

Abstract
Members of the SEPALLATA (SEP) MADS-box subfamily are required for specifying the “floral state” by contributing to floral organ and meristem identity. SEP genes have not been detected in gymnosperms and seem to have originated since the lineage leading to extant angiosperms diverged from extant gymnosperms. Therefore, both functional and evolutionary studies suggest that SEP genes may have been critical for the origin of the flower. To gain insights into the evolution of SEP genes, we isolated nine genes from plants that occupy phylogenetically important positions. Phylogenetic analyses of SEP sequences show that several gene duplications occurred during the evolution of this subfamily, providing potential opportunities for functional divergence. The first duplication occurred prior to the origin of the extant angiosperms, resulting in the AGL2/3/4 and AGL9 clades. Subsequent duplications occurred within these clades in the eudicots and monocots. The timing of the first SEP duplication approximately coincides with duplications in the DEFICIENS/GLOBOSA and AGAMOUS MADS-box subfamilies, which may have resulted from either a proposed genome-wide duplication in the ancestor of extant angiosperms or multiple independent duplication events. Regardless of the mechanism of gene duplication, these pairs of duplicate transcription factors provided new possibilities of genetic interactions that may have been important in the origin of the flower.
MOLECULAR genetic studies over the past 2 decades have identified a large number of regulatory genes that control early floral development (Ma 1994; Weigel and Meyerowitz 1994; Zhao et al. 2001; Jack 2004). In particular, genetic studies in Arabidopsis thaliana and Antirrhinum majus have led to the proposal of the genetic “ABC model” for specifying floral organ identity (Coen and Meyerowitz 1991). Most of the genes required for the ABC functions are MADS-box genes that encode putative transcription factors (Theissen et al. 1996; Becker and Theissen 2003). In Arabidopsis, the A function requires the APETALA1 (AP1) gene, B function needs both the APETALA3 (AP3) and the PISTILLATA (PI) genes, and the AGAMOUS (AG) gene is necessary for C function. In Antirrhinum, the functional homologs of AP3, PI, and AG are DEF, GLO, and PLE, respectively (Coen and Meyerowitz 1991; Ma 1994; Ma and Depamphilis 2000). These ABC MADS-box genes are expressed in the floral meristem before or at the time of organ primordia initiation in regions corresponding to the genetically defined functional domains, indicating that the expression patterns of these genes are good predictors of functional domains (Ma and Depamphilis 2000). Phylogenetic analyses indicate that the ABC genes and their close homologs form several well-defined subfamilies, whose evolutionary histories have been, or are currently being, studied phylogenetically (Kramer et al. 1998, 2004; Becker and Theissen 2003; Litt and Irish 2003; Kim et al. 2004; Soltis et al. 2005).
Although ABC genes are critical for floral organ identity, they are not sufficient to convert leaves into floral organs, indicating that other flower-specific genes are needed. More recent genetic and molecular studies in Arabidopsis indicate that four additional MADS-box genes, SEPALLATA1/2/3/4 (SEP1/2/3/4; formerly AGL2/4/9/3) (Ma et al. 1991; Huang et al. 1995; Mandel and Yanofsky 1998), are required for the specification of the identity of all four whorls of floral organs and for floral meristem determinacy (Honma and Goto 2000; Pelaz et al. 2000, 2001a,b; Ditta et al. 2004). The Arabidopsis SEP1, -2, and -4 genes are expressed throughout the floral meristem at stage 2, slightly earlier than SEP3, which is expressed in a region corresponding to the inner three whorls just before the initiation of floral organ primordia (Flanagan and Ma 1994; Savidge et al. 1995; Mandel and Yanofsky 1998; Ditta et al. 2004). Subsequently, SEP1 and SEP2 expression persist in all floral organ primordia and SEP3 is expressed in the inner three whorls, whereas SEP4 expression becomes more highly expressed in the central dome than in the sepals. Genetic studies indicate that the SEP genes are functionally redundant in the control of all floral organ identities, as all organs are replaced by leaf-like organs in the quadruple mutant, as well as have redundant roles in promoting floral meristem determinacy (Pelaz et al. 2000; Ditta et al. 2004).
One or more Arabidopsis SEP proteins can form protein complexes with one or more ABC proteins in vitro and in yeast two-hybrid assays (Fan et al. 1997; Honma and Goto 2000; Pelaz et al. 2001a; Favaro et al. 2002; Immink et al. 2002, 2003; Ferrario et al. 2003), suggesting that these MADS-box proteins form multimeric complexes that control various transcriptional programs in specific organ types (Theissen and Saedler 2001). The “floral quartet model” integrates the genetic ABC model with the recent addition of known protein-protein interactions (Theissen 2001). The recent finding of SEP function in the first whorl (Ditta et al. 2004) supports a revised quartet model: AP1-AP1-SEP-SEP, AP1-AP3-PI-SEP, AP3-PI-AG-SEP, and AG-AG-SEP-SEP complexes are required for the formation of sepals, petals, stamens, and carpels, respectively, highlighting the importance of the SEP proteins in specifying all four whorls of the flower.
