

Abstract
Many commercial inbred lines are available in crops. A large amount of genetic variation is preserved among these lines. The genealogical history of the inbred lines is usually well documented. However, quantitative trait loci (QTL) responsible for the genetic variances among the lines are largely unexplored due to lack of statistical methods. In this study, we show that the pedigree information of the lines along with the trait values and marker information can be used to map QTL without the need of further crossing experiments. We develop a Monte Carlo method to estimate locus-specific identity-by-descent (IBD) matrices. These IBD matrices are further incorporated into a mixed-model equation for variance component analysis. QTL variance is estimated and tested at every putative position of the genome. The actual QTL are detected by scanning the entire genome. Applying this new method to a well-documented pedigree of maize (Zea mays L.) that consists of 404 inbred lines, we mapped eight QTL for the maize male flowering trait, growing degree day heat units to pollen shedding (GDUSHD). These detected QTL contributed >80% of the variance observed among the inbred lines. The QTL were then used to evaluate all the inbred lines using the best linear unbiased prediction (BLUP) technique. Superior lines were selected according to the estimated QTL allelic values, a technique called marker-assisted selection (MAS). The MAS procedure implemented via BLUP may be routinely used by breeders to select superior lines and line combinations for development of new cultivars.
IN line-crossing experiments, the prerequisite for mapping quantitative trait loci (QTL) is a segregating population derived from the crosses of some carefully chosen inbred lines. The mapped QTL largely depend on the parental lines selected, leading to inconsistent results from one experiment to another. However, many commercial inbred lines are available in crops (Cui et al. 1999). Genetic variance among these lines is largely unexplored due to lack of appropriate statistical methods. To harvest the entire genetic variation among lines using current QTL mapping procedures, one may need to design a diallel crossing experiment that includes all lines as parents. This would be extremely difficult in terms of space, time, funds, and analytical methods.
Is it possible to use all the existing lines to map QTL without use of segregating progeny? The answer is yes, but not with the conventional QTL-mapping procedures. Grupe et al. (2001) proposed a method known as in silico QTL mapping. The method is a simple correlation analysis with one variable defined as the indicator of an identity-by-state (IBS) allele shared by a pair of inbred lines (x) and the other variable as the phenotypic difference between the two lines (y). If two lines share IBS at a particular locus, x is defined as 1 and otherwise 0. The total number of observations (data points) is n(n − 1)/2, where n is the total number of inbred lines included in the analysis. Using this method, Grupe et al. (2001) identified numerous QTL responsible for the variation of 10 traits in 15 inbred lines of laboratory mice (Mus musculus L.). Although Chesler et al. (2001) and Darvasi (2001) have questioned the above in silico QTL-mapping method, Chesler et al. (2001) still believe that detecting QTL from inbred lines may indeed be possible. Recently, Parisseaux and Bernardo (2004) explored the usefulness of in silico mapping via a mixed-model approach and found that their method can detect QTL highly repeatable across different populations. The method of Parisseaux and Bernardo (2004) assumed that the marker effects are fixed, whereas in this article the effects of QTL linked to markers are assumed random. In this study, we propose a variance-component-based method for QTL mapping using data from multiple commercial inbred lines. The proposed method is a variant of association mapping (Risch and Merikangas 1996) except that the response and explanatory variables are defined differently.
The advantages of using inbred lines for QTL mapping over backcross (BC) and F2 designs may be summarized as follows: (1) the phenotypic value of each inbred line can be measured in replicated experiments across environments, which results in reduced environmental and measurement errors; (2) the genotypes of inbred lines are constant across generations (breeding true); (3) cumulative historical recombination events are used so that QTL can be mapped at a fine scale; (4) experimental hybrids and their segregating progeny are no longer needed; and (5) after QTL mapping, the allelic values of QTL for each inbred line can be predicted using the best linear unbiased prediction (BLUP) so that breeders can select superior lines and line combinations for development of new cultivars.
The star phylogeny of the inbred lines may be a first-order approximation if no historical records of the inbred lines are available. In laboratory mice, partial information is available about the genealogy of the strains (Beck et al. 2000) and this information should be incorporated into the mapping program. In plant breeding, most crop varieties of self-pollinated crops are inbred lines and their parentages are well documented. These inbred lines were usually generated from repeated selfings of a hybrid derived from two parents. So, each line is literally a recombinant inbred line with respect to its parents. The progeny carry mosaic segments of the founder chromosomes. Using molecular markers, one can trace each chromosome segment of a progeny back to the origin of the founder chromosome. If two lines are traced back to the same founder for the chromosome segment in question, the two segments are said to be identity by descent (IBD), which is the building block of the random-model methodology of genetic mapping (Elston and Stewart 1971; Lander and Green 1987; Xu and Atchelly 1995; Sobel and Lange 1996). In contrast to the IBS method, the IBD analysis can eliminate spurious association due to factors other than physical linkage. We infer the IBD values shared by all pairs of lines and construct the IBD matrix for each locus. The IBD matrix varies from one locus to another, which provides the power to separate different loci in terms of genetic variances contributed by the loci.
Our approach is similar to the two-step IBD-based method of George et al. (2000), who first estimated the locus-specific IBD matrices using existing software (Heath 1997) and then incorporated these IBD matrices into a mixed-model program for variance component analysis. The difference between our method and that of George et al. (2000) is that our pedigrees are made of all inbred lines whereas their method handles pedigrees initiated from outbred founders.
