

Abstract
We investigated genetic polymorphism in the Caenorhabditis elegans srh and str chemoreceptor gene families, each of which consists of ∼300 genes encoding seven-pass G-protein-coupled receptors. Almost one-third of the genes in each family are annotated as pseudogenes because of apparent functional defects in N2, the sequenced wild-type strain of C. elegans. More than half of these “pseudogenes” have only one apparent defect, usually a stop codon or deletion. We sequenced the defective region for 31 such genes in 22 wild isolates of C. elegans. For 10 of the 31 genes, we found an apparently functional allele in one or more wild isolates, suggesting that these are not pseudogenes but instead functional genes with a defective allele in N2. We suggest the term “flatliner” to describe genes whose functional vs. pseudogene status is unclear. Investigations of flatliner gene positions, dN/dS ratios, and phylogenetic trees indicate that they are not readily distinguished from functional genes in N2. We also report striking heterogeneity in the frequency of other polymorphisms among these genes. Finally, the large majority of polymorphism was found in just two strains from geographically isolated islands, Hawaii and Madeira. This suggests that our sampling of wild diversity in C. elegans is narrow and that identification of additional strains from similarly isolated regions will greatly expand the diversity available for study.
CAENORHABDITIS elegans has ∼1300 predicted genes that encode members of putative chemosensory receptors (Troemel et al. 1995; Robertson 1998, 2000, 2001). Together, these genes define the SR superfamily. On the basis of sequence alignment and phylogenetic trees, SR superfamily members fall into at least a dozen families (H. Robertson, personal communication, and our unpublished data). These families range in size from the large srh and str families (>300 genes each) to the modestly sized sra and srv families (∼30 genes each). The SR superfamily belongs to the broader class of G-protein-coupled receptors (GPCRs). The separation of the putative chemoreceptor families from other GPCR families is well defined phylogenetically and is supported by expression pattern and functional studies (Sengupta et al. 1994; Troemel et al. 1995). Each SR family appears to have arisen by gene duplication and divergence from a founder gene. These duplications have occurred sporadically over a long evolutionary period, giving rise to complex phylogenetic relationships. Near one extreme, str-5 and str-6 result from a very recent duplication and differ by only two nucleotides in their coding sequence. Near the other extreme, aligned STR-1 and STR-47 proteins have only 19% amino acid identity and presumably diverged from a much older duplication. Members of different SR families are even more distantly related, with the most distant proteins nearly unalignable.
Investigation of the srh and str families (Robertson 2000, 2001) revealed a curious feature of their functional status. Nearly one-third of the genes in each family are annotated as putative pseudogenes, because they contain obvious functional defects in the sequence of the N2 isolate of C. elegans. This observation is not peculiar to nematodes: the olfactory gene families in mammals also contain many apparently defective genes (Glusman et al. 2001; Zozulya et al. 2001; Young and Trask 2002; Young et al. 2002; Zhang and Firestein 2002). In a gene family with the potential for extensive functional redundancy and relaxed selective pressure on individual genes, this finding is not surprising and indeed might be expected. The curious feature of the nematode genes is that more than half of the “pseudogenes” have only a single apparent defect, usually a stop codon, deletion, or frameshift that would be expected to abrogate function completely (Robertson 2000, 2001). For the 31 genes analyzed here, we resequenced the alleles in N2 from genomic PCR products and ruled out the trivial possibility that the defect was due to a cloning artifact or sequencing error by the genome project (C. elegans Sequencing Consortium 1998). In theory, such a defective gene should lose purifying selection and become a neutrally evolving sequence (Ohno et al. 1968). Over time, the sequence should accumulate additional easily recognized mutational defects (such as stop codons) and a high frequency of nonsynonymous codon changes.
The existence of a high frequency of singly defective genes is consistent with at least two explanations. First, if the mutational spectrum in C. elegans has a high frequency of large deletions relative to point mutations and small deletions, then genes that have lost purifying selection would disappear from the genome before they had the opportunity to acquire multiple additional defects. In this case, the existing pattern of defects in the N2 sequence would result from a dynamic equilibrium between generation of new genes by duplication and loss of defective genes by deletion. An estimate of the frequency and size spectrum of deletions relative to point mutations has been made for neutrally evolving mariner elements in C. elegans (Witherspoon and Robertson 2003). That study estimated that the evolutionary half-life of neutrally evolving sequences is ∼0.1 substitutions per nucleotide, a value consistent with the fact that most existing mariner elements have multiple apparent functional defects (similar to classical pseudogenes). Assuming that an SR gene that has suffered a null mutation will evolve neutrally, these measurements are inconsistent with the observed pattern of SR mutations. The second explanation is that SR pseudogenes remain subject to purifying selection. In principle, this might result from unexpected residual function of the allele found in the N2 sequence. Alternatively, the N2 allele might result from a recent mutation and functional alleles might be present in other individuals in C. elegans wild populations. We report a variety of analyses that support this latter possibility.
MATERIALS AND METHODS
Genomic positions of srh and str genes:
Custom software was implemented to graph the genomic positions of arbitrary sets of genes as specified in a text list of their names. Position information was derived from GFF data sets provided by the WormBase download site (ftp://ftp.wormbase.org/pub/wormbase/elegans-current_release/GENE_DUMPS/).
Closest C. briggsae srh relatives:
Annotation of the SR family genes in C. briggsae is weak and undertaking the complete annotation of the srh and str gene families is difficult. For the srh family we carried out an annotation of all putative functional C. briggsae genes by a combination of tblastn searches and implementation of a novel motif-searching method described elsewhere (J. H. Thomas, J. L. Kelley, H. Robertson, K. Ly and W. Swanson, unpublished results). For the str family a less complete analysis was undertaken to ensure that the closest C. briggsae genes were identified. Each flatliner protein was used as query in a tblastn search of the C. briggsae genome (release cb25). A tblastn postprocessor script was used to cluster local search hits into probable gene clusters and the 10 best clusters were annotated in detail. We also used a blastp search of the predicted C. briggsae protein set (release cb25.hybrid) and similarly updated annotations for the best hits. Because of gross inaccuracy in gene prediction this approach was ineffective: many best matches found by tblastn were missed entirely and those that were found tended to be more complete gene predictions (rather than better homologs) because they matched across a longer region. In all, 9 entirely new srh genes were identified, 64 existing srh gene predictions were modified, and 15 srh gene predictions were retained as correct. In the str family, 46 existing gene predictions were modified and 4 entirely new genes were identified. In comparative analyses, all C. briggsae genes were included for which a plausible gene model could be derived that encoded a protein that aligned well with other family members. One-to-one orthologs were assessed by full pairwise alignments; mutual best matches were identified and analyzed for Table 2. Data are available as supplementary material (SM) at http://calliope.gs.washington.edu/flatlinerdata/index.html (SM 8).