The SEP genes form a separate subfamily in the MADS-box gene family (Becker and Theissen 2003), designated here as the SEPALLATA (SEP) subfamily. SEP1 and SEP2 are believed to be the result of a recent gene duplication (Ermolaeva et al. 2003) but their relationships to SEP3 and SEP4 are not clear. Some analyses place SEP1 and SEP2 more closely to SEP4 than to SEP3 (Yu and Goh 2000; Lemmetyinen et al. 2004), whereas other studies concluded that SEP3 is the closest relative of SEP1 and SEP2 (Purugganan 1998; Lawton-Rauh et al. 2000; Sung et al. 2000; Becker and Theissen 2003; Parenicova et al. 2003; Vandenbussche et al. 2003b; Nam et al. 2004). This uncertainty has made it hard to understand the function of ancestral SEP homologs and to study functional changes during gene evolution.
Homologs of the SEP genes have been isolated from other plants and are referred to as “SEP homologs” here for such genes whose closest relative in Arabidopsis is SEP1, -2, -3, or -4, regardless of function. SEP homologs from petunia and rice (Oryza sativa) have functions similar to those of the Arabidopsis SEP genes (Ferrario et al. 2003; Vandenbussche et al. 2003b). The petunia FBP2 gene can partially rescue the Arabidopsis sep1 sep2 sep3 triple-mutant phenotype (Ferrario et al. 2003) and can form multimeric complexes with other floral homeotic MADS-box proteins, suggesting that FBP2 and SEP are functional counterparts (Ferrario et al. 2003). However, in the composite Gerbera hybrida, the SEP homolog GRCD2 not only is important for floral meristem identity, but also promotes inflorescence meristem determinacy (Uimari et al. 2004). Furthermore, because the expression patterns of rice SEP homologs, such as members of the LEAFY HULL STERILE1 (LHS1) clade, are highly variable among taxa (Malcomber and Kellogg 2004), it is possible that other SEP homologs may also have diverse expression patterns and functions.
The fact that multiple SEP homologs are present in distant angiosperm lineages suggests that this subfamily has experienced several gene duplication events. However, the lack of information about SEP homologs in basal angiosperms makes it uncertain whether the first gene duplication occurred before or after the initial diversification of extant angiosperms. Phylogenetic analysis suggests that the closest relatives of the SEP subfamily are members of the AGL6 subfamily, which contains both angiosperm and gymnosperm genes (Becker and Theissen 2003; De Bodt et al. 2003; Martinez-Castilla and Alvarez-Buylla 2003; Nam et al. 2003). It is possible that either the extant gymnosperms have lost their SEP homologs or the SEP subfamily is angiosperm specific. Alternatively, it is also possible that gymnosperm homologs of SEP genes have not yet been isolated or identified despite the recovery of the AG, DEF/GLO, and AGL6 subfamily members from multiple gymnosperm taxa.
Phylogenetic analyses of ABC genes and their angiosperm and gymnosperm homologs suggest that most MADS-box subfamilies evolved from precursors in the common ancestors of extant seed plants (Becker and Theissen 2003). Experimental evidence supports that members of the SEP subfamily are “flower-specific” factors. The fact that SEP proteins are required for all ABC functions, along with the possibility that SEP genes are angiosperm specific, suggests that the evolution of SEP homologs may be intimately associated with the evolution of the angiosperms and may even be partly responsible for the emergence and subsequent explosion of angiosperms. In this article, we have identified SEP homologs from angiosperms that occupy phylogenetically important positions. By sequencing floral cDNAs from six species, we found and analyzed nine new SEP homologs from two basal-most extant angiosperms (Amborella trichopoda and Nuphar advena), as well as two magnoliids (Persea americana and Liriodendron tulipifera), one basal monocot (Acorus americanus), and one basal eudicot (Eschscholzia californica) (Figure 1). Extensive phylogenetic analyses of these genes, along with all other publicly available SEP homologs, suggest that this subfamily originated and underwent a gene duplication event before the diversification of the extant angiosperms. In addition, investigation of protein sequences identified many lineage-specific amino acid changes that may have potentially affected the biochemical features of the proteins. On the basis of these results, the evolution of the SEP subfamily and its possible role in the origin and diversification of floral developmental programs are discussed.
Figure 1.—

A generalized phylogenetic tree of angiosperms.
MATERIALS AND METHODS
Taxon selection and plant materials:
Recent phylogenetic studies suggest that all extant angiosperms fall into the eudicots, monocots, or magnoliids, or one of several small lineages near the base of the angiosperm tree (Qiu et al. 1999; Soltis et al. 1999, 2000; Angiosperm Phylogeny Group 2003) (Figure 1). Among basal angiosperms, Amborellaceae and Nymphaeaceae (waterlilies) were resolved as the sister clade(s) of all other angiosperms, followed by Austrobaileyales and all other lineages (Mathews and Donoghue 1999; Qiu et al. 1999; Soltis et al. 1999; Barkman et al. 2000; Graham and Olmstead 2000; Zanis et al. 2002; Aoki et al. 2004). To maximize the coverage of the base of the angiosperm tree, we have sampled the basal-most angiosperm A. trichopoda, the water lily N. advena, the magnoliids L. tulipifera and P. americana, the basal monocot A. americanus, and the basal eudicot E. californica. Additionally, the SEP homologs Eu.be.AGL9 (AY821780), Ma.gr.AGL2 (AY821781), and Ma.gr.AGL9 (AY821782) were isolated from Eupomatia bennettii and Magnolia grandiflora, as described previously (Kim et al. 2005). The sources of plant materials are A. trichopoda (DL8346, TF6481, DHL8350, HAW, Kauai, HI), N. advena (L. Landherr, PAC 95537, University Park, PA), L. tulipifera (S. Schlarbaum, MWPCC 00-027, NA, Washington, DC), P. americana (S. Kim, 1125, FLAS, Gainesville, FL), Eschscholzia californica (S. Chiorean, PAC 95526–99531, University Park, PA), and A. americanus (J. Leebens-Mack, Cus.Crk.1-12, PAC 95538).