QTL mapping is the first step toward marker-assisted selection. The mixed-model methodology provides all the machinery for evaluation of the inbred lines in terms of the allelic values of the identified QTL. Once the elite genes are identified, they can be used for marker-assisted selection for development of superior cultivars carrying all the desirable genes. It has been demonstrated that the phenotype-based BLUP is useful for identifying superior single crosses (Bernardo 1996a,b). The phenotype- and marker-based BLUP is even more useful for identifying superior lines for plant breeding (Bernardo 1998).
METHODS
Mixed-model analysis:
Let n be the number of inbred lines in a pedigree. Denote the number of founder lines by n0 and the number of nonfounders by n1, where n0 + n1 = n. Let 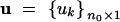 be a vector for the effects of the QTL of all founders and
be a vector for the effects of the QTL of all founders and 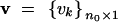 be a vector of polygenic effects of all the founders. The phenotypic value of the jth line may be described by the following mixed model,
be a vector of polygenic effects of all the founders. The phenotypic value of the jth line may be described by the following mixed model,
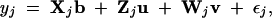 |
1 |
where Xj is an incidence matrix for the fixed (nongenetic) effects; b is a vector of the fixed effects; εj is the residual error assumed to be normally distributed with mean zero and variance σ2, denoted by εj ∼ N(0, σ2); and uk ∼ N0, σ2u, vk ∼ N0, σ2v, and σ2u and σ2v are the variances of the QTL and the polygene, respectively. The remaining symbols are defined as follows. Zj is an incidence matrix for the QTL effects and defined as a 1 × n0 vector with all elements being zero except one element. The nonzero element is unity, which occurs at the position corresponding to the founder whose allele has been transmitted to the jth line. Wj is an incidence matrix for the polygenic effects and defined as an 1 × n0 vector with the kth element being the probability that the kth founder allele has been passed to the jth line. Because all lines in the pedigree are inbred (homozygous for all loci), dominance effects cannot be modeled. Theoretically, epistatic effects can be included in the model, but we decided to exclude them in this study to simplify the method. Therefore, we are exclusively dealing with an additive model in this study. The polygenic effects are the collective effects of all loci affecting the quantitative trait that are unlinked to the QTL. The entire data array may be expressed by the following model in matrix notation,
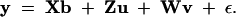 |
2 |
The expectation and variance matrix of the above model are
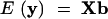 |
3 |
and
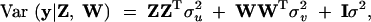 |
4 |
respectively. Note that these variances are defined as the genetic variances among the inbred lines (homozygotes), and as such they are twice the genetic variances defined in outbred populations. The variance matrix defined this way is conditional on Z and W. In genetic mapping, these incidence matrices are not observable but estimated from marker information. Therefore, the actual variance matrix is defined as
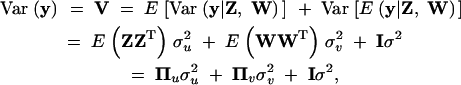 |
5 |
where Πu = E(ZZT) is called the IBD matrix for the QTL and Πv = E(WWT) is the additive relationship matrix for the polygene. It should be mentioned that Var[E(y|Z, W)] = 0 because E(y|Z, W) = Xb is a constant in the mixed model. The additive relationship matrix depends on the pedigree information and the IBD matrix of the QTL depends on the QTL position and the marker information. Methods to estimate these matrices are described in the next section. We now focus on the variance component analysis and significance test.
We take a genome-scan approach to searching for QTL from one end of the genome to the other end. At each putative position, we calculate the IBD matrix and plug in this matrix to PROC MIXED of SAS (SAS Institute 1999), which allows us to input unstructured variance matrices. PROC MIXED also calculates the likelihood value, which is required for the significance test. To test 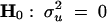 , we need to run the program twice, once to obtain the likelihood value under the full model,
, we need to run the program twice, once to obtain the likelihood value under the full model,
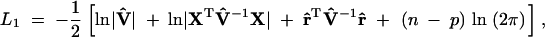 |
6 |
where 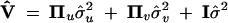 , r̂ = y − X(XTV̂−1X)− XTV̂−1y, and p is the rank of X and the other to obtain the likelihood value under the reduced model,
, r̂ = y − X(XTV̂−1X)− XTV̂−1y, and p is the rank of X and the other to obtain the likelihood value under the reduced model,
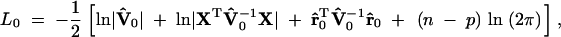 |
7 |
where 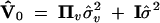 and
and 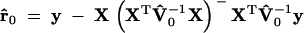 . The method is called the restricted maximum likelihood (REML) in which the vector of fixed effects has been integrated out. The likelihood-ratio test statistic is defined as
. The method is called the restricted maximum likelihood (REML) in which the vector of fixed effects has been integrated out. The likelihood-ratio test statistic is defined as
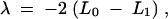 |
8 |
which is compared to a critical value for declaration of statistical significance. The critical value was calculated by the quick method developed by Piepho (2001). The genome-wide type I error for the analysis was set at 5%. Note that the relationship between λ and the logarithm of odds (LOD) score in the likelihood-ratio test is LOD = λ/(2 ln 10).
IBD matrix of QTL and additive relationship matrix:
The IBD matrix of a QTL is a function of the incidence matrix Z. However, this incidence matrix is not observable and must be estimated from information of markers linked with the putative QTL. There is no explicit form for the probability distribution of Z. However, we can take a Monte Carlo approach to simulating Z and use the average of ZZT over the replicated Monte Carlo simulations to approximate Πu = E(ZZT). We simulate Z one row (a vector) at a time from the top (founders) to the bottom (descendants) of the pedigree. As usual in pedigree analysis, individual lines are required to be listed according to their chronological order; i.e., parental lines must be listed before their progeny. This requirement will guarantee that the incidence matrices of the parents are sampled before those of their progeny. First, we order the founders from 1 to n0 and the progeny from n0 + 1 to n0 + n1. The Z vectors for the founders are actually given and no simulation is required. For example, the Z vector for the kth founder is simply a vector with all elements equal to zero except that the kth element is 1. Essentially, each founder is given a unique label from 1 to n0, from which the Z vector can be constructed. Each progeny is also given a label from 1 to n0, but this label is unknown. For example, if the jth line (progeny) received the ith founder allele, the label for line j is i and thus Zj is a vector with all elements equal to zero except that the ith element is one. In other words, Zj will be the same as the Z vector of the ith founder. Therefore, the labels serve as the blueprint for all the progeny from which the Z vectors are reconstructed.
