and then 40 cycles of 95° for 15 sec and 60° for 60 sec). Effects of varying primer and probe concentrations (in the range of 0.2 and 0.9 μm for primers and 0.2 and 0.4 μm for probes) were tested on N. uncinatum total RNA (5 ng reaction−1), and no effects were detected. Therefore, 0.4 μm of each primer and 0.2 μm probe was used together with 50 ng (fungal) or 100 ng (plant-fungus symbiotum) total RNA in each reaction. The threshold cycle number (CT) for the fungal tub2 gene was used to correct for differences in RNA concentration between samples. The average coefficient of variation of the CT in duplicate measurements was <1% for all genes tested. To relate CT to template concentration, standards for each lolC ortholog were generated by PCR (95° for 9 min and then 35 cycles of 95° for 25 sec, 64° for 30 sec, and 72° for 30 sec) with primer lolC forw and lolC-1 rev or lolC-2 rev, using purified lolC-1 and lolC-2 cDNA as template and AmpliTaq Gold enzyme (Applied Biosystems). The products were purified as described above, quantified in a fluorometer (Hoefer; Amersham Pharmacia Biotech, Piscataway, NJ), and used as standards in real-time PCR to generate standard curves (r2 > 0.99) in the same CT range as the RNA samples. Specificity was tested by using primers and probe of lolC-1 or lolC-2 on the matching or mismatching lolC DNA standard at concentrations ranging from 0.5 to 500 fg reaction−1. Differences in CT between matching and mismatching probe-standard combinations showed >10-fold higher specificity of the matching combinations, indicating that each assay was highly discriminative in the quantification of lolC-1 or lolC-2 expression.
RESULTS
Gene clusters associated with loline-alkaloid production:
We previously identified lolC cDNA and cDNA clones of two lolA homologs, lolA-1 and lolA-2, whose expression correlated with loline-alkaloid production (Spiering et al. 2002). Secondary metabolite pathways are often clustered in fungal genomes (Zhang et al. 2004), so we hypothesized that lolA and lolC may also be clustered. Long-range PCR with primers, lolA-3′ and lolC-5′a (see supplementary Table 1 at http://www.genetics.org/supplemental/), gave an 8-kb product. This product was purified and sequenced, verifying the presence of lolA and lolC sequences at the ends. Submission of the 8-kb sequence to BLASTX search of the nr database revealed two additional genes, lolD and lolO, having similarities to genes for ornithine decarboxylase and oxidoreductases, respectively (Table 1).
TABLE 1.
Sizes of the predictedlol-gene products, relationships to known enzymes, and protein signature patterns
| Genea | Predicted function | Sizeb | Closest enzyme matchc (GenBank accession no.) |
Identityd | E-value | Signature patterns in predicted proteins (Prosite or pfam pattern)e |
|---|---|---|---|---|---|---|
| lolF-1 | FAD-containing monooxygenase |
540 | 1,2-Cyclopentanone monooxygenase (CAD10798) |
35 (482) | 7e-89 | VIVVGAGFSGILAV (pfam00743.11, FAD-containing monooxygenases; probable FAD-binding site) |
| lolF-2 | FAD-containing monooxygenase |
540 | 1,2-Cyclopentanone monooxygenase (CAD10798) |
35 (482) | 2e-89 | AIVVGAGFSGILAV (see lolF-1) |
| lolC-1 | γ-type PLP enzyme | 473 | O-Acetylhomoserine(thiol)-lyase (P50125) | 54 (431) | 1e-126 | DIVVHSATKWIGGHG (Prosite PS00868, γ-type pyridoxal phosphate (PLP) enzymes; PLP-attachment site) |
| lolC-2 | γ-type PLP enzyme | 473 | O-Acetylhomoserine(thiol)-lyase (XP381593) | 53 (426) | 1e-124 | DIVVHSATKWIGGHG (see lolC-1) |
| lolD-1 | PLP enzyme/decarboxylase | 420 | Ornithine decarboxylase (CAC80209) |
35 (401) | 3e-69 | FAVKSSYDRRLIQTLATCG (Prosite PS00878, α-type PLP enzymes; decarboxylases; family 2 PLP-attachment site) ARRVGLNPTVLDIGGGYT (Prosite PS00879, family 2 signature 2) |
| lolD-2 | PLP enzyme/decarboxylase | 415 | Ornithine decarboxylase (P27121) |
36 (428) | 6e-69 | FAVKSSYDRRLIQTLATCG (Prosite PS00878; see lolD-1) ARQVGLNPTVLDIGGGYT (Prosite PS00879 see lolD-1) |
| lolO-1 | Oxidoreductase/ dioxygenase |
362 | Probable oxidoreductasef (NP248837) |
25 (359) | 2e-21 | ND |
| lolO-2 | Oxidoreductase/ dioxygenase |
362 | Probable oxidoreductasef (NP248837) |
25 (359) | 2e-23 | ND |
| lolA-1 | Amino acid binding | 209 | Aspartate kinase (KIBYD; C-terminal domain) |
28 (160) | 7e-08 | ND |
| lolA-2 | Amino acid binding | 210 | Aspartate kinase (KIBYD; C-terminal domain) |
28 (160) | 5e-08 | ND |
| lolU-1 | Possible DNA-binding protein |
495 |
A. nidulans predicted protein (EAA61586) |
21 (484) | 7e-04 | WTRSEDGSL (Prosite PS00037, Myb transcription factor; DNA-binding domain repeat signature 1) |
| lolU-2 | Possible DNA-binding protein |
506 |
A. nidulans predicted protein (EAA61586) |
20 (489) | 5e-04 | WTTSEDGTL (see lolU-1) |
| lolP-1 | P450 monooxygenase | 496 | Pisatin demethylase (Q12645) |
28 (477) | 1e-47 | FGLGRWQCAG (Prosite PS00086, cytochromes P450; cysteine heme-iron ligand signature) |
| lolP-2g | P450 monooxygenase | 184 | Pisatin demethylase (Q12645) |
28 (152) | 1e-12 | FGLGRWQCAG (see lolP-1) |
| lolT-1 | Class V-aminotransferase PLP-enzyme |
454 | Isopenicillin N epimerase (P18549) |
24 (388) | 1e-13 | PDFFVSDCHKWLFVPRPCAV (Prosite PS00595, α-type PLP enzymes: amino-transferases and related enzymes; class-V PLP-attachment site) |
| lolT-2 | Class V-aminotransferase PLP-enzyme |
464 | Isopenicillin N epimerase (P18549) |
25 (400) | 2e-13 | PDFFVSDCHKWLFVPRPCAF (see lolT-1) |
| lolE-1 | Epoxidase/hydroxylase | 256 | Epoxidase subunit A (BAA75924) |
39 (242) | 2e-42 | ND |
| lolE-2 | Epoxidase/hydroxylase | 256 | Epoxidase subunit A (BAA75924) |
39 (242) | 2e-42 | ND |
The letters in the gene designations were assigned according to the predicted functions of known proteins related to the lol-gene products: lolF, FAD-containing monooxygenases; lolC, CYSD (homocysteine synthase) in A. nidulans; lolD, ornithine decarboxylase; lolO, oxidoreductases; lolA, aspartate kinase; lolU, unknown (no significant match); lolP, P450 monooxygenases; lolT, amino transferases; and lolE, epoxidase.