TABLE 2.
Closest functional relatives for various sets ofC. elegans srh genes
| Family | Gene set | N | Mean identity (SD) to C. briggsae |
Mean identity (SD) in C. elegans |
|---|---|---|---|---|
| SRH | Deletions—functional | 4 | 0.369 (0.019) | 0.753 (0.159) |
| All other deletions | 17 | 0.404 (0.090) | 0.670 (0.161) | |
| Stops—functional | 5 | 0.381 (0.079) | 0.794 (0.145) | |
| All other stops | 6 | 0.395 (0.063) | 0.704 (0.119) | |
| Frameshift and splice | 9 | 0.414 (0.043) | 0.633 (0.178) | |
| No apparent defects | 216 | 0.442 (0.133) | 0.625 (0.170) | |
| STR | Deletion—functional | 1 | ND | 0.591 (NA) |
| All other deletions | 28 | ND | 0.593 (0.147) | |
| All other stops | 19 | ND | 0.614 (0.50) | |
| Frameshift and splice | 9 | ND | 0.579 (0.106) | |
| No apparent defects | 228 | ND | 0.608 (0.170) |
N, number of cases available for analysis (all available cases were analyzed). ND, not determined; NA, not applicable. Mean identity was computed from the fraction of identical amino acids in each pair alignment. None of the gene sets was significantly different from “no apparent defects” by both a t-test and a Mann-Whitney U-test. STR proteins were not compared to C. briggsae because annotation of the genes is inadequate. Full data sets for all matches are available in supplemental materials.
dN/dS analysis:
For each flatliner gene of interest, several closest functional paralogs in C. elegans were identified. A protein multiple alignment was generated using ClustalX (BLOSUM matrices, otherwise default settings) and from this alignment a corresponding codon alignment and a maximum-likelihood (ML) phylogenetic tree were constructed (Felsenstein 1993). The codon alignment and tree were supplied to the codeml program in the PAML package (Yang 1997) to obtain the Nei-Gojobori pairwise dN and dS values (Nei and Gojobori 1986) among the closest paralog pairs. In addition, we used codeml to perform a ML assessment of the dN/dS ratio on the terminal phylogenetic tree branch leading to the flatliner gene. This test was done by comparing the ML value with model 0 (one dN/dS ratio on all tree branches) to the ML value with a free dN/dS ratio on the flatliner branch (Yang 1998). Significance was evaluated by a χ2 test of twice the ML difference with 1 d.f. (Yang 1997).
Inferred functional gene products for nonfunctional alleles:
In the srh family, 9 genes with putative functional alleles were found in one or more wild C. elegans isolates. In addition to these, there were 15 other genes with single stop codons or frameshift mutations whose sequence was sufficiently complete to permit inference of a putative functional product. For each of these cases, we aligned the nearly complete prediction with the closest srh relatives from C. elegans and replaced the single mutant amino acid site with the amino acid that aligned from the closest relative. For the stop codon cases, this amino acid could be encoded by the N2 allele with a single-nucleotide change in the premature stop codon.
DNA preparation and amplification:
Twenty-one of the 22 C. elegans strains analyzed were obtained from the Caenorhabditis Genetics Center. The final strain, JT11362, was recently isolated by John Kemner (in our lab) from a compost bin in Seattle, Washington. Strains were grown to near starvation on 10-cm agarose plates for DNA extraction. Genomic DNA was purified either by lysis with proteinase K and extraction with phenol/chloroform/isoamyl alcohol or with the DNeasy tissue kit (QIAGEN, Valencia, CA). Regions to be amplified were selected according to Hugh Robertson's protein predictions for the srh and str chemoreceptor families (Robertson 2000, 2001; WormBase; and H. Robertson, personal communication). Primers for amplification were designed on the basis of flanking N2 sequence. All PCR products obtained for stop-codon flatliners were sequenced; for deletion flatliners, we sequenced only those PCR products that showed a size difference from N2. PCR was performed using 25-μl reactions with 1.5 mm MgCl2, 10 mm Tris-HCl, pH 8.3, 0.3 mm dNTPs, 0.3 μm primers, Taq DNA Polymerase (Promega, Madison, WI), and ∼1 μg of genomic DNA. A 5-μl aliquot of PCR product was tested for size and purity on a 1% agarose gel. For nine loci, we were unable to obtain a PCR product from 1–3 strains. We attempted at least twice to amplify each fragment on the basis of conditions that amplified the product reproducibly from other strains. This problem occurred most frequently in strain JU258 (see Table 3). On the basis of other data, JU258 is highly divergent from N2 at the nucleotide level. It is likely that our failure to amplify these fragments reflects polymorphism in the primer sites.
TABLE 3.
Summary of sequencing results for 31 flatliner genes in 22C. elegans wild isolates
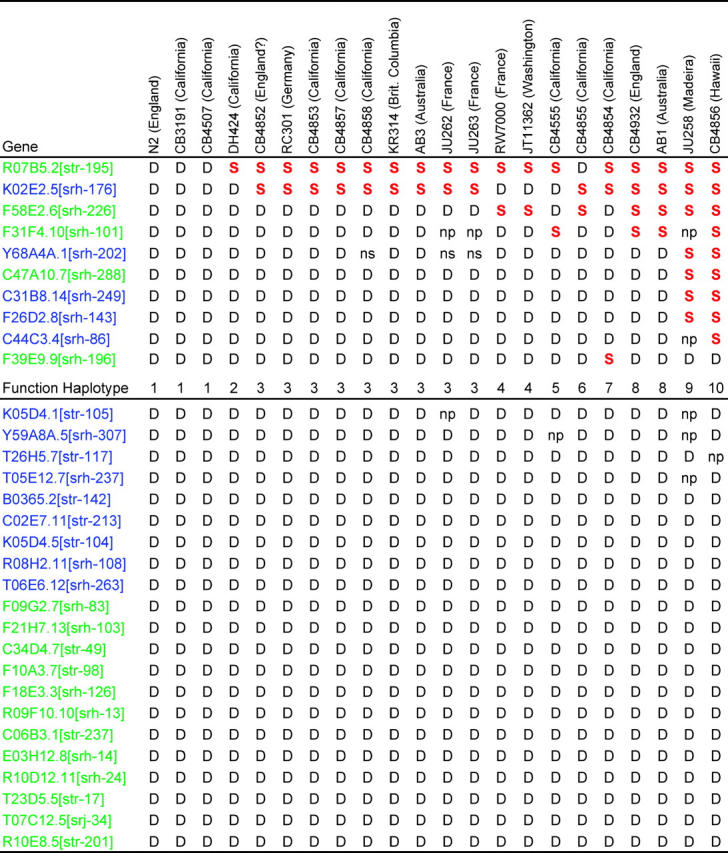
Genes with putative functionally defective alleles are marked D (stop codons or deletions), and those with putative functional alleles are marked with a red S (sense codon or insertion alleles). Genes are named both by genome project names and by their three letter srh or str names according to Robertson's system (Robertson 2000, 2001). Genes with a stop codon defect are labeled in purple and those with deletion alleles are labeled in green. Strains are listed by their CGC or lab strain designation and their approximate site of isolation from the wild. A few gene-strain combinations repeatedly failed to give PCR products and are labeled np (no product). One gene gave mixed sequence from three strains, suggesting amplification of mixed products. The mixed sequence was not resolved, so these cases are labeled ns (no sequence). Genes and strains are ordered by the number of putative functional alleles found. Below the 10 genes that had putative functional alleles, a summary of the function haplotypes is listed, with the np cases interpreted as defective alleles and the ns cases interpreted as ambiguous.