Let lj be the label for line j for j = 1, … , n. If j is one of the founders, say founder k, then lj = k for k = 1, … , n0. If j is not a founder, the parental lines of j must be known. Let m and f be the male and female lines from which line j is derived. Note that in plants m and f are used simply to distinguish the two parents. The labels for the two parents and the progeny are denoted by lm, lf, and lj, respectively. Note that line j is not the direct progeny of the two parents. It is a recombinant inbred line (RIL) derived from the two parents. Therefore, lj takes either lm or lf but not both. We can use the following equation to describe the recurrent relationship,
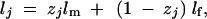 |
9 |
where zj is an indicator variable defined as
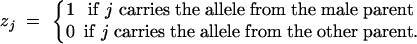 |
For a random locus without any marker information, zj takes either 1 or 0 with an equal chance. With marker information, the probability will be p(zj = 1|Im) = pj, which may be different from 1/2, where Im stands for marker information. Once pj is calculated, we can sample the value of zj from a Bernoulli distribution with parameter pj. These sampled labels are used to reconstruct the Z matrix and thus the IBD matrix. The expected IBD matrix is then approximated by repeated simulations using
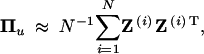 |
10 |
where N is the total number of repeated simulations and Z(i) is the simulated Z matrix in the ith replicate.
The conditional probability, p(zj = 1|Im) = pj, is calculated using a multipoint method (Rao and Xu 1998). The interval-mapping procedure (Lander and Botstein 1989) for RIL has an identical formula to that for a BC design except that the recombination fraction used in BC, r, is replaced by
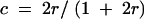 |
11 |
in the RIL type of pedigree analysis.
The IBD (additive relationship) matrix for the polygene Πv is obtained similarly except that the simulation does not depend on markers. In other words, we simulate the W matrix in the same way as we simulate the Z matrix except that the indicator variables, zj, for a polygene is simulated from a Bernoulli distribution with parameter 1/2. The IBD matrix calculated this way (using N replicated simulations) is identical to that calculated from the average of N independent loci. In fact Πv calculated this way is also the same as that obtained from the tabular method. The reason for using the Monte Carlo method to calculate Πv is that a new subroutine is not required for Πv calculation. Note that when we search for QTL of the entire genome, Πv is calculated once but Πu is calculated as many times as the number of putative positions evaluated.
BLUP estimation of QTL effects of individual lines:
To facilitate marker-assisted selection, we need to know the allelic values of each line at the detected QTL. BLUP is the appropriate tool for evaluating the inbred lines (Henderson 1975). Theoretically, we need only to predict the QTL values for the founders because the progeny carry the combination of all founder alleles. However, the incidence matrix Z is not observable and has been integrated into the IBD matrix. As a result, we are unable to predict the values of founder lines alone. Instead, we can predict the allelic values of QTL for all the inbred lines, including both the founders and the progeny. To do this, the mixed model must be rewritten as
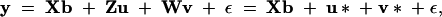 |
12 |
where u* = Zu and v* = Wv are n × 1 vectors for the QTL values and polygenic values of all the inbred lines (including both the founders and the progeny). The mixed-model equation for this kind of “animal model” is
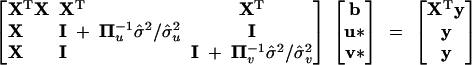 |
13 |
If the IBD matrix for QTL is singular, the PROC MIXED of SAS uses the Cholesky decomposition to handle this problem so that the inverse of the IBD matrix for QTL is no longer included in the mixed-model equation (Henderson 1984; SAS Institute 1999). Once the QTL effects are evaluated, these inbred lines can be ranked and selected.
APPLICATIONS
QTL mapping in maize:
We applied this method to a maize (Zea mays L.) pedigree consisting of 404 inbred lines with 103 founders and developed over 70 years of pedigree breeding. The experiments were carried out at Pioneer Hybrid International breeding stations located in the United States corn belt and south-central Canada (Woodstock, Ontario) during 1985–1997 with a minimum of three locations and a maximum of eight environments per year. The days to flowering for all inbred lines from early to late maturities were between June 30 and August 15 and the range of maturities was from 980 growing degree day heat units to pollen shedding (GDUSHD) to 2090 GDUSHD. Note that there was an overlap between ∼20–25% of the inbreds grown at locations from Union City, Tennessee, in the southern U.S. corn belt to Woodstock, Ontario in Canada. The average number of environments in which the inbred lines were evaluated was 9.5 with a range from 5 to 50 environments. The trait we analyzed is the male flowering trait named GDUSHD. This trait is related to corn adaptation to latitude change and has been one of the target traits for corn improvement. The trait values used in this analysis were the best linear unbiased estimates of GDUSHD of all lines calculated from unbalanced data. None of the founders have phenotypic records. Of the 301 nonfounders, only 282 have phenotypic records. Therefore, only the 282 lines with phenotypic records were subjected to mixed-model analysis.
