

Abstract
A geostatistical perspective on spatial genetic structure may explain methodological issues of quantifying spatial genetic structure and suggest new approaches to addressing them. We use a variogram approach to (i) derive a spatial partitioning of molecular variance, gene diversity, and genotypic diversity for microsatellite data under the infinite allele model (IAM) and the stepwise mutation model (SMM), (ii) develop a weighting of sampling units to reflect ploidy levels or multiple sampling of genets, and (iii) show how variograms summarize the spatial genetic structure within a population under isolation-by-distance. The methods are illustrated with data from a population of the epiphytic lichen Lobaria pulmonaria, using six microsatellite markers. Variogram-based analysis not only avoids bias due to the underestimation of population variance in the presence of spatial autocorrelation, but also provides estimates of population genetic diversity and the degree and extent of spatial genetic structure accounting for autocorrelation.
METHODS for the analysis of spatial genetic structure have mostly been developed for single-locus, diploid genotypic data such as provided by isozymes (Smouse and Peakall 1999). In contrast to this latter marker type, microsatellite data also contain information on repeat numbers of individual gene copies. Microsatellite markers are often highly variable, and differences in allele size are interpreted in the light of alternative evolutionary models. Under the infinite allele model (IAM), any mutation is assumed to lead to a new allele, whereas under the stepwise mutation model (SMM), mutation increases or decreases the number of repeats at a microsatellite locus most likely by one (Balloux and Goudet 2002). Neither of these two extreme mutation models seems to fit perfectly to microsatellite loci, so that measures based on IAM and SMM are often reported together (Balloux and Lugon-Moulin 2002). The difference between statistical measures (see below) under the two models is assumed to indicate the relative importance of mutation and drift (Hardy 2003).
Population genetic analyses are based on gene diversity under the IAM (cf. FST) and on molecular variance under the SMM (cf. RST). FST and RST quantify the differentiation of isolated populations assuming random mating within and restricted gene flow among populations. Both FST and RST can be adapted to pairwise comparisons, and Mantel tests are used to test the correlation with geographic distance between pairs of populations (Hardy and Vekemans 2002). However, limited gene movement can cause isolation-by-distance effects even within continuous populations. The resulting spatial genetic structure within a population can be summarized by kinship for IAM (Loiselle et al. 1995) or relationship coefficients for SMM (Streiff et al. 1998). Kinship and relationship coefficients assess the similarity of homologous alleles between individuals and may be expressed as a function of geographic distance. Statistical tests for isolation-by-distance within continuous populations often involve either a Mantel permutation test of Moran's I (or related correlation coefficients, e.g., Smouse and Peakall 1999) or join-count statistics (Epperson 2003).
When assessing genetic diversity, it may be necessary to exclude comparisons of gene copies within individuals if they cannot be assumed to be independent. For organisms with variable ploidy levels within populations such as Taraxacum sp. (Meirmans et al. 2003; Van der Hulst et al. 2003), individuals with a high ploidy level will receive more weight in the estimation of the population genetic diversity than, e.g., diploid individuals unless ploidy level is accounted for. A similar problem arises for clonal organisms, where the multiple sampling of ramets from the same genetic individuum (genet) can bias any measure of genetic structure of a population (Parks and Werth 1993; Balloux et al. 2003; Hämmerli and Reusch 2003). This is commonly taken into account by retaining a single sample per genet, either assuming the center of a clonal patch to be its origin or randomly selecting one sample per genet (Reusch et al. 1999; Chung and Epperson 2000; Hämmerli and Reusch 2003). Both approaches may, however, lead to a considerable loss of information and increased error in the description of the spatial genetic structure within populations.
Vekemans and Hardy (2004) identified some important problems and common misuses of spatial analysis in population genetics.
The spatial genetic structure is often described in terms of a maximum distance to which such structure extends. The common practice of assessing the extent of spatial genetic structure by the distance at which a Moran's I correlogram reaches zero (e.g., Epperson 2003) is misleading, as this estimate depends strongly on the sampling design (Vekemans and Hardy 2004).
The presence of nonrandom spatial genetic structure can be tested using Mantel permutation tests for a series of distance classes, and a Bonferroni correction is applied to account for multiple tests. Vekemans and Hardy (2004) caution that while the uncorrected test is too liberal, the correction makes it too conservative, and they argue that this approach should not be used to determine the scales of spatial genetic structure, as the null hypothesis is only the overall absence of spatial genetic structure.
The amount of spatial genetic structure should not be assessed from the value (e.g., of Moran's I) for the first distance class, as this absolute value depends strongly on the sampling design (Fenster et al. 2003; Vekemans and Hardy 2004).
Estimating biological parameters, such as dispersal distances, is valid only if the observed spatial genetic structure represents a true isolation-by-distance pattern at dispersal-drift equilibrium (Vekemans and Hardy 2004), thus assuming that the patterning results only from limited dispersal, that it has reached a stationary phase, and that the scale of the study is appropriate (Vekemans and Hardy 2004).
Moran's I, Mantel tests, and join-count statistics were borrowed from the general field of spatial statistics, originally developed, e.g., in geography, and adapted to population genetic data and questions as necessary. Other measures of spatial genetic structure, such as kinship or relationship coefficients, were developed specifically for genetic data and are little integrated with spatial statistical theory. However, many of the above problems are of a general nature and not specific to population genetics. Particularly, variogram modeling as developed in geostatistics may provide explanations and alternatives for the problems raised by Vekemans and Hardy (2004). The term variogram refers to a plot of the semivariance (see below) against distance. The well-known Geary's c correlogram is actually a standardized variogram (Legendre and Legendre 1998). Several population genetic measures and methods rely on the semivariance, namely the genetic distance measure by Goldstein et al. (1999) and the RST statistic (Slatkin 1995). Nonetheless, variogram modeling is rare in population genetics. Piazza and Menozzi (1983) proposed a variogram of differences in allele frequencies between populations, and Monestiez and Goulard (1997) provided an application of multivariate geostatistical analysis to genetic data, but neither approach found much resonance in the population genetic literature.
Wagner (2003)(2004) developed a formal integration of multivariate analysis and geostatistics in the context of plant community ecology. The crucial point of such an integration of spatial and nonspatial analysis is that the semivariance partitions the estimate of the population variance by distance class (Wagner 2003). Hence, the semivariance can be used to partition the results of nonspatial analyses, such as population estimates of genetic diversity, by distance (multiscale ordination), and variograms can be interpreted in an ecologically more meaningful way.
This article extends the spatial partitioning of variance to population genetic data and problems. a geostatistical perspective introduces key geostatistical concepts and methods and discusses the sensitivity of commonly used measures of autocorrelation and population variance. development of methods pursues three specific objectives: (i) to derive a spatial partitioning of measures of genetic diversity compatible with IAM and SMM, (ii) to develop a method for weighting sampling units to reflect different ploidy levels or multiple sampling of ramets within genets without data reduction, and (iii) to show how variogram modeling can be used for estimating population genetic parameters and summarizing the spatial genetic structure within populations. The methods are illustrated with a worked example (appendix) and with an application to empirical microsatellite data from a population of the haploid, tree-colonizing (epiphytic) lichen Lobaria pulmonaria. We conclude with considerations for the robust estimation of the spatial genetic structure of continuous populations.