DNA sequencing and analysis:
One microliter of each PCR product was cycle sequenced using the BigDye Terminator v3.1 system (Applied Biosystems, Foster City, CA). We used internal primers to sequence some genes, but when the PCR product was clean of extraneous bands, we found that reusing one of the PCR primers gave high-quality sequence and that purification of the PCR product on a sizing column resulted in little or no improvement in sequence quality. Sequencing reactions were cleaned on a Sephadex G-50 column, evaporated in an Eppendorf Vacufuge without heat, and submitted to the University of Washington Biochemistry DNA Sequencing Facility. Most sequence runs gave high-quality sequence of 500–800 nt. Nucleotide sequences were aligned using MacVector (ClustalW); proteins were aligned with ClustalX and Bonsai 1.1 (http://calliope.gs.washington.edu/software/index.html; Thompson et al. 1994, 1997).
Two strain/gene combinations presented particular analysis challenges: CB4854/F39E9.9(srh-196) and JU258/T23D5.5(str-17). In both cases we amplified unexpectedly large fragments due to substantial sequence differences from N2. Exon 1 of F39E9.9 in CB4854 includes an insertion (112 bp) and an additional 43 SNPs with respect to N2. We conducted a number of blastn searches with short segments of the CB4854 sequence against N2 genomic sequence to be sure that the CB4854 locus is indeed allelic to F39E9.9. We also translated the CB4854 gene: no in-frame stop codons are introduced by the sequence differences from N2, and all splice sites are intact. Therefore we included this allele as a putative functional protein in our analysis of genotypic haplotypes. In the case of JU258/T23D5.5(str-17), the forward primer appears to have annealed to str-18, the upstream gene in N2, and the reverse primer annealed at the intended site. The targeted str-17 insertion is present in JU258, but the upstream region of the gene has multiple defects (presumably accounting for failure of the primer to anneal there). We conclude that this allele is unlikely to encode a functional protein and we did not include it in our genetic haplotype analysis. This was one of two instances in JU258 where the strain was apparently functional for the targeted mutation but was disqualified as a putative functional protein by other defects not found in N2. The other case was K05D4.5, in which the stop codon in N2 is changed to a cysteine codon but a small upstream deletion puts most of the gene out of frame.
RESULTS
Pseudogene prevalence in the str and srh families:
Among the 305 genes in the srh family and the 321 genes in the str family, 89 and 93, respectively, were annotated as pseudogenes in the sequenced genome of the standard laboratory isolate of C. elegans, called N2 (Robertson 2000, 2001). Only loci that could encode at least half of an SR protein were included in these counts; ∼50 more gene fragments are in each family (data not shown; H. Robertson, personal communication). Only obvious stop codons, frameshifts, splice defects, and deletions were counted as functionally defective; an unknown fraction of the remaining genes presumably have amino acid changes, promoter defects, or other undiagnosed defects that prevent function. These observations suggest that more than one-third of all the loci in the srh and str families are nonfunctional in the N2 strain. Curiously, 102 of the 182 putative srh and str pseudogenes have only a single apparent defect (e.g., a stop codon). Evolutionary theory indicates that, once purifying selection for a gene has been lost due to a null mutation, additional defects should accumulate by drift and fixation (Ohno et al. 1968). One possible explanation is that the single-defect alleles are specific to the sequenced N2 strain and that these genes have functional alleles in other wild isolates of C. elegans. A pseudogene is defined classically as a gene that lacks function throughout a species, usually indicated by the presence of multiple defects (Proudfoot and Maniatis 1980). To avoid confusion, we refer to genes with a single putative defect in N2 as “flatliner” genes (Schumacher 1990), indicating an unclear functional status in C. elegans as a species (Merriam-Webster, Ninth Edition: flat·line: 1a: to register on an electronic monitor as having no brain waves or heartbeat). As we show below, many of these genes have apparently functional alleles in one or more wild isolates of C. elegans and therefore are not pseudogenes. Annotated sets of proteins used in this article are available at http://calliope.gs.washington.edu/flatlinerdata/index.html (SM 1–4).
Flatliner genes are unexceptional compared to other family members:
We investigated whether there were features peculiar to flatliner genes in the srh and str families that distinguish them from functional genes in the same families. First, we determined the chromosomal positions of genes with varying numbers of apparent genetic defects. As previously described (Robertson 2000, 2001), str and srh genes are heavily concentrated on chromosome V, mostly on the two arms of the chromosome. As shown in Figure 1, the positions of defective genes are similar to the positions of functional genes. To the extent that the number of genes was sufficient to analyze, similar patterns were also seen on other chromosomes (data not shown). We also analyzed 15 probable gene fragments in the srh gene family, whose positions were also similar to the more complete genes (data not shown). These observations indicate that defective genes from these families do not occupy any special genomic position when compared to functional genes.
Figure 1.—

Chromosome V positions of srh and str genes. The positions of srh (top half) and str (bottom half) genes on chromosome V were graphed in 200-kb bins. Genes were grouped according to the number of apparent functional defects in the fully sequenced N2 strain: solid bar, no defects; shaded bar, one defect; open bar, multiple defects. The entire length of chromosome V is shown. Notable features include strong clustering of both gene families, a notable paucity of genes in the central third of the chromosome, and the lack of obvious differences in the positions of different defect classes.
We also found that flatliner genes occupy unexceptional positions in the phylogenetic trees of their close relatives in C. elegans. Various phylogenetic trees for large sets of flatliner and functional genes in the srh and str families are available at http://calliope.gs.washington.edu/flatlinerdata/index.html (SM 5A–5C and 6A–6C). Figure 2 shows local trees for a few examples that are typical of the general patterns. In each tree, a single flatliner gene is shown with its eight or nine closest functional relatives from C. elegans. The trees in Figure 2 demonstrate the range of divergence patterns seen in the families as a whole. For example, flatliner gene K02E2.5 and functional gene F37B4.6 each have diverged a relatively long time from their closest surviving relative, whereas flatliner gene C31B8.14 and functional gene F20E11.4 each have a close relative in the genome. In their broader phylogenetic trees, flatliner genes are scattered widely and show no obvious general pattern of clustering or degree of divergence.
