

Abstract
The persistence phenotype is an epigenetic trait exhibited by a subpopulation of bacteria, characterized by slow growth coupled with an ability to survive antibiotic treatment. The phenotype is acquired via a spontaneous, reversible switch between normal and persister cells. These observations suggest that clonal bacterial populations may use persister cells, whose slow division rate under growth conditions leads to lower population fitness, as an “insurance policy” against antibiotic encounters. We present a model of Escherichia coli persistence, and using experimentally derived parameters for both wild type and a mutant strain (hipQ) with markedly different switching rates, we show how fitness loss due to slow persister growth pays off as a risk-reducing strategy. We demonstrate that wild-type persistence is suited for environments in which antibiotic stress is a rare event. The optimal rate of switching between normal and persister cells is found to depend strongly on the frequency of environmental changes and only weakly on the selective pressures of any given environment. In contrast to typical examples of adaptations to features of a single environment, persistence appears to constitute an adaptation that is tuned to the distribution of environmental change.
A large part of evolutionary theory is devoted to understanding population structure and dynamics in a constant environment. For many applications, such theory is reasonable because the timescales of interest (e.g., the time to fixation of a beneficial allele) are shorter than those over which the environment changes in an essential way. Some questions, however, require explicit consideration of environmental change (Lewontin and Cohen 1969; Cooper and Kaplan 1982; Seger and Brockmann 1987; Haccou and Iwasa 1995; Jablonka et al. 1995; McNamara 1995; Sasaki and Ellner 1995; Grafen 1999; Robson et al. 1999). Understanding the long-term survival of species is one area in which such consideration is essential.
Many examples of bacterial mechanisms are thought to be adaptations for survival in changing environments, some of which have recently been reviewed, including the mutator phenotypes (Giraud et al. 2001; de Visser 2002), sporulation (Eichenberger et al. 2004), adaptive mutation (Rosenberg 2001), and phase variation (Hallet 2001). A conclusive demonstration of the adaptive value of any one of these mechanisms, however, has not been made. In this article we provide a quantitative connection between experiment and theory for the bacterial persistence phenotype (Moyed and Bertrand 1983; Wolfson et al. 1990; Korch et al. 2003; Balaban et al. 2004; Keren et al. 2004) and show that this particular physiological mechanism could be an adaptation to changing environments.
It has been known since the 1940s (Bigger 1944) that growing bacteria cultures cannot be completely killed by antibiotic treatment—a small fraction of cells “persist.” Remarkably, the insensitivity to antibiotics exhibited by these persister cells is nonheritable: cultures grown up from persister cells are as sensitive to the drug as the parent culture from which the persisters were derived (Moyed and Broderick 1986). Persister cells do not remain persisters indefinitely, but spontaneously switch back to the normal, nonpersistent state and regain the usual sensitivity to antibiotics. Upon switching back, they appear, according to all available experimental observation, indistinguishable from normal cells.
Bacteria can protect themselves from various stresses, including many antibiotics, at the cost of suspending their growth. In nutrient-rich environments, in the absence of such stresses, bacteria face a crucial choice between two strategies: proliferate and risk death if stress conditions are encountered or suppress growth and be protected under such conditions. Persistence represents a risk-reducing solution to the above dilemma, a kind of “insurance policy” wherein the majority of the population does proliferate quickly under growth conditions, but a small fraction significantly suppresses growth. These slow-growing persister cells can save the population from extinction during times of stress. The actual investment in such an insurance policy, namely the number of persister cells, is known to be genetically determined and could therefore be subject to evolutionary adaptation. Here, we study the trade-offs associated with changes in the fraction of persister cells.
Persistence has been a difficult phenotype to study experimentally, both because membership in the persister subpopulation changes continually and more significantly because the subpopulation is only a tiny fraction of the total population of bacteria: only 1 in 105–106 cells of wild-type Escherichia coli are persister cells (Moyed and Bertrand 1983). An additional complication stems from the observation in Balaban et al. (2004) that there are at least two types of persistent cells. The type I persisters are cells that exhibit a very slow exit from stationary phase. The size of the type I subpopulation depends on the number of cells that have passed through stationary phase and does not increase during growth phase. The type II persisters, on the other hand, are formed via a phenotype-switching mechanism whereby a normal cell spontaneously becomes a type II persister, and the type II persister cell can spontaneously switch back to the normal phenotype. This switch appears to be unrelated to the exit from stationary phase. Single-cell observation of persisters has been carried out in Balaban et al. (2004) where the spontaneous switch of single persister cells has been observed under the microscope.