Library construction and DNA sequencing:
As part of our effort to identify floral genes from divergent angiosperm taxa and study their evolutionary relationships, the Floral Genome Project (FGP) includes the determination of several thousand EST sequences (Soltis et al. 2002). cDNA libraries were constructed from premeiotic floral buds from the aforementioned species, following the procedure described by Albert et al. (2005) and at the website http://www.floralgenome.org/fgp/. EST sequencing was performed as described (Albert et al. 2005) and sequences were stored in the FGP EST database (http://pgn.cornell.edu/). BLAST searches of the FGP EST database identified cDNA clones with significant matches to SEP1, -2, -3, or -4, with a cut-off E-value set at e−10. Full-length sequences of these cDNAs were then obtained by sequencing the clones from both the 5′- and 3′-ends using T3 and T7 primers. Internal primers were designed and used to complete sequencing, as needed (data not shown).
New genes were named with the first two letters of the genus name, in uppercase, and the first two letters of the specific epithet, in lowercase, followed by the name of the closest Arabidopsis homolog, in uppercase (Ma et al. 1991; Mandel and Yanofsky 1998). If more than one homolog of the same Arabidopsis gene was isolated, they were designated with a period followed by the number 1, 2, etc. For example, the AGL2 and AGL9-like genes from A. trichopoda were named AMtrAGL2 and AMtrAGL9, respectively, and the two AGL9-like genes from P. americana were named PEamAGL9.1 and PEamAGL9.2, respectively. The GenBank accession numbers for the new sequences are AMtrAGL9, AY850178; AMtrAGL2, AY850179; EScaAGL9, AY850180; EScaAGL2, AY850181; LItuAGL9, AY850182; NUadAGL2, AY850183; ACamAGL2, AY850184; PEamAGL9.1, AY850185; and PEamAGL9.2, AY850186.
Data retrieval:
In addition to the new genes described in this study, sequences of all other SEP homologs were retrieved from previously published studies (Becker and Theissen 2003; Litt and Irish 2003; Malcomber and Kellogg 2004) and from BLAST searches against publicly available databases (see supplementary material at http://www.genetics.org/supplemental/). BLAST searches of the NCBI and The Institute for Genomic Research protein databases were performed using each of the SEP homologs as queries. Sequences retrieved this way were then used as queries to search for their closest relative in the Arabidopsis genome. A sequence was not considered a SEP homolog unless its best hit in the Arabidopsis protein database was SEP1, -2, -3, or -4. Sequences from the same species were treated as duplicates if they were >95% identical at the DNA level, as suggested by Zhang et al. (2001). Duplicate sequences and sequences with poor quality (i.e., those with obvious sequencing errors) were excluded from further analyses. Twenty-six AGL6 subfamily members, 19 from angiosperms and 7 from gymnosperms, were also included, because several recent phylogenetic studies suggest that this subfamily is the closest relative of the SEP subfamily (Becker and Theissen 2003; Litt and Irish 2003; Nam et al. 2003). Five SQUA-like genes were used as outgroups to root the phylogenetic trees.
Sequence alignment:
Because phylogenetic topology can be alignment dependent, to maximize the reliability of our phylogenetic analyses, we generated three different alignments for our data set. For the first alignment (alignment I), full-length amino acid sequences were initially aligned using CLUSTALX version 1.81, with the “gap opening” penalty (GOP) set to 8.0 and the “gap extension” penalty (GEP) to 0.3. A preliminary phylogenetic tree was produced on the basis of the highly conserved M and K domains, and the order of the sequences was then rearranged according to their phylogenetic placements. When the closely related sequences were listed close to each other, it became easier to improve the alignment manually within less conserved regions. After manual adjustments, a second phylogenetic tree was generated using the most conserved M and K domains as well as the less conserved I domain and part of the C-terminal region. Again, the order of sequences in the matrix was changed according to their new placements in the phylogenetic tree, and further adjustments were made to align the remaining residues in the C-terminal region. By repeating these steps, we were able not only to align the least conserved C-terminal region confidently, but also to obtain a better understanding of the differences in amino acid substitution pattern between the conserved and the less conserved regions.
For the second alignment (alignment II), we applied different GOP and GEP values for different regions in the matrix. The range of GEP for the pairwise alignment was 5–70. Especially in the C-domain region, we applied a maximum GEP and then adjusted manually. Note that not as many manual adjustments were used in the second alignment as in the first. Because both the first and second alignments involve human judgment and thus could be arbitrary, we also generated a third alignment (alignment III), using only a computer program, to compare to the other two alignments. We used a recently developed alignment program, MUSCLE (Edgar 2004), because benchmark alignment tests indicate that this program performs consistently better than all other tested multiple sequence alignment programs, including CLUSTALX and T-Coffee (Edgar 2004). For each amino acid alignment, alignments of corresponding DNA sequences were also generated and analyzed (Nicholas et al. 1997).