Figure 2.—

Local phylogenetic trees for four flatliner genes. For each flatliner gene, the closest putative functional relatives in C. elegans were identified and a neighbor-joining distance tree was constructed among them, rooted by their relation to the entire family tree (see supplemental materials). The flatliner genes shown all have putative functional alleles in other wild strains, and these are the sequences used in the tree. Flatliners are marked in red, those with stop codons are marked by *, and deletions are shown by #.
If flatliner genes were truly pseudogenes, they should evolve neutrally because they would no longer be subject to purifying selection. One indicator of neutrally evolving coding regions is an equal frequency of nonsynonymous codon changes (dN) compared to synonymous codon changes (dS). To investigate this possibility, we determined dN/dS ratios between flatliner genes and their closest relatives in C. elegans and compared these with dN/dS ratios among putative functional genes. As shown in Table 1, these comparisons indicate no apparent differences, with dN/dS ratios of ∼0.2–0.3 in most cases for both flatliner and functional pairs. We also used a more sensitive maximum-likelihood method to test whether the phylogenetic tree branch leading to the flatliner gene had a significantly different dN/dS ratio when compared to other nearby branches (see materials and methods). In all cases, there was no significant difference (data not shown). These results argue that srh and str flatliner genes are currently under purifying selection, or that they were under purifying selection until very recently. dN/dS ratios are higher in the chemoreceptor families than in the average gene (Stein et al. 2003), reflecting a relatively high tolerance for amino acid changes; the key result is that flatliner genes in the srh and str families are no different from functional genes in this regard.
TABLE 1.
dN/dS values among flatliner and nonflatliner relatives
| Gene A | Gene B | dN/dS | dN | dS |
|---|---|---|---|---|
| C31B8.14a | C31B8.13 | 0.192 | 0.168 | 0.875 |
| C31B8.14a | H05B21.2 | 0.193 | 0.082 | 0.427 |
| C31B8.13 | H05B21.2 | 0.194 | 0.164 | 0.848 |
| F26D2.8a | F26D2.4 | 0.138 | 0.137 | 0.997 |
| F26D2.8a | T08F3.5 | 0.237 | 0.364 | 1.536 |
| F26D2.4 | T08F3.5 | 0.254 | 0.366 | 1.440 |
| C44C3.4a | W03F9.6 | 0.104 | 0.304 | 2.923 |
| C44C3.4a | C04F2.1 | (0.107) | (0.407) | (3.814) |
| W03F9.6 | C04F2.1 | 0.125 | 0.294 | 2.361 |
| C47A10.7a | C47A10.10 | 0.230 | 0.146 | 0.635 |
| C47A10.7a | Y94A7B1.1 | 0.224 | 0.383 | 1.714 |
| C47A10.10 | Y94A7B1.1 | 0.286 | 0.390 | 1.361 |
| F31F4.10a | F21H7.7 | 0.239 | 0.386 | 1.615 |
| F31F4.10a | F21H7.11 | 0.181 | 0.432 | 2.384 |
| F21H7.7 | F21H7.11 | (0.083) | (0.433) | (5.191) |
| F58E2.6a | C35D6.1 | 0.167 | 0.074 | 0.444 |
| F58E2.6a | F47C12.5 | 0.209 | 0.457 | 2.181 |
| C35D6.1 | F47C12.5 | 0.270 | 0.445 | 1.652 |
| K02E2.5a | K02E2.3 | 0.160 | 0.311 | 1.947 |
| K02E2.5a | D1054.12 | 0.126 | 0.344 | 2.739 |
| K02E2.3 | D1054.12 | (0.107) | (0.380) | (3.568) |
| F39E9.9a | R52.7 | 0.302 | 0.287 | 0.949 |
| F39E9.9a | F47D2.9 | 0.261 | 0.420 | 1.608 |
| R52.7 | F47D2.9 | 0.166 | 0.371 | 2.239 |
| R07B5.2a | ZK285.1 | 0.183 | 0.322 | 1.761 |
| R07B5.2a | F20E11.4 | 0.210 | 0.290 | 1.380 |
| ZK285.1 | F20E11.4 | 0.231 | 0.232 | 1.005 |
| Y6A4A.1a | F20E11.10 | 0.096 | 0.024 | 0.246 |
| Y6A4A.1a | E03D2.3 | 0.167 | 0.269 | 1.608 |
| F20E11.10 | E03D2.3 | 0.194 | 0.273 | 1.406 |
| Mean (SD) flatliner— functional: |
0.186 (0.055) |
0.280 (0.128) |
1.589 (0.899) |
|
| Mean (SD) functional— functional: |
0.191 (0.067) |
0.329 (0.069) |
2.043 (1.229) |
dN and dS were computed by the Nei-Gojobori method using codeml. Parentheses mark three cases in which the divergence was high enough (dS > 3) that the dN and dS are difficult to compute accurately. The functional gene pairs may be slightly more divergent from each other (see averages) because they were selected as the two closest functional genes to the flatliner and thus are not necessarily the closest to each other.
Flatliner genes.
Finally, we investigated the closest relative in C. briggsae for functional genes and genes with various types of defects in C. elegans N2. For this analysis to be valid, we needed to reannotate the C. briggsae gene predictions because we found that available SR family gene predictions are of low quality in C. briggsae. This is a major undertaking and we completed it systematically only for the srh family. We made a partial reannotation of the str family (see materials and methods), and it is likely that the general findings are similar to those for srh (data not shown). As with other SR families (Stein et al. 2003), it appears that the srh family in C. briggsae is substantially smaller than that in C. elegans. Specifically, we found 88 putative functional srh genes in C. briggsae. These included corrections of 64 srh gene models and the addition of 9 previously unpredicted srh genes. We also identified 39 srh genes with defects of the same sorts as found in C. elegans, 5 srh genes with incomplete available sequence, and several srh gene fragments. Many of the defective srh genes in C. briggsae have a single putative defect, similar to the situation in C. elegans. This result suggests that flatliner genes in SR families are also prevalent in C. briggsae. Lists of proteins from these annotations are available at http://calliope.gs.washington.edu/flatlinerdata/index.html (SM 3 and 4).