To understand the potential advantage of spontaneous switching, we restrict our discussion to type II persistence. We fully develop the model of persistence given in Balaban et al. (2004), and using this framework we explain when and how persistence can confer an adaptive advantage in changing environments. To this end, we compare the population dynamics of the hipQ mutant strain of E. coli (Wolfson et al. 1990), which exhibits a 1000-fold higher rate of switching from normal type to persister cells, with that of wild-type E. coli. We find that certain types of environmental fluctuations are selective for the hipQ mutant, while others favor the wild type. Thus we show that persistence is an example of a biology whose adaptive advantage depends strongly on environmental changes. Finally, we discuss biological aspects that our model does not incorporate and their relation to current understanding of persistence.
MODEL AND METHODS
Modeling persistence:
As shown in Balaban et al. (2004), type II persistence can be described using a simple population genetics model. The total population of bacteria cells is divided into two subpopulations: normal cells and persister cells. At any time t, the number of normal cells, n(t), and persister cells, p(t), forms the population vector x(t) = (n(t), p(t)). This vector changes in time due to the growth or decline of the two subpopulations. Such growth or decline depends on the state of the environment. When exposed to antibiotics, for example, normal cells die quickly, while persisters do not. Under typical growth conditions, on the other hand, persisters hardly grow at all, while normal cells rapidly proliferate. The characteristics of the environment are therefore an essential ingredient of our model.
The environment itself is a tremendously complex entity, and we generally ignore most of this complexity. We lump all of the intricacies of nutrient supply, toxicity, temperature, and media into a single number representing the fitness of a given type of cell in a particular environment. To simplify matters further, we consider cases where there are only two possible environments, corresponding to the presence or absence of antibiotic. We label the antibiotic environment S for “stress” and the antibiotic-free environment G for “growth.” The environment is then defined by a variable, E(t), which may take either the value S or G at any given time.
The equations governing the population vector, x(t), are straightforward. Cells of each type (normal or persister) are assigned a growth rate that depends on the environment, E(t). These growth rates are labeled μn and μp, for normal and persister cells, respectively. Cells of each type are allowed to switch to the other type spontaneously. The rate of switching from normal to persister is labeled a, and the rate of switching from persister to normal is labeled b. The dynamics are therefore described by the following first-order, linear differential equations for the growth of and switching between the two subpopulations
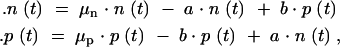 |
1 |
where the dot denotes differentiation with respect to t. In matrix notation, these equations are written more compactly as
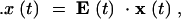 |
2 |
where the environment variable E(t) appearing above is a matrix of the form
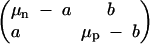 |
3 |
The switching rates a and b were experimentally found to be independent of the presence or absence of antibiotic (Balaban et al. 2004), so we assume that these values remain constant in time. The growth rates μn and μp depend on conditions: μn is positive under growth conditions and negative under antibiotic conditions, while μp is small in both environments.
The two possible values of E(t), namely S and G, are matrices of the form given in Equation 3, with the appropriate values of μn, μp, a, and b. The switching rates a and b are markedly different between wild-type E. coli and the mutant hipQ strain. The experimentally measured values of the matrices S and G for each strain are given in Table 1. When discussing a particular strain, we label the matrices S and G with subscripts corresponding to the wild-type or hipQ strain: Swt, Gwt, Ship, and Ghip.
TABLE 1.
Growth and switching rates (per hour) for wild type andhipQ in the presence and absence of antibiotic
| Strain | Conditions | E(t) | μn | μp | a | b |
|---|---|---|---|---|---|---|
| Wild type | Growth | Gwt | 2 | 0 | 1.2 × 10−6 | 0.1 |
| Antibiotic | Swt | −4 | −0.4 | 1.2 × 10−6 | 0.1 | |
| hipQ | Growth | Ghip | 2 | 0.2 | 0.001 | 10−6–10−4 |
| Antibiotic | Ship | −4 | −0.4 | 0.001 | 10−6–10−4 |
Parameters are taken in agreement with experiments described in Balaban et al. (2004). For hipQ, a wide range of values of b was consistent with experiments and is indicated.
