

Abstract
The Mus101 family of chromosomal proteins, identified initially in Drosophila, is widely conserved and has been shown to function in a variety of DNA metabolic processes. Such functions include DNA replication, DNA damage repair, postreplication repair, damage checkpoint activation, chromosome stability, and chromosome condensation. Despite its conservation and widespread involvement in chromosome biogenesis, very little is known about how Mus101 is regulated and what other proteins are required for Mus101 to exert its functions. To learn more about Mus101, we have initiated an analysis of the protein in C. elegans. Here, we show that C. elegans mus-101 is an essential gene, that it is required for DNA replication, and that it also plays an important role in the DNA damage response. Furthermore, we use RNA interference (RNAi)-mediated reverse genetics to screen for genes that modify a mus-101 partial loss-of-function RNAi phenotype. Using a systematic approach toward modifier gene discovery, we have found five chromosome I genes that modify the mus-101 RNAi phenotype, and we go on to show that one of them encodes an E3 SUMO ligase that promotes SUMO modification of MUS-101 in vitro. These results expand our understanding of MUS-101 regulation and show that genetic interactions can be uncovered using screening strategies that rely solely on RNAi.
THE combination of RNA interference (RNAi)-mediated gene knockdown methods and functional genomics has provided an alternative to classic forward genetics for gene discovery efforts in many organisms (Fraser et al. 2000; Gonczy et al. 2000; Berns et al. 2004; Boutros et al. 2004; Paddison et al. 2004). RNAi is triggered when double-strand RNA (dsRNA) is processed in a manner that results in the destruction of mRNAs that are homologous to the dsRNA (reviewed by Novina and Sharp 2004). In the nematode Caenorhabditis elegans dsRNA is delivered to the organism by one of three methods: soaking (Maeda et al. 2001), injection (Fire et al. 1998), or feeding (Timmons and Fire 1998). The availability of a complete genome sequence in C. elegans has allowed systematic, high-throughput RNAi screens to be routinely performed, and screens have been reported using all three dsRNA delivery methods. While not absolute, a trend has emerged from these studies that indicates that RNAi by feeding is less robust and less penetrant than the other methods, suggesting that for many genes RNAi by feeding is phenotypically closer to the hypomorphic than to the null condition.
When using RNAi to analyze the properties of genes that encode essential functions, a hypomorphic condition can be useful. Partial depletion of an essential gene could, for example, reveal additional, nonessential functions of the gene. In addition, the hypomorphic condition can be exploited to search for genes that modify the phenotype of an essential gene and may therefore act in a common pathway. This last assertion is based on considerable evidence from budding yeast where synthetic lethal screens have been used to define and order genetic pathways. Synthetic lethal phenotypes result when two mutations, neither of which affects viability on its own, cause a decrease in viability when combined (reviewed by Hartman et al. 2001). This can occur by one of three ways: by combining null alleles of two nonessential genes, by combining a hypomorphic allele of an essential gene with a null allele of a nonessential gene, and by combining hypomorphic alleles of two different essential genes. That such genetic interactions are specific, and that they suggest inclusion of the genes in common or similar pathways, has been addressed through systematic genetic analysis (SGA) of the complete collection of nonessential yeast deletion mutants. SGA involves making double mutants between a mutant of interest and all 5100 nonessential deletion mutants and then scoring for effects on fitness (Tong et al. 2001). A recent study reported the results of 132 SGA screens totaling ∼4000 interactions (Tong et al. 2004). The average number of interactions per target was 34, demonstrating that such screens are reasonably specific in the interactions that are revealed. Another indication of specificity was the correlation in function between targets and hits uncovered in the screen: 27% of the interactions involved pairs of genes that were previously known to function in the same or similar pathways. These data reveal that systematic genetic modifier screens can be effective tools for gene discovery.
We have been studying the DNA damage response in C. elegans and have been particularly interested in the mus-101 locus. mus-101 (F37D6.1) encodes a 1227-amino-acid protein that is characterized by the presence of six copies of the BRCA1 carboxyl-terminal (BRCT) repeat. BRCT repeats are commonly found in proteins involved in DNA metabolism and are thought to mediate protein-protein interactions between BRCT-repeat-containing proteins and phosphorylated protein-binding partners (Manke et al. 2003; Yu et al. 2003). The mus-101 gene is highly conserved throughout evolution, with homologs present in humans (TopBP1; Yamane et al. 1997), Xenopus (Xmus101; Van Hatten et al. 2002), Drosophila (mus101; Boyd et al. 1976), Arabidopsis (MEI1; Grelon et al. 2003), and fission and budding yeast (cut5 and DPB11, respectively; Saka and Yanagida 1993; Araki et al. 1995). Mus101 family members have been linked to a variety of chromosomal pathways, including DNA replication, S-phase checkpoint activation, postreplication repair of damaged DNA, chromosome condensation, chromosome stability, and meiosis. An important question regarding Mus101 function is how the protein is regulated so that it can participate in multiple chromosomal pathways without inducing inappropriate cross-talk between different pathways. To help answer this question, we have initiated an analysis of the C. elegans mus-101 gene. RNAi by soaking resulted in robust depletion of mus-101, a failure to replicate DNA, and embryonic lethality. RNAi using the feeding protocol resulted in less penetrant depletion, viability, and sensitivity to the DNA-damaging agent methyl methanesulfonate (MMS). We took advantage of the hypomorphic condition caused by mus-101 RNAi by feeding to systematically screen for genes that, when codepleted with mus-101, modified the mus-101 phenotype. From a library of 2425 chromosome I genes we isolated five modifiers. Three of the five displayed MMS sensitivity, suggesting a common function with mus-101. Additionally, one of the identified modifiers, gei-17, encodes a presumptive E3 SUMO ligase and was shown to stimulate SUMO modification of MUS-101 in vitro. These results shed new light on the regulation of mus-101 and demonstrate that systematic codepletion by RNAi is a useful method for identification of genetic modifiers.
MATERIALS AND METHODS
C. elegans strains and culturing:
The N2 Bristol strain was used in all experiments. Worms were maintained as described (Brenner 1974). Embryonic lethality was determined by counting the percentage of eggs that failed to hatch 20 hr after laying.