For comparison with the reannotated srh genes in C. briggsae, we divided the srh genes from C. elegans into groups according to their type of defect. For genes in each group we identified the best putative functional srh match in C. briggsae and generated protein alignments for each best match pair. We performed a similar test for each group compared to their closest functional match in C. elegans. The average pair amino acid identity for each group of genes is summarized in Table 2. Full data sets are available at http://calliope.gs.washington.edu/flatlinerdata/index.html (SM 7). Proteins from each group of C. elegans genes matched their closest C. briggsae homologs with approximately the same quality. There may be a slight tendency for functional C. elegans genes to match better, but none of the comparisons were statistically significant. Similar results were seen for the str family compared to their closest relatives in C. elegans (Table 2). Investigation of one-to-one orthologs weakly supported the possibility of a slight difference between functional and flatliner genes from the N2 strain. Specifically, of the 88 putative functional srh genes in C. briggsae, 44 could be assigned one-to-one orthologs in C. elegans; 43 of these were orthologous to a functional gene and 1 to a flatliner gene. Full lists of ortholog assignments and supporting data are available at http://calliope.gs.washington.edu/flatlinerdata/index.html (SM 8). The C. elegans protein sets used in this analysis included the products of 216 functional genes and inferred functional products from 24 flatliner genes (see materials and methods). Fisher's exact test (43/216 compared to 1/24) indicates that functional genes may be enriched in the one-to-one ortholog set (P = 0.056). A plausible explanation of these results is that srh flatliner genes have a slight tendency to belong to more dynamic parts of their phylogenetic tree; for example, these parts of the tree might have a higher frequency of deletion since the elegans-briggsae speciation event. In any case, the trends are slight and statistically marginal. The general picture is that flatliner and functional genes in the srh and str families from N2 are difficult to distinguish from each other on the basis of genome position, dN/dS ratios, and phylogenetic divergence pattern.
Flatliner alleles are likely to be null:
Although difficult to address rigorously, the sequences of flatliner genes indicate that they are unlikely to retain residual function that confers purifying selection on the flatliner allele. A general analysis of this point was made by H. Robertson (Robertson 2000, 2001). Figures 3 and 4 show more detailed information that supports a null defect for specific flatliner alleles. In brief, the stop codons analyzed occur well inside conserved coding exons and cause protein truncations missing large parts of the receptor protein. Protein and gene model alignments between three specific flatliner genes and their closest functional C. elegans relative are shown in Figure 3. For each, the intron positions are conserved and the stop codon is embedded in well-aligned protein sequence. The possibility that an alternative splice or an unidentified nearby exon could produce a functional protein was ruled out by tblastn searches (data not shown). The deletion protein alignments shown in Figure 4 are equally clear; each flatliner has a substantial section of conserved protein deleted. In addition, most of the deletions either cause a frameshift or extend into introns, causing probable splicing defects.
Figure 3.—

Stop codon flatliners are probable null alleles. Three examples of gene structure and protein alignments are shown for three flatliner genes and their closest putative functional relatives in the N2 genome. On the gene models, stop codons are marked with a black bar. On the protein alignments, stop codons are marked with a red asterisk and codons with splice junctions are marked with a red arrowhead. Protein alignments are truncated to make the sequence visible and alignment positions are marked above each alignment (cropped from an alignment with 1 at the N terminus). The entire alignment in each case is ∼350 amino acids in length.
Figure 4.—
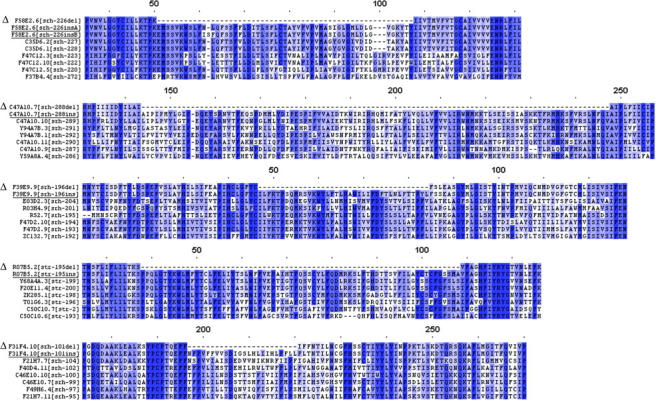
Alignments of insertion alleles with their closest functional relatives from C. elegans. For each of the five cases of insertion alleles from wild strains, the protein sequence of the N2 deletion, the insertion, and the seven closest putative functional genes are shown. In one case, there were two insertion variants; both are shown. Alignments were cropped to show the insertion region; the rest of the proteins align well for their entire length. The degree of similarity in the insertion region varies; the key observation is that the insertion aligns to other family members approximately as well as they align to each other. The N2 deletion allele is marked by Δ and the insertion alleles are underlined. Alignments were made with ClustalX using default settings except using Blosum score matrices. Clustal alignments were imported to Bonsai 1.2 for figure generation. The darkness of the purple background is proportional to the alignment score for each residue to the sum of other residues in the same position, as determined by the sum of pairs method using the Blosum62 score matrix. Alignment positions are marked above each alignment (cropped from an alignment with 1 at the N terminus). The entire alignment in each case is ∼350 amino acids in length. Gene names are as in Figure 3.
As expected for random mutations, a few defects were not as obviously null as the examples shown. We excluded from our sequence analysis a few of the most marginal cases, mostly small in-frame deletions that might retain function. Nevertheless, alignment with large numbers of closely related protein sequences indicates that most of the small in-frame deletions occur in highly conserved blocks and are likely to disrupt function. Finally, we note that the positions of stop codons and deletions in the various flatliner genes bear no obvious relationship to splicing structure or protein sequence; they are scattered evenly through the genes (available at http://calliope.gs.washington.edu/flatlinerdata/index.html, SM 9; Robertson 2000, 2001).
Sequence of flatliners:
To determine whether flatliner genes might have functional sequences in other wild isolates of C. elegans, we investigated a set of loci for which a single clearly defined defect was present in N2. We restricted our attention to genes with stop codons and sizeable deletion alleles, with the logic that a functional allele would be readily interpretable. From ∼60 candidate loci with these properties, 31 were selected arbitrarily for sequencing (19 from the srh family and 12 from the str family). Of the 31 loci sequenced, 14 have premature stop codons and 17 have deletions varying from ∼10 to 170 codons (on the basis of alignment with closely related genes). On the basis of the N2 sequence, we designed PCR primers flanking the region of the defect and used these primers to amplify and analyze the corresponding genomic region in N2 and 21 other wild isolates of C. elegans. The results are summarized in Table 3. In every case the resequenced region from N2 confirmed the genome project sequence. The 21 other wild isolates were chosen to represent efficiently the available genetic diversity; a few other isolates were not analyzed because existing evidence suggests that they are identical or nearly identical to one of the 22 strains analyzed (Hodgkin and Doniach 1997). Indeed, the results in Tables 3 and 4 suggest that the available genetic diversity in C. elegans could be largely covered with about half the strains we analyzed. For genes with stop codons, all PCR products were sequenced; for genes with deletions, only PCR products whose size indicated the possibility of a function-restoring insertion were sequenced. Unsequenced deletion alleles had PCR products indistinguishable in size from N2, indicating that the locus must encode a defective gene. In a few cases, we repeatedly failed to generate a PCR product from specific strains, despite reliable amplification of the product from all other strains and reliable amplification of other products from the same strain. In these cases, we tentatively conclude that the strain differs from N2 at one or both amplification primer sites. In three cases, all with the same gene, the PCR product repeatedly gave garbled sequence that appeared to be a mixture of two or more sequences, suggesting that these strains have multiple loci that amplify with the primers. Preliminary attempts to resolve these ambiguities were not successful and we chose not to expend excessive energy investigating them.