In our model, the changing environment is characterized by a pair of numbers (tg, ts), where tg is the duration of growth and ts is the duration of stress. The pair (2, 1), for example, corresponds to environmental change in which bacteria are allowed to grow for 2 hr, followed by 1 hr of antibiotic, followed again by 2 hr of growth and 1 hr of antibiotic, and so forth indefinitely. We thus consider periodically changing (cyclic) environments and do not allow for true randomness in their durations. As explained in the discussion, the periodic case is sufficiently general for our purposes.
To compare persistence of the wild-type and hipQ strains, we need to solve Equation 2 and obtain the dependence of the population size on the environmental changes. In a cycling environment with durations (tg, ts) the solution of Equation 2 is obtained using the matrix exponential of the environment variable E(t). The result takes the form
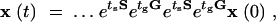 |
4 |
where the dots indicate that the pattern repeats a certain number of times that depend on the value of t. It can be shown (see below) that the population vector eventually grows (or decays) at a single rate given by the highest eigenvalue of the matrix etsSetgG. We call this rate the long-term fitness of the strain and denote it by λ1etsSetgG. Note that the order of the matrices, λ1etsSetgG or λ1etgGetsS, does not matter since the eigenvalues are the same. When two strains coexist in a changing environment, each strain eventually grows at the rate given by its long-term fitness. The strain with the larger long-term fitness grows faster and would take over the population.
Our model assumes that all growth (or decay) is exponential. Incorporation of nonexponential growth would render the analysis more difficult, but would allow the inclusion of stationary phase in the description, as well as the type I persistence mechanism (this type of persistence is associated with the exit from stationary phase). Such modeling awaits deeper biological understanding about the interaction between the mechanisms governing the two known types of persistence, as well as a more detailed physiology of persistence.
Computation of long-term fitness:
Suppose by time t exactly q cycles of growth and stress have occurred. Then the solution of Equation 2, given by Equation 4, can be rewritten as follows:
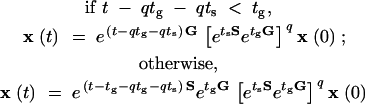 |
5 |
Naturally if the order of environments is initially reversed, we would have a slightly different expression in which etgG and etsS are exchanged. As q becomes large, the growth rate of the population vector will be completely dominated by the highest eigenvalue of the matrix etsSetgG. To see this, we diagonalize the bracketed matrix in Equation 5, letting λ1 and λ2 be the two eigenvalues and M be the matrix of eigenvectors:
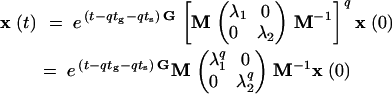 |
6 |
If λ1 is the larger of the two eigenvalues, then for large q the population vector grows exponentially as (λ1)q. Note that the quantity t − qtg − qts is always less than tg + ts, so it does not grow with q and therefore does not affect the population growth rate. The long-term fitness is thus given by λ1etsSetgG.
Stochastic simulation:
The stochastic description of persistence considers Equation 1 as a discrete Markov process in continuous time (Bharucha-Reid 1960). For each strain, wild type or hipQ, there are four independent processes, each with a prescribed rate: (1) growth of normal cells, μn; (2) growth of persisters, μp; (3) normal cell to persister switching, a; and (4) persister to normal cell switching, b. The population vector for each strain is (n(t), p(t)) as before, except that population sizes are now whole numbers. Since each cell can undergo growth or switching independently, the rate of each process at the population level is given by the single-cell rate times the population size. Simulation is performed by iterated cycles of four steps (Gillespie 1977):
The time of the next event is computed. This is done by calculating the total rate of all processes, which is λtot = n(t)(μn + a) + p(t)(μp + b), and then randomly choosing the time of the next event, tnext, from the exponential distribution, λtote−tλtotdt.