RNA interference assays:
mus-101 RNAi by soaking was performed as described (Maeda et al. 2001). RNAi by feeding was performed as described (Timmons and Fire 1998). For feeding RNAi, all bacteria were cultured for 24 hr at 37° in Terrific Broth containing 50 μg/ml ampicillin, seeded onto NGM plates containing 5 mm isopropyl-β-d-thiogalactopyranoside (IPTG), and allowed to dry overnight.
Hoechst's 33258 staining of embryos and gonads:
Worms were dissected on glass microscope slides and permeabilized by freeze cracking. Slides were fixed for 10 min in methanol/formaldehyde fixative at −20° and washed in PBST. DNA staining was accomplished by adding 10 μl of 10 μg/μl Hoechst's 33258.
Antibody production:
PCR primers were designed to amplify fragments corresponding to the C-terminal 333 amino acids of mus-101. The fragments were subcloned into the pET30 bacterial expression vector (Novagen) and used to produce recombinant protein. Six-histidine-tagged recombinant protein was purified on a nickel agarose column under denaturing conditions. Purified proteins were then used as antigens to immunize rabbits. Polyclonal antibodies were obtained and affinity purified according to standard procedures.
Propidium iodide and tubulin staining of embryos:
Worms were mounted, fixed, and washed as described for Hoechst's staining. Slides were incubated in 200 μg/ml RNAse for 30 min at 37° followed by a 5-min incubation in 0.1 mg/ml propidium iodide solution (Sigma, St. Louis). Embryos were then incubated with an antibody against α-tubulin (MAb DM1A 1:100; Sigma) for 2 hr at room temperature, followed by incubation with donkey anti-mouse, FITC-labeled secondary antibody. Embryos were visualized on an Olympus BX51 microscope. Pictures were captured using a SPOT RT monochrome camera (Diagnostic Instruments).
Embryo culture assays:
Embryos were prepared for culture experiments on the basis of published protocols (Edgar and McGhee 1988) with some deviations. Briefly, dissected adult worms were collected and incubated for 2 min in a 1:9 solution of 6% hypochlorite (Fisher). Worms were then pelleted and resuspended in minimal egg growth media (EGM; Edgar and McGhee 1988), followed by another 2-min hypochlorite treatment. The remaining eggs were pelleted, resuspended a final time in EGM, transferred to a gelatin-subbed slide (2%), and covered with a coverslip supported by half-inch adhesive transfer tape. Pressure was applied to the coverslip to attach the embryos to the slide and permeabilize the vitelline membrane. Additional EGM was added to fill the chamber. Finally, three washes of 30 μl EGM with cytochalasin B were flushed through the chamber and embryos were incubated for 1 hr. Exposure to the replication inhibitor hydroxyurea (HU) was accomplished by incubating permeabilized embryos in EGM containing 5 mm HU and cytochalasin B. Embryos were fixed by flushing the chamber with three 30-μl washes of fixative (Edgar and McGhee 1988), followed by PBS. DNA staining was accomplished by flushing the chamber with three 30-μl washes of a solution containing 10 μg/μl Hoechst's 33258, followed by PBS and, finally, 30 μl of mounting medium (2% NPG in 80% glycerol).
MMS sensitivity assays:
L4 F1 worms grown on plates containing the appropriate bacterial expression vectors were transferred to plates containing 0.05 mg/ml MMS (Sigma) at 25°. Eggs laid by these worms were collected over time and scored for survival.
Chromosome I RNAi modifier screen:
The chromosome I RNAi library was purchased from MRC geneservice. Bacteria were amplified as described and mixed in a 1:1 ratio with either mus-101 RNAi bacteria or control RNAi bacteria (bacteria expressing dsRNA against the exogenous green fluorescent protein, GFP). Additionally, mus-101 RNAi bacteria were mixed at a 1:1 ratio with GFP RNAi bacteria to establish baseline mus-101 RNAi lethality (which was typically <5%). Bacterial mixes were then plated on media containing 5 mm IPTG and allowed to dry overnight. P0 worms were seeded as L1s and cultured at 25°. Approximately 100 F2 embryos were collected for each mixture and scored for survival. Genes were scored as modifiers when a higher percentage of lethality was observed in the gene X/mus-101 RNAi combination than in the gene X/GFP RNAi or mus-101/GFP RNAi combinations after multiple rounds of screening.
SUMOylation assay:
A cDNA encoding full-length mus-101 was transcribed and translated in the presence of [35S]methionine according to the manufacturer's instructions (Promega, Madison, WI). The sumoylation kit was purchased from LAE Biotech International and in vitro sumoylation reactions were performed according to the manufacturer's instructions with the following exceptions. Recombinant C. elegans his-tagged UBC-9 was substituted for the human protein supplied with the kit. Twenty-microliter reactions contained 5 μl of the translation product, 0.36 μg C. elegans UBC-9, 0.68 μg C. elegans GEI-17, 1 μg human SUMO-I, 1 μg human SUMO III, and 150 ng human E1. To generate recombinant C. elegans UBC-9 and GEI-17, full-length cDNAs were amplified from a cDNA library and cloned into the pET-28A expression vector (Novagen). Recombinant protein was purified using standard his-tag purification protocols. Induction of GEI-17 was performed for 4 hr at 16°.
RESULTS
Differential depletion of mus-101 after soaking or feeding RNAi:
To initiate an analysis of mus-101 function in C. elegans, we depleted the gene product using RNAi by both soaking and feeding. To assess the relative effectiveness of feeding and soaking RNAi on mus-101 depletion, we probed whole-worm lysates derived from treated animals with an affinity-purified anti-MUS-101 antibody. RNAi by soaking resulted in a robust depletion of MUS-101 protein, whereas RNAi by feeding was less effective (Figure 1). Quantification of the band intensities using National Institutes of Health Image revealed that soaking RNAi reduced MUS-101 levels to 10% of wild type while feeding RNAi reduced the level to 53% of wild type. When embryonic viability of the progeny of treated animals was assessed, we found that RNAi by soaking caused a high (78%) level of embryonic lethality whereas RNAi by feeding resulted in only a very low level of embryonic lethality (4%). We conclude that while mus-101 is an essential gene, a partial decrease in mus-101 expression does not dramatically compromise viability.