Phylogeny reconstruction:
More than 20 different phylogenetic analyses were conducted to understand the evolution of the SEP subfamily. All three alignments were examined phylogenetically using both an amino acid matrix and a corresponding nucleotide matrix. Each matrix was analyzed both with and without the highly variable C-terminal region. An additional matrix including all amino acid residues with higher-than-12 column scores as indicated in CLUSTALX and its corresponding nucleotide matrix were also analyzed. We refer to this alignment as “alignment I with a partial C terminus.”
Phylogenetic analyses for each matrix were carried out using maximum-parsimony and maximum-likelihood (ML) methods in PAUP* version 4.10b (Swofford 1993) and PHYML version 2.4 (Guindon and Gascuel 2003), respectively. For parsimony analyses, heuristic searches were conducted with 1000 random addition replicates, with tree bisection-reconnection (TBR) branch swapping and saving all most parsimonious trees (MulTree on). Support for the placement of branches was assessed using bootstrap analyses with 250 bootstrap replicates (Felsenstein 1985), each with 100 random stepwise additions and TBR branch swapping, saving 10 trees per replicate. Likelihood analyses were performed in PHYML (Guindon and Gascuel 2003) using a general time reversibility (GTR) substitution model with invariant sites and additional among-site rate variation modeled as a discrete gamma distribution (Yang 1994). On the basis of MODELTEST v3.06 (Posada and Crandall 1998), we selected the model of molecular evolution using GTR + I + Γ, which assumes GTR, a certain proportion of invariable sites (I), and a gamma approximation of the rate variation among sites (Γ). Likelihood analyses were performed with PHYML (Guindon and Gascuel 2003). ML parameter values were then optimized, with a BIONJ tree as a starting point (Gascuel 1997) with the appropriate parameters. Support values for nodes on the ML tree were estimated with 250 bootstrap replicates (Felsenstein 1985). In addition to the maximum-parsimony and maximum-likelihood methods, Bayesian analyses were conducted for alignment I, alignment II, and alignment I with a partial C terminus using MrBayes version 3.0b4. For each Bayesian analysis, we ran four chains of the Markov chain Monte Carlo, sampling one tree every 100 generations for 1,000,000 (or more, if needed) generations, starting with a random tree. After excluding the trees generated during the “burn-in” period, all other trees were imported to PAUP to generate the consensus tree.
We examined the amino acid matrix of residues with quality scores of >12 in CLUSTALX from alignment I with partial C terminus using MacClade (Maddison and Maddison 1989) to determine which residues are more like AGL2/3/4 and which are more like AGL9. Additionally, we reconstructed the ancestral states of members of the AGL2/3/4 and AGL9 clades using the trace character function of MacClade showing all most parsimonious states at each node (Maddison and Maddison 1989).
Expression studies:
For RNA in situ hybridizations, poppy plants (E. californica cv. Aurantiaca Orange) were grown in the greenhouse from seed purchased from J. L. Hudson Seedsman. Fresh tissue was collected and fixed as described in the Meyerowitz protocol (http://www.its.caltech.edu/plantlab/protocols/insitu.html). One modification to this protocol was the omission of the denaturation and postfixation steps. Plasmid DNA was digested with restriction enzymes to remove the highly conserved MADS-box region and probes were synthesized using T7 RNA polymerase. A control sense probe was prepared and hybridized for each experiment from an EcGLO clone by digesting the 3′-end of the clone and synthesizing the probe using T3 RNA polymerase (data not shown). For RT-PCR experiments, total RNAs were extracted from Nuphar floral organs and young leaves using the RNeasy plant mini kit (QIAGEN, Stanford, CA). RT-PCR was performed as described previously (Kim et al. 2005).
RESULTS
Sequences of SEPALLATA homologs:
Nine new SEP homologs were identified in this study, i.e., AMtrAGL2 and AMtrAGL9 from A. trichopoda, NUadAGL2 from N. advena, PEamAGL9.1 and PEamAGL9.2 from P. americana, LItuAGL9 from L. tulipifera, ACamAGL2 from A. americanus, and EScaAGL2 and EScaAGL9 from E. californica. The fact that the basal-most angiosperm Amborella, the magnoliid Magnolia, and the basal eudicot Eschscholzia each contain two SEP homologs suggests that all angiosperms have more than one member of the SEP subfamily, as observed in the core eudicots and monocots.
Alignment of predicted amino acid sequences from these genes, along with previously published subfamily members, demonstrates that overall the MIK domains are highly conserved, with many positions nearly invariant throughout the angiosperms. Within the K domain we observed the previously recognized three putative α-helices. The expected a and d positions of the (abcdefg)n repeats identified by Yang et al. (2003) are all very highly conserved, although the first d and the third a sites in the K1, the second d site in the K2, and the second and third a sites in the K3 regions are occupied by hydrophilic (i.e., S, Q, S, E, and N, respectively) rather than hydrophobic amino acids (see supplemental materials at http://www.genetics.org/supplemental/).