The type of event is computed. One of the four events given above occurs at the time tnext. The probability of each event is proportional to the rate of its process. The total rate, λtot, is therefore the appropriate normalization, giving the following probabilities for the occurrence of each type of event: μn · n(t)/λtot, μp · p(t)/λtot, a · n(t)/λtot, and b · p(t)/λtot. These probabilities are computed, and the event type is randomly selected from this distribution.
The population vector is updated to reflect the change. If one of the growth processes occurs, then n(t) or p(t) is incremented by one. If a switching process occurs, then one component is decreased, and the other is increased, by one.
Population size is rescaled, if necessary. If the total population size, Ntot, which counts all cells of both strains, exceeds some upper limit, Nmax, the population is rescaled to a smaller size, Ni. The rescaling is done by assigning to n(t) and p(t) a random value sampled from a Poisson distribution having mean n(t)Ni/Ntot and p(t)Ni/Ntot, respectively.
In addition to these four steps, the environment obeys the periodicity fixed by tg and ts, so the rates of the four processes at a given time depend, as in Table 1, on the conditions occurring at time t. In all simulations we used the parameter values given in Table 1, with bhip = 10−4.
RESULTS
Large populations—the deterministic limit:
The long-term fitness of the wild-type strain and of the hipQ mutant is computed using λ1etgGwtetsSwt and λ1etgGhipetsShip, respectively. These fitness values depend on the changes of environment, manifested by the duration of growth, tg, and the duration of antibiotic stress, ts. For a given type of changing environment specified by (tg, ts), we wish to know which strain has higher fitness. We therefore compute the curve in the (tg, ts) plane for which wild type and hipQ have equal fitness. The curve is given by the equation
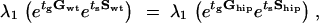 |
7 |
which implicitly gives the dependence of ts on tg. We plot the curve of equal fitness in Figure 1. The curve separates the plane into two parts. Below the curve, wild type has higher fitness than hipQ, while above the curve the situation is reversed.
Figure 1.—

Phase diagram for competition between wild type and hipQ in periodic environments. Growth and stress durations in hours are given by tg and ts, respectively. The strain with the higher fitness is indicated in each of the two regions. All parameter values used for this computation are given in Table 1. Extreme possible values of the switching parameter b of the hipQ mutant were considered: main figure uses bhip = 10−4; inset uses bhip = 10−6.
By performing an asymptotic expansion of the fitness values, for large values of tg, we found that the curve asymptotically grows linearly with tg. We do not provide the details of this computation as it is lengthy and not particularly illuminating, giving the result
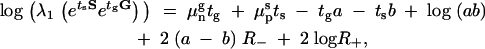 |
8 |
where 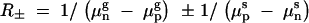 . Here, we have used superscripts g and s to indicate the environment; for example μsn refers to the killing rate of normal cells under stress conditions. Using this expression in the equation of the equal fitness curve (7), we find that Equation 7 is linear in tg and ts, for large durations, and that the slope of the line is −μgn,wt − μgn,hip − awt + ahip/μsp,wt − μsp,hip − bwt + bhip, which is ∼ahip/bwt = 0.01 (when values in Table 1 are used).
. Here, we have used superscripts g and s to indicate the environment; for example μsn refers to the killing rate of normal cells under stress conditions. Using this expression in the equation of the equal fitness curve (7), we find that Equation 7 is linear in tg and ts, for large durations, and that the slope of the line is −μgn,wt − μgn,hip − awt + ahip/μsp,wt − μsp,hip − bwt + bhip, which is ∼ahip/bwt = 0.01 (when values in Table 1 are used).
As noted in Table 1, the value of the switching rate bhip is poorly determined. To assess the effect of this parameter on the fitness, we used the upper-bound value of bhip when plotting the curve of equal fitness in Figure 1 and the lower bound in the Figure 1 inset. The main effect of decreasing the value of bhip is to increase the size of the region of wild-type superiority. Using Equation 8, we see that when bhip is small, changing its value by an order of magnitude will effect a change of the hipQ fitness mainly through the log(b) term. Since this term is positive, decreasing bhip will decrease the fitness of hipQ and therefore increase the region in which wild type wins.