A brief description about the measurement of the trait is given here. Growing degree day heat units (GDU), which are the same as growing degree day (GDD), were measured as accumulated heat units and calculated as
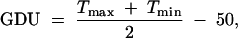 |
14 |
where Tmax and Tmin are maximum and minimum temperature per day, respectively, a value of Tmax > 86°F being entered as 86°F and a value Tmin < 50°F being entered as 50°F in the formula. GDUSHD is an accumulated GDU from seedling emergence until pollen shed rounded to the nearest 10 GDUSHD and recorded as GDUSHD/10. The calculation method most commonly used in the United States for determining heat unit accumulation relative to corn phenology was first suggested by the National Oceanic and Atmospheric Administration in 1969 and labeled as the “modified GDD” formula in 1971.
A total of 189 microsatellite markers were included in the analysis. These markers covered 22.587 M of the corn genome with an average marker interval of 15.80 cM. Of the 189 markers, 152 have been assigned to the 10 linkage groups and the remaining 37 markers that have not been assigned to any of the linkage groups were analyzed independently. The IBD matrices were obtained by taking the averages of N = 3000 independent simulations.
Figure 1 shows the LOD score profile of the genome scan with a 2-cM increment. The threshold value used to declare statistical significance at the genome level was 3.77, which was calculated using the approximate method of Piepho (2001). We detected eight QTL, six of which were mapped to five linkage groups (1, 4, 5, 8, and 9), and two were located to independent markers M097 and M028. Of the eight detected QTL, the smallest one contributes 43% of the total phenotypic variance and the largest one contributes 80% of the variance (Table 1). The large QTL variances relative to the total phenotypic variance are due to (1) the small error variance (Table 1) and (2) small sample size. Recall that the phenotypic value of a line actually reflects the genotypic value of the line and thus the environmental variance is virtually zero. This has clearly demonstrated the advantage of QTL mapping using multiple inbred lines over line-crossing experiments.
Figure 1.—

The LOD score profile of the maize genome scan. The genome is divided into 10 linkage groups (separated by the reference lines on the horizontal axis). The LOD scores of the 37 markers that have not been assigned to any of the linkage groups are plotted in the last block with 10 cM between consecutive markers.
TABLE 1.
Estimated parameters of QTL identified in the genome scan for the corn pedigree data
| Single QTL model
|
Multi-QTL model
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| QTL | Linkage group |
Position (cM) |
Flanking markers |
Confidence interval |
σ2u | σ2v | σ2 | ĥ2u (%) | LOD | Variance | Heritability (%) |
| qtl1 | 1 | 55.8 | M098–M188 | 54.8–60.8 | 95.51 | 47.13 | 2.29 | 65.90 | 4.66 | 4.72 | 2.62 |
| qtl2 | 4 | 142.0 | M153–M178 | 140.0–144.0 | 124.74 | 64.18 | 2.30 | 65.23 | 5.05 | 97.18 | 53.85 |
| qtl3 | 5 | 140.9 | M156–M157 | 138.9–146.9 | 93.01 | 58.12 | 3.23 | 60.26 | 3.81 | 7.73 | 4.28 |
| qtl4 | 5 | 175.9 | M076–M007 | 172.9–191.9 | 81.79 | 50.56 | 2.08 | 60.84 | 5.80 | 13.48 | 7.47 |
| qtl5 | 8 | 86.8 | M035–M174 | 86.3–88.2 | 237.09 | 56.52 | 1.49 | 80.34 | 4.84 | 6.14 | 3.40 |
| qtl6 | 9 | 122.9 | M133–M015 | 120.9–126.9 | 99.38 | 52.05 | 1.94 | 64.80 | 5.78 | 16.16 | 8.95 |
| qtl7 | — | M097 | M097 | — | 57.66 | 77.29 | 0.00 | 72.73 | 3.94 | 1.69 | 0.94 |
| qtl8 | — | M028 | M028 | — | 79.29 | 63.41 | 0.90 | 55.22 | 5.44 | 2.81 | 1.56 |
| Polygene | 27.86 | 15.44 | |||||||||
| Residual | 2.69 | 1.49 | |||||||||
σ2u is the estimated genetic variance for QTL, σ2v is the polygenic variance, σ2 is the residual variance, and ĥ2u is the proportion of the total variance contributed by the QTL and is expressed as 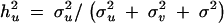 .
.
The results from Table 1 showed that on average, each of the eight detected QTL explains ∼62% of the total phenotypic variance, and the overall proportion of the variance contributed by all the QTL is thus >100%. This phenomenon may be ascribed to both the small sample size (pedigree) and the small residual variance. The results cannot be combined in a simple way due to the fact that each QTL was detected using a different model. We treated the result of the genome scan as the first step to identify the chromosomal regions and then used a mixed model that included all the eight detected QTL simultaneously to reevaluate the variance components. The reestimated variances are given in Table 1. Note that the “large” QTL identified in the one-dimensional scan were not necessarily large when reevaluated in a multiple-effect model. This may be partly explained by random associations between the locus-specific IBD matrices and the polygenic IBD matrix caused by the limited sample size (small pedigree). After the reevaluation, the largest QTL explained 54% of the variance whereas the smallest QTL explained only 1% of the total variance. The overall proportion of the QTL variance was then 83% (Table 1).
QTL values of the inbred lines were evaluated using BLUP for all the eight detected loci. The mixed-model equation was simply an extension of Equation 13 for multiple QTL effects. The summary statistics of the estimated QTL values are given in Table 2. The QTL are ranked in a descending order according to the size of their variance: qtl2, qtl6, qtl4, qtl3, qtl5, qtl1, qtl8, and qtl7. Therefore, marker-assisted selection may focus on the large QTL first. The extreme lines for each of the eight QTL are determined. For example, if we want to increase the trait value, we should design a strategy of marker-assisted selection that combines the allele of line 90 for qtl2, the allele of line 37 for qtl6, alleles from line 100 for qtl4 and qtl3, and so on into a single line. Such a line is considered to be a super line that carries all the good alleles. If decreasing the trait value is our selection objective, we need to combine the allele of line 22 for qtl2, the allele of line 24 for qtl6, alleles from line 23 for qtl4 and qtl3, and so on.