Figure 1.—

Immunoblot analysis of MUS-101 protein levels in wild-type animals and after soaking or feeding RNAi. Below each RNAi lane is the percentage embryonic lethality (emb) in the F1 (soaking) or F2 (feeding) progeny of RNAi-treated animals. For soaking RNAi, N2 worms were submerged in a solution of concentrated dsRNA according to published procedures (Maeda et al. 2001). After a recovery period whole-worm lysates were prepared for immunoblots. For feeding RNAi, animals were cultured on plates seeded with bacteria expressing the dsRNA for two generations at 25° prior to preparation of whole-worm lysates. Each lane on the blot corresponds to 10 adult worms.
DNA replication problems in embryos depleted of mus-101 by soaking RNAi:
A conserved function of the Mus101 protein family is DNA replication (Saka and Yanagida 1993; Araki et al. 1995; Van Hatten et al. 2002); thus one likely explanation for the embryonic lethality induced by mus-101 RNAi by soaking is a failure to replicate. To investigate this further, either wild-type or mus-101 RNAi by soaking embryos were treated with RNAse and then fixed and stained with the nucleic acid stain propidium iodide to assess DNA content. As shown in Figure 2A, the propidium iodide signal (“DNA”) in mus-101 RNAi by soaking embryos is far less intense than that in the wild type, while the signal resulting from an anti-α-tubulin antibody (“tubulin”) is qualitatively equivalent. The mus-101 soaking RNAi embryos therefore contain substantially less DNA than do wild-type embryos. A similar observation has been reported for embryos depleted of the DNA replication factor CDT-1 (Zhong et al. 2003).
Figure 2.—

DNA replication is attenuated after mus-101 RNAi by soaking. (A) Wild-type or mus-101 soaking RNAi embryos were fixed, treated with RNase I, and stained with propidium iodide to visualize the DNA (“DNA”) and with anti-α-tubulin to visualize tubulin (“tubulin”). (B) Schematic of the uncoupling of DNA replication from cell division that occurs upon treatment of permeabilized embryos with the cytokinesis inhibitor cytochlasin B. (C) Permeabilized embryos were fixed and stained with Hoechst's 33258 stain either immediately after permeabilization (I) or after a 1-hr incubation in culture media (II–IV). The embryos shown in II and IV are wild type, while the embryo depicted in III was depleted of mus-101 gene product by soaking RNAi. The embryo in IV was treated with HU to block replication. The embryos depicted in I–IV are representative of a larger (>25) sample set that was analyzed for each condition.
The data in Figure 2A suggest a role for mus-101 in DNA replication. To examine more directly this possibility, we took advantage of previous observations that DNA synthesis occurs normally in embryos treated with the actin poison cytochlasin (Edgar and McGhee 1988). Cytochlasin blocks cell division but does not prevent DNA replication; thus cytochlasin-treated embryos will perform multiple rounds of DNA replication and accumulate masses of DNA within individual cells (Figure 2B). This allows a more direct assessment of the involvement of a given gene in DNA replication, as embryos that cannot replicate DNA will simply fail to accumulate masses of DNA over time. For this experiment, four-cell-stage embryos were collected, the vitelline membranes were permeabilized, and the samples were treated with cytochalasin B. After a 60-min incubation period, the embryos were fixed and the DNA was visualized by the application of Hoechst's 33258. As shown in Figure 2C, and consistent with previous reports (Edgar and McGhee 1988), wild-type embryos accumulated large amounts of DNA within individual cells after the hour-long incubation (Figure 2C, compare I to II). By contrast, mus-101 RNAi by soaking embryos accumulated very little DNA during the incubation period (Figure 2C, III). The amount of DNA synthesized in mus-101 RNAi by soaking embryos was qualitatively similar to the amount synthesized by wild-type embryos that had been treated with the replication inhibitor HU (Figure 2C, compare III to IV). From this, we conclude that mus-101 is required for DNA replication in the early C. elegans embryo.
Depletion of mus-101 by feeding RNAi mimics a hypomorphic condition:
By contrast to RNAi by soaking, RNAi by feeding did not cause extensive embryonic lethality (Figure 1). It did, however, result in 20% sterility in the F1 progeny of treated animals. Staining of the germ lines of F1 fertile or sterile adults revealed that the sterile animals had an abnormal gonad morphology characterized by necrotic-looking germ cell nuclei (Figure 3). This demonstrates that, at low frequency, mus-101 feeding RNAi affects proliferation of the germ line. Although the basis for this proliferation defect is not known, the morphology of affected nuclei is reminiscent of wild-type germ-cell nuclei that are blocked for replication with HU (MacQueen and Villeneuve 2001) or treated with ionizing radiation (Gartner et al. 2000). Consistent with a replication problem causing this phenotype is the observation that RNAi by feeding depletion of the replication initiation factor ORC-2 causes an identical sterility/abnormal nuclear morphology defect (A. Holway, unpublished data).
Figure 3.—

Abnormal nuclear morphology in animals sterilized by mus-101 feeding RNAi. Gonads from F1 progeny of depleted animals were dissected and stained with Hoechst's 33258 to visualize the DNA. Roughly 80% of these animals are fertile and show normal nuclear morphology in the germ line (I), whereas the remainder are sterile and display abnormal morphology (II). A single gonad arm is shown, with the distal tip up and to the right in each case.
In Drosophila, hypomorphic alleles of mus101 that retain the DNA replication function but not the DNA damage response function of the protein exist (Boyd et al. 1976). In C. elegans we have shown that feeding RNAi allows embryonic viability, and thus DNA replication, and it was therefore of interest to determine if the DNA damage response function of mus-101 was also retained after feeding RNAi. To examine this, we tested for MMS sensitivity. F1 L4 stage animals were plated on media containing 0.05 mg/ml MMS and allowed to lay eggs for 16 hr before being transferred to a fresh MMS plate for an additional 16 hr of egg laying. Embryonic lethality in the F2 was then determined by counting the number of eggs that failed to hatch. As shown in Figure 4, and by contrast to wild type, mus-101 RNAi by feeding resulted in MMS sensitivity. This result makes two important points: (1) mus-101 is required for the DNA damage response in C. elegans embryos and (2) a separation of the DNA replication from the DNA damage response function of mus-101 can be achieved through RNAi by feeding.