It is also interesting to plot the ratio of growth rates of the two strains as a function of tg and ts. This is shown in Figure 2, plotted on a logarithmic scale. This quantity indicates how quickly the fitter strain would win a hypothetical competition between the two strains. We see that in the region below the line of equal fitness in Figure 2A, wild type wins, but only by a bit, while above the line, hipQ wins by an order of magnitude or more, depending on the environmental durations. This is because in this region of very short antibiotic durations, the fitness difference between the two strains is well approximated by the difference in growth rates of normal cells in the absence of antibiotic. This difference is ahip − awt, which is therefore only 10−3 in favor of wild type.
Figure 2.—

Ratio of growth rates of hipQ to wild type in the (tg, ts) plane. Time is given in hours. (A) bhip = 10−4; (B) bhip = 10−6. The black line in A and B is the curve of equal fitness, shown in Figure 1, along which the z-coordinate is identically zero.
Small populations—the stochastic limit:
The fitness ratio shown in Figure 2 plays an important role in determining the sensitivity of the results to changes in population size. To determine how small population sizes affect the deterministic results, we ran stochastic simulations in which the growth and switching rates given by the matrices G and S determined the stochastic replication and switching times for a finite population of cells. The simulation is described in model and methods. At every time t, the simulation records the number of normal and persister cells for each strain. The population size is always kept smaller than a given size, consistent with the idea of a limiting resource.
In Figure 3A, we show a sample stochastic trajectory, for a population of size 105, in the changing environment given by (tg, ts) = (20, 2.5). The simulation begins with equal numbers of wild-type and hipQ cells. In this particular simulation, after the fourth round of stress, the normal cells of both wild type and hipQ are completely killed (Figure 3A, event b). The hipQ mutant, however, has generated a handful of persisters during the growth phase, while wild type has not. These persisters are able to grow slowly during the subsequent growth phase and are not completely killed during stress. Eventually the hipQ persisters reach a population size of 104, at which point the total rate of switching back to the normal cell type is 104 bhip, which is 1. We see that a switch occurs at this point (t = 125 hr), and the hipQ normal cell population thereafter reestablishes itself.
Figure 3.—

Competition of wild type against hipQ at (tg, ts) = (20, 2.5), starting from 50,000 wild-type and 50,000 hipQ cells: (A) stochastic dynamics; (B) deterministic dynamics. Gray bars indicate periods of growth, while spaces between bars are periods of antibiotic stress. When the red and blue lines overlap completely, we use a dashed line to resolve them. Circled events are as follows: a is the formation of a single wild-type persister cell, which dies soon after; b is the complete extinction of normal cells of both wild type and hipQ; and c is the switch of a single hipQ persister cell to the normal type. In these simulations, Nmax = 110,000 and Ni = 100,000. Parameter values are given in Table 1, taking bhip = 10−4. Deterministic dynamics correspond to numerical solution of Equations 1, with population size rescaling, as in the stochastic simulation.
Due to stochastic fluctuations, the result of competitions between wild type and hipQ for small population sizes may differ from the deterministic case. In the stochastic simulation of Figure 3A, hipQ wins because it is able to generate a few persisters that allow it to survive a particularly calamitous antibiotic episode (event b), while wild type does not. In the deterministic case, shown in Figure 3B, wild type wins every time because its population is never completely wiped out by antibiotic, and therefore its small fitness advantage over hipQ guarantees eventual victory. To study the deviation of finite populations from the deterministic limit, we ran stochastic competitions at tg = 20 for various values of ts. Each competition was repeated 100 times, and the fraction of wild-type wins was computed. The results, for three different population sizes, are shown in Figure 4.
Figure 4.—

Stochastic competitions between wild type and hipQ at tg = 20 for various values of ts. For each data point, 100 simulations were run, starting from a 1:1 ratio of wild type to mutant. Ni was taken to be the given population size, while Nmax was set to 1.1 Ni. Due to long simulation times for N = 106, the points on this curve for ts < 3 are averages over 20 runs. The dotted line indicates the result in the large population (deterministic) limit.