TABLE 2.
Summary statistics of the BLUPs of QTL effects among the 404 inbred lines
| QTLa | Mean | Standard deviation |
Minimum | Maximum | Range |
|---|---|---|---|---|---|
| qtl2 | 0.49 | 4.53 | −16.22 | 16.37 | 32.59 |
| qtl6 | −0.60 | 1.68 | −5.34 | 7.34 | 12.68 |
| qtl4 | −0.02 | 1.62 | −6.44 | 3.20 | 9.64 |
| qtl3 | −0.02 | 0.93 | −3.69 | 1.83 | 5.29 |
| qtl5 | −0.88 | 1.24 | −4.86 | 2.03 | 6.89 |
| qtl1 | −0.38 | 0.96 | −3.52 | 3.26 | 6.78 |
| qtl8 | 0.09 | 0.58 | −2.01 | 1.30 | 3.31 |
| qtl7 | −0.10 | 0.46 | −2.13 | 0.77 | 2.90 |
QTL are sorted by variance in a descending order. For example, qtl2 is the largest QTL and qtl7 is the smallest QTL.
Simulation studies:
We took the maize pedigree with 404 inbred lines. We used the existing marker maps for the five chromosomes (chromosome 1, 4, 5, 8, and 9) and the two unlinked markers (M097 and M028) that have shown evidence of QTL in the real data analysis. The marker maps and marker genotypes remained the same as that reported in the real data analysis. We then simulated eight QTL at positions exactly the same as reported in the real data analysis. In the simulation experiment, we simply simulated the genotypes of the eight QTL and genotypic values of the QTL according to the true parameter values under our control. We then simulated a small residual variance of 2.5 to generate the phenotypic values of all the inbred lines. The true parameter values used in the simulation are given in Table 3 along with the estimated values using the single-QTL model. The simulation was replicated 50 times to obtain a rough estimate of the statistical power for detection of each QTL.
TABLE 3.
Results of the simulation experiment under the single-QTL model (50 replications)
| Variance
|
Heritability
|
||||||
|---|---|---|---|---|---|---|---|
| QTL | Power (%) | Position (cM) |
QTL(σ2u) | Polygene(σ2v) | Residual (σ2) | QTL(h2u) | Polygene(h2v) |
| 1 | |||||||
| True value | — | 56.00 | 50.0000 | — | 2.5000 | 0.3000 | — |
| Estimate | 94 | 56.28 (2.68) |
67.1193 (29.8300) |
92.8245 (28.7283) |
3.3223 (4.5495) |
0.4042 (0.1201) |
0.5766 (0.1272) |
| 2 | |||||||
| True value | — | 142.00 | 33.3333 | — | 2.5000 | 0.2000 | — |
| Estimate | 86 | 141.92 (5.08) |
59.0012 (30.1816) |
101.2365 (28.0739) |
4.8061 (4.5128) |
0.3509 (0.1336) |
0.6204 (0.1321) |
| 3 | |||||||
| True value | — | 141.00 | 33.3333 | — | 2.5000 | 0.2000 | — |
| Estimate | 84 | 142.58 (4.39) |
53.4422 (27.2483) |
108.9404 (35.5669) |
3.9165 (4.1938) |
0.3215 (0.1349) |
0.6558 (0.1394) |
| 4 | |||||||
| True value | — | 176.00 | 16.6667 | — | 2.5000 | 0.1000 | — |
| Estimate | 82 | 178.67 (9.87) |
50.7666 (26.4947) |
101.6241 (34.4699) |
4.4681 (4.6572) |
0.3249 (0.1442) |
0.6486 (0.1509) |
| 5 | |||||||
| True value | — | 87.00 | 16.6667 | — | 2.5000 | 0.1000 | — |
| Estimate | 44 | 83.15 (14.02) |
38.6235 (23.8458) |
122.2050 (44.2634) |
4.8012 (5.1568) |
0.2473 (0.1573) |
0.7250 (0.1618) |
| 6 | |||||||
| True value | — | 123.00 | 8.3333 | — | 2.5000 | 0.0500 | — |
| Estimate | 18 | 118.97 (6.31) |
28.1781 (21.9769) |
129.0334 (41.1543) |
4.2142 (4.9946) |
0.1783 (0.1354) |
0.7967 (0.1401) |
| 7 | |||||||
| True value | — | 0.00 | 3.3333 | — | 2.5000 | 0.0200 | — |
| Estimate | 6 | 0.00 (0.00) |
5.4262 (7.5327) |
144.1987 (38.1987) |
4.3689 (5.1116) |
0.0379 (0.0563) |
0.9352 (0.0643) |
| 8 | |||||||
| True value | — | 0.00 | 2.5000 | — | 2.5000 | 0.0150 | — |
| Estimate | 2 | 0.00 (0.00) |
7.6491 (9.8328) |
142.9929 (38.4481) |
4.2194 (4.9936) |
0.0524 (0.0717) |
0.9217 (0.0761) |
The estimated parameters were obtained from the average of 50 replicated simulations with the standard deviations among the replicates given in parentheses.