Inspection of the SEP protein sequences revealed two short, relatively conserved motifs, referred to here as SEP I and SEP II motifs, near and at the very C termini of most proteins from the SEP subfamily (Figure 2; supplemental Figure 1 at http://www.genetics.org/supplemental/), even though the C-terminal region of SEP proteins appears to be less conserved than that of the AG and DEF/GLO MADS-box subfamilies (Kramer et al. 1998, 2004). Some SEP proteins from the grasses lack the SEP II motif in this region because they are the result of a recent gene duplication followed by a frameshift mutation in the LHS clade (Vandenbussche et al. 2003a). Structurally, the SEP I and SEP II motifs primarily contain hydrophobic and polar residues and do not resemble any motif with known function. Nevertheless, they may be functionally important because they are located in positions similar to the AG I and AG II motifs in AG proteins and the PI-derived and AP3 motifs in AP3 proteins (Kramer et al. 1998, 2004; Kim et al. 2004).
Figure 2.—
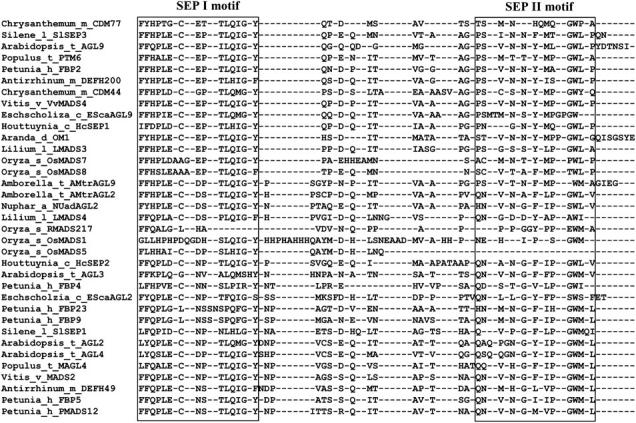
A manual alignment of the terminal region of the C terminus of representative members of the SEP subfamily. The SEP I and SEP II motifs are boxed. A larger number of representative samples can be observed in supplemental Figure 1 at http://www.genetics.org/supplemental/.
A preangiosperm gene duplication event within the SEPALLATA subfamily:
The major topological aspects of phylogenetic trees of the SEP subfamily are consistent regardless of alignment (alignment I and alignment II), data type (AA or DNA), portion of the sequence analyzed (with C, without C, or with partial C terminus), and methods of analysis (parsimony, likelihood, or Bayesian) used (Figure 3; supplemental Figures 2 and 3; see also supplemental materials at http://www.genetics.org/supplemental/). In general, the more characters included in our analyses, the greater the resolution. For example, analyses of the nucleotide matrices yielded trees with greater resolution and higher support values than the corresponding amino acid matrices, and matrices with the C-terminal region yielded better results than matrices without this region. Analyses of alignment I with a partial C terminus, from which those characters defined by CLUSTALX as highly variable, as defined by quality scores in CLUSTALX, resulted in more highly resolved trees, although the bootstrap support for some nodes was not as high as in other analyses (Figure 3; supplemental Figures 2 and 3 at http://www.genetics.org/supplemental/).
Figure 3.—
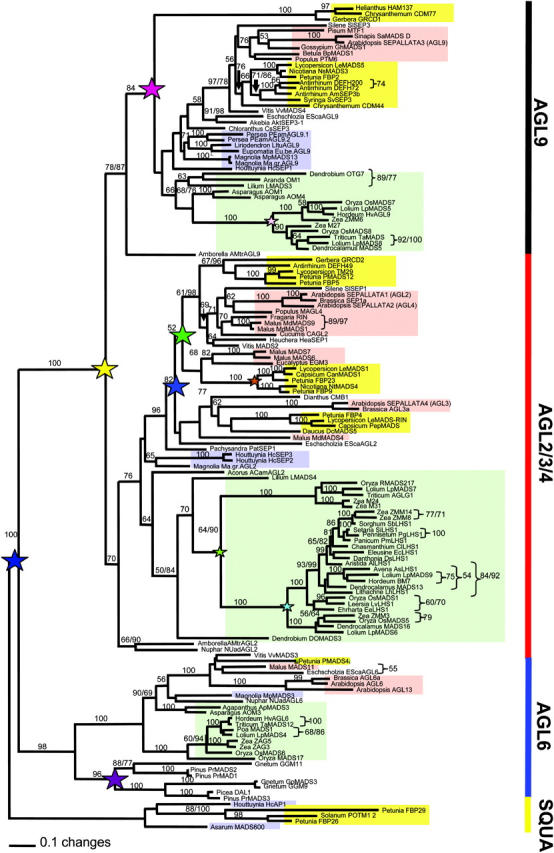
Maximum-likelihood tree of 113 representative SEP genes, 26 AGL6 genes, and 5 SQUA genes. Analyses and 250 bootstrap replicates were each performed with 100 random stepwise additions and TBR branch swapping, saving 10 trees per replicate of alignment I. Positions with two numbers indicate nodes where resulting maximum-parsimony bootstrap (250 bootstrap replicates, each with 100 random stepwise additions and TBR branch swapping, saving 10 trees per replicate) differed from the maximum-likelihood analyses by >5%. Stars indicate hypothesized gene duplication events; small stars represent duplication at a family level and large stars represent duplications at larger hierarchical levels. Note that the placement of the GRCD1 lineage near the base of the AGL9 clade appears to be spurious and that other analyses (supplemental Figures 2 and 3 at http://www.genetics.org/supplemental/) place this clade within the core eudicots. Background colors represent major angiosperm lineages: red, rosids; yellow, asterids; green, monocot; blue, magnoliid.