TABLE 4.
Other polymorphisms by strain
| Strain | Frequency (%) |
No. polymorphisms |
Sequenced nt |
Notes |
|---|---|---|---|---|
| CB4856 | 0.47 | 25 | 5,375 | Hawaii |
| JU258 | 0.46 | 14 | 3,029 | Madeira |
| RW7000 | 0.09 | 4 | 4,654 | Bergerac |
| CB4555 | 0.07 | 4 | 5,370 | |
| DH424 | 0.06 | 3 | 4,654 | |
| CB4507 | 0.04 | 2 | 4,756 | |
| AB1 | 0.04 | 2 | 5,335 | |
| CB4932 | 0.04 | 2 | 5,370 | |
| JU262 | 0.05 | 2 | 3,646 | |
| JU263 | (0.03) | 1 | 3,596 | |
| V8 | (0.03) | 1 | 3,844 | |
| CB4854 | (0) | 0 | 4,649 | Excludes hyperdivergent allele |
| AB3 | (0) | 0 | 4,928 | |
| CB4852 | (0) | 0 | 5,096 | |
| CB4853 | (0) | 0 | 5,101 | |
| CB391 | (0) | 0 | 2,855 | |
| CB4857 | (0) | 0 | 5,101 | |
| CB4858 | (0) | 0 | 2,326 | |
| RC301 | (0) | 0 | 5,101 | |
| KR314 | (0) | 0 | 5,101 |
Flatliner changes were not included in this set. Frequency estimates from JU263 to the end of the table are weak because of small numbers of mutations, as indicated by parentheses.
Remarkably, for 10 of the 31 genes tested, we found that one or more of the wild isolates had an apparently functional allele for the gene. In Table 3 these genes are marked S, indicating the presence of sense codon or an insertion relative to N2. Five of the 10 cases involved a stop codon that was changed to a sense codon, and the other 5 cases involved an insertion that appeared to restore a normal gene structure. As shown in Figure 4, the insertion cases were particularly compelling: in each case, the insertion restored a segment of protein sequence of an appropriate length that aligned well with other functional members of the gene family from N2. For the 5 cases with stop codons in N2, the functional allele had a sense codon that matches other close family members. These analyses also produced several hundred nucleotides of sequence flanking the targeted site. In all but 2 cases, no obvious defects were found in this flanking sequence (e.g., stop codons, frameshifts, etc.). As described below, a variety of other polymorphisms were found but none would be expected to abrogate function. Without more information about the normal functions of members of these gene families, it is challenging to test critically whether the putative functional alleles encode active receptors. Nevertheless, for reasons elaborated in the discussion, we think it is reasonable to infer that the alleles are functional. For simplicity, we refer to these alleles as functional for the remainder of this article. If this inference is correct, many of the wild isolates have a complement of functional chemoreceptors distinct from other wild isolates. Among the 22 strains there were at least 10 function haplotypes, as marked in Table 3. Since our analysis was restricted to a small subset of candidate flatliner genes, it is likely that the range of functional diversity is even broader.
Sequence of ancestral alleles:
For genes with putative functional polymorphism, the following results argue that the functional allele is ancestral. First, the dN/dS ratio of flatliner genes is not significantly different from that of other members of the family, as shown in Table 1. This means that the flatliner genes are currently subject to purifying selection or were so until recently. If the defective alleles are functionally null, a functional allele of the gene must have been responsible for this selection. Second, for the stop codon cases, the nonfunctional alleles all had the same stop codon sequence as N2. The most parsimonious explanation is that the ancestral sequence codes for an amino acid and the stop codon allele arose once and was inherited by some wild strains and not others. Third, and most compellingly, in all five of the deletion cases, the putative functional allele carries an insertion relative to N2 that has the clear potential to encode a typical member of the protein family. This result is evident in the protein alignments among close relatives in Figure 4. All five cases clearly demonstrate that the insertion allele must be ancestral and that the nonfunctional sequence in N2 arose by deletion. All of the wild isolates with the deletion allele produced a PCR product of the same size, arguing that the deletion arose once and spread in the C. elegans population.
Other polymorphisms:
In the process of sequencing flatliner regions, we identified a number of polymorphisms other than those we targeted. Tables 4 and 5 summarize the frequencies of these polymorphisms by gene and by strain. To avoid ascertainment bias, the stop codon and deletion polymorphisms are not included in these counts, since they were targeted as probable sites of mutation. Apart from simply identifying additional polymorphisms in most wild isolates, these results were interesting when compared with previous findings based on random sequence reads from a few wild strains (Koch et al. 2000). When viewed by gene (Table 5), it appears that SR genes have widely differing frequencies of polymorphism. At the extremes, two genes with adequate sample size (C31B8.14 and Y68A4A.1) had 15 polymorphisms each, yet one gene (R07B5.2) had no polymorphisms. Fisher's exact test shows that the frequencies of polymorphisms in C31B8.14, Y68A4A.1, F31F4.10, K02E2.5, and C44C3.4 are significantly greater than those in R07B5.2 (P < 0.001). These results strongly support the previous conclusion that polymorphisms are heterogeneously distributed in the genome (Koch et al. 2000) and suggest that SR genes are common among polymorphism hotspots. Despite our inclusion of many strains with low polymorphism, the average frequency of polymorphism in some SR genes is much higher than the genome average for the most divergent wild isolate previously analyzed (Koch et al. 2000). In addition, when averaged over all loci (Table 4), the frequency of polymorphism in the Hawaii and Madeira strains is much higher than the highest genome-average polymorphism previously analyzed (0.07% in strain CB4857, Koch et al. 2000). These results suggest that the SR genes as a whole are more polymorphic than the genome average, perhaps as much as 5- to 10-fold higher. Although we cannot rule out the possibility that this high frequency is restricted to flatliner genes, there is no specific reason to expect this to be the case.