Figure 4.—
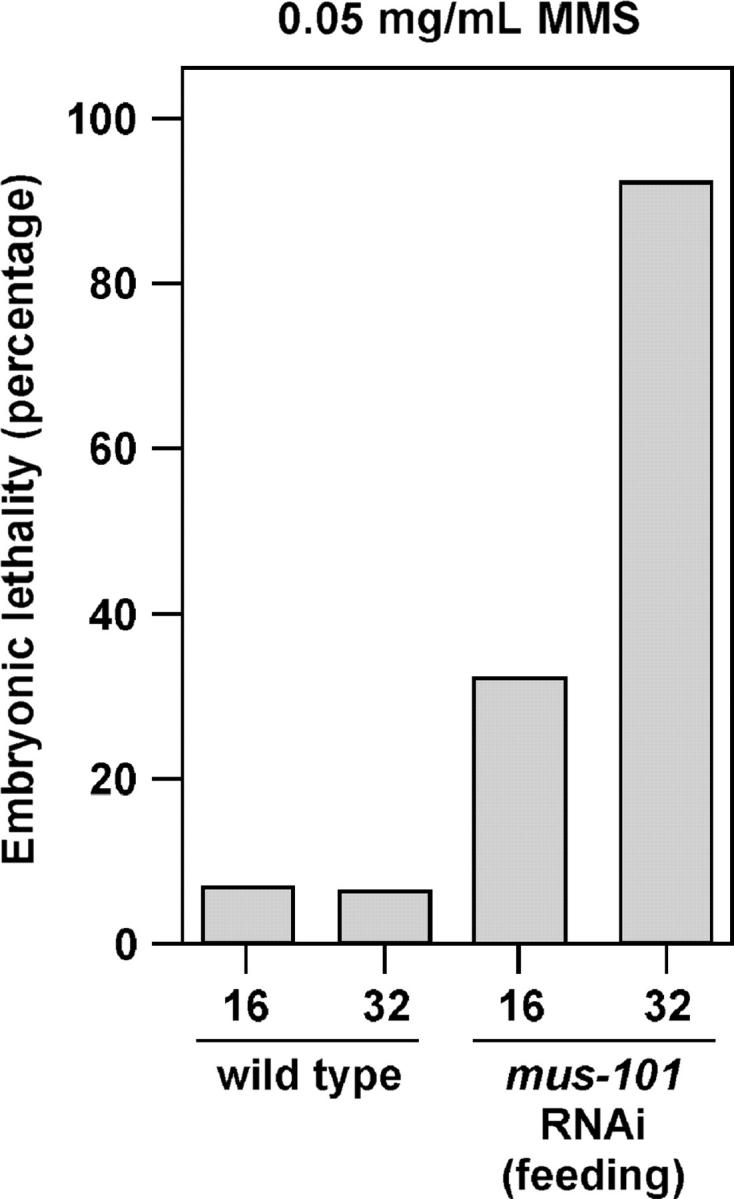
MMS sensitivity after mus-101 feeding RNAi. Either wild-type or mus-101 feeding RNAi animals were plated as L4s on media containing 0.05 mg/ml MMS. Embryonic lethality in the progeny of these animals was determined after 16 or 32 hr of egg laying as described in materials and methods.
Identification of mus-101 modifiers:
The immunoblot in Figure 1 shows that mus-101 RNAi by feeding reduces the steady-state level of MUS-101 protein to roughly half of what is normally present. The experiments shown in Figures 3 and 4 reveal that while this reduction is generally tolerable, there are phenotypic consequences in both the germ line and the embryo. RNAi by feeding therefore mimics a hypomorphic allele of mus-101. We sought to exploit this hypomorphic condition to identify other genes that function with mus-101 by systematically searching for genes that, upon codepletion, modified the mus-101 feeding RNAi phenotype. In particular, we screened for genes that when codepleted with mus-101 caused an increase in embryonic lethality. Codepletions were performed by mixing, at a 1:1 ratio, bacteria expressing dsRNA against mus-101 with bacteria expressing dsRNA against the gene to be screened. The mixture was then seeded onto media plates prior to plating of L1 stage worms. After two generations on the selective media embryonic viability was determined and compared to that observed for a control codepletion that substituted mus-101 for the irrelevant gene GFP. Two criteria were imposed to weed out weak and/or nonspecific interactions (Figure 5). One, the percentage embryonic lethality produced upon codepletion of mus-101 and a candidate gene, must be at least twice that observed when the candidate was codepleted with GFP. Two, the embryonic lethality produced upon codepletion of mus-101 and a candidate gene must be at least 10% as pilot experiments had shown that reproducibility suffered when the cutoff was set beneath this value.
Figure 5.—

Screening strategy for isolation of mus-101 modifiers by dual RNAi. Well A is the control plate that contains bacteria expressing dsRNA against the gene to be screened with dsRNA against an irrelevant protein (GFP). This produces N% embryonic lethality (emb). When dual RNAi is performed against the gene to be screened and mus-101 (well B), then the percentage of emb must be >10 and at least twice N for the gene to be pursued further.
To isolate mus-101 modifiers, we screened every gene present in a library containing 2425 chromosome I genes (representing 89% of the chromosome; see Fraser et al. 2000 for details on the library). One concern raised as we were performing this screen was that mixing two different feeding vectors in the same depletion experiment might weaken or eliminate the ability of either feeding vector alone to effectively silence its target gene. This did not occur. Fraser et al. (2000) have reported a chromosome I RNAi by feeding screen using a full dose of the chromosome I feeding vectors. Our screen, which used a half dose of the chromosome I vectors, produced outcomes that were consistent with those reported by Fraser et al. (2000) 95% of the time. The 5% of the cases where the outcomes did not agree were equally divided between examples where Fraser et al. (2000) reported a phenotype for a given gene and we did not and examples where we could observe a phenotype and Fraser et al. (2000) did not. Thus this 5% discrepancy likely reflects the noise inherent in any large-scale functional genomics screen. The high degree of correlation between phenotypes reported by Fraser et al. (2000) using a full complement of feeding vector and those reported here using a half complement shows unambiguously that RNAi codepletion by feeding allows silencing of both target genes in the codepletion.