On the basis of our analyses the SEP subfamily is monophyletic. Within the SEP subfamily, there are two major clades, the AGL9 group and the AGL2/3/4 group (Figure 3; supplemental Figures 2 and 3 at http://www.genetics.org/supplemental/). Note that these names were adopted because these clades contain the Arabidopsis AGL9 (SEP3) gene and AGL2, -3, and -4 (SEP1, SEP4, and SEP2) genes, respectively. Each of these two clades contains sequences from the basal-most angiosperm Amborella, magnoliids, monocots, and eudicots, suggesting that the first gene duplication event within the SEP subfamily occurred before the origin of extant angiosperms.
Additional duplication events within the monocot and eudicot lineages:
Within the AGL2/3/4 and AGL9 clades, several additional gene duplication events were detected within the monocot and eudicot lineages (Figure 3; supplemental Figures 2 and 3 at http://www.genetics.org/supplemental/). In monocots, we detected at least five distinct grass clades, three in the AGL2/3/4 lineage and two in the AGL9 lineage, suggesting that the grasses may have experienced at least three gene duplication events. In the AGL2/3/4 clade, the evolutionary patterns of SEP homologs in eudicots are complicated, and the relative timing of some of the duplication events cannot be determined. At least two gene duplications occurred after the origin of the eudicots but before the diversification of the core eudicots. These duplication events resulted in three distinct clades, i.e., AGL2 (which contains both SEP1 and SEP2), AGL3 (which contains SEP4), and FBP9, each of which contains sequences from the rosid and asterid clades. The monophyly of the AGL2 and FBP9 groups is well supported in some analyses, with bootstrap values >60% (Figure 3) and >75% in additional analyses (supplemental Figures 2 and 3 at http://www.genetics.org/supplemental/). In most of our phylogenetic analyses, the FBP9 clade is sister to the AGL2 clade, although the position of the FBP9 clade is not strongly supported. These results are similar to what was observed in the SQUA/AP1 MADS-box subfamily, where multiple gene duplication events have occurred within the core eudicots (Litt and Irish 2003). The AGL3 and FBP9 clades share slightly more derived residues than do the AGL2 and FBP9 clades. Moreover, members of both the AGL3 and FBP9 clades are highly variable in the C-terminal region, making it difficult to determine the relationship between these clades. The absence of an Arabidopsis gene in the FBP9 clade suggests either that this clade is part of the AGL2 (or AGL3) clade or that the Arabidopsis lineage lost a FBP9 ortholog.
Within the AGL9 clade a single gene duplication seems to have occurred either before the origin of the sunflower family (Asteraceae) or during its diversification. However, the exact timing of this gene duplication is difficult to determine, because the placement of the clade formed by Helianthus HAM137, Chrysanthemum CDM77, and Gerbera GRCD1 genes is not well resolved. In this clade, 1 fixed amino acid change in the MADS-box domain and 22 fixed amino acid changes in the K-box domain were detected (Figure 4A). In some trees this clade is placed near the base of the AGL9 clade (Figure 3), while in other analyses its placement is within the core eudicots (supplemental Figures 2 and 3 at http://www.genetics.org/supplemental/). These findings suggest that this clade warrants further study. Because the protein sequences of these three asterid genes are so divergent from all other AGL9 clade members, it is possible that they may have acquired novel functions during evolution.
Figure 4.—

(A) A generalized phylogeny of the SEP subfamily with mapped unique amino acid state changes from the MADS and K domains. (B) A generalized AGL2/3/4 lineage from monocots illustrating the duplication events and amino acid state changes within the grasses. (C) A generalized AGL9 lineage from monocots illustrating the duplication events and amino acid state changes within the grasses.
Conserved sequence characteristics vary among clades:
The members of the AGL9 and AGL2/3/4 clades have several clade-specific changes within the K-box and C-terminal regions (supplemental Figure 1 at http://www.genetics.org/supplemental/; Figure 4; also see other supplemental materials at http://www.genetics.org/supplemental/). Figure 4 shows the clade-specific amino acid residues in the MADS-box and K-box in most major lineages in the SEP subfamily. The GRCD1 asterid clade in the AGL9 lineage and grass clades in both the AGL9 and AGL2/3/4 lineages have accumulated a number of clade-specific differences in the K domain. The K domain is highly conserved among MIKC-type (MADS, intervening region, K domain, C-terminal region) MADS-box proteins with a hypothesized coiled-coil domain proposed to be involved in protein-protein interactions (Davies and Schwarz-Sommer 1994; Riechmann and Meyerowitz 1997). The high number of clade-specific changes evidenced here suggests that the presence of duplicate copies may have allowed for sub- and neofunctionalization in these clades.