A GEOSTATISTICAL PERSPECTIVE
Geostatistical concepts and methods:
Spatial autocorrelation and stationarity:
Spatial autocorrelation refers to the common phenomenon that nearby observations tend to be more similar than distant ones. Positive spatial autocorrelation is assumed to result from any kind of spatial process, such as pollen flow or seed dispersal in plants. When a variable is studied in space, the observed spatial autocorrelation can be quantified for various purposes (Fortin et al. 2001), such as: (i) testing for the presence of autocorrelation, e.g., to meet assumptions for estimating population characteristics; (ii) assessing the range of autocorrelation, i.e., the distance beyond which observations are spatially independent; (iii) fitting a theoretical model to summarize the observed spatial structure; and (iv) inferring about the underlying spatial process, such as dispersal distances and differences among populations. However, geostatistical analysis requires some assumption of stationarity, i.e., the structure of spatial autocorrelation must be the same throughout the study area. Specifically, it is common to assume weak stationarity, where the mean and the variance are constant and the autocorrelation depends only on the geographic distance between sampling units (Burrough 1995).
Correlograms and the empirical variogram:
Geostatistics considers four statistical moments of a random variable: (i) its mean, (ii) variance, (iii) covariance, and (iv) semivariance (Burrough 1995). Spatial autocorrelation can be quantified on the basis of covariance (Moran's I) or semivariance (empirical variogram and Geary's c correlogram). Bertorelle and Barbujani (1995) derived alternative versions of Moran's I and Geary's c for binary-coded data on the basis of, e.g., DNA sequences or RFLP patterns. Correlograms are standardized through division by the sample variance (Moran's I) or population variance (Geary's c; Cliff and Ord 1981):
Moran's I:
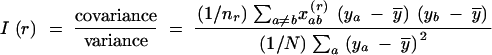 |
1 |
Geary's c:
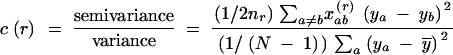 |
2 |
Empirical variogram:
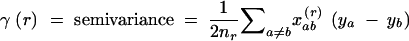 |
3 |
Given N samples, these coefficients are calculated on the basis of pairs of samples a and b falling into a series of distance classes r. The Kronecker weight xrab for the pair of observations a and b takes the value 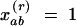 if a pair of samples belongs to distance class r and
if a pair of samples belongs to distance class r and 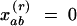 otherwise, and nr is the sum of the weights xrab for the given distance class, i.e., the number of pairs of gene copies a and b from two samples separated by a distance falling into distance class r. However, nr decreases for large distance classes r, and bias may arise from the fact that only the observations from the edge of the sampled population can contribute to the estimates for larger distances. It is therefore customary to limit the description of the spatial structure to half the maximum distance between sampling units (Cressie 1993).
otherwise, and nr is the sum of the weights xrab for the given distance class, i.e., the number of pairs of gene copies a and b from two samples separated by a distance falling into distance class r. However, nr decreases for large distance classes r, and bias may arise from the fact that only the observations from the edge of the sampled population can contribute to the estimates for larger distances. It is therefore customary to limit the description of the spatial structure to half the maximum distance between sampling units (Cressie 1993).
Figure 1A shows the empirical variogram and Figure 1B shows Moran's I and Geary's c correlograms of an artificial, spatially autocorrelated random variable. Geary's c correlogram is a rescaled version of the empirical variogram, and Moran's I correlogram resembles, but is not identical to, 1 − c(r) (Legendre and Legendre 1998). In the absence of spatial autocorrelation, the expected value of Geary's c is E[c(r)] = 1, whereas the expected value of Morans's I is E[I(r)] = −1/(N − 1), which approaches zero for large sample sizes N (Sokal and Wartenberg 1983; Epperson 2003).
Figure 1.—

Empirical variogram (A) and correlograms (B) for an artificial, spatially autocorrelated random variable simulated on a grid of 30 × 30 cells. Each symbol denotes the semivariance γ(r) (circles), Geary's c(r) (squares), or Moran's I(r) (triangles) calculated from all pairs of samples falling into each distance class r. The value for the last distance class of each series contains all pairs separated by >20 units and is drawn at the mean of the respective distances. (A) The solid line represents the fitted exponential variogram model. The dotted line (sill) indicates the population variance as estimated accounting for autocorrelation. The dashed line indicates the practical range, where the curve reaches 95% of the sill. The intercept (nugget variance) is the variance component that is not spatially structured. (B) The solid line indicates the expected value of Moran's I(r), which is very close to zero, whereas the dashed line marks the expected value of Geary's c(r), which equals one.
Variogram modeling:
The autocorrelation structure can be modeled by fitting a theoretical variogram model to the empirical variogram. The elementary theoretical variograms suitable for modeling patterns due to a single, stationary spatial process are defined by the following parameters: (i) model family, such as exponential, spherical, or Gaussian; (ii) nugget variance, i.e., the variance among adjacent samples; (iii) range, or the distance beyond which observations are spatially independent; and (iv) sill, the constant variance among spatially uncorrelated samples (Figure 1A; Isaaks and Srivastava 1989).
Sensitivity of measures of autocorrelation and population variance:
Sensitivity to nonstationarity:
The assumption of weak stationarity can be violated in several ways, including (i) nonstationarity of the mean in the population, e.g., in the presence of clinal structure, (ii) nonstationarity of the variance, e.g., if the variability of a microsatellite locus increases with increasing number of repeats, or (iii) anisotropy, where the autocorrelation structure depends on direction, e.g., if mean seed dispersal distances are larger than average in the predominant wind direction. Strictly speaking, the stationarity assumption concerns the underlying process and not the observed pattern, so that it cannot be tested directly (Fortin et al. 2003). However, the empirical variogram can be used to check for problems with nonstationarity. A finite, constant variance will always result in the presence of a sill, whereas a continued increase of the semivariance with distance may indicate a spatial trend in the mean, possibly coupled with dependence of the variance on the mean. Separate empirical variograms can be calculated for different directions and compared to check for anisotropy. In theory, the same visual inspections could be performed with correlograms, but only the variogram offers the possibility of modeling different components of variance and, thus, accounting for them. For instance, an exponential variogram function could be used to model the autocorrelation due to a stationary spatial process, and a linear variogram function could be used to model the increase in variance with distance due to a cline.
Sensitivity to the sampling design:
The variogram can be interpreted as a distance-dependent estimate of the population variance (Wagner 2003). The commonly used “unbiased” estimator of population variance, 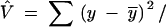 (N − 1), assumes independent, spatially uncorrelated observations, which would correspond to a strictly horizontal empirical variogram. In essence, this requires the assumption of a panmictic population with random dispersal, which is likely to be violated in most natural systems.
(N − 1), assumes independent, spatially uncorrelated observations, which would correspond to a strictly horizontal empirical variogram. In essence, this requires the assumption of a panmictic population with random dispersal, which is likely to be violated in most natural systems.
Spatial autocorrelation reduces the variance between closely spaced pairs of observations. The following simulation illustrates the consequences of spatial autocorrelation for estimating the population variance and thus for rescaling Moran's I and Geary's c correlograms. An artificial, autocorrelated variable was sampled in different ways and the estimates of the population variance, averaged over many replicate simulations, were compared to the true value. We compared three sampling strategies: (i) systematic sampling, with a spacing known to be larger than the range of spatial autocorrelation to obtain spatially uncorrelated, independent sampling units; (ii) random sampling; and (iii) stratified or clustered sampling, selecting groups of nearby locations to obtain an appropriate representation of short distance classes for spatial analysis. The criteria for comparison were accuracy, i.e., the absence of bias so that the mean of all replicate estimates is close to the true population value, and precision, i.e., low variability of replicate estimates (Palmer 1990). For details of the simulation experiment, see the Table 1 legend.