Table 3 shows that the estimated QTL variances were larger than the corresponding true values, so were the estimated proportions of phenotypic variance explained by the QTL. This is consistent with what was observed in the real data analysis under the single-QTL model. The estimated positions of QTL and residual variance were quite close to the true values. The estimated polygenic variance was well over the true value of zero. This was expected because the polygenic variance in the single-QTL model actually absorbed the variances of all other QTL not included in the single-QTL model. For the power evaluation, the result did show the expected trend, power increasing as the size of QTL increased.
As done in the real data analysis, we included all the eight QTL in a single mixed model and reevaluated the variances. The result is shown in Table 4. Clearly, the biases for all variance component estimates have been reduced. The polygenic variance and residual variance estimates, however, were still biased slightly. This may be acceptable given the small sample size and the small number of replicates.
TABLE 4.
Reevaluation of the eight QTL simulated under the multiple-QTL model (50 replications)
| QTL
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Parameters | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | Polygene | Residual |
| Variance | ||||||||||
| True value | 50.0000 | 33.3333 | 33.3333 | 16.6667 | 16.6667 | 8.3333 | 3.3333 | 2.5000 | 0.0000 | 2.5000 |
| Mean | 46.8222 | 36.3070 | 29.3333 | 19.1388 | 14.9889 | 8.2659 | 3.3545 | 4.1599 | 2.3156 | 1.6648 |
| SD | 25.5695 | 20.2418 | 24.8239 | 13.2557 | 11.8921 | 6.1652 | 4.9980 | 4.2488 | 5.8230 | 1.5677 |
| Heritability | ||||||||||
| True value | 0.3000 | 0.2000 | 0.2000 | 0.1000 | 0.1000 | 0.0500 | 0.0200 | 0.0150 | 0.0000 | 0.0150 |
| Mean | 0.2738 | 0.2194 | 0.1710 | 0.1194 | 0.0940 | 0.0523 | 0.0212 | 0.0256 | 0.0132 | 0.0101 |
| SD | 0.1226 | 0.1052 | 0.1118 | 0.0843 | 0.0725 | 0.0397 | 0.0310 | 0.0257 | 0.0320 | 0.0088 |
SD, standard deviation. The estimates were obtained from the average of 50 replicated simulations. Heritability of QTL is the proportion of the total variance contributed by the QTL and is expressed as 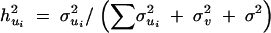 .
.
DISCUSSION
Inbred lines are the most common forms of crop cultivars for self-pollinated crops. Therefore, the method presented in this study is more suitable for rice (Oryza sativa L.), soybean [Glycine max (L.) Merrill], wheat (Triticum aestivum L.), and other self-pollinated crops than for open-pollinated crops such as corn. The maize pedigree happened to be available to us and we took advantage of the data to demonstrate the application of the method. A very small percentage of the markers (<0.01) in a few lines of the maize pedigree were still heterozygous. These markers were simply treated as missing values in the study. If the heterozygosity in the pedigree were sufficiently high, we would have to take them into account so that dominance variance components would have to be included in the model. The model also ignored the epistatic variance components for simplicity. Therefore, the key assumption of the variance component analysis of QTL presented in this study is the additivity of QTL effects. Additional work is needed if dominance and epistatic effects are deemed to be important and should be included in the model.
As demonstrated in the corn pedigree analysis, naturally occurring genetic variance among commercial inbred lines is large and it has not been fully explored due to lack of appropriate statistical methods. Conventional QTL mapping that uses intercrosses of a chosen pair of lines is able to detect only a minute fraction of the existing genetic variance. We have successfully applied the IBD method implemented via the mixed model methodology to a maize data set and detected QTL explaining a large proportion of the phenotypic variance. The method provides a general machinery to explore naturally occurring genetic variation among inbred lines for other plant species with well-documented pedigrees, e.g., rice, soybean, wheat, etc. It also can be used for genetic mapping in inbred mice, rats (Rattus rattus L.), and other laboratory animals. There are >400 inbred strains of mice with well-documented pedigrees (Beck et al. 2000). Almost all of them have multiple phenotypic records and 10% of the strains have saturated marker data (Beck et al. 2000). Genetic mapping in laboratory animals is mainly for the purpose of seeking candidate loci that may be responsible for complex diseases in humans. Results from intercross mapping using a pair of strains certainly have limited value in comparative genomic analysis. The pedigree analysis that includes many strains should have a much broader inference space and thus be more relevant to human genetic studies.
Statistical estimation of the IBD matrices is pivotal to the success of QTL mapping with multiple lines. Currently, three methods are used to estimate the IBD matrices: the Elston-Stewart algorithm (Elston and Stewart 1971), the Lander-Green algorithm (Lander and Green 1987), and Markov chain Monte Carlo methods (Sobel and Lange 1996; Heath 1997; Yi and Xu 2000). Unfortunately, none of them can be used here for inbred lines, which forced us to develop a new Monte Carlo algorithm particularly suitable for inbred lines. The basic assumptions of the method are that every inbred line was derived from the hybrid of two parental lines and the genetic variance among the inbred lines is not generated by mutation but preserved from the original variance among the founders. These assumptions are valid for most inbred lines in plants because the breeding history of the pedigree is typically <100 years. Some of the inbred strains in laboratory mice, however, were not generated from crosses; rather, they were derived from independent founders by new mutations. Therefore, the model requires some modification to take into account mutation to be applied to some of the current mouse (M. musculus L.) pedigrees. This is an on going project of our laboratory.
Genetic mapping of maize flowering traits, including male anthesis, female silking, and the anthesis-silking interval, has been extensively studied (Ribaut et al. 1996; Jiang et al. 1999; Vladutu et al. 1999; Austin et al. 2001). In most cases, six to eight QTL were identified for the above flowering traits (Ribaut et al. 1996; Jiang et al. 1999; Austin et al. 2001). These mapped QTL account for ∼40% of the phenotypic variation. We also searched the maize genetic database (http://www.maizegdb.org/) to see if we could find genes similar to what we found. The two QTL mapped to linkage groups 5 and 9 in this article have also mapped to the same positions in Berke and Rocheford (1995) and Koester et al. (1993), respectively. The QTL located to linkage group 4 in this article may be different from the QTL mapped to the other side of linkage group 4 by Beavis et al. (1994).