Lineage-specific changes were also noted for other clades. For example, within the K-box domain, there are changes supporting the monophyly of the eudicot AGL9, the monocot AGL9, the core eudicot AGL2, the core eudicot FBP9, and the monocot AGL 2/3/4 clades, respectively (Figure 4A). Although many monocot- and grass-specific changes were identified in the K-box region in the AGL2 clade, the number of residues that were conserved within the grasses, but differed from other lineages, remained high in the C-terminal region (see supplementary materials at http://www.genetics.org/supplemental/). Notably, the OsMADS5 clade contains genes that apparently have lost the final 12–15 amino acids relative to all other genes from the SEP subfamily. Additionally, a previously identified frameshift mutation occurred within the C-terminal end of genes from the LHS grass lineage (Vandenbussche et al. 2003a).
In general, the similarity between a member of the AGL9 clade and a member of the AGL2/3/4 clade in a basal angiosperm is greater than that between such paralogs in a eudicot or monocot. For example, the two Amborella genes, AMtrAGL2 and AmtrAGL9, are 67% identical at the amino acid sequence level and share features with members of both the AGL9 and AGL2/3/4 clades. The level of conservation observed between an AGL2/3/4-type gene and an AGL9-type gene decreased in Houttuynia and is even less in the monocot and eudicot lineages. Of the 44 amino acid residues in the C-terminal region that are variable among AGL2/3/4 and AGL9 clade members, 17 are identical between the two Amborella proteins. Of these 17 residues, 13 were more like AGL9-type proteins and 4 were more like AGL2/3/4-type proteins. Furthermore, 51 of 60 residues of the C terminus had the same ancestral state for both the AGL9 and AGL2/3/4 lineages (Figure 5 and see supplemental materials at http://www.genetics.org/supplemental/).
Figure 5.—

A color-coded map of the evolution of amino acid residues within the aligned C-terminal regions of representative basal taxa with a quality score >12, as calculated by CLUSTALX. The colors show clade-specific conservation as determined using the trace character function in MacClade (Maddison and Maddison 1989) and are not dependent on individual amino acid residues at the position shown. In this color-coded alignment, purple designates ancestral residues across the AGL2/3/4 clade, compared to the core eudicot AGL2/3/4 clade, and red designates ancestral residues across the AGL9 clade, compared to the core eudicot AGL9 clade, as determined using the trace changes option in MacClade4.03 (Maddison and Maddison 1989). In several instances, conserved AGL2/3/4-like residues are found in the AGL9 lineage in basal lineages and, vice versa, AGL9-like residues extend into the AGL2/3/4 clade. The ancestral traits for each clade, as calculated, are presented by the Amborella genes with a question mark (?) designating equivocal character states.
In addition to clade-specific changes,we observed several incidents of apparent convergence in amino acid residues (see supplemental materials at http://www.genetics.org/supplemental/). One example of convergence occurred in the K-domain where the AGL9 core eudicot clade evolved a tyrosine at site 126, compared to valine, while the AGL2 clade evolved a shift toward tyrosine, with some taxa with either a cytosine or phenylalanine at that position. Likewise, a residue in the C-terminal domain in the AGL2 clade is predominantly glycine, but is predominantly asparagine in the AGL9 clade with the sole exception of the grasses, which are also predominantly glycine. These shifts may represent codon biases inherent in the taxa but hypothetically they also may represent directional selection on protein function as partners of specific members of other MADS-box families. The evolution of these genes and the selective forces that may have caused this apparent convergence presents an interesting line of future inquiry.
Expression of SEP homologs in a basal eudicot and basal-most angiosperm:
Expression of the AGL9 homolog, EScaAGL9 from the basal eudicot, California poppy (E. californica), was analyzed by RNA in situ hybridization (Figure 6) and found to be similar to those of AGL9/SEP3. Expression was not observed in an early floral meristem (Figure 6A) prior to sepal primordia initiation, which corresponds to stage 3 in Arabidopsis (Smyth et al. 1990; Buzgo et al. 2004). After sepal primordia were initiated, EScaAGL9 signal was detected in the regions of the floral meristem that will become the petals, stamens, and carpels (Figure 6B) and was maintained throughout floral development in the inner three whorls. In addition, a low level of expression was detected in the upper portions of sepals at the stage after the ovule primordia began to form from the carpel (Figure 6, C and D). At this stage, EScaAGL9 expression appears to be higher in the developing petals and stamens than in the carpel and ovule primordia. Expression is high in the developing seeds (Figure 6E), but not in the walls of the developing capsule. In the water lily Nuphar, the expression of an AGL9 homolog was detected in all floral whorls but not in the leaves (Figure 7). These experiments suggest that SEP homologs in basal eudicots and basal angiosperms are expressed in the floral meristem and multiple floral organs, similar to their eudicot homologs.