TABLE 5.
Other polymorphisms by gene
| Gene | Frequency (%) | No. polymorphisms | Sequenced nt | Notes |
|---|---|---|---|---|
| C31B8.14 | 0.158 | 15 | 9,468 | 12 distinct, 2 deletions |
| Y68A4A.1 | 0.142 | 15 | 10,580 | 8 distinct, 1 deletion |
| F31F4.10 | 0.426 | 7 | 1,644 | 3 distinct, all SNPs |
| K02E2.5 | 0.086 | 6 | 7,014 | All same SNP |
| C44C3.4 | 0.084 | 6 | 7,152 | 2 distinct, 1 deletion |
| R08H2.11 | 0.032 | 3 | 9,437 | 2 distinct |
| F26D2.8 | 0.033 | 3 | 9,051 | 2 distinct |
| T06E6.12 | 0.032 | 2 | 6,258 | 2 distinct |
| T05E12.7 | (0.012) | 1 | 8,721 | |
| C02E7.11 | (0.009) | 1 | 11,117 | 1 deletion |
| R07B5.2 | (0) | 0 | 16,989 |
Flatliner changes were not included in this set. Frequency estimates for T05E12.7, C02E7.11, and R07B5.2 are weak because of small numbers of mutations, as indicated by parentheses.
Attempts to organize the polymorphisms into a population history indicated that the strains analyzed have exchanged varying degrees of genetic material subsequent to their primary isolation event. Surprisingly, the strains from Hawaii and Madeira were not only the most divergent from N2, but they also shared many specific polymorphisms not observed in any other strain. We speculate in the discussion about how such geographically isolated island strains could share so much genetic material.
srh-196 in strain CB4854 is hyperdivergent:
In the process of sequencing flatliner alleles we encountered one case in which the sequence was highly divergent. The sequence of F39E9.9 (srh-196) from strain CB4854 had an insertion in exon 1 that putatively restores function to the deletion present in N2. In addition to this insertion, the CB4854 sequence has 43 SNPs (Figure 5) and a second insertion inside intron 1. None of these changes would obviously compromise the function of the gene, so we tentatively conclude that CB4854 has a functional allele of srh-196. However, the number of additional polymorphisms was unique among our sequence data. The polymorphic region appears to be allelic to F39E9.9 from N2 rather than to a distinct gene: the PCR product from CB4854 produced a single band on agarose gels, the sequence quality was high, and blastn searches and other analyses showed that the sequence from CB4854 clearly corresponds to F39E9.9 from N2. The pattern of polymorphism was also peculiar: it may form a gradient along the chromosome (see Figure 5). We hypothesize that a site of ancient divergence exists in or near exon 1 of srh-196 and that recombination between two divergent alleles has produced this phenomenon. We cannot exclude the possibility that there were two distinct but related genes in the last common ancestor of N2 and CB4854 and that each strain subsequently suffered complementary deletions of one of the two genes. If this were the case, the high degree of polymorphism would simply reflect an older ancestral relationship between the two copies currently present in these two strains.
Figure 5.—

Polymorphism in F39E9.9(srh-196) from strain CB4854. The numbers of single-nucleotide polymorphisms in CB4854 relative to N2 are shown in blocks on the gene model for F39E9.9. The intron marked “divergent” was too divergent to align except near the ends where the splice junctions are conserved. The numbers of amino acid changes resulting from the polymorphism are marked below each block. The function-restoring insertion in CB4854 is at the left.
DISCUSSION
Genetic diversity in C. elegans:
Ten of the 31 flatliner genes analyzed had putative functional alleles in one or more wild C. elegans isolates. The following considerations argue that a much larger fraction of flatliner genes have functional alleles present somewhere in wild C. elegans populations. Our sampling of wild diversity was limited to 22 individual isolates in a species with worldwide distribution and presumably large natural populations. Because C. elegans is propagated in the lab as self-fertilizing hermaphrodites, there is a strong drive to homozygosity, so our sampling was effectively of 22 wild haplotypes. Furthermore, the pattern of diversity apparent in the existing isolates strongly implies that much diversity remains to be tapped. Specifically, 5 of the 10 cases of putative functional alleles were found in only 2 wild isolates. These 2 isolates were obtained from Hawaii and Madeira, ocean islands well separated from any other sampled populations. Although the worldwide population structure of C. elegans remains puzzling, this pattern of island diversity strongly suggests that additional diversity will be evident in other geographically isolated sites. The close relatedness of currently available strains of C. elegans may be a consequence of their isolation from gardens and compost heaps in areas of human cultivation. We suggest that these strains arose recently from a single source that spread with human agriculture and gardening in recent times. These observations imply that isolation of new C. elegans strains from isolated geographic sites and areas that are relatively unperturbed by human cultivation would be highly productive for future population genetics studies. The wild isolates from Hawaii and Madeira are unexpectedly similar to each other in sequence, despite their divergence from N2 and their geographic separation from each other. It seems unlikely that there is active genetic exchange between these two populations, but a recent common origin is plausible. Madeira and the Azores were common staging sites for European trans-oceanic explorations, and in the 1870s Hawaiian sugar plantations attracted a substantial emigration of Portuguese workers from Madeira (Daws 1974). It is possible that C. elegans was introduced from Madeira to Hawaii by one of these routes.
Functional diversity in chemoreceptors:
Specificity in chemosensory response derives largely from the specificity of primary sensory receptor proteins. Our results suggest extensive wild diversity in the genetic repertoire of receptors is available in different C. elegans strains. The observed diversity seems likely to have prominent sensory consequences, although proof will require experimental evidence linking the functional receptor repertoire to differences in sensory responses. Diversity in chemosensory receptor alleles has also recently been described for human odorant receptors (Menashe et al. 2002, 2003). The human diversity is strikingly similar to that described here, including putative functional diversity at stop codons and small deletions and insertions in genes previously described as pseudogenes. In addition, comparisons of chemoreceptor paralogs within species and homologs in closely related species have suggested that functional polymorphism is common and might extend to diversity within the species (e.g., Robertson et al. 2003; Parry et al. 2004).