Table 1 lists the results of this screen. Five genes that fit the criteria outlined in Figure 5 were isolated. Three of the genes, gei-17, arx-7, and let-49, have been previously characterized, but not in significant detail. The other two, W01A8.1 and F26B1.2, have not been characterized. W01A8.1 encodes a protein with no strong homology to anything in the current databases, excepting its presumptive ortholog in C. briggsae. F26B1.2 encodes a protein containing a heterogeneous nuclear ribonucleoprotein K homology (KH) domain. gei-17 encodes a protein with homology to the mammalian protein inhibitor of activated STAT (PIAS) family of transcriptional regulators and E3 SUMO ligases (reviewed by Schmidt and Muller 2003). arx-7 encodes a component of the Arp2/3 complex, which regulates actin dynamics, and let-49 is a transcriptional regulator that controls expression of genes required for germ-line formation and postembryonic development (Kwon et al. 2001).
TABLE 1.
Genes that interact withmus-101 after codepletion by feeding RNAi
| Gene | emb (GFP) (%) | emb (mus-101) (%) | Presumed function |
|---|---|---|---|
| W01A8.1 | 3.3 (9/269) | 14.7 (58/395) | Unknown |
| F26B1.2 | 7.7 (29/379) | 14.5 (62/427) | KH domain |
| gei-17 | 12.8 (20/156) | 28.9 (43/149) | PIAS SUMO ligase |
| arx-7 | 4.6 (11/239) | 12.6 (17/135) | Arp 2/3 complex |
| let-49 | 1.7 (6/356) | 13.5 (21/156) | Transcription factor |
Emb was determined by counting the number of eggs that failed to hatch. Both the percentage and the actual counts are shown. Emb (GFP) refers to embryonic lethality after codepletion with GFP and emb (mus-101) refers to embryonic lethality after codepletion with mus-101.
The five genes identified in the screen all caused an increase in embryonic lethality when they were codepleted with mus-101, relative to GFP. To make sure that the enhanced embryonic lethality was due to synergy with mus-101 RNAi and was not due to an unexpected suppression of embryonic lethality by the GFP RNAi, we performed full-dose feeding RNAi experiments on all five genes. No significant differences were observed in the embryonic lethality after a full dose of feeding RNAi relative to that observed for the GFP codepletions for four of the five genes. For let-49, a full dose of feeding RNAi resulted in 100% sterility in the F1, thus precluding analysis of the F2. We conclude that none of the genes identified in the screen were false positives resulting from interactions with GFP RNAi.
MMS sensitivity of mus-101 modifiers:
Four of the screen hits were next tested for MMS sensitivity. We did not score let-49 in this assay owing to the sterility phenotype. Three of the four genes tested showed sensitivity (Figure 6). One of the genes, gei-17, was exceptionally sensitive to MMS, with embryonic lethality of 100% at the first time point. The other two genes, F26B1.2 and arx-7, were more modestly sensitive. For arx-7, we did note a weak eggshell phenotype; therefore it is unclear whether the MMS sensitivity in the arx-7 RNAi embryos was due to direct involvement in the DNA damage response or to the weakened eggshell resulting in the embryos experiencing a higher effective concentration of MMS during the experiment.
Figure 6.—

MMS sensitivity in mus-101 modifiers. MMS sensitivity assays were performed as described in Figure 4 after feeding RNAi against the indicated genes.
To understand more about why mus-101, gei-17, and F26B1.2 depleted embryos were MMS sensitive, we stained MMS-exposed embryos with the DNA stain Hoechst's 33258 to examine the morphology of the nuclear DNA. As shown in Figure 7, nuclear morphology in F26B1.2 depleted embryos did not differ from wild type after MMS exposure. By contrast, nuclear morphology in both mus-101 and gei-17 depleted embryos was affected by MMS. Specifically, many nuclei were of discontinuous size and shape, some nuclei appeared to be torn or fragmented, and numerous anaphase bridges were also observed. Such nuclear morphology aberrations were not observed in either mus-101 or gei-17 depleted embryos in the absence of MMS exposure. Thus, in addition to MMS sensitivity, mus-101 and gei-17 share a similar nuclear morphology defect upon MMS exposure. This suggests that mus-101 and gei-17 may be components of a common pathway that responds to MMS-induced DNA damage in the embryo.
Figure 7.—

MMS induces nuclear morphology abnormalities after mus-101 and gei-17 feeding RNAi. Wild-type (I), mus-101 (II), gei-17 (III), and F26B1.2 (IV) feeding RNAi embryos were exposed to MMS and then fixed and stained with Hoechst's 33258 to visualize the DNA. V and VI show enlarged examples of the anaphase bridges that were commonly observed in the mus-101 and gei-17 feeding RNAi samples, respectively.
GEI-17 stimulates SUMO modification of MUS-101 in vitro:
To strengthen the connection between mus-101 and gei-17, we sought biochemical evidence for an interaction between the two gene products. The PIAS domain family of proteins, of which GEI-17 is a member, has been shown to stimulate SUMO modification of substrate proteins (Johnson and Gupta 2001). SUMO is a small, ubiquitin-like polypeptide that is covalently attached to substrate proteins (reviewed by Johnson 2004). SUMO modification of substrates can alter their function, and chromosomal proteins in particular are subject to regulation by the SUMO system. Attachment of SUMO to the substrate is similar to ubiquitination in that attachment is mediated through sequential transfer of SUMO from an E1 enzyme to an E2 and then on to substrate. The last step, transfer of SUMO from the E2 to substrate, is often facilitated by an E3 ligase such as a PIAS domain protein. Because gei-17 modifies the mus-101 feeding RNAi phenotype, we speculated that GEI-17 may stimulate SUMO modification of MUS-101. We therefore asked if GEI-17 protein could stimulate SUMO modification of MUS-101 in a reconstituted in vitro SUMOylation assay. MUS-101 was radiolabeled through transcription and translation in the presence of [35S]methionine to produce MUS-101 TnT. MUS-101 TnT was mixed with the core components of the SUMOylation machinery (E1, E2, and SUMO) as well as recombinant GEI-17, and the reaction products were resolved on an SDS-PAGE gel. This resulted in the appearance of high-molecular-weight bands that ran above MUS-101 TnT on SDS-PAGE (Figure 8). The appearance of these bands was dependent on the presence of both GEI-17 and SUMO in the reaction mixture, demonstrating that they were derived from SUMO modification of MUS-101 and that GEI-17 facilitates this. We conclude that GEI-17 stimulates SUMO modification of MUS-101 in vitro, suggesting that GEI-17 may control MUS-101 function in vivo.