Missing marker information is one of the major problems in pedigree analysis. In the maize pedigree analyzed here, ∼5% of the markers were missing. Two approaches may be used for handling missing markers. One approach is the multipoint method (Jiang and Zeng 1997; Goldgar 1990) in which all markers in the linkage group are used simultaneously to infer the genotype of the putative QTL. The other approach is to impute the missing marker genotypes via Monte Carlo simulations. Once the missing marker genotypes are simulated, the standard interval mapping approach will apply. We took the second approach. For each missing genotype, we evaluated all the possible genotypes compatible with the pedigree information. We then randomly selected one compatible genotype. The expected IBD matrix was calculated on the basis of a large number of independent simulations. As the number of replicated simulations increases, all possible genotypes have a probability of being sampled. Fortunately, our method does not require evaluation of all possible genotypes. Most compatible genotypes may lead to the same IBD values. The number of replicated simulations was chosen as N = 3000 in our study. We actually tried several different N and found that when N < 3000, the results were not stable, but when N > 3000, the gain was not dramatic. In practice, N may depend on the size of the pedigree and the marker information content. If computing time is not a major concern, one can always try a large N. The mixed-model analysis itself is extremely fast. The majority of the computing time of the pedigree analysis is actually spent on computing the IBD matrix. Because we adopted the independent Monte Carlo imputation approach, we can stop at any number of simulations and store the data. Later on if more simulations are used, we can simply add the new simulations to the old data set to increase N.
We have taken an interval mapping approach to scan the entire genome. The model is a single-QTL model. Multiple QTL are implied if multiple peaks are present in the test-statistic profile. Given the positions of the detected QTL, we reevaluated the QTL variances using a multiple-QTL model. This two-step approach has been used previously (Lander and Botstein 1989; Yano et al. 1997; Hunt et al. 1999; Bunyamin et al. 2002). The single-QTL model in line-crossing experiments is being replaced by the multi-QTL model via either the maximum-likelihood method (Kao et al. 1999) or the Bayesian method (Sillanpaa and Arjas 1998, 1999; Bink et al. 2002; Kilpikari and Sillanpaa 2003; Xu 2003). Theoretically, similar extensions can be made here for pedigree analysis. Unfortunately, a multiple-QTL model under the variance component framework is difficult to implement. Therefore, the single-QTL model is still the best available model in pedigree analysis of this kind. We plan to develop a multiple-QTL model under the Bayesian framework. However, such a multiple-QTL model will provide only a practically convenient tool and not necessarily devalue the conceptual and theoretical contribution of this study.
Acknowledgments
We thank two anonymous reviewers and the associate editor for their comments on the first version of this article. This research was supported by the National Institutes of Health grant R01-GM55321 to S.X.
References
- Austin, D. F., M. Lee and L. R. Veldboom, 2001. Genetic mapping in maize with hybrid progeny across testers and generations: plant height and flowering. Theor. Appl. Genet. 102: 163–176. [Google Scholar]
- Bernardo, R., 1996. a Best linear unbiased prediction of maize single-cross performance. Crop Sci. 36: 50–56. [DOI] [PubMed] [Google Scholar]
- Bernardo, R., 1996. b Testcross additive and dominance effects in best linear unbiased prediction of maize single-cross performance. Theor. Appl. Genet. 93: 1098–1102. [DOI] [PubMed] [Google Scholar]
- Bernardo, R., 1998 Predicting the performance of untested single crosses: trait and marker data, pp. 117–127 in Concepts and Breeding of Heterosis in Crop Plants, Pub. 25, edited by K. R. Lamkey and J. E. Staub. Crop Science Society of America, Madison, WI.
- Beavis, W. D., O. S. Smith, D. Grant and R. Fincher, 1994. Identification of quantitative trait loci using a small sample of top crossed and F4 progeny from maize. Crop Sci. 34: 882–896. [Google Scholar]
- Beck, J. A., S. Lloyd and M. Hafeparast, 2000. Genealogies of mouse inbred strains. Nat. Genet. 24: 23–25. [DOI] [PubMed] [Google Scholar]
- Berke, T. G., and T. R. Rocheford, 1995. Quantitative trait loci for flowering, plant and ear height, and kernel traits in maize. Crop Sci. 35: 1542–1549. [Google Scholar]
- Bink, M., P. Uimari, M. J. Sillanpaa, L. Janss and R. Jansen, 2002. Multiple QTL mapping in related plant populations via a pedigree-analysis approach. Theor. Appl. Genet. 104: 751–762. [DOI] [PubMed] [Google Scholar]
- Bunyamin, T., T. E. Michaels and K. P. Pauls, 2002. Genetic mapping of agronomic traits in common bean. Crop Sci. 42: 544–556. [Google Scholar]
- Chesler, E. J., S. L. Rodriguez and J. S. Mogil, 2001. In silico mapping of mouse quantitative trait loci. Science 294: 2423. [DOI] [PubMed] [Google Scholar]
- Cui, Z. L., J. Y. Gai, T. E. CARTER, Jr, J. X. Qiu and T. J. Zhao, 1999 The Released Chinese Soybean Cultivars and Their Pedigree Analysis (1923–1995). China Agriculture Publishing House, Beijing, China.