We find that for finite population sizes, the line of equal fitness shifts in favor of hipQ. That is, the amount of stress at which wild type has a 50% chance of winning is strictly less than that in the deterministic case shown by the dotted line in Figure 4. To understand this effect, note that a population of size 10n will be completely killed by antibiotic within 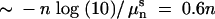 hr. Without persisters, a population of size 104, for example, cannot survive >2.4 hr of stress. During the growth phase, the wild type will have produced ∼104awt = 0.01 persisters, meaning that 99% of the time there will be no persisters, while hipQ will produce 104ahip = 10 persisters. In this case hipQ is far more likely to survive than wild type, as seen in Figure 4.
hr. Without persisters, a population of size 104, for example, cannot survive >2.4 hr of stress. During the growth phase, the wild type will have produced ∼104awt = 0.01 persisters, meaning that 99% of the time there will be no persisters, while hipQ will produce 104ahip = 10 persisters. In this case hipQ is far more likely to survive than wild type, as seen in Figure 4.
At what population size can we expect the line of equal fitness to coincide with the deterministic line at ts = 3.9? We note that persisters in antibiotic die at a rate of μsp per hour, meaning that we need at least exp−μspts persisters to survive ts hr of antibiotic. Wild type will produce this number of persisters when its population size is on the order of exp−μspts/awt ≈ 106−107, while hipQ produces this number already at smaller sizes. We thus expect population sizes of order 107 to be nearly deterministic. We can see in Figure 4 that this estimate is in good agreement with the rate of approach of the point of equal fitness on the curves with N = 104, 105, and 106 toward the dotted line. Stochastic simulations with N = 107 are, unfortunately, prohibitively long for us to gather meaningful statistics.
For population sizes <104, both wild type and hipQ have a high probability of extinction even when antibiotic stress is relatively short. Competitions at such small sizes are not particularly meaningful because both populations are dying on average, unless very short durations of stress are used. In such cases ts is close to 0, and we can roughly assume that both populations are under conditions of growth, so that the fitness of normal wild-type cells is μgn − awt and of normal hipQ cells is μgn − ahip. The fitness difference is thus ∼10−3 in favor of wild type. We know from the diffusion approximation in population genetics (Roughgarden 1996) that stochastic effects become important when population size times the fitness difference is smaller than one. As population size decreases to order of 103, the fitness difference between wild type and hipQ will become insignificant, and we will find that each strain wins ∼50% of the time. This trend is seen clearly in Figure 4 for the N = 104 curve at low values of ts. When we ran 100 simulations using N = 103 at ts = 0, wild type won 51 times.
Optimal switching rates:
Having established that certain types of environmental change can be selective for one strain over another, we would like to know in which type of cycling environment each strain is optimal. Optimality, here, refers solely to the persistence mechanism and, more specifically, to the switching parameters a and b. We consider a specific changing environment, fixed by the pair (tg, ts), and we tune the parameters a and b to maximize the long-term fitness. The optimization is carried out by setting to zero the partial derivatives of the long-term fitness with respect to a and b, 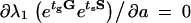 and
and 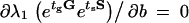 , and solving for a and b. Using the approximation given in Equation 8, we obtain the following result:
, and solving for a and b. Using the approximation given in Equation 8, we obtain the following result:
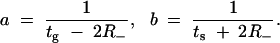 |
9 |
We checked this solution numerically and found that in the region of parameters relevant to E. coli (Table 1), the error in the approximate optimal values of a and b given in Equation 9 is 5%. Since R− ≈ 0.22 for wild type, and R− ≈ 0.27 for hipQ, we find that for times longer than a few hours, approximate 1/t relations hold: a ≈ 1/tg and b ≈ 1/ts. This type of result was seen also in different models, analytically in Lachmann and Jablonka (1996) and numerically in Thattai and van Oudenaarden (2004).
DISCUSSION
We have analyzed the model of persistence first presented in Balaban et al. (2004) in the deterministic, large-population dynamics of Equation 2, using the concept of long-term fitness, as well as in the stochastic regime of small population size. Our central conclusion is that the type of environmental change, more than the specifics of any given environment, selects one strain over another. If persistence is an adaptation whose particulars, such as switching rates, have been significantly evolved, then it appears that the switching rates are largely determined by the way in which the environment changes. This is clear from the result that the optimal values of a and b are determined mostly by the durations tg and ts, which specify how often the environment changes, and only negligibly by the parameters μn and μp.