Figure 8.—

GEI-17 stimulates SUMO modification of MUS-101. An in vitro SUMOylation assay was performed as described in materials and methods. (+) indicates that the given component was included in the reaction and (−) indicates that it was omitted.
DISCUSSION
In this article we have used RNAi to reduce expression of the mus-101 gene. Soaking RNAi cleared 90% of MUS-101 protein, whereas feeding RNAi removed roughly half. Accordingly, soaking RNAi caused high embryonic lethality, and this was likely through a reduction in DNA replication. For the remainder of this work we have focused on the mus-101 RNAi by feeding phenotype.
The mus-101 RNAi by feeding phenotype:
Feeding RNAi caused a low level (20%) of sterility in the F1, and DNA staining of the germ lines of sterile animals revealed a defect in germ-cell proliferation. We have observed this phenotype after RNAi against other DNA replication proteins, such as subunits of the origin recognition complex (A. Holway, unpublished data), and thus we suspect that the phenotype is connected to a defect in the initiation of DNA replication. It is also possible that an inability to repair replication-induced damage may be responsible for this phenotype.
Unlike the sterility phenotype, which was modestly penetrant, the MMS sensitivity phenotype after feeding RNAi was highly penetrant with embryonic lethality of >90% as compared to <10% for wild type. This suggests that a 50% reduction in MUS-101 protein is incompatible with an efficient embryonic DNA damage response. This, in turn, suggests that the replication and damage response functions of mus-101 can be uncoupled by feeding RNAi. Interestingly, such uncoupling has also been observed for the Drosophila and the budding and fission yeast homologs of mus-101. Drosophila mus101 hypomorphic alleles that are MMS sensitive and defective in postreplication repair but are nonetheless viable exist (Boyd et al. 1976). In fission yeast, separation-of-function alleles of cut5 that allow replication but not activation of the DNA damage checkpoint have been isolated (McFarlane et al. 1997). Finally, in budding yeast, the dpb11-1 allele is MMS sensitive at the permissive temperature for DNA replication (Wang and Elledge 2002). Thus a common theme in this gene family is the availability of hypomorphic alleles. The data presented here shed light on this by showing that alterations to the mus-101 coding sequences are not necessary to generate hypomorphic alleles as limiting the expression of an otherwise wild-type copy of the gene is sufficient to do this. One interpretation of this is that the DNA damage response function of the Mus101 family requires more protein on a per-cell basis than does the replication function.
The modifier screen:
Having established a hypomorphic condition using mus-101 feeding RNAi, we exploited this to screen for genes that modified the phenotype. This resulted in the isolation of five genes that displayed enhanced embryonic lethality when codepleted with mus-101. Interestingly, none of the genes identified are known to encode DNA replication factors. Indeed, when directed codepletion experiments were performed with mus-101 and a number of different DNA replication factors, we failed to detect any interactions (A. Holway and C. Hung, unpublished data). This is in contrast to a synthetic lethal screen performed in budding yeast using the dpb11-1 allele (Kamimura et al. 1998). This screen identified numerous sld genes, many of which are DNA replication factors. One explanation for this is that in C. elegans MUS-101 is in excess, even after feeding RNAi for embryonic DNA replication, thus making it difficult to further weaken this pathway. If so, we would not expect to find many replication genetic interactions, given that embryonic lethality was the scored phenotype.
Interestingly, three of the five genes tested (gei-17, arx-7, and F26B1.2) showed MMS sensitivity after a full dose of feeding RNAi, implying participation in the DNA damage response. F26B1.2 encodes a KH-domain-containing protein. KH proteins bind single-strand nucleic acids, and while roles for these proteins in the DNA damage response are not common, we do note that the MCG10 KH protein has been shown to be a transcriptional target of p53 and to promote cell cycle arrest and apoptosis after DNA damage in human cells (Zhu and Chen 2000). Our finding that F26B1.2 RNAi embryos are MMS sensitive further strengthens the connection between KH proteins and the DNA damage response.
The gene from the screen that showed the most robust MMS sensitivity was gei-17. The gei-17 gene was initially identified as 1 of 26 genes recovered in a yeast two-hybrid screen using the transcription factor gex-3 as bait (Tsuboi et al. 2002) and has not been further characterized. gei-17 is highly conserved and shares homology with the Su(var)2-10 chromosomal regulator in Drosophila (Hari et al. 2001) and the PIAS family of mammalian SUMO ligases (reviewed in Seeler and Dejean 2003). Three findings reported here provide strong evidence that gei-17 and mus-101 act together in a common pathway:
The gei-17 gene was selected by our screen as a mus-101 modifier.
Like mus-101, gei-17 RNAi embryos show MMS sensitivity and an MMS-induced nuclear morphology defect. We have observed MMS sensitivity after RNAi for a number of DNA damage response genes and yet the only two genes to date that produce this nuclear morphology defect are mus-101 and gei-17. Thus the nuclear morphology defect is relatively rare.
We have demonstrated a biochemical interaction between MUS-101 and GEI-17 by showing in an in vitro SUMOylation assay that GEI-17 stimulates SUMO modification of MUS-101.
Taken together, these results all suggest that SUMO modification of MUS-101 by GEI-17 regulates MUS-101 function during the DNA damage response. The connections between Mus101 and PIAS proteins are not limited to those reported here. We note that the Drosophila gei-17 homolog Su(var)2-10 shares a chromosomal phenotype with fly mus101 in that both genes are required for condensation of heterochromatic regions of the chromosome (Gatti et al. 1983; Yamamoto et al. 2000; Hari et al. 2001). How might this phenotype be connected to the MMS sensitivity reported here for the C. elegans orthologs? We propose that both mus-101 and gei-17 are required to promote DNA replication under conditions where replication is difficult. MMS-mediated damage of DNA is known to stress replication (Tercero and Diffley 2001), and replication through heterochromatin is also thought to be difficult (reviewed by Maison and Almouzni 2004). Thus the heterochromatin condensation defect may be an indirect result of a failure to replicate heterochromatic DNA. Consistent with this hypothesis is a recent study from budding yeast that isolated DPB11 in a screen for chromosome integrity genes (Huang and Koshland 2003). This work showed that DPB11 is required during the elongation phase of replication to maintain replication fork progression. Hypomorphic alleles of DPB11 caused elongation failures at higher frequencies than did wild type when elongation was challenged by increasing the distance between active origins on mini-chromosomes. While much further work is required to confirm this hypothesis, it is intriguing to speculate that GEI-17-mediated SUMO modification of MUS-101 allows MUS-101 to promote replication under stressful conditions.