Figure 6.—
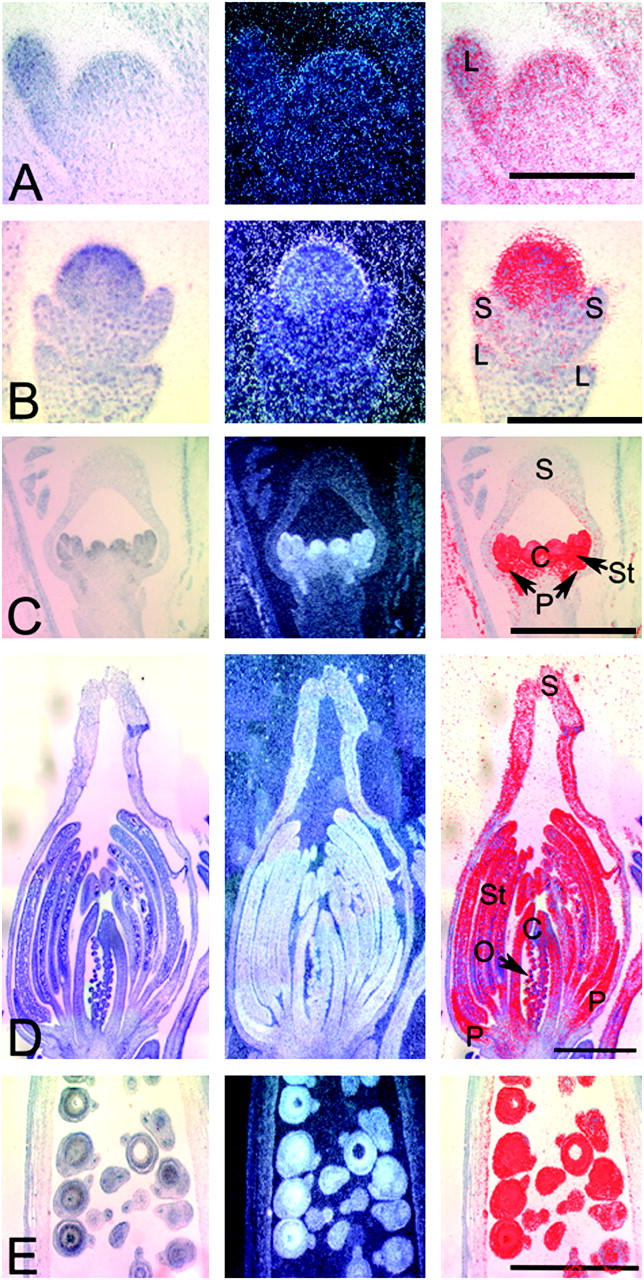
RNA in situ hybridization of EScaAGL9 in Eschscholzia. (A) A floral bud before sepal initiation. Bar, 0.2 mm. (B) A floral bud just after sepal initiation, with expression in the floral meristem just inside the sepals. Bar, 0.1 mm. (C) A floral bud at a later stage showing expression in the petal, stamen, and carpel primordia. Bar, 1 mm. (D) A floral bud at a stage when the ovule primordia have initiated, with strong expression in petals and stamens, moderate levels in the carpel and ovules, and possibly weak expression in the upper sepals. Bar, 1 mm. (E) A developing fruit showing strong expression in the developing seeds. Bar, 1 mm. L, leaf; S, sepal; P, petal; St, stamen; C, carpel; and O, ovule.
Figure 7.—

RT-Q-PCR of NUadAGL2 showing expression in all floral whorls. OTE, outer tepals; ITE, inner tepals; SD, staminodes; SN, stamens; CA, carpels; LE, leaves.
DISCUSSION
A possible origin of the SEP subfamily and its implications:
Previous studies have identified SEP homologs from eudicots and monocots, but the present study is the first report of SEP homologs from basal angiosperms. In addition, phylogenetic and molecular clock studies suggest that the SEP homologs belong to one of the most recently originated MADS-box subfamilies (Nam et al. 2003). Therefore, it was not clear whether the SEP subfamily originated before or after the emergence of angiosperms. SEP homologs occur in A. trichopoda and N. advena, representing the basal-most lineages of extant angiosperms, and our phylogenetic analyses indicate that SEP homologs are present in all major lineages of extant angiosperms. Furthermore, these results, coupled with the fact that SEP homologs have not been detected in gymnosperms, suggest that the SEP gene(s) originated in the ancestor of extant angiosperms.
The SEP subfamily has been hypothesized to be sister to the AGL6 clade, which has both gymnosperm and angiosperm members (Becker and Theissen 2003; De Bodt et al. 2003; Martinez-Castilla and Alvarez-Buylla 2003; Nam et al. 2003). AGL6 homologs from angiosperms and gymnosperms form strongly supported sister clades, which together are sister to the SEP subfamily, implying that an ancestor of the SEP genes may have existed in the common ancestor of angiosperms and gymnosperms and was lost in the ancestor of the gymnosperms (Becker and Theissen 2003), or at least in the lineages examined to date. Regardless of the precise timing of the origin of the SEP subfamily, it is clear that it originated prior to the origin of extant angiosperms, as evidenced by the phylogenetic placement of the two Amborella SEP homologs in this study. Furthermore, the presence of SEP homologs in all major lineages of angiosperms and their apparent absence in the gymnosperms strongly suggests that they may have played a critical role in the origin of the flower. Their role in floral development is directly demonstrated by genetic studies in the eudicots Arabidopsis and petunia and strongly supported by expression studies in a number of other species in both the eudicot and monocot clades. Among the floral MADS-box genes (Theissen et al. 2000), both the DEF/GLO and the AG subfamilies have homologs in gymnosperms that appear to play important roles in reproductive development. Therefore, these genes are likely examples of existing genes that contributed to the evolution of the flower and the angiosperms (Irish 2003; Kim et al. 2004).
The coincidence between the origin and diversification of the SEP subfamily and the emergence of angiosperms suggests that the origin of the SEP subfamily may be part of a hypothesized “molecular innovation” that made possible the morphological invention of the flower. Others have proposed that