In particular, we find that the hipQ strain appears to be optimal for conditions in which antibiotic stress is frequent and intense (at least 10 times as frequent as growth conditions), while wild type is optimal when antibiotics are rarely encountered (∼1 day of antibiotic every 100 years). It is important to note that these numbers are only rough estimates and are probably best considered as order-of-magnitude estimates. The reason for this is that the measurements of switching rates are all made under laboratory conditions, rather than in the natural environments of E. coli. The type of antibiotic, the nature of its presentation to the cells, as well as many other external factors may affect the switching rates of the persistence mechanism. For this reason we do not believe that any inferences about the natural environment of E. coli may be made from the optimal durations determined above.
In modeling environmental change, we limited our treatment to regular, periodic changes. It would be more realistic to model the environmental durations as random variables with some characteristic probability distribution. Calculation of the population fitness in such a setting is significantly more complex and will be presented in a future publication. We find that when environmental durations are long enough [specifically, for tg, ts ≫ 1/(μn − μp)], the long-term fitness does not depend on the particular form of randomness of the environment (E. Kussell and S. Leibler, unpublished results), but rather, for the case of two environments, it depends simply on their mean durations. Our conclusions for periodically changing environments therefore remain valid even when environmental durations are taken to be random variables, provided that these durations are not too short. The fixed values of tg and ts can then simply be replaced by average durations, 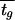 and
and 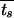 .
.
Practically nothing is known about interactions between the mechanism of persistence and other survival mechanisms such as sporulation, adaptive mutation, heat shock, phase variation, and the mutator phenotypes. Is there cross-talk between these networks? Is the persistence mechanism upregulated in reponse to certain cues? Our model does not account for these possibilities, nor is there presently enough understanding of the relevant physiology to allow meaningful modeling. The model presented here incorporates all of the known physiology of persistence and remains simple enough for a complete analysis.
The stochastic treatment of the model identified the range of population sizes for which the deterministic results hold. We showed that the deterministic conclusions are valid for population sizes >107. If the difference between the long-term fitnesses of the two strains, given in Figure 2, is large, then the deterministic results will hold for much smaller population sizes. Since the fitness difference shown in Figure 2 is a strictly increasing function of the environmental durations tg and ts (when these durations are not too short), we conclude that 107 is a rough upper bound on the population size above which a deterministic description should hold. As this number is not an unreasonable population size for E. coli, it appears that the deterministic description of persistence should be sufficient for the purpose of quantifying its adaptive advantage over long timescales.
We have examined the bacterial persistence mechanism within a theoretical framework to demonstrate that its adaptive value is directly related to the distribution of environmental changes. As such, it is a rather unique example in which the particular spectrum of environments is selective, rather than the particular features of any given environment. The ability to test rigorous theory in this system leads us to believe that a deeper understanding of adaptations to changing environments, and phenotype switching in particular, will emerge from its study.
Acknowledgments
We thank Gerard Ben Arous, Andrew Murray, and Jack Merrin for many fruitful discussions, as well as members of our laboratory. E.K. thanks the Alfred P. Sloan foundation for support.
References
- Balaban, N. Q., J. Merrin, R. Chait and S. Leibler, 2004. Bacterial persistence as a phenotypic switch. Science 305: 1622–1625. [DOI] [PubMed] [Google Scholar]
- Bharucha-Reid, A. T., 1960 Elements of the Theory of Markov Processes and Their Applications. Dover Publications, Mineola, NY.
- Bigger, J. W., 1944 Treatment of staphylococcal infections with penicillin. Lancet ii: 497–500.