TABLE 1.
Accuracy and precision of estimates of the population variance for different sampling designs
| N | Systematic | Random | Clustered | |
|---|---|---|---|---|
| Estimated population variance | 10 | 1.019 | 1.026 | 0.584 |
| 20 | 1.003 | 1.013 | 0.757 | |
| 50 | 1.011 | 1.033 | 0.904 | |
| 100 | 1.007 | 0.997 | 0.969 | |
| Standard deviation of estimates | 10 | 0.477 | 0.478 | 0.810 |
| 20 | 0.288 | 0.330 | 0.616 | |
| 50 | 0.151 | 0.192 | 0.401 | |
| 100 | 0.024 | 0.131 | 0.284 | |
| Proportion of autocorrelated pairs | 10 | 0.00 | 0.016 | 0.448 |
| 20 | 0.00 | 0.016 | 0.218 | |
| 50 | 0.00 | 0.016 | 0.089 | |
| 100 | 0.00 | 0.016 | 0.048 |
The autocorrelated data were generated by dividing an ordered vector of 500 random values from a standard normal distribution into groups of five consecutive values. The entire groups and the values within each group were reordered at random, representing a hypothetical transect where always five neighboring locations would show very similar values, with random steps between groups. The data set has an expected variance of 1.0 and the proportion of (autocorrelated) within-group comparisons is 0.016. Three types of samples were taken from the transect: (i) a random sample from all locations, (ii) a systematic sample selecting every fifth location, and (iii) a clustered sample, where entire groups of five neighboring locations were selected at random. Data simulation and sampling were repeated 1000 times for each sample size of 10, 20, 50, or 100.
The systematic and the random samples provided unbiased estimates, independent of sample size (Table 1). Precision increased with sample size; i.e., the standard deviation of the estimates was reduced. For small sample sizes, where the chances of randomly selecting autocorrelated samples were small, the systematic and the random samples reached a similar precision. With increasing sample size, however, the random samples provided a lower precision than the systematic samples. This effect was due to the increasing number of comparisons between autocorrelated samples, not their proportion. Parametric statistical tests assume spatially uncorrelated samples, which in this simulation corresponds to the systematic sampling design. For a spatially autocorrelated variable, the increased variability of estimates from a random sample may render such tests too liberal. This means that the actual probability of rejecting the null hypothesis when it is true (type I error) may be larger than the stated significance level α.
On average, the clustered samples strongly underestimated the population variance (Table 1). This negative bias was reduced with increasing sample size, as more and more clusters of samples were selected, thus reducing the proportion of comparisons between autocorrelated pairs of samples. In fact, the variance of the estimates based on clustered samples was comparable to the variance for systematic samples with a five times smaller sample size, which can be explained by the sampling of clusters of five strongly autocorrelated locations. However, the clustered samples provided biased estimates, whereas the corresponding systematic samples were unbiased. Hence, the unbiased variance estimator may be negatively biased due to spatial autocorrelation. The magnitude of this systematic bias will depend on the spatial autocorrelation structure and the proportion of autocorrelated samples, which are functions of the spatial configuration of the sample rather than sample size. One may argue that the spatial autocorrelation structure is an inherent characteristic of a population. However, because the estimate of the population variance depends on the sampling design, it should be based on independent samples.
Correlograms imply division by the sample or population variance (see Equations 1 and 2). Because of this, it follows that (i) the actual values of Moran's I(r) and Geary's c(r) depend on the spatial configuration of the sample, and (ii), for a stationary process, I(r) reaches a value slightly below zero, and c(r) a value above one, for distances beyond the range of spatial autocorrelation. The exact deviation cannot be predicted without knowing the spatial autocorrelation structure and the details of the sampling design. This is not accounted for by subtracting E[I(r)] = −1/(N − 1) for Moran's I(r). On the other hand, an empirical variogram can be used to estimate the real population variance accounting for autocorrelation, usually by fitting a theoretical variogram model. Hence, the above exemplified problem of Moran's I and Geary's c can be avoided.
DEVELOPMENT OF METHODS
Definition of genetic variograms:
This paragraph shows how variance-based measures of genetic distance can be estimated from pairwise comparisons, so that variograms can be defined that provide an estimate of genetic diversity as a function of geographic distance.
Variogram of molecular variance:
The univariate definition of a variogram (Equation 3) can be extended to multivariate data. Thus, under the SMM, ya and yb are not two observations of the allele size yl of a single locus l, but vectors Ya and Yb of two observations of the number of repeats at L loci. The empirical semivariance γ̂(r) becomes half the squared Euclidean distance between Ya and Yb and is equal to the sum of the empirical semivariances γ̂l(r) of the number of repeats yl (Wagner 2003):
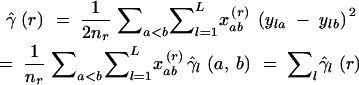 |
4 |
More generally, a multivariate variogram can be defined as a weighted average 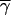 r of the component variograms γ̂l(r), with Equation 4 as the special case of wl = 1:
r of the component variograms γ̂l(r), with Equation 4 as the special case of wl = 1:
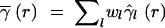 |
5 |
Most often, the variograms of the L loci will be weighted by wl = 1/L.
Under SMM, genetic diversity is related to differences in allele size, where allele size yla is defined as the number of repeats of gene copy a at locus l. The molecular variance of a single locus l with k alleles can be defined as
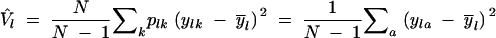 |
(Renwick et al. 2001), where
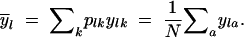 |
6 |
Equations 4 and 6 provide a distance-dependent estimate V̂(r) of the molecular variance V̂, averaged over L loci, which can be used as a within-population analog to RST to investigate isolation-by-distance effects within a continuous population:
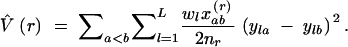 |
7 |
The statistical significance of a departure of V̂(r) from its expected value under the null hypothesis of no spatial autocorrelation can be tested in a Mantel permutation test (Legendre and Legendre 1998). If the alternative hypothesis is positive spatial autocorrelation at short distances, a one-sided test with a progressive Bonferroni correction can be applied, where the significance level for the kth distance class is α/k (Hewitt et al. 1997; Legendre and Legendre 1998; Lichstein et al. 2002). This correction is appropriate when significant autocorrelation is hypothesized to occur in the smallest distance classes and the aim is to determine the extent of spatial structure (Legendre and Legendre 1998).
Variogram of gene diversity:
The analysis of the genetic structure of a locus l under the IAM is often based on join-count statistics. The proportion of unlike joins between observations is equivalent to the sum of the variograms of a set of dummy variables zk, where zka = 1 if gene copy a is of allele k, and zka = 0 otherwise:
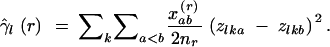 |
8 |
Due to the inherent correlation between the dummy variables, γ̂l(ab) will equal 1 if gene copies a and b are different alleles and 0 if they are the same allele.
Gene diversity or expected heterozygosity of a locus is a key parameter in population genetics under IAM. Gene diversity Hl is the probability that two gene copies sampled with replacement differ at locus l. The unbiased estimator of gene diversity, Ĥl, at locus l for a sample of N gene copies of k different alleles is
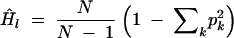 |
9 |
(Nei 1978).
On the basis of Equations 8 and 9, the variogram of multilocus gene diversity Ĥ can be defined as
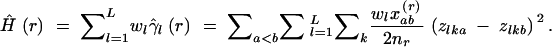 |
10 |
Ĥ(r) provides a within-population analog to FST. As with V̂(r), the significance of an observed autocorrelation in Ĥ(r) can be tested with a Mantel permutation test.