- Darvasi, A., 2001. In silico mapping of mouse quantitative trait loci. Science 294: 2423. [PubMed] [Google Scholar]
- Elston, R. C., and J. Stewart, 1971. A general model for the genetic analysis of pedigree data. Hum. Hered. 21: 523–542. [DOI] [PubMed] [Google Scholar]
- George, A. W., P. M. Visscher and C. S. Haley, 2000. Mapping quantitative trait loci in complex pedigrees: a two-step variance component approach. Genetics 156: 2081–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldgar, D. E., 1990. Multipoint analysis of human quantitative genetic variation. Am. J. Hum. Genet. 47: 957–967. [PMC free article] [PubMed] [Google Scholar]
- Grupe, A., S. Germer, J. Usuka, D. Aud, J. K. Belknap et al., 2001. In silico mapping of complex disease-related traits in mice. Science 292: 1915–1918. [DOI] [PubMed] [Google Scholar]
- Heath, S., 1997. Markov chain Monte Carlo segregation and linkage analysis for oligogenic models. Am. J. Hum. Genet. 61: 748–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson, C. R., 1975. Best linear unbiased estimation and prediction under a selection model. Biometrics 31: 423–447. [PubMed] [Google Scholar]
- Henderson, C. R., 1984 Application of Linear Models in Animal Breeding. University of Guelph, Guelph, Ontario, Canada.
- Hunt, G. J., A. M. Collins, R. Rivera, R. E. Page, Jr. and E. Guzman-Novoa, 1999. Quantitative trait loci influencing honeybee alarm pheromone levels. J. Hered. 90: 585–589. [DOI] [PubMed] [Google Scholar]
- Kao, C. H., Z-B. Zeng and R. D. Teasdale, 1999. Multiple interval mapping for quantitative trait loci. Genetics 152: 1203–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilpikari, R., and M. J. Sillanpaa, 2003. Bayesian analysis of multilocus association in quantitative trait and qualitative traits. Genet. Epidemiol. 25: 122–135. [DOI] [PubMed] [Google Scholar]
- Koester, R. P., P. H. Sisco and C. W. Stuber, 1993. Identification of quantitative trait loci controlling days to flowering and plant height in two near isogenic lines of maize. Crop Sci. 33: 1209–1216. [Google Scholar]
- Jiang, C., and Z-B. Zeng, 1997. Mapping quantitative trait loci with dominant and missing markers in various crosses from two inbred lines. Genetica 101: 47–58. [DOI] [PubMed] [Google Scholar]
- Jiang, C., G. O. Edmeades, I. Armstead, D. Hoisington, H. R. Lafitte et al., 1999. Genetic analysis of adaptation differences between highland and lowland tropical maize using molecular markers. Theor. Appl. Genet. 99: 1106–1119. [DOI] [PubMed] [Google Scholar]
- Lander, E. S., and D. Botstein, 1989. Mapping Mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics 121: 185–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander, E. S., and P. Green, 1987. Construction of multilocus genetic linkage maps in humans. Proc. Natl. Acad. Sci. USA 84: 2363–2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisseaux, B., and R. Bernardo, 2004. In silico mapping of quantitative trait loci in maize. Theor. Appl. Genet. 109: 508–514. [DOI] [PubMed] [Google Scholar]
- Piepho, H. P., 2001. A quick method for computing approximately thresholds for quantitative trait loci detection. Genetics 157: 425–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao, S., and S. Xu, 1998. Mapping quantitative trait loci for ordered categorical traits in four-way crosses. Heredity 81: 214–224. [DOI] [PubMed] [Google Scholar]
- Ribaut, J. M., D. A. Hoisington, J. A. Deutsch, C. Jiang and D. Gonzalez-De-Leon, 1996. Identification of quantitative trait loci under drought conditions in tropical maize. 1. Flowering parameters and the anthesis-silking interval. Theor. Appl. Genet. 92: 905–914. [DOI] [PubMed] [Google Scholar]
- Risch, N., and K. Merikangas, 1996. The future of genetic studies of complex human diseases. Science 273: 1516–1517. [DOI] [PubMed] [Google Scholar]
- SAS Institute, 1999 SAS/STAT User's Guide, Version 8. SAS Institute, Cary, NC.
- Sillanpaa, M. J., and E. Arjas, 1998. Bayesian mapping of multiple quantitative trait loci from incomplete inbred line cross data. Genetics 148: 1373–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sillanpaa, M. J., and E. Arjas, 1999. Bayesian mapping of multiple quantitative trait loci from incomplete outbred offspring data. Genetics 151: 1605–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobel, E., and K. Lange, 1996. Descent graphs in pedigree analysis: applications to haplotyping, location scores, and marker-sharing statistics. Am. J. Hum. Genet. 58: 1323–1337. [PMC free article] [PubMed] [Google Scholar]
- Vladutu, G., J. Mclaughlin and R. L. Phillips, 1999. Fine mapping and characterization of linked quantitative trait loci involved in the transition of the maize apical meristem from vegetative to generative structures. Genetics 153: 993–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, S., 2003. Estimating polygenic effects using markers of the entire genome. Genetics 163: 789–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, S., and W. R. Atchelly, 1995. A random model approach to interval mapping of quantitative trait loci. Genetics 141: 1189–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano, M., Y. Harushima, Y. Nagamura, N. Kurata, Y. Minobe et al., 1997. Identification of quantitative trait loci controlling heading date in rice using a high density linkage map. Theor. Appl. Genet. 95: 1025–1032. [Google Scholar]
- Yi, N., and S. Xu, 2000. Bayesian mapping of quantitative trait loci under the identity-by-descent-based variance component model. Genetics 156: 411–422. [DOI] [PMC free article] [PubMed] [Google Scholar]