Dual RNAi modifier screens:
This article presents, to our knowledge, the first large-scale screen for genetic modifiers that is based entirely on RNAi. We screened a chromosome I library that covers 13% of the C. elegans genome and isolated five modifiers. Through extrapolation we can estimate that a total of 38 modifier genes would be isolated under these conditions by screening the entire genome. This is, interestingly, in line with the average number of 34 modifiers uncovered in budding yeast SGA. Whether or not dual RNAi modifier screens are universally useful in isolating genetic modifiers remains to be determined. The drawbacks include the possibility of low penetrance that can occur using feeding RNAi. Additionally, the method does appear to isolate nonspecific interactors as it is unclear how arx-7, which encodes a cytoplasmic component of the actin polymerization regulator Arp2/3 complex, would participate directly with mus-101. Nonetheless, the method does offer two advantages. First, when working with essential genes, feeding RNAi can produce the hypomorphic conditions essential for screening. Thus the relative inefficiency of feeding RNAi can be exploited to perform modifier screens that would otherwise require the laborious and imperfect isolation of hypomorphic alleles. This allows modifier analysis to be performed on a systematic level. Second, as is the case in all functional genomic screens, once an interaction is uncovered, then the identification of the relevant gene is immediate. We have shown here that, by combining careful analysis of a feeding RNAi phenotype of an essential gene with dual RNAi modifier screening, specific and novel genetic interactions can be discovered.
Acknowledgments
We thank Craig Hunter and his colleagues for generous and expert advice on working with C. elegans. We are also grateful to Tim Schedl for thoughtful discussion and for advice and assistance during the early stages of this work. We thank Mike Matunis for help and advice on SUMO modification. The N2 Bristol strain and HT115(DE3) cells used in this work were provided by the Caenorhabditis Genetics Center, which is funded by the National Institutes of Health National Center for Research Resources. The chromosome I RNAi library was created by Julie Ahringer and distributed by MRC geneservice. A.H.H. was supported by funds from a Genetics and Genomics training grant from the National Institute of General Medical Sciences (NIGMS) and a Don Wiley Award for Excellence in Graduate Studies (funded by Merck). Additional support for this work was provided by research grants from the NIGMS (R01GM67735), the American Cancer Society (RSG-03-153), and a Searle Scholar Award to W.M.M.
References
- Araki, H., S. H. Leem, A. Phongdara and A. Sugino, 1995. Dpb11, which interacts with DNA polymerase II(epsilon) in Saccharomyces cerevisiae, has a dual role in S-phase progression and at a cell cycle checkpoint. Proc. Natl. Acad. Sci. USA 92: 11791–11795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berns, K., E. M. Hijmans, J. Mullenders, T. R. Brummelkamp, A. Velds et al., 2004. A large-scale RNAi screen in human cells identifies new components of the p53 pathway. Nature 428: 431–437. [DOI] [PubMed] [Google Scholar]
- Boutros, M., A. A. Kiger, S. Armknecht, K. Kerr, M. Hild et al., 2004. Genome-wide RNAi analysis of growth and viability in Drosophila cells. Science 303: 832–835. [DOI] [PubMed] [Google Scholar]
- Boyd, J. B., M. D. Golino, T. D. Nguyen and M. M. Green, 1976. Isolation and characterization of X-linked mutants of Drosophila melanogaster which are sensitive to mutagens. Genetics 84: 485–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner, S., 1974. The genetics of Caenorhabditis elegans. Genetics 77: 71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar, L. G., and J. D. Mcghee, 1988. DNA synthesis and the control of embryonic gene expression in C. elegans. Cell 53: 589–599. [DOI] [PubMed] [Google Scholar]
- Fire, A., S. Xu, M. K. Montgomery, S. A. Kostas, S. E. Driver et al., 1998. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391: 806–811. [DOI] [PubMed] [Google Scholar]
- Fraser, A. G., R. S. Kamath, P. Zipperlen, M. Martinez-Campos, M. Sohrmann et al., 2000. Functional genomic analysis of C. elegans chromosome I by systematic RNA interference. Nature 408: 325–330. [DOI] [PubMed] [Google Scholar]
- Gartner, A., S. Milstein, S. Ahmed, J. Hodgkin and M. O. Hengartner, 2000. A conserved checkpoint pathway mediates DNA damage–induced apoptosis and cell cycle arrest in C. elegans. Mol. Cell 5: 435–443. [DOI] [PubMed] [Google Scholar]
- Gatti, M., D. A. Smith and B. S. Baker, 1983. A gene controlling condensation of heterochromatin in Drosophila melanogaster. Science 221: 83–85. [DOI] [PubMed] [Google Scholar]
- Gonczy, P., C. Echeverri, K. Oegema, A. Coulson, S. J. Jones et al., 2000. Functional genomic analysis of cell division in C. elegans using RNAi of genes on chromosome III. Nature 408: 331–336. [DOI] [PubMed] [Google Scholar]
- Grelon, M., G. Gendrot, D. Vezon, G. Pelletier, G. Mathilde et al., 2003. The Arabidopsis MEI1 gene encodes a protein with five BRCT domains that is involved in meiosis-specific DNA repair events independent of SPO11-induced DSBs. Plant J. 35: 465–475. [DOI] [PubMed] [Google Scholar]
- Hari, K. L., K. R. Cook and G. H. Karpen, 2001. The Drosophila Su(var)2–10 locus regulates chromosome structure and function and encodes a member of the PIAS protein family. Genes Dev. 15: 1334–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman, J. L. T., B. Garvik and L. Hartwell, 2001. Principles for the buffering of genetic variation. Science 291: 1001–1004. [DOI] [PubMed] [Google Scholar]
- Huang, D., and D. Koshland, 2003. Chromosome integrity in Saccharomyces cerevisiae: the interplay of DNA replication initiation factors, elongation factors, and origins. Genes Dev. 17: 1741–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, E. S., 2004. Protein modification by SUMO. Annu. Rev. Biochem. 73: 355–382. [DOI] [PubMed] [Google Scholar]
- Johnson, E. S., and A. A. Gupta, 2001. An E3-like factor that promotes SUMO conjugation to the yeast septins. Cell 106: 735–744. [DOI] [PubMed] [Google Scholar]
- Kamimura, Y., H. Masumoto, A. Sugino and H. Araki, 1998. Sld2, which interacts with Dpb11 in Saccharomyces cerevisiae, is required for chromosomal DNA replication. Mol. Cell. Biol. 18: 6102–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon, J. Y., J. Kim-Ha, B. J. Lee and J. Lee, 2001. The MED-7 transcriptional mediator encoded by let-49 is required for gonad and germ cell development in Caenorhabditis elegans. FEBS Lett. 508: 305–308. [DOI] [PubMed] [Google Scholar]
- MacQueen, A. J., and A. M. Villeneuve, 2001. Nuclear reorganization and homologous chromosome pairing during meiotic prophase require C. elegans chk-2. Genes Dev. 15: 1674–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda, I., Y. Kohara, M. Yamamoto and A. Sugimoto, 2001. Large-scale analysis of gene function in Caenorhabditis elegans by high-throughput RNAi. Curr. Biol. 11: 171–176. [DOI] [PubMed] [Google Scholar]
- Maison, C., and G. Almouzni, 2004. HP-1 and the dynamics of heterochromatin maintenance. Nat. Rev. Mol. Cell Biol. 5: 296–304. [DOI] [PubMed] [Google Scholar]
- Manke, I. A., D. M. Lowery, A. Nguyen and M. B. Yaffe, 2003. BRCT repeats as phosphopeptide-binding modules involved in protein targeting. Science 302: 636–639. [DOI] [PubMed] [Google Scholar]
- McFarlane, R. J., A. M. Carr and C. Price, 1997. Characterisation of the Schizosaccharomyces pombe rad4/cut5 mutant phenotypes: dissection of DNA replication and G2 checkpoint control function. Mol. Gen. Genet. 255: 332–340. [DOI] [PubMed] [Google Scholar]
- Novina, C. D., and P. A. Sharp, 2004. The RNAi revolution. Nature 430: 161–164. [DOI] [PubMed] [Google Scholar]
- Paddison, P. J., J. M. Silva, D. S. Conklin, M. Schlabach, M. Li et al., 2004. A resource for large-scale RNA-interference-based screens in mammals. Nature 428: 427–431. [DOI] [PubMed] [Google Scholar]
- Saka, Y., and M. Yanagida, 1993. Fission yeast cut5+, required for S phase onset and M phase restraint, is identical to the radiation-damage repair gene rad4+. Cell 74: 383–393. [DOI] [PubMed] [Google Scholar]
- Schmidt, D., and S. Muller, 2003. PIAS/SUMO: new partners in transcriptional regulation. Cell. Mol. Life Sci. 60: 2561–2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeler, J. S., and A. Dejean, 2003. Nuclear and unclear functions of SUMO. Nat. Rev. Mol. Cell Biol. 4: 690–699. [DOI] [PubMed] [Google Scholar]
- Tercero, J. A., and J. F. Diffley, 2001. Regulation of DNA replication fork progression through damaged DNA by the Mec1/Rad53 checkpoint. Nature 412: 553–557. [DOI] [PubMed] [Google Scholar]
- Timmons, L., and A. Fire, 1998. Specific interference by ingested dsRNA. Nature 395: 854. [DOI] [PubMed] [Google Scholar]
- Tong, A. H., M. Evangelista, A. B. Parsons, H. Xu, G. D. Bader et al., 2001. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science 294: 2364–2368. [DOI] [PubMed] [Google Scholar]
- Tong, A. H., G. Lesage, G. D. Bader, H. Ding, H. Xu et al., 2004. Global mapping of the yeast genetic interaction network. Science 303: 808–813. [DOI] [PubMed] [Google Scholar]
- Tsuboi, D., H. Qadota, K. Kasuya, M. Amano and K. Kaibuchi, 2002. Isolation of the interacting molecules with GEX-3 by a novel functional screening. Biochem. Biophys. Res. Commun. 292: 697–701. [DOI] [PubMed] [Google Scholar]
- Van Hatten, R. A., A. V. Tutter, A. H. Holway, A. M. Khederian, J. C. Walter et al., 2002. The Xenopus Xmus101 protein is required for the recruitment of Cdc45 to origins of DNA replication. J. Cell Biol. 159: 541–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, H., and S. J. Elledge, 2002. Genetic and physical interactions between DPB11 and DDC1 in the yeast DNA damage response pathway. Genetics 160: 1295–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto, R. R., J. M. Axton, Y. Yamamoto, R. D. Saunders, D. M. Glover et al., 2000. The Drosophila mus101 gene, which links DNA repair, replication and condensation of heterochromatin in mitosis, encodes a protein with seven BRCA1 C-terminus domains. Genetics 156: 711–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamane, K., M. Kawabata and T. Tsuruo, 1997. A DNA-topoisomerase-II-binding protein with eight repeating regions similar to DNA-repair enzymes and to a cell-cycle regulator. Eur. J. Biochem. 250: 794–799. [DOI] [PubMed] [Google Scholar]
- Yu, X., C. C. Chini, M. He, G. Mer and J. Chen, 2003. The BRCT domain is a phospho-protein binding domain. Science 302: 639–642. [DOI] [PubMed] [Google Scholar]
- Zhong, W., H. Feng, F. E. Santiago and E. T. Kipreos, 2003. CUL-4 ubiquitin ligase maintains genome stability by restraining DNA-replication licensing. Nature 423: 885–889. [DOI] [PubMed] [Google Scholar]
- Zhu, J., and X. Chen, 2000. MCG10, a novel p53 target gene that encodes a KH domain RNA-binding protein, is capable of inducing apoptosis and cell cycle arrest in G(2)-M. Mol. Cell. Biol. 20: 5602–5618. [DOI] [PMC free article] [PubMed] [Google Scholar]