Variogram of genotypic diversity:
Genotypic diversity measured by Simpson's diversity D is similar to single-locus gene diversity Ĥl, but, instead of allele k, the multilocus genotype g is used, so that D is the probability of sampling two individuals of different multilocus genotypes. The unbiased estimator of genotypic diversity is
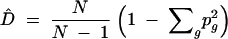 |
11 |
The variogram of genotypic diversity D̂ (Simpson diversity) is obtained by coding each multilocus genotype by a dummy variable zg, which takes the value zg = 1 if individual a is of genotype g and zg = 0 if it is not. For a haploid organism, the analysis is based on gene copies, whereas for diploid organisms, genotype coding would normally reflect diploid genotypes. The variogram of genotypic diversity, D̂(r), estimates the probability of sampling two individuals of different multilocus genotypes as a function of their distance in space and is calculated as
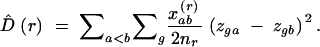 |
12 |
Accounting for ploidy levels and clonality:
This paragraph introduces a weighting scheme that accommodates different ploidy levels or multiple sampling of genets. If the different gene copies of the same diploid or polyploid organism are not assumed to be independent, e.g., due to inbreeding, one may want to restrict comparisons to gene copies from different individuals. In organisms with various ploidy levels, one may want to give equal weight to each individual independent of its ploidy level. Both problems can be solved by modifying the weights xrab,
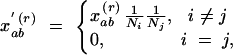 |
13 |
where Ni is the number of gene copies of individual i with gene copy a and Nj is the number of gene copies of individual j with gene copy b. The same type of weighting can be applied to account for multiple sampling of genets in clonal organisms. In that case, Ni is the number of gene copies from genet i, etc.
The permutation test needs to be adapted so that, instead of permuting gene copies, the individuals or genotypes are permuted.
Modeling of genetic variograms:
Expected shape of spatial genetic structure:
Theoretical models of isolation-by-distance predict that, in a two-dimensional space and if certain conditions are met, kinship or relationship coefficients between individuals, as well as pairwise FST or RST, vary approximately linearly with the logarithm of distance (Rousset 1997; Hardy and Vekemans 1999; Hardy 2003). Thus, with some assumptions concerning the drift-dispersal-mutation equilibrium and the dispersal function, the observed spatial genetic structure can be quantified to infer gene dispersal parameters (Vekemans and Hardy 2004). The general approach, as described by Vekemans and Hardy (2004), is to estimate the probability of identity in state as a function of the spatial distance between individuals. Because this function depends on the variability and thus the mutation rate of the locus, it needs to be standardized, for instance, by reference to random genes from a sample of individuals (Rousset 2000, 2002). The standardized values F(r) for each distance class r are regressed against spatial distance (one-dimensional case with linear relationship) or against the logarithm of distance (two-dimensional case with exponential relationship) to estimate the slope parameter b̂F. As b̂F is negative and depends somewhat on the sampling design, Vekemans and Hardy (2004) proposed quantifying spatial genetic structure by a new statistic, Sp = b̂F/(1 − FN), where FN is the relatedness of immediate neighbors competing for the same resources and may be estimated by F(1), the value of F(r) for the first distance class. If the observed, two-dimensional spatial genetic structure results solely from isotropic limited gene dispersal, if a dispersal-drift equilibrium has been reached, and if the sampling scale is appropriate for the dispersal distance of the organism, then dispersal parameters can be estimated from Sp (Vekemans and Hardy 2004).
Exponential variogram model:
Assuming an exponential relationship in a two-dimensional case (see above), the spatial genetic structure of a population can be summarized by fitting an exponential variogram model (Figure 1),
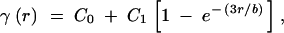 |
14 |
where C0 is the nugget variance, or the proportion of the variance that is not spatially structured, and C1 is the spatially structured variance component (Legendre and Legendre 1998). The sill C = C0 + C1 provides an estimate of the population variance based on spatially independent samples, i.e., accounting for spatial autocorrelation. The relative size of the nugget provides an estimate of FN: 1 − C0/C = C1/C = F̂N. This can be set to F(1) by fitting a fixed-nugget model, constraining the nugget variance to the observed semivariance for the first distance class.
The exponential model approaches the sill C asymptotically. Therefore, the range or slope parameter b indicates the practical range of the exponential variogram, i.e., the distance at which the curve reaches 95% of the sill (Journel and Huijbregts 1978). It can be shown that b = −3/b̂F. Directional dispersal or migration may lead to anisotropy, where the genetic structure depends on direction. If there is reason to expect anisotropy, directional variograms can be fitted, providing estimates of the slope parameter b for different compass directions.
A confidence interval for the slope parameter b may be estimated using the permutation method for the confidence interval for the matrix regression coefficient proposed by Manly (1997). The residuals of the exponential model are randomly permuted many times to obtain the reference distribution of the correlation of the residuals with distance. A series of exponential models with varying range parameters is derived, and the critical values are determined at which the correlation of the residuals of these new models with distance is as strong as for the α/2 and the (1 − α)/2 quantiles of the reference distribution. The two critical values provide the lower and upper limits of the confidence interval for the range parameter b.
APPLICATION TO THE GENETIC STRUCTURE OF L. PULMONARIA
Model organism:
L. pulmonaria is a foliose epiphytic lichen species of humid temperate and boreal regions of the northern hemisphere and cooler parts of the tropics (Yoshimura 1971). This clonal and recombinant species (Walser et al. 2004), which produces both vegetative and sexual diaspores, is considered endangered in most parts of Central Europe (Wirth et al. 1996) and in other industrialized regions. It is used as an indicator of ecological continuity (Rose 1992) in natural forests and traditionally managed agro-forestry landscapes, such as wooded pastures and chestnut orchards (Scheidegger et al. 2002).
Data:
We studied the spatial genetic structure of a continuous population of L. pulmonaria from the Swiss Jura Mountains. A hierarchical random sample of 461 thalli was collected from a pasture-woodland landscape. In a first step, 100 circular plots of 1 ha were randomly selected from the wooded parts of the study area. Within each plot, all suitable trees exceeding 5 cm in diameter at breast height were searched for L. pulmonaria. A maximum of 24 thalli were randomly selected from different trees in each of the 24 plots where the lichen was present. If there were <24 colonized trees, multiple thalli were sampled from the same tree, and if there were <24 thalli in a plot, every thallus found was included. This results in a heterogeneous data set that could exhibit spatial autocorrelation at varying scales.
DNA extraction and fragment length determination at six microsatellite loci (LPu03, LPu09, LPu15, LPu16a, LPu20a, and LPu27a), specific to the haploid mycobiont, using an ABI 3100-Avant automated sequencer (Applied Biosystems, Foster City, CA), followed Walser et al. (2003). Allele assignment was performed using GENOTYPER 2.5 software (Applied Biosystems).
Statistical analyses:
Omni-directional variograms of molecular variance V̂(r) and gene diversity Ĥ(r) were calculated according to Equations 7 and 10, giving equal weight to each of the six loci. The first distance class of r = 0 contained pairs of thalli from the same tree. The lag distance was 50 m. The last distance class contained all sample comparisons at distances >450 m. Autocorrelation was tested per distance class using a one-sided Mantel test with 500 permutations of the thalli and a progressive Bonferroni correction of α = 0.05/k for the kth distance class up to the first nonsignificant value.