In amino acids; predicted on the basis of longest contiguous ORF in cDNAs and FGENESH HMM-based gene structure predictions (N. crassa) at http://www.softberry.com.
Including only genetically or functionally characterized activities/enzymes, unless indicated otherwise.
Identity (%) with best match determined by BLASTP; number of aligned amino acids is given in parentheses (note: number of aligned amino acids can be greater than the number in lol-gene products due to gaps in the BLASTP alignment).
Functionally conserved amino acids in the signature patterns are underlined. ND, none detected.
The predicted lolO gene products gave several highly significant matches to putative/probable oxidoreductases/dioxygenases; matches to enzymes with known activity were less significant and included gibberellin 7-oxidase (T09683; E < 2e-14) and isopenicillin N synthetase (P05326; E < 1e-04).
The lolP-2 gene appeared to be truncated due to a 469-bp intragenic deletion (determined by comparison with lolP-1 and lolP in N. coenophialum).
A partial genomic library of N. uncinatum was screened for lolA, and a single positive clone was identified and sequenced. The cosmid was a chimeric clone that included a 10,138-bp insert with lolA sequence within a region of 95% identity to the 8-kb LA-PCR fragment. Thus, the LA-PCR fragment and the cosmid insert were derived from similar but distinct gene clusters, which we designated LOL-1 and LOL-2, respectively (Figure 2). Additional sequences from the two clusters were determined by genome walking into unknown regions with primers specific to LOL-1 and LOL-2. Genome walking was stopped when several PCR attempts with different primer sets failed to yield distinct products. A total of 25 kb was sequenced from LOL-1 and 16 kb was sequenced from LOL-2.
Figure 2.—

Two orthologous gene clusters, LOL-1 and LOL-2, associated with loline-alkaloid production in N. uncinatum. Inferred genes are indicated by arrows, indicating direction of transcription. The lol genes are indicated as F (lolF), C (lolC), etc. The predicted products of the lol genes and similarities to known enzymes are listed in Table 1. Linkage of lolF-2 and lolC-2 is hypothesized but unconfirmed.
Surprisingly, fragments generated by attempted genome walking from lolC-1 exhibited single-nucleotide polymorphisms in sequencing traces, suggesting that some product was also generated from the LOL-2 cluster in the same PCR reactions. These sequences revealed two alleles of another likely gene, designated lolF. Primers were designed for locus-specific amplification of the lolF to lolC regions, but product was obtained only for lolF-1 to lolC-1. Nevertheless, the inferred lolF-2 region was further sequenced by primer walking until, again, no specific PCR products were obtained. Although lolF-2 and lolC-2 were not definitively joined in one contig, we have tentatively assigned them to the same locus, LOL-2, as a working hypothesis (Figure 2).
Approximately 74% of the DNA sequences in LOL-1 and LOL-2, including all inferred genes (described below), gave significant alignment with each other, averaging 93% identity between aligned sequences. The LOL-1 and LOL-2 clusters had 46% and 49% G + C content, respectively.
Identification of genes in LOL-1 and LOL-2:
Open reading frames (ORFs) in LOL-1 and LOL-2 (Table 1; Figure 2) were identified by gene-prediction searches with the FGENESH program. To test whether the identified ORFs corresponded to expressed genes, primers specific to each putative ORF were designed (all primers suffixed “cDNA” in supplementary Table 1 at http://www.genetics.org/supplemental/) and used in PCR with total cDNA from loline-alkaloid-producing N. uncinatum cultures (Figure 3A). All of the ORFs predicted by FGENESH were expressed.
Figure 3.—

Expression of the lol genes in (A) N. uncinatum and (B) N. coenophialum. Expression was monitored for both fungi in minimal medium (MM) and in planta; expression in N. uncinatum was also assessed in complex medium (CM). The lol genes analyzed are indicated above each lane as F (lolF), C (lolC), etc., and for the N. uncinatum genes the ortholog of each was analyzed separately as indicated by numeral 1 or 2 above each lane. Molecular sizes of some marker (m) bands are indicated in kilobases. Loline-alkaloid expression was detected in planta (+); in culture, lolines were detected at either high (>100 mg liter−1; +) or low (∼10 mg liter−1; ±) levels or not detected (−).
The LOL-1 cluster had an arrangement of four pairs of divergently transcribed genes, with only one unpaired gene, lolD (Figure 2). LOL-2 exhibited the identical order and orientations of genes as in LOL-1, with the possible exception of lolF-2, whose linkage with lolC-2 was unconfirmed. Near lolF-2 was a putative tenth gene, designated seqX, which was also identified by FGENESH, and was transcribed under loline-alkaloid-producing conditions (Figure 3, A and B). No homolog of seqX was identified in or near LOL-1.