We hypothesize that different wild strains of C. elegans have distinct patterns of response to environmental chemical stimuli on the basis of their complement of functional chemoreceptors. Though the calculation is surely naïve, it is interesting to consider how these findings might extend to the entire chemoreceptor gene family. The srh and str families together contain 102 flatliner genes on the basis of N2 sequence. These families constitute about one-third of the entire SR superfamily in C. elegans. More limited analysis of other SR families strongly suggests that a similar frequency of defective genes will characterize the SR superfamily as a whole (H. Robertson, personal communication; our unpublished results). In addition, there is every reason to expect that at least as many genes will be functional in N2 but have defective alleles in other wild strains (i.e., there is no evidence that N2 holds a privileged position among current isolates). Together, these inferences suggest that several hundred chemoreceptor family members will have common functional polymorphism among wild strains. We also note that this calculation includes only the most extreme case of polymorphism—a functional allele in one strain and an obviously defective one in another strain. Other functional polymorphisms, resulting from differences in amino acid sequence, are presumably common as well. Even with the roughness of these inferences in mind, it is difficult to escape the conclusion that C. elegans is characterized by an extraordinary degree of functional chemoreceptor polymorphism. We hypothesize that this polymorphism is an important determinant of wild behavioral diversity in this species.
Acknowledgments
We thank Hugh Robertson for inspiring this analysis and John Kemner for isolating and providing a new C. elegans isolate from Seattle, Washington. Evan Galloway was supported by a Howard Hughes Medical Institute Undergraduate Research Internship. This work was supported by National Institutes of Health grant RO1GM48700.
References
- C. elegansSequencing Consortium, 1998. Genome sequence of the nematode C. elegans: a platform for investigating biology. Science 282: 2012–2018. [DOI] [PubMed] [Google Scholar]
- Daws, G., 1974 Shoal of Time: A History of the Hawaiian Islands. University of Hawaii Press, Honolulu.
- Felsenstein, J., 1993 PHYLIP (Phylogeny Inference Package) Version 3.6a2. Department of Genome Sciences, University of Washington, Seattle.
- Glusman, G., I. Yanai, I. Rubin and D. Lancet, 2001. The complete human olfactory subgenome. Genome Res. 11: 685–702. [DOI] [PubMed] [Google Scholar]
- Hodgkin, J., and T. Doniach, 1997. Natural variation and copulatory plug formation in Caenorhabditis elegans. Genetics 146: 149–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch, R., H. G. van Luenen, M. van der Horst, K. L. Thijssen and R. H. Plasterk, 2000. Single nucleotide polymorphisms in wild isolates of Caenorhabditis elegans. Genome Res. 10: 1690–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menashe, I., O. Man, D. Lancet and Y. Gilad, 2002. Population differences in haplotype structure within a human olfactory receptor gene cluster. Hum. Mol. Genet. 11: 1381–1390. [DOI] [PubMed] [Google Scholar]
- Menashe, I., O. Man, D. Lancet and Y. Gilad, 2003. Different noses for different people. Nat. Genet. 34: 143–144. [DOI] [PubMed] [Google Scholar]
- Nei, M., and M. Gojobori, 1986. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol. 3: 418–426. [DOI] [PubMed] [Google Scholar]
- Ohno, S., U. Wolf and N. B. Atkin, 1968. Evolution from fish to mammals by gene duplication. Hereditas 59: 169–187. [DOI] [PubMed] [Google Scholar]
- Parry, C. M., A. Erkner and J. le Coutre, 2004. Divergence of T2R chemosensory receptor families in humans, bonobos, and chimpanzees. Proc. Natl. Acad. Sci. USA 101: 14830–14834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proudfoot, N. J., and T. Maniatis, 1980. The structure of a human alpha-globin pseudogene and its relationship to alpha-globin gene duplication. Cell 21: 537–544. [DOI] [PubMed] [Google Scholar]
- Robertson, H. M., 1998. Two large families of chemoreceptor genes in the nematodes Caenorhabditis elegans and Caenorhabditis briggsae reveal extensive gene duplication, diversification, movement, and intron loss. Genome Res. 8: 449–463. [DOI] [PubMed] [Google Scholar]
- Robertson, H. M., 2000. The large srh family of chemoreceptor genes in Caenorhabditis nematodes reveals processes of genome evolution involving large duplications and deletions and intron gains and losses. Genome Res. 10: 192–203. [DOI] [PubMed] [Google Scholar]
- Robertson, H. M., 2001. Updating the str and srj (stl) families of chemoreceptors in Caenorhabditis nematodes reveals frequent gene movement within and between chromosomes. Chem. Senses 26: 151–159. [DOI] [PubMed] [Google Scholar]
- Robertson, H. M., C. G. Warr and J. R. Carlson, 2003. Molecular evolution of the insect chemoreceptor gene superfamily in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 100(Suppl. 2): 14537–14542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher, J., 1990 Flatliners (motion picture). Columbia Pictures, Los Angeles.
- Sengupta, P., H. A. Colbert and C. I. Bargmann, 1994. The C. elegans gene odr-7 encodes an olfactory-specific member of the nuclear receptor superfamily. Cell 79: 971–980. [DOI] [PubMed] [Google Scholar]
- Stein, L. D., Z. Bao, D. Blasiar, T. Blumenthal, M. R. Brent et al., 2003. The genome sequence of Caenorhabditis briggsae: a platform for comparative genomics. PLoS Biol. 1: E45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, J. D., D. G. Higgins and T. J. Gibson, 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22: 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, J. D., T. J. Gibson, F. Plewniak, F. Jeanmougin and D. G. Higgins, 1997. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25: 4876–4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troemel, E. R., J. H. Chou, N. D. Dwyer, H. A. Colbert and C. I. Bargmann, 1995. Divergent seven transmembrane receptors are candidate chemosensory receptors in C. elegans. Cell 83: 207–218. [DOI] [PubMed] [Google Scholar]
- Witherspoon, D. J., and H. M. Robertson, 2003. Neutral evolution of ten types of mariner transposons in the genomes of Caenorhabditis elegans and Caenorhabditis briggsae. J. Mol. Evol. 56: 751–769. [DOI] [PubMed] [Google Scholar]
- Yang, Z., 1997. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. 13: 555–556. [DOI] [PubMed] [Google Scholar]
- Yang, Z., 1998. Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution. Mol. Biol. Evol. 15: 568–573. [DOI] [PubMed] [Google Scholar]
- Young, J. M., and B. J. Trask, 2002. The sense of smell: genomics of vertebrate odorant receptors. Hum. Mol. Genet. 11: 1153–1160. [DOI] [PubMed] [Google Scholar]
- Young, J. M., C. Friedman, E. M. Williams, J. A. Ross, L. Tonnes-Priddy et al., 2002. Different evolutionary processes shaped the mouse and human olfactory receptor gene families. Hum. Mol. Genet. 11: 535–546. [DOI] [PubMed] [Google Scholar]
- Zhang, X., and S. Firestein, 2002. The olfactory receptor gene superfamily of the mouse. Nat. Neurosci. 5: 124–133. [DOI] [PubMed] [Google Scholar]
- Zozulya, S., F. Echeverri and T. Nguyen, 2001 The human olfactory receptor repertoire. Genome Biol 2: RESEARCH0018. [DOI] [PMC free article] [PubMed]