A second set of variograms, V̂′(r) and Ĥ′(r), was calculated weighting each thallus by the number of occurrences of its multilocus genotype within the population, using modified weights x′rab (Equation 13). Autocorrelation was again tested per distance class using a one-sided Mantel test with 500 permutations of the multilocus genotypes, using the same settings as above.
An isotropic and four-directional variograms of genotypic diversity D̂(r) were also calculated according to Equation 12, giving equal weight to all samples. Exponential variogram models were fitted to all variograms, using the weighted least-squares algorithm (Cressie 1993) that minimizes the expression
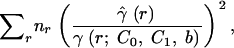 |
15 |
where γ(r; C0, C1, b) is the fitted semivariance for distance class r on the basis of the exponential model with parameters C0, C1, and b.
All calculations were performed in R (Ihaka and Gentleman 1996). The exponential variograms were fitted using the R library “GSTAT” (Pebesma and Wesseling 1998; Pebesma 2004).
Results and discussion:
On the basis of the six microsatellite markers, we found 92 multilocus genotypes of the haploid mycobiont of L. pulmonaria. All but 9 multilocus genotypes occurred in single 1-ha plots, and only 1 was spread over >210 m. The probability of origin by recombination was <0.003 for all recurrent multilocus genotypes, suggesting that they arose from clonal propagation. Weighting for recurrent genotypes drastically reduced effective sample size from 461 thalli to 92 multilocus genotypes. For recurrent genotypes, pairwise comparisons were distributed over several distance classes: the first distance class contained an equivalent of 36.8 pairs (instead of 1588 for all samples) and the other distance classes up to 450 m comprised 47.6–148.6 pairs (instead of >2000).
The spatial genetic structure of the studied L. pulmonaria population consisted of two patterns caused by clonal reproduction [variogram of genotypic diversity, D̂(r)] and sexual reproduction [variograms of molecular variance, V̂(r), and gene diversity, Ĥ(r); Figure 2]. Weighting for recurrent genotypes reduced the autocorrelation of the first distance class and the range estimate both for molecular variance and for gene diversity (Table 2). After weighting for clones, the range parameters b of the fitted exponential variogram models were smaller for molecular variance, V̂′(r), and gene diversity, Ĥ′(r), than for genotypic diversity, D̂(r), suggesting larger dispersal distances or several steps of clonal dispersal (Table 2). On the other hand, genotypic diversity showed a higher degree of autocorrelation for the first distance class, F̂(1), which consisted of pairs of samples from the same tree. The fitting of directional variograms for genotypic diversity revealed that spatial genetic structure extended further in the main wind direction (WSW–ENE; Vittoz 1998) than in the other directions (Table 2).
Figure 2.—

Variogram of gene diversity Ĥ(r) for a population of Lobaria pulmonaria. Each symbol denotes the mean semivariance over six microsatellite loci averaged over all pairs of thalli within each distance class. The semivariance is unweighted (circles) or weighted for recurrent genotypes (squares). The lines indicate the corresponding fitted exponential models; the dashed line shows the nonspatial model. Values below the dashed line correspond to positive, and values above the dashed line to negative, autocorrelation. Solid symbols indicate statistically significant positive autocorrelation based on a one-sided Mantel permutation test with progressive Bonferroni correction (α = 0.05).
TABLE 2.
Variogram parameters forLobaria pulmonaria
| Variogram modeling
|
|||||
|---|---|---|---|---|---|
| Diversity measure | Weighting | 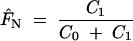 |
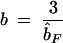 |
C = C0 + C1 | Conventional estimation: V̂, Ĥ, D̂ |
| Molecular variance V | All samples | 0.93 | 121.5 | 25.1 | 24.3 |
| Weighted for clones | 0.66 | 58.2 | 25.7 | 25.5 | |
| Gene diversity H | All samples | 0.87 | 135.9 | 0.67 | 0.64 |
| Weighted for clones | 0.55 | 69.3 | 0.68 | 0.68 | |
| Genotypic diversity D | All samples | 0.71 | 106.5 | 1.00 | 0.98 |
| WSW–ENE | — | 158.7 | 1.00 | — | |
| WNW–ESE | — | 77.7 | 1.00 | — | |
| SSW–NNE | — | 89.7 | 0.99 | — | |
| NNW–SSE | — | 110.7 | 1.00 | — | |
Estimated parameters of the exponential variogram model were fitted to the variograms of molecular variance and gene diversity for a population of Lobaria pulmonaria (N = 461) assessed with six microsatellite markers, with and without accounting for recurrent genotypes, and for genotypic diversity (clonal structure), with and without accounting for compass direction. The relatedness between immediate neighbors, FN, is estimated from the autocorrelation of the first distance class of thalli taken from the same tree. The range parameter b denotes the distance at which the curve reaches 95% of the sill and provides an estimate of the extent of spatial genetic structure. The total sill C estimates the population diversity (molecular variance, gene diversity, or genotypic diversity) accounting for spatial autocorrelation, whereas the conventional estimators V̂, Ĥ, and D̂ do not account for autocorrelation and are, therefore, susceptible to bias.
The conventional estimates of the population variance, V̂, Ĥ, and D̂, slightly underestimated the variance for spatially independent samples, i.e., the total sill C for all three measures of diversity (Table 2). In this specific example, however, weighting for recurrent genotypes largely compensated this bias.
DISCUSSION
Advantages of variogram analysis:
A geostatistical perspective on spatial genetic structure can provide explanations to many of the issues raised by Vekemans and Hardy (2004) and suggest new approaches to address them. First, the sampling design has a strong influence on the absolute values of Moran's I or other coefficients of relatedness, and this may severely limit comparability between studies. The sampling design also affects the distance at which these measures reach their expected value in the absence of spatial structuring. Therefore, this distance provides only a somewhat arbitrary estimate of the extent of spatial genetic structure (Vekemans and Hardy 2004). This problem affects the analysis of kinship structure with Moran's I or relationship coefficients, where empirical values for larger distances tend to be slightly below zero, whereas in theory negative kinship coefficients are not allowed (Barbujani 1987). Our simulation experiment showed that this effect is not simply due to sample size, but relates to the inclusion of autocorrelated samples in the estimation of the population variance, which is commonly used as a reference for rescaling correlograms and other measures of relatedness. Variogram modeling, on the other hand, provides an estimate of the population variance accounting for spatial autocorrelation, and its model parameters are scaled by this corrected estimate.
Second, Vekemans and Hardy (2004) suggested that if a plot of F(r) against distance r, e.g., a Moran's I correlogram, decreases steadily until some distance x and shows no further trend, this distance may be interpreted as the extent of spatial genetic structure. In geostatistical terms, this means that if the variogram represents a stationary spatial process as indicated by the presence of a sill, the range b can be estimated. Rather than visually identifying a critical distance at which the sill is reached, one would fit an exponential variogram model and estimate the practical range, where the curve reaches 95% of the sill. This provides an estimate of the extent of spatial genetic structure. A confidence interval for the range parameter can be constructed using a method developed for matrix regression coefficients by Manly (1997).
Third, the interpretation of Moran's I as a correlation coefficient or of other measures as the absolute degree of kinship or relationship is jeopardized by the dependence of the empirical values on the sampling design through an implicit rescaling (see above). The proposed empirical variograms, however, have direct interpretations independent of the sampling design, as they provide distance-dependent estimates of molecular variance V̂, gene diversity Ĥ, and genotypic diversity D̂, providing within-population analogs to the population pairwise RST and FST statistics.
The variogram of molecular variance, V̂(r), for microsatellite data has a straightforward interpretation as the variance in the number of repeats expected for samples at a given distance in geographic space. It corresponds directly to a plot of (half) the sum of squared size differences as used in AMOVA of microsatellite data (Schneider et al. 2000) or of D0/2, where D0 is the average squared difference in repeat numbers for two alleles drawn from the same population (Goldstein et al. 1995). The variogram of genotypic diversity, D̂(r), can be interpreted as the probability of sampling two different multilocus genotypes as a function of their spatial distance. The interpretation of the variogram of gene diversity, Ĥ(r), is the probability of sampling two different alleles given their distance in geographic space, averaged over different loci. For a single locus, it corresponds exactly to a plot of the proportion of unlike links against distance, but the variogram definition is computationally simpler than the explicit coding of links. This method is related to the cc-correlogram proposed by Bertorelle and Barbujani (1995), which involves division by the population variance, and could easily be adapted to dominant markers such as randomly amplified polymorphic DNA (RAPD), intersimple sequence repeat (ISSR), or amplified fragment length polymorphism (AFLP) markers.
Fourth, Vekemans and Hardy (2004) proposed a new statistic for quantifying spatial genetic structure, Sp = −bF/FN, which arguably is more robust than either of the two component measures that are sensitive to the sampling design (see above). Rather than taking the ratio of two potentially biased quantities, variogram modeling accounts for the source of this potential bias by estimating the variance between uncorrelated samples. Hence, the variogram parameters nugget variance, range, and sill can be directly compared between studies. Furthermore, FN can be estimated in two different ways. If the first distance class contains the direct neighboring samples, thus representing the smallest possible distance, the semivariance for this distance class can be used as an estimate of FN (fixed nugget-effect model). Alternatively, if the first distance class also contains not directly adjacent samples, the nugget effect needs to be fitted, providing an estimate of FN.
Robust estimation of spatial genetic structure:
Weighting for clonality and ploidy levels:
The lichen example illustrated the importance of distinguishing between spatial patterns of clonality and of genetic diversity resulting from sexual recombination. Specifically, it is crucial to account for clonal patterns when analyzing patterns of genetic diversity within a population. The confounded pattern does not represent the average of the two component patterns, but their multiplication, so that the degree and extent of spatial genetic structure may be severely overestimated.
For diploid organisms, the weighting results in measures similar to the kinship coefficient by Loiselle et al. (1995) and the relationship coefficient of Streiff et al. (1998), as links within individuals are excluded. The weighting proposed here is more general and can equally be used for organisms with variable ploidy levels and applied to the correlation coefficient r by Smouse and Peakall (1999) or to join-count statistics (Epperson 2003). The proposed weighting of clones solves the problem of arbitrary resampling of recurrent genotypes, which may bias the analysis of spatial genetic structure within continuous populations (Reusch et al. 1999; Hämmerli and Reusch 2003). Whether to weight for recurrent genotypes or not will depend on the research question (e.g., dispersal distances vs. distances between mates) and the type of organism under study (e.g., clonal organisms with physically connected or detached ramets).
Deviation from exponential relationship:
Simulations showed that under isolation-by-distance on a two-dimensional grid, Moran's I typically drops from positive values at short distances to negative values at intermediate distances before reaching values just below zero for larger distances in the absence of a cline (Epperson 2003). In our L. pulmonaria example, the variograms of molecular variance (not shown) and gene diversity showed evidence for such a humped distribution. This type of nonmonotonic autocorrelation structure is often encountered in geostatistical analysis and may arise from a periodic structure (Pyrcz and Deutsch 2003) or as a sampling artifact (Journel and Huijbregts 1978; Palmer and White 1994). It may be modeled by a dampened sine hole-effect model (Journel and Huijbregts 1978; Legendre and Legendre 1998). In addition, a clinal structure may cause a linear change of variance with distance, which can be modeled by a linear variogram model. Anisotropic variograms can help identify clinal structure, e.g., in the case of directional dispersal.
Robust variogram estimation:
Testing of differences between spatial patterns from different populations is notoriously difficult, as the hypothesis concerns the underlying process and not its observed realization (Fortin et al. 2003). Many measures of spatial genetic structure suffer from a high sampling variance (Vekemans and Hardy 2004). Thus, rigorous statistical testing requires a large number of replicate populations, each with a large internal sample. Several robust variogram estimators exist (Cressie and Hawkins 1980; Cressie 1993). While Cavalli-Sforza (1984) used a robust variogram estimator based on the median variance, the modulus variogram (Cressie and Hawkins 1980) has been shown to perform better than the basic variogram estimator in situations with at least 50% nugget variance (Cressie 1993). We will perform simulations to assess to what degree robust variogram estimators may help reducing sample size within populations.
Conclusions:
Most measures of spatial genetic structure are rescaled with reference to random samples from the population. This reference is itself estimated from the data set and subject to bias unless spatial autocorrelation is accounted for. Such bias limits the interpretation of absolute values of various measures of spatial genetic structure and poses problems to the comparison between studies and to the estimation of biological parameters (Vekemans and Hardy 2004). Variogram modeling, on the other hand, estimates its reference value accounting for spatial autocorrelation, thus providing parameter estimates that are comparable between studies. Furthermore, the proposed variograms of molecular variance, gene diversity, and genetic diversity are directly interpretable without rescaling, as they provide a partitioning of genetic diversity by the distance between samples. While this article focuses on microsatellite data as interpreted under either IAM or SMM, the approach may be adapted to other types of genetic data. The formal integration with variograms makes the theory and tools of geostatistics available for population genetics, which may help to address some important challenges in bridging the gap between empirical studies of spatial genetic structure and theoretical approaches to isolation-by-distance.
Acknowledgments
We thank Magnus Nordborg and two anonymous reviewers for their helpful comments on this and an earlier version of the manuscript. This research is part of a project funded by the Swiss National Science Foundation under the National Centre of Competence in Research (NCCR) Plant Survival and through grant 3100A0-105830/1.
APPENDIX:
WORKED EXAMPLE
Example data:
The example data set consists of two artificial variables y1 and y2 that describe the fragment lengths x of two loci in N = 6 haploid individuals A–F along a transect t. There are three multilocus genotypes g with differing frequencies:
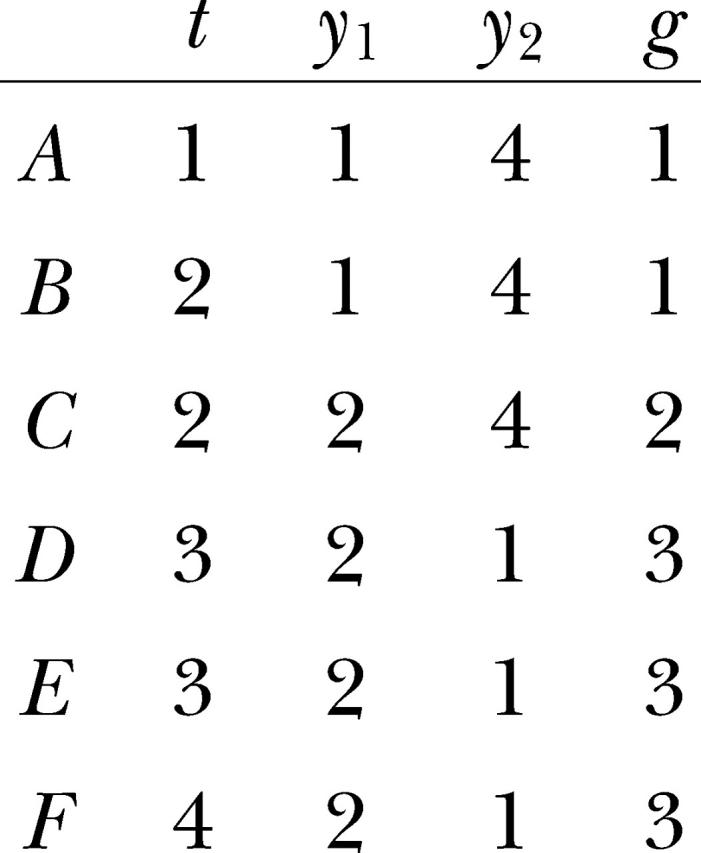 |
For instance, the comparison of gene copy A to gene copies B and D provides
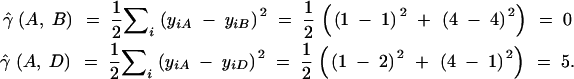 |
The semivariance γ̂i(a, b) for each pair of gene copies is tabulated in the following matrix:
 |
Matrix of distances r:
Common geostatistical analysis omits distances of r = 0, so that an object is never compared to itself. For organisms such as the epiphytic lichen L. pulmonaria, however, individuals may share the same two-dimensional geographic coordinates if they grow on the same tree. Therefore, it may be important to distinguish between different individuals separated by a distance of zero in two-dimensional space and the comparison of an individual with itself:
 |
Variogram of molecular variance:
The empirical variogram of molecular variance is calculated using Equation 7, as is illustrated here for distance class r = 2 on the basis of unique pairs only [top or bottom triangle of matrices γ(a, b) and r]:
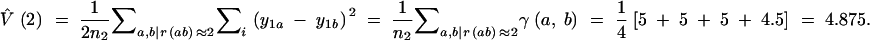 |
The variance estimates V̂(r) for all distance classes r are listed below. A weighted average of the molecular variance per distance class V̂(r), weighted by nr, the number of pairs of gene copies, provides the global variance V̂.
 |
The total number of pairwise comparisons, n, is given by n = ∑rnr = N(N − 1)/2.
Variogram of gene diversity:
For the variogram of gene diversity, a dummy variable zlk is defined for each allele k at each locus l. The semivariance is calculated from the matrix of dummy variables following Equation 8:
 |
For each pair of observations a and b, the semivariance γ̂(a, b) equals the number of loci at which they differ. The variogram of gene diversity is
 |
Variogram of genotypic diversity:
For the variogram of genotypic diversity following Equation 12, a dummy variable zg is defined for each genotype g, and the semivariance is calculated from the matrix of dummy variables:
 |
As a computational shortcut, the same result can be obtained by setting all values γ̂(a, b) > 0 to 1.
The variogram of genotype diversity is
 |
Weighting for recurrent genotypes:
Each gene copy a receives a weight wa which is inverse to the number of gene copies of the same multilocus genotype ga. According to Equation 13, the matrix of weights wawb is
 |
If G = 3 is the number of genotypes, the sum of all weights is
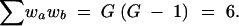 |
The variogram of molecular variance between genotypes is derived as
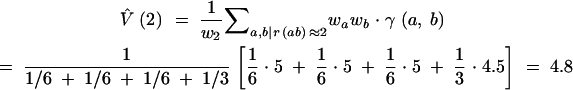 |
 |
References
- Balloux, F., and J. Goudet, 2002. Statistical properties of population differentiation estimators under stepwise mutation in a finite island model. Mol. Ecol. 11: 771–783. [DOI] [PubMed] [Google Scholar]
- Balloux, F., and N. Lugon-Moulin, 2002. The estimation of population differentiation with microsatellite markers. Mol. Ecol. 11: 155–165. [DOI] [PubMed] [Google Scholar]
- Balloux, F., L. Lehmann and T. de Meeus, 2003. The population genetics of clonal and partially clonal diploids. Genetics 164: 1635–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbujani, G., 1987. Autocorrelation of gene frequencies under isolation by distance. Genetics 117: 777–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertorelle, G., and G. Barbujani, 1995. Analysis of DNA diversity by spatial autocorrelation. Genetics 140: 811–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrough, P. A., 1995 Spatial aspects of ecological data, pp. 213–251 in Data Analysis in Community and Landscape Ecology, edited by R. H. G. Jongman, C. J. F. ter Braak and O. F. R. van Tongeren. Cambridge University Press, Cambridge, UK.
- Cavalli-Sforza, L. L., 1984 Isolation by distance, pp. 229–248 in Human Population Genetics: The Pittsburgh Symposium, edited by A. Chakravarti. Van Nostrand Reinhold, New York.
- Chung, M. G., and B. K. Epperson, 2000. Clonal and spatial genetic structure in Eurya emarginata (Theaceae). Heredity 84: 170–177. [DOI] [PubMed] [Google Scholar]
- Cliff, A. D., and J. K. Ord, 1981 Spatial Processes: Models and Applications. Pion, London.
- Cressie, N. A. C., 1993 Statistics for Spatial Data. John Wiley & Sons, New York.
- Cressie, N., and D. M. Hawkins, 1980. Robust estimation of the variogram. J. Int. Assoc. Math. Geol. 12: 115–125. [Google Scholar]
- Epperson, B. K., 2003 Geographical Genetics. Princeton University Press, Princeton, NJ.
- Fenster, C. B., X. Vekemans and O. J. Hardy, 2003. Quantifying gene flow from spatial genetic structure data in a metapopulation of Chamaecrista fasciculata (Leguminosae). Evolution 57: 995–1007. [DOI] [PubMed] [Google Scholar]
- Fortin, M.-J., M. R. T. Dale and J. ver Hoef, 2001 Spatial analysis in ecology, pp. 2051–2058 in The Encyclopedia of Environmetrics, edited by A. H. El-Shaarawi and W. W. Piegorsch. John Wiley & Sons, Chichester, UK.
- Fortin, M. J., B. Boots, F. Csillag and T. K. Remmel, 2003. On the role of spatial stochastic models in understanding landscape indices in ecology. Oikos 102: 203–212. [Google Scholar]
- Goldstein, D. B., A. R. Linares, L. L. Cavalli-Sforza and M. W. Feldman, 1995. An evaluation of genetic distances for use with microsatellite loci. Genetics 139: 463–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein, D. B., G. W. Roemer, D. A. Smith, D. E. Reich, A. Bergman et al., 1999. The use of microsatellite variation to infer population structure and demographic history in a natural model system. Genetics 151: 797–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hämmerli, A., and T. B. H. Reusch, 2003. Genetic neighbourhood of clone structures in eelgrass meadows quantified by spatial autocorrelation of microsatellite markers. Heredity 91: 448–455. [DOI] [PubMed] [Google Scholar]
- Hardy, O. J., 2003. Estimation of pairwise relatedness between individuals and characterization of isolation-by-distance processes using dominant genetic markers. Mol. Ecol. 12: 1577–1588. [DOI] [PubMed] [Google Scholar]
- Hardy, O. J., and X. Vekemans, 1999. Isolation by distance in a continuous population: reconciliation between spatial autocorrelation analysis and population genetics models. Heredity 83: 145–154. [DOI] [PubMed] [Google Scholar]
- Hardy, O. J., and X. Vekemans, 2002. SPAGEDi: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol. Ecol. Notes 2: 618–620. [Google Scholar]
- Hewitt, J. E., P. Legendre, B. H. McArdle, S. F. Thrush, C. Bellehumeur et al., 1997. Identifying relationships between adult and juvenile bivalves at different spatial scales. J. Exp. Mar. Biol. Ecol. 216: 77–98. [Google Scholar]
- Ihaka, R., and R. Gentleman, 1996. R: a language for data analysis and graphics. J. Comp. Graph. Stat. 5: 299–314. [Google Scholar]
- Isaaks, E. H., and R. M. Srivastava, 1989 Applied Geostatistics. Oxford University Press, New York.
- Journel, A. G., and C. J. Huijbregts, 1978 Mining Geostatistics. Academic Press, New York.
- Legendre, P., and L. Legendre, 1998 Numerical Ecology. Elsevier, Amsterdam.
- Lichstein, J. W., T. R. Simons, S. A. Shriner and K. E. Franzreb, 2002. Spatial autocorrelation and autoregressive models in ecology. Ecol. Monogr. 72: 445–463. [Google Scholar]
- Loiselle, B. A., V. L. Sork, J. Nason and C. Graham, 1995. Spatial genetic structure of a tropical understory shrub, Psychotria officinalis (Rubiaceae). Am. J. Bot. 82: 1420–1425. [Google Scholar]
- Manly, B. F. J., 1997 Randomization, Bootstrap and Monte Carlo Methods in Biology. Chapman & Hall, London.
- Meirmans, P. G., E. C. Vlot, J. C. M. Den Nijs and S. B. J. Menken, 2003. Spatial ecological and genetic structure of a mixed population of sexual diploid and apomictic triploid dandelions. J. Evol. Biol. 16: 343–352. [DOI] [PubMed] [Google Scholar]
- Monestiez, P., and M. Goulard, 1997 Analysing spatial genetic structures by multivariate geostatistics: study of wild populations of perennial ryegrass (Lolium perenne), pp. 1197–1208 in Geostatistics Wollongong ‘96, edited by E. Y. Baafi and N. A. Schofield. Kluwer, Dordrecht, The Netherlands.
- Nei, M., 1978. Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89: 583–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer, M. W., 1990. The estimation of species richness by extrapolation. Ecology 71: 1195–1198. [Google Scholar]
- Palmer, M. W., and P. S. White, 1994. Scale dependence and the species-area relationship. Am. Nat. 144: 717–740. [Google Scholar]
- Parks, J. C., and C. R. Werth, 1993. A study of spatial features of clones in a population of bracken fern, Pteridium aquilinum (Dennstaedtiaceae). Am. J. Bot. 80: 537–544. [DOI] [PubMed] [Google Scholar]
- Pebesma, E. J., 2004. Multivariable geostatistics in S: the gstat package. Comput. Geosci. 30: 683–691. [Google Scholar]
- Pebesma, E., and C. G. Wesseling, 1998. Gstat, a program for geostatistical modelling, prediction and simulation. Comput. Geosci. 24: 17–31. [Google Scholar]
- Piazza, A., and P. Menozzi, 1983 Geographic variation in human gene frequencies, pp. 444–450 in Numerical Taxonomy, edited by J. Felsenstein. Springer, Berlin.
- Pyrcz, M., and C. Deutsch, 2003. The whole story on the hole effect. Newsl. Geostat. Assoc. Australas. 18: 3–5. [Google Scholar]
- Renwick, A., L. Davison, H. Spratt, J. P. King and M. Kimmel, 2001. DNA dinucleotide evolution in humans: fitting theory to facts. Genetics 159: 737–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reusch, T. B. H., W. Hukriede, W. T. Stam and J. L. Olsen, 1999. Differentiating between clonal growth and limited gene flow using spatial autocorrelation of microsatellites. Heredity 83: 120–126. [DOI] [PubMed] [Google Scholar]
- Rose, F., 1992 Temperate forest management: its effects on bryophyte and lichen floras and habitats, pp. 211–233 in Bryophytes and Lichens in a Changing Environment, edited by J. W. Bates and A. Farmer. Clarendon Press, Oxford.
- Rousset, F., 1997. Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics 145: 1219–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousset, F., 2000. Genetic differentiation between individuals. J. Evol. Biol. 13: 58–62. [Google Scholar]
- Rousset, F., 2002. Inbreeding and relatedness coefficients: What do they measure? Heredity 88: 371–380. [DOI] [PubMed] [Google Scholar]
- Scheidegger, C., P. Clerc, M. Dietrich, M. Frei, U. Groner et al., 2002 Rote Liste der gefährdeten Arten der Schweiz: Baum-und Erdbewohnende Flechten. BUWAL, Bern, Switzerland.
- Schneider, S., D. Roessli and L. Excoffier, 2000 ARLEQUIN Version 2.000: A Software for Population Genetic Data Analysis. Genetics and Biometry Laboratory, University of Geneva, Geneva.
- Slatkin, M., 1995. A measure of population subdivision based on microsatellite allele frequencies. Genetics 139: 457–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smouse, P. E., and R. Peakall, 1999. Spatial autocorrelation analysis of individual multiallele and multilocus genetic structure. Heredity 82: 561–573. [DOI] [PubMed] [Google Scholar]
- Sokal, R. R., and D. E. Wartenberg, 1983. A test of spatial autocorrelation analysis using an isolation-by-distance model. Genetics 105: 219–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streiff, R., T. Labbe, R. Bacilieri, H. Steinkellner, J. Glössl et al., 1998. Within-population genetic structure in Quercus robur L. and Quercus petraea (Matt.) Liebl. assessed with isozymes and microsatellites. Mol. Ecol. 7: 317–328. [Google Scholar]
- Van der Hulst, R. G. M., T. H. M. Mes, M. Falque, P. Stam, J. C. M. Den Nijs et al., 2003. Genetic structure of a population sample of apomictic dandelions. Heredity 90: 326–335. [DOI] [PubMed] [Google Scholar]
- Vekemans, X., and O. J. Hardy, 2004. New insights from fine-scale spatial genetic structure analyses in plant populations. Mol. Ecol. 13: 921–935. [DOI] [PubMed] [Google Scholar]
- Vittoz, P., 1998 Flore et végétation du Parc jurassien vaudois: typologie, écologie et dynamiqu des milieux. Ph.D. Thesis, Université de Lausanne, Lausanne, Switzerland.
- Wagner, H. H., 2003. Spatial covariance in plant communities: integrating ordination, geostatistics, and variance testing. Ecology 84: 1045–1057. [Google Scholar]
- Wagner, H. H., 2004. Direct multiscale ordination with canonical correspondence analysis. Ecology 85: 342–351. [Google Scholar]
- Walser, J. C., C. Sperisen, M. Soliva and C. Scheidegger, 2003. Fungus-specific microsatellite primers of lichens: application for the assessment of genetic variation on different spatial scales in Lobaria pulmonaria. Fungal Genet. Biol. 40: 72–82. [DOI] [PubMed] [Google Scholar]
- Walser, J. C., F. Gugerli, R. Holderegger, D. Kuonen and C. Scheidegger, 2004. Recombination and clonal propagation in different populations of the lichen Lobaria pulmonaria. Heredity 93: 322–329. [DOI] [PubMed] [Google Scholar]
- Wirth, V., H. Schöller, P. Scholz, G. Ernst, T. Feuerer et al., 1996. Rote Liste der Flechten (Lichenes) der Bundesrepublik Deutschland. Schriftenreihe für Vegetationskunde 28: 307–368. [Google Scholar]
- Yoshimura, I., 1971. The genus Lobaria of Eastern Asia. J. Hattori Bot. Lab. 34: 231–364. [Google Scholar]