Complete cDNA sequences, including the 5′- and 3′-ends, were obtained by RACE for both lolO gene homologs. The 5′-end of lolE-2 cDNA was likewise mapped by RACE, and sequence containing its full-length ORF was obtained. cDNA sequences including the complete putative ORFs were also obtained for lolC-2, lolD-1, lolD-2, lolE-1, lolF-2, lolP-1, lolP-2, lolT-2, lolU-1, and lolU-2. The cDNA sequence for lolF-1 was nearly complete except for the 3′-end of its ORF. No cDNA sequence was obtained from lolT-1, due to apparently very low expression of this gene homolog both in planta and in culture (Figure 3). The seqX sequence was predicted by FGENESH to encode a gene, and was transcribed, but the cDNA contained several small ORFs, suggesting that it may be an expressed pseudogene. Complete cDNA sequences of lolA-1, lolA-2, and lolC-1 were determined in the previous study (Spiering et al. 2002).
The lolD-1 ORF appeared to be ∼15 bp longer than the lolD-2 ORF, due to sequence differences near the 3′-ends. The lolP homologs showed an even more dramatic difference, namely, a 469-bp deletion within lolP-2 that shifted the reading frame such that a stop codon truncated its ORF to 555 bp (the lolP-1 ORF was 1491 bp long). A further indication that lolP-2 had a deletion was that the lolP sequence in N. coenophialum had high similarity (>99% identity) with lolP-2, but encoded an ORF identical in length to that of lolP-1.
Most lol genes contained between one and five introns, and in all homologous pairs the genes had identical intron positions. Genes lolU-1 and lolU-2 apparently had no introns. Comparisons of the cDNA sequences with gene structures predicted by FGENESH identified some concordances and differences. FGENESH correctly assigned the ATG start codon in >80% of the genes. Moreover, FGENESH predicted the exact locations of 52% of the exons (total number, 54) and 58% of introns (total number, 36). All of the introns had 5′-GT and 3′-AG splice junctions common to most spliceosomal introns in Neotyphodium and Epichloë species [an exception being the 5′-GC intron boundary in one of the β-tubulin gene introns (Byrd et al. 1990)].
Expression of lol genes:
Loline-alkaloid production in N. uncinatum cultures can be controlled by culture conditions (Blankenship et al. 2001; Spiering et al. 2002). To determine whether the LOL-cluster genes show coordinated expression under loline-alkaloid-inducing or suppressing conditions, we extracted RNA from these cultures and from N. uncinatum-symbiotic plants for use in reverse-transcription PCR with the cDNA primers (see supplementary Table 1 at http://www.genetics.org/supplemental/). Expression of all genes from both LOL clusters was detected in loline-alkaloid-producing cultures (total lolines >100 mg liter−1 culture medium), as well as in planta (Figure 3A), as was expression of seqX. Reverse-transcription PCR of lolT-1 sequence consistently gave low amounts of product, suggesting lower expression levels from lolT-1 compared with the other genes. Several lol genes, especially lolC, lolA, and lolT, appeared to be expressed at lower levels or unexpressed in cultures low (≤10 mg liter−1) in lolines.
Amplified cDNA products from lolP-1 and lolP-2 differed in size (Figure 3A), due to the 469-bp deletion in the lolP-2 sequence described above. In RT-PCR analyses of some lol-gene transcripts, multiple products were observed or there were differences in product sizes from different RNA samples (most prominently observed for lolP-1, but also for lolT-2 and lolD). Sequencing of these bands suggested alternative splicing; e.g., in some lolT-2 cDNAs only the second of the two introns in lolT-2 had been spliced out.
We also profiled lol-gene expression in N. coenophialum, an endophyte of tall fescue (L. arundinaceum). This endophyte contained sequences highly similar (∼99% identity) to LOL-2 in N. uncinatum, but lacked LOL-1. Although N. coenophialum did not produce lolines under the MM culture conditions that induced N. uncinatum to produce lolines (Blankenship et al. 2001), plants symbiotic with N. coenophialum accumulated lolines. Expression of all lol genes was detected in tall fescue plants with N. coenophialum, but only lolF and lolU transcripts were detectable in N. coenophialum cultures (Figure 3B). Thus, loline-alkaloid production was associated with expression of the lol genes in both N. uncinatum and N. coenophialum.
Relationships of predicted lol genes to known metabolic genes:
The predicted LolC, LolD, and LolT sequences gave highly significant BLASTP matches to pyridoxal phosphate (PLP)-containing enzymes involved in amino acid metabolism/interconversion and secondary metabolite pathways (Table 1; Figure 4). All of the predicted PLP enzymes had a lysine residue for PLP binding within a conserved signature region. In addition, LolD had a substrate-binding signature site typical of ornithine decarboxylase.
Figure 4.—



Intron locations in relation to amino acid sequences of putative lol-gene products and paralogues. Indicated are alignments of amino acid sequences deduced from (A) lolC-1, (B) lolD-1, and (C) lolE-1 cDNAs with protein sequences of their closest putative paralogues (the corresponding LOL-2 orthologs gave very similar alignments with both paralogues). The putative paralogues of the lol-gene products were O-acetylhomoserine (thiol)-lyase (CysD) from A. nidulans (U19394), ornithine decarboxylase (ODC) from N. crassa (BX842618), and epoxidase subunit A from Penicillium decumbens (D73371). Locations of introns are indicated as solid triangles. Triangles between letters indicate introns between codons, and those above or below letters indicate introns within the corresponding codons. Sequences similar to protein signatures in Prosite (see Table 1) are underlined. Conserved residues in LolE-1 (His125, Asp127, His162), possibly forming a 2-His-1-carboxylate facial triad typical of many non-heme-iron(II) enzymes (Costas et al. 2004), are underlined in C.
Four genes were predicted to encode enzymes for oxidation or oxygenation reactions (Table 1). One, lolP, was predicted to encode a cytochrome P450 enzyme, having a heme-iron-binding motif. LolF showed similarity to FAD-containing monooxygenases, including the putative FAD-binding site in the N-terminal segment. The lolO and lolE products showed relationships to non-heme-iron oxidoreductases. Alignment of LolO with isopenicillin N synthetase from A. nidulans (GenBank accession no. P05326) indicated conservation of the 2-His-1-carboxylate facial triad motif (His222, Asp224, His280) implicated in metal (iron) binding (Roach et al. 1997; Costas et al. 2004). The LolE sequences gave significant similarities with a fungal epoxidase and several dioxygenases/ hydroxylases. CD searches with LolE indicated significant similarity (E-value = 7e-16) to domains (PhyH; pfam05721.3) of phytanoyl-CoA dioxygenase. Alignment of LolE with the pfam05721.3 consensus also indicated a likely 2-His-1-carboxylate facial triad (Figure 4C).
There was no significant match of LolU with known enzymes, but Prosite searches identified a potential DNA-binding site in LolU (Table 1), suggesting that it may be a transcription activation or regulatory protein. No BLASTP or CD match was identified for seqX.
Structures of the lol genes were compared with those of the known genes to which they were related. In lolC, the numbers of exons and introns were identical to its closest match, the O-acetylhomoserine (thiol)-lyase (cysD) gene from A. nidulans, and all of the intron positions appeared to be identical between lolC and cysD (Figure 4A). Similarly, the first intron in lolD was at the same position as the intron in two fungal genes for ornithine decarboxylase (odc), although lolD had two additional introns not present in the known fungal odc genes (Figure 4B). In lolE, the intron was at the same position as the second intron in its closest match, a fungal epoxidase gene (Figure 4C). Comparison of lolP with one of its closest matches, PDA (L20976, encoding pisatin demethylase), indicated that the genes differed in the numbers of introns and exons; lolP had four exons, whereas PDA had five exons, and amino acid sequences did not align at the intron-exon boundaries.
RNA interference of lolC:
A lolC-RNAi construct, pKAES178 (see Figure 1), was introduced by transformation into N. uncinatum protoplasts, and empty vector was introduced into another batch of protoplasts to generate vector-only transformants as controls. Among the protoplasts transformed with the lolC-RNAi construct, two independent hygromycin-resistant transformants were obtained (designated NUMS1 and NUMS2). PCR with combinations of vector- and lolC-specific primers verified genomic integration of the lolC-RNAi construct (data not shown). The presence of both insert-vector junctions in the lolC-RNAi transformants suggested that the constructs were integrated at ectopic positions.
Expression of the lolC genes, growth, and loline-alkaloid production by the lolC-RNAi transformants and two vector-only control transformants (designated NUMS3 and NUMS4) were assessed in loline-alkaloid-inducing cultures. The lolC-RNAi transformants and vector-only controls showed no detectable difference in growth (DW accumulation per culture volume). Expression of both lolC-1 and lolC-2 was quantified by real-time PCR, and expression of the β-tubulin gene (tub2) was measured as a standard to correct for differences in mRNA concentration between reactions (very similar results were obtained whether lolC expression was normalized to tub2 or to total RNA in each reaction). Transformants with the same construct (lolC-RNAi or vector) showed similar levels of lolC expression and loline-alkaloid production, so their data were combined for statistical analysis. Expression of both lolC homologs in the lolC-RNAi transformants was ∼25% of lolC expression in the vector-only controls (significant at P < 0.05 for lolC-1 and P < 0.01 for lolC-2, Mann-Whitney U-test; Figure 5A). In cultures of both lolC-RNAi transformants, loline-alkaloid levels were significantly lower than the levels in the two vector-only cultures (∼50%; P < 0.001, t-test; Figure 5B). Thus, introduction of the lolC-RNAi construct into N. uncinatum significantly decreased expression of both lolC homologs and significantly decreased production of lolines.
Figure 5.—

(A) Expression of lolC and (B) loline-alkaloid production in N. uncinatum transformed with pKAES178 or empty vector (vector-only). The graphs show the means of five independent experiments; expression of the housekeeping gene, tub2, in the lolC-RNAi and vector-only transformants is indicated in A. Loline-alkaloid production (based on total lolines per gram fungal DW) is expressed as percentage of the mean in the vector-only controls in each experiment. Error bars indicate the standard error of the mean.
lol-gene presence in loline-alkaloid-producing endophytes:
We used diagnostic PCR to test several endophytes with known alkaloid profiles (Spiering et al. 2002) for orthologs of lolF and lolE, the genes at the ends of the known LOL-1 sequence. The PCR primers used in this test were not allele specific, but were predicted to anneal to the lolF or lolE sequences from both N. uncinatum clusters. Both genes were detected in all loline-alkaloid producers tested, namely, N. coenophialum ATCC 90664, N. siegelii ATCC 74483, Epichloë festucae CBS 102475, and E. festucae × E. typhina isolate Tf18. There was no indication of lolE or lolF in any of the non-producers tested, namely, E. festucae CBS 102477, E. typhina ATCC 200736, N. lolii isolate 138, and N. lolii × E. typhina isolate Lp1 (data not shown).
In tests with two different sets of PCR primers, seqX sequences were identified in the loline producers, N. coenophialum ATCC 90664, E. festucae × E. typhina isolate Tf18, and a loline nonproducer, Neotyphodium sp. 269 (MYA2503), but not in the loline producers E. festucae CBS 102475 and N. siegelii ATCC 74483.
DISCUSSION
We identified nine genes associated with loline biosynthesis by the fungal endophyte, N. uncinatum, and obtained experimental evidence that most or all of these genes encode enzymes or regulatory proteins for loline-alkaloid biosynthesis. Two homologs of each gene were present in N. uncinatum, and (with the possible exception of lolF) the homologs were arranged similarly in the two clusters, LOL-1 and LOL-2. Among the LOL-cluster genes were lolA and lolC, associated with loline-alkaloid production in a prior study (Spiering et al. 2002). Here, we have shown that knocking down lolC expression in N. uncinatum significantly decreases loline-alkaloid production in culture, providing direct evidence for involvement of lolC in loline-alkaloid biosynthesis. Furthermore, all nine lol genes are expressed in N. uncinatum under culture conditions conducive to loline-alkaloid production and in planta, whereas expression of many lol genes is apparently reduced under culture conditions with low loline levels. The association was strengthened by investigating N. coenophialum, which expressed lolines and all lol genes in planta, but expressed no lolines and only lolU and lolF in culture. Endophyte isolates differing in their capabilities to produce lolines were screened for orthologs of lolF and lolE—the genes located at the 5′- and 3′-ends of the known LOL-1 sequence—and orthologs of these genes were detectable only in the loline-alkaloid producers.
Roles of the lol genes in loline-alkaloid biosynthesis were suggested by the putative functions of their products: LolF, LolO, LolP, and LolE were predicted to carry out redox reactions; LolC, LolD, and LolT were predicted to possess a PLP cofactor and, therefore, to act on primary amine-containing substrates; LolU was a possible regulatory protein; and the role of LolA was unclear except that it likely binds amino acids.
Due to a high AT content and several repeat sequences within the DNA flanking the clusters, we have so far been unable to walk outward from LOL-1 or LOL-2. However, there was some evidence that lolF and lolE homologs might be at the ends of the regions uniquely possessed by loline-alkaloid-producing endophytes. Preliminary results of long-range PCR suggest that lolE might be located next to an acetamidase gene, which is also present in loline-alkaloid nonproducers (M. J. Spiering, H. H. Wilkinson and C. L. Schardl, unpublished data). This gene arrangement is also indicated in E. festucae (H. H. Wilkinson and B. L. Kutil, unpublished data). Also, the putative gene or pseudogene that we designated seqX, located near lolF-2, was not detected in a loline-alkaloid-producing isolate of E. festucae and in the loline producer N. siegelii. Therefore, at present we consider it a reasonable hypothesis that lolF and lolE demarcate the boundaries of the LOL clusters, but further studies will be required to test this.
Clustering of secondary-metabolism genes is very common in fungi (Zhang et al. 2004). Such pathways often involve activities similar to those predicted for some lol-gene products—especially monooxygenases, oxidoreductases, and dioxygenases—though PLP enzymes are much less common in secondary product pathways. The sequence relationships of LolC to biosynthetic enzymes are of particular interest in light of feeding studies that have identified loline-alkaloid precursors (Blankenship et al. 2005). LolC includes a conserved PLP-binding site and has highly significant similarity to O-acetylhomoserine (thiol)-lyase (homocysteine synthase), which is involved in methionine biosynthesis (Sienko et al. 1998). A further indication of this relationship was the correspondence of all five intron positions in lolC with those in the A. nidulans cysD gene encoding this enzyme. However, expression of the lolC ORF in an A. nidulans cysD− mutant did not restore methionine prototrophy (M. J. Spiering and C. L. Schardl, unpublished results), suggesting that the activity of LolC differs from homocysteine synthase in terms of substrate specificity or catalytic activity. Homocysteine synthase uses activated l-homoserine as a substrate in PLP-mediated γ-substitutions (Sienko et al. 1998; Steegborn et al. 1999). The amino acids l-proline and l-homoserine have been identified as loline-alkaloid precursors, and observations by Blankenship et al. (2005) raise a possibility that an early step in the loline pathway might be a novel biochemical reaction in which the l-proline amine is condensed with the 4-carbon of l-homoserine by a LolC-catalyzed γ-substitution (Figure 6).
Figure 6.—
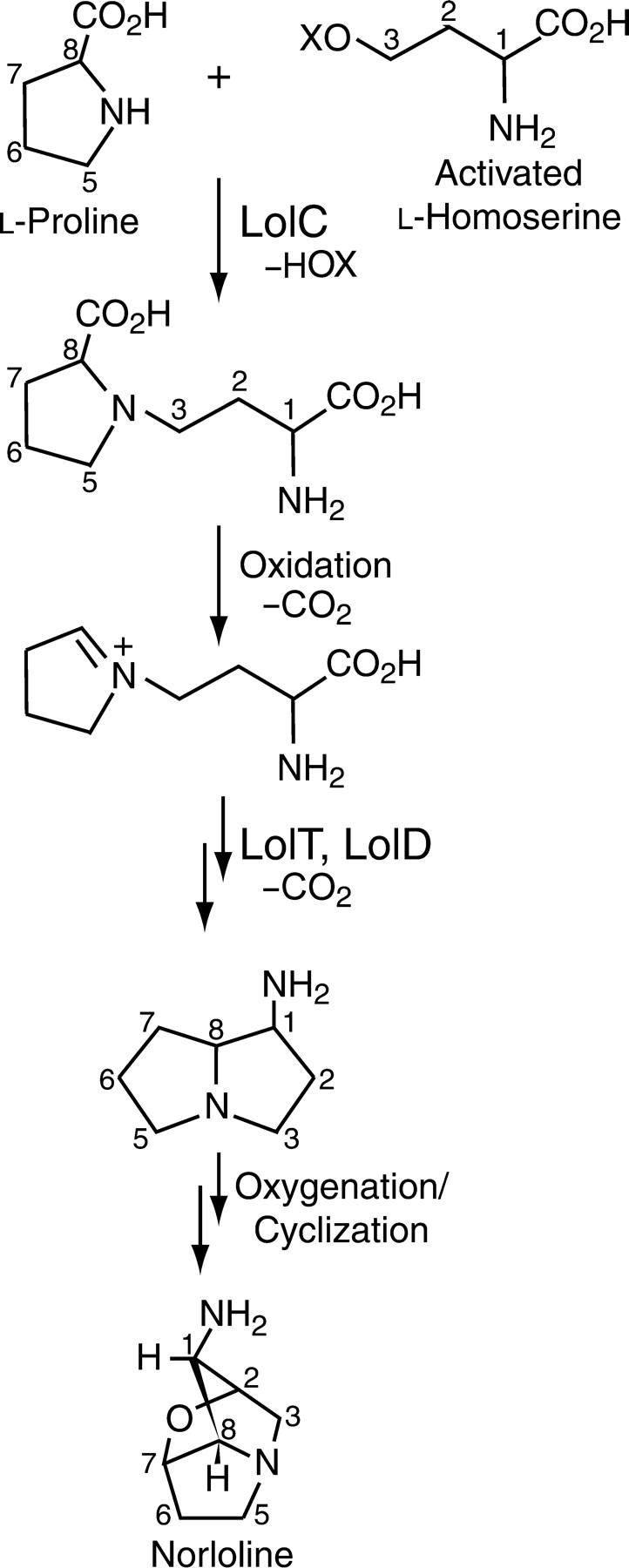
Summary of a possible biosynthetic route to norloline, with hypothesized roles of some lol-gene products indicated.
Reasonable conjectures can be made about the roles of other lol-gene products (Figure 6). One of the oxidizing enzymes (LolE, LolF, LolO, or LolP) might oxidatively decarboxylate the l-proline moiety, generating a pyrrolinium ion, a reasonable intermediate in A-ring formation. One of the PLP enzymes (LolD or LolT) could act on the l-homoserine-derived primary amine to facilitate decarboxylation, and the other could promote cyclization to complete the pyrrolizidine ring system. Other oxidases/oxygenases would be involved in ether bridge formation, although the strained ether linkage characteristic of lolines is highly unusual, and there appears to be no known precedent to suggest the mechanism of its formation. In total it is conceivable that the simplest loline alkaloid, norloline, could be synthesized with the three predicted PLP enzymes plus oxidases/oxygenases sufficient for six electron transfers. We predict that 8–12 electron transfers would be catalyzed by LolF, LolP, LolO, and LolE. Therefore, we hypothesize that the products of the lol genes identified in this study may be sufficient for biosynthesis of the entire loline-alkaloid three-ring structure.
The lolU gene product is a candidate lol regulatory protein, since it has a potential DNA-binding site, but further studies will be required to test this possibility. lolU was also expressed in nonproducing cultures when most other lol genes were not expressed (Figure 3). Thus, if LolU is involved in transcriptional regulation of the lol genes, it may require activation or deactivation (e.g., via phosphorylation) in a regulatory cascade.
The possible role of LolA is also of interest, considering that it has similarity only to the C-terminal domain of aspartate kinases, excluding the kinase active site (Spiering et al. 2002). The C-terminal domain likely contains an allosteric binding site for amino acids produced from aspartate (Arévalo-Rodríguez et al. 1999). In fungi, l-homoserine is an intermediate in the pathway from aspartate 4-phosphate to l-threonine, l-methionine, and l-isoleucine. We speculate that LolA might interact with aspartate kinase to prevent feedback inhibition of its activity by these end products (Arévalo-Rodríguez et al. 1999; Azevedo and Lea 2001) and thereby promote production of l-homoserine from l-aspartate.
The gene orders in LOL-1 and LOL-2 and their orientations were highly conserved between the two clusters, and most genes showed head-to-head arrangements with one of their proximal neighbors, suggesting that genes located next to each other may share 5′-regulatory sequences as has been shown for some clustered genes in other systems (Liu and Xiao 1997). However, not all of the proximal neighbors showed identical patterns of expression. For example, although lolU was expressed in N. coenophialum in culture, expression of its 5′ neighbor, lolP, was not detected, suggesting more complex regulation for at least some of the lol genes.
There are two possible reasons for the presence of two LOL clusters in N. uncinatum: duplication of an ancient LOL cluster in N. uncinatum or inheritance of the clusters from two ancestors. Like many Neotyphodium species, N. uncinatum is a heteroploid interspecific hybrid (Craven et al. 2001). We consider its hybrid origin the most plausible reason, on the basis of phylogenetic analyses of the lolC intron sequences in different loline-producing endophyte species and isolates (M. J. Spiering and C. L. Schardl, unpublished results), suggesting that N. uncinatum has inherited lolC from its two likely ancestors, E. typhina and E. bromicola (Craven et al. 2001).
Functional tests of genes in the Neotyphodium species can be conducted by marker-exchange mutagenesis (Panaccione et al. 2001; Wang et al. 2004), but because of their slow growth each transformation experiment takes several months, frequencies of knockouts are low (<1%), and repeated knock-outs would be required for N. uncinatum. In an attempt to knock out lolO in N. uncinatum, PCR screening of >350 hygromycin-resistant N. uncinatum transformants indicated no replacement of lolO (M. J. Spiering and C. L. Schardl, unpublished observations). Yet, N. uncinatum is the only endophyte species so far known to produce lolines in culture, and in other species time-consuming reintroduction into plants would be required. Given these considerations, we used the alternative approach of gene silencing by RNAi, performed for the first time with a mutualistic fungus. A potential disadvantage of RNAi is that it usually reduces but does not completely abolish expression of the target gene (Smith et al. 2000; Ullu et al. 2002). In the lolC-RNAi transformants, expression of both lolC-1 and lolC-2 was significantly reduced to ∼25%, and total lolines were also significantly reduced to ∼50% of the vector-only controls. The lack of strict correspondence between the relative levels of lolC mRNA and the relative amounts of lolines produced was unsurprising. Enzyme expression is regulated at various steps of transcription, translation, and enzyme modification, and in a multiple-enzyme metabolic process other regulated steps and kinetic effects that would lead to such a difference are likely. Nevertheless, the observed effect of reduced lolC expression on loline-alkaloid production indicates that LolC is involved in biosynthesis of lolines.
Although it is likely that the effect of the lolC-RNAi construct was dsRNA-mediated gene silencing, other effects are conceivable. In particular, because the lolC-2 promoter was used for expression—on the basis of the very high level of lolC expression in loline-alkaloid-producing cultures (Spiering et al. 2002)—it is possible that competition for regulatory factors between native and introduced promoters might also have influenced lolC expression in the lolC-RNAi transformants.
In summary, the RNAi results indicating a role for LolC, ample precedence in fungi for clustering of genes for biosynthetic pathways, expression studies indicating that all LOL-cluster genes are expressed under conditions of loline-alkaloid production, and the presence of the cluster genes in all loline-alkaloid producers, all indicate that the genes identified in the LOL-1 and LOL-2 clusters encode biosynthetic and regulatory functions for the lolines.
Acknowledgments
We thank Kuey-Chu Chen for valuable advice in quantitative real-time PCR and Daniel J. Ebbole and Brandi L. Kutil for critical reading of the manuscript. We are thankful to Andrej Paszewski of the Polish Academy of Science for providing an A. nidulans cysB− cysD− strain and to Peter M. Mirabito, University of Kentucky, for valuable assistance with the complementation experiments. We appreciate the technical support provided by Walter Hollin, Alfred D. Byrd, Amy G. Goins, Oriaku A.-K. Njoku, and LaTasha S. Williams. This work was supported by U.S. National Science Foundation grants 9808554 and 0213217 and U.S. Department of Agriculture National Research Initiatives grant 2003-35319-13562. Sequence analyses were conducted in the University of Kentucky Advanced Genetic Technologies Center, managed by Karl G. Lindstrom and supported by U.S. Department of Agriculture Special grant 2002-34457-11844. This is Kentucky Agricultural Experiment Station publication number 04-12-176, published with the approval of the director.
References
- Ahn, S. C., B. S. Baek, T. Oh, C. S. Song and B. Chatterje, 2000. Rapid mini-scale plasmid isolation for DNA sequencing and restriction mapping. Biotechniques 29: 466–468. [PubMed] [Google Scholar]
- Al-Samarrai, T. H., and J. Schmid, 2000. A simple method for extraction of fungal genomic DNA. Lett. Appl. Microbiol. 30: 53–56. [DOI] [PubMed] [Google Scholar]
- Altschul, S. F., T. L. Madden, A. A. Schäffer, J. Zhang, Z. Zhang et al., 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25: 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arévalo-Rodríguez, M., I. L. Calderón and S. Holmberg, 1999. Mutations that cause threonine sensitivity identify catalytic and regulatory regions of the aspartate kinase of Saccharomyces cerevisiae. Yeast 15: 1331–1345. [DOI] [PubMed] [Google Scholar]
- Azevedo, R. A., and P. J. Lea, 2001. Lysine metabolism in higher plants. Amino Acids 20: 261–279. [DOI] [PubMed] [Google Scholar]
- Blankenship, J. D., M. J. Spiering, H. H. Wilkinson, F. F. Fannin, L. P. Bush et al., 2001. Production of loline alkaloids by the grass endophyte, Neotyphodium uncinatum, in defined media. Phytochemistry 58: 395–401. [DOI] [PubMed] [Google Scholar]
- Blankenship, J. D., J. B. Houseknecht, S. Pal, L. P. Bush, R. B. Grossman et al., 2005. Biosynthetic precursors of fungal pyrrolizidines, the loline alkaloids. ChemBioChem (in press). [DOI] [PubMed]
- Bullock, W. O., J. M. Fernandez and J. M. Short, 1987. XL1-Blue: a high efficiency plasmid transforming recA Escherichia coli strain with beta-galactosidase selection. Biotechniques 5: 376–379. [Google Scholar]
- Bush, L. P., F. F. Fannin, M. R. Siegel, D. L. Dahlman and H. R. Burton, 1993. Chemistry, occurrence and biological effects of saturated pyrrolizidine alkaloids associated with endophyte-grass interactions. Agric. Ecosyst. Environ. 44: 81–102. [Google Scholar]
- Bush, L. P., H. H. Wilkinson and C. L. Schardl, 1997. Bioprotective alkaloids of grass-fungal endophyte symbioses. Plant Physiol. 114: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrd, A. D., C. L. Schardl, P. J. Songlin, K. L. Mogen and M. R. Siegel, 1990. The β-tubulin gene of Epichloë typhina from perennial ryegrass (Lolium perenne). Curr. Genet. 18: 347–354. [DOI] [PubMed] [Google Scholar]
- Carroll, A. M., J. A. Sweigard and B. Valent, 1994. Improved vectors for selecting resistance to hygromycin. Fungal Genet. Newsl. 41: 22–23. [Google Scholar]
- Costas, M., M. P. Mehn, M. P. Jensen and L. Que, 2004. Dioxygen activation at mononuclear nonheme iron active sites: enzymes, models, and intermediates. Chem. Rev. 104: 939–986. [DOI] [PubMed] [Google Scholar]
- Craven, K. D., J. D. Blankenship, A. Leuchtmann, K. Hignight and C. L. Schardl, 2001. Hybrid fungal endophytes symbiotic with the grass Lolium pratense. Sydowia 53: 44–73. [Google Scholar]
- Dougherty, C. T., F. W. Knapp, L. P. Bush, J. E. Maul and J. Van Willigen, 1998. Mortality of horn fly (Diptera: Muscidae) larvae in bovine dung supplemented with loline alkaloids from tall fescue. J. Med. Entomol. 35: 798–803. [DOI] [PubMed] [Google Scholar]
- Hartmann, T., and L. Witte, 1995 Chemistry, biology and chemoecology of the pyrrolizidine alkaloids, pp. 155–231 in The Alkaloids: Chemical and Biological Perspectives, edited by S. W. Pelletier. Springer-Verlag, New York.
- Jackson, J. A., D. R. Varney, R. J. Petroski, R. G. Powell, L. P. Bush et al., 1996. Physiological responses of rats fed loline and ergot alkaloids from endophyte-infected tall fescue. Drug. Chem. Toxicol. 19: 85–96. [DOI] [PubMed] [Google Scholar]
- Kutyavin, I. V., I. A. Afonina, A. Mills, V. V. Gorn, E. A. Lukhtanov et al., 2000. 3′-minor groove binder-DNA probes increase sequence specificity at PCR extension temperatures. Nucleic Acids Res. 28: 655–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y., and W. Xiao, 1997. Bidirectional regulation of two DNA-damage-inducible genes, MAG1 and DDI1, from Saccharomyces cerevisiae. Mol. Microbiol. 23: 777–789. [DOI] [PubMed] [Google Scholar]
- Malinowski, D. P., and D. P. Belesky, 2000. Adaptations of endophyte-infected cool-season grasses to environmental stresses: mechanisms of drought and mineral stress tolerance. Crop Sci. 40: 923–940. [Google Scholar]
- Orbach, M. J., 1994. A cosmid with a HyR marker for fungal library construction and screening. Gene 150: 159–162. [DOI] [PubMed] [Google Scholar]
- Panaccione, D. G., R. D. Johnson, J. H. Wang, C. A. Young, P. Damrongkool et al., 2001. Elimination of ergovaline from a grass-Neotyphodium endophyte symbiosis by genetic modification of the endophyte. Proc. Natl. Acad. Sci. USA 98: 12820–12825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petroski, R. J., S. G. Yates, D. Weisleder and R. G. Powell, 1989. Isolation, semi-synthesis, and NMR spectral studies of loline alkaloids. J. Nat. Prod. 52: 810–817. [Google Scholar]
- Riedell, W. E., R. E. Kieckhefer, R. J. Petroski and R. G. Powell, 1991. Naturally occurring and synthetic loline alkaloid derivatives: insect feeding behavior modification and toxicity. J. Entomol. Sci. 26: 122–129. [Google Scholar]
- Roach, P. L., I. J. Clifton, C. M. H. Hensgens, N. Shibata, C. J. Schofield et al., 1997. Structure of isopenicillin N synthase complexed with substrate and the mechanism of penicillin formation. Nature 387: 827–830. [DOI] [PubMed] [Google Scholar]
- Salamov, A. A., and V. V. Solovyev, 2000. Ab initio gene finding in Drosophila genomic DNA. Genome Res. 10: 516–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer, B. C., 1995. Revolutions in rapid amplification of cDNA ends: new strategies for polymerase chain reaction cloning of full-length cDNA ends. Anal. Biochem. 227: 255–273. [DOI] [PubMed] [Google Scholar]
- Schardl, C. L., A. Leuchtmann and M. J. Spiering, 2004. Symbioses of grasses with seedborne fungal endophytes. Annu. Rev. Plant Biol. 55: 315–340. [DOI] [PubMed] [Google Scholar]
- Sienko, M., J. Topczewski and A. Paszewski, 1998. Structure and regulation of cysD, the homocysteine synthase gene of Aspergillus nidulans. Curr. Genet. 33: 136–144. [DOI] [PubMed] [Google Scholar]
- Smith, N. A., S. P. Singh, M. B. Wang, P. A. Stoutjesdijk, A. G. Green et al., 2000. Gene expression—total silencing by intron-spliced hairpin RNAs. Nature 407: 319–320. [DOI] [PubMed] [Google Scholar]
- Spiering, M. J., H. H. Wilkinson, J. D. Blankenship and C. L. Schardl, 2002. Expressed sequence tags and genes associated with loline alkaloid expression by the fungal endophyte Neotyphodium uncinatum. Fungal Genet. Biol. 36: 242–254. [DOI] [PubMed] [Google Scholar]
- Steegborn, C., A. Messerschmidt, B. Laber, W. Streber, R. Huber et al., 1999. The crystal structure of cystathionine γ-synthase from Nicotiana tabacum reveals its substrate and reaction specificity. J. Mol. Biol. 290: 983–996. [DOI] [PubMed] [Google Scholar]
- Tofern, B., M. Kaloga, L. Witte, T. Hartmann and E. Eich, 1999. Phytochemistry and chemotaxonomy of the Convolvulaceae part 8—occurrence of loline alkaloids in Argyreia mollis (Convolvulaceae). Phytochemistry 51: 1177–1180. [Google Scholar]
- Tsai, H.-F., M. R. Siegel and C. L. Schardl, 1992. Transformation of Acremonium coenophialum, a protective fungal symbiont of the grass Festuca arundinacea. Curr. Genet. 22: 399–406. [DOI] [PubMed] [Google Scholar]
- Ullu, E., A. Djikeng, H. Shi and C. Tschudi, 2002. RNA interference: advances and questions. Philos. Trans. R. Soc. Lond. B 357: 65–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J., C. Machado, D. G. Panaccione, H.-F. Tsai and C. L. Schardl, 2004. The determinant step in ergot alkaloid biosynthesis by an endophyte of perennial ryegrass. Fungal Genet. Biol. 41: 189–198. [DOI] [PubMed] [Google Scholar]
- Wilkinson, H. H., M. R. Siegel, J. D. Blankenship, A. C. Mallory, L. P. Bush et al., 2000. Contribution of fungal loline alkaloids to protection from aphids in a grass-endophyte mutualism. Mol. Plant-Microbe Interact. 13: 1027–1033. [DOI] [PubMed] [Google Scholar]
- Zhang, Y., H. H. Wilkinson, N. Keller and D. Tsitsigiannis, 2004 Secondary metabolite gene clusters, pp. 355–385 in Handbook of Industrial Mycology, edited by Z. An. Marcel Dekker, New York.