- Cooper, W. S., and R. H. Kaplan, 1982. Adaptive coin-flipping—a decision—theoretic examination of natural-selection for random individual variation. J. Theor. Biol. 94: 135–151. [DOI] [PubMed] [Google Scholar]
- de Visser, J., 2002. The fate of microbial mutators. Microbiology 148: 1247–1252. [DOI] [PubMed] [Google Scholar]
- Eichenberger, P., M. Fujita, S. T. Jensen, E. M. Conlon, D. Z. Rudner et al., 2004. The program of gene transcription for a single differentiating cell type during sporulation in Bacillus subtilis. PLoS. Biol. 2: 1664–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie, D. T., 1977. Exact stochastic simulation of coupled chemical reactions. J. Phys. Chem. 81: 2340–2361. [Google Scholar]
- Giraud, A., M. Radman, I. Matic and F. Taddei, 2001. The rise and fall of mutator bacteria. Curr. Opin. Microbiol. 4: 582–585. [DOI] [PubMed] [Google Scholar]
- Grafen, A., 1999. Formal Darwinism, the individual-as-maximizing-agent analogy and bet-hedging. Proc. R. Soc. Lond. Ser. B Biol. Sci. 266: 799–803. [Google Scholar]
- Haccou, P., and Y. Iwasa, 1995. Optimal mixed strategies in stochastic environments. Theor. Popul. Biol. 47: 212–243. [Google Scholar]
- Hallet, B., 2001. Playing Dr Jekyll and Mr Hyde: combined mechanisms of phase variation in bacteria. Curr. Opin. Microbiol. 4: 570–581. [DOI] [PubMed] [Google Scholar]
- Jablonka, E., B. Oborny, I. Molnar, E. Kisdi, J. Hofbauer et al., 1995. The adaptive advantage of phenotypic memory in changing environments. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 350: 133–141. [DOI] [PubMed] [Google Scholar]
- Keren, I., N. Kaldalu, A. Spoering, Y. P. Wang and K. Lewis, 2004. Persister cells and tolerance to antimicrobials. FEMS Microbiol. Lett. 230: 13–18. [DOI] [PubMed] [Google Scholar]
- Korch, S. B., T. A. Henderson and T. M. Hill, 2003. Characterization of the hipA7 allele of Escherichia coli and evidence that high persistence is governed by (p)ppGpp synthesis. Mol. Microbiol. 50: 1199–1213. [DOI] [PubMed] [Google Scholar]
- Lachmann, M., and E. Jablonka, 1996. The inheritance of phenotypes: an adaptation to fluctuating environments. J. Theor. Biol. 181: 1–9. [DOI] [PubMed] [Google Scholar]
- Lewontin, R. C., and D. Cohen, 1969. On population growth in a randomly varying environment. Proc. Natl. Acad. Sci. USA 62: 1056–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara, J. M., 1995. Implicit frequency dependence and kin selection in fluctuating environments. Evol. Ecol. 9: 185–203. [Google Scholar]
- Moyed, H. S., and K. P. Bertrand, 1983. Hipa, a newly recognized gene of Escherichia-coli K-12 that affects frequency of persistence after inhibition of murein synthesis. J. Bacteriol. 155: 768–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyed, H. S., and S. H. Broderick, 1986. Molecular-cloning and expression of hipA, a gene of Escherichia-coli K-12 that affects frequency of persistence after inhibition of murein synthesis. J. Bacteriol. 166: 399–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robson, A. J., C. T. Bergstrom and J. K. Pritchard, 1999. Risky business: sexual and asexual reproduction in variable environments. J. Theor. Biol. 197: 541–556. [DOI] [PubMed] [Google Scholar]
- Rosenberg, S. M., 2001. Evolving responsively: adaptive mutation. Nat. Rev. Genet. 2: 504–515. [DOI] [PubMed] [Google Scholar]
- Roughgarden, J., 1996 The Theory of Population Genetics and Evolutionary Ecology. Prentice Hall, Upper Saddle River, NJ.
- Sasaki, A., and S. Ellner, 1995. The evolutionarily stable phenotype distribution in a random environment. Evolution 49: 337–350. [DOI] [PubMed] [Google Scholar]
- Seger, J., and H. J. Brockmann, 1987 What is bet-hedging?, pp. 182–211 in Oxford Surveys in Evolutionary Biology, edited by P. H. Harvey and L. Partridge. Oxford University Press, Oxford.
- Thattai, M., and A. van Oudenaarden, 2004. Stochastic gene expression in fluctuating environments. Genetics 167: 523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfson, J. S., D. C. Hooper, G. L. McHugh, M. A. Bozza and M. N. Swartz, 1990. Mutants of Escherichia coli K-12 exhibiting reduced killing by both quinolone and beta-lactam antimicrobial agents. Antimicrob. Agents Chemother. 34: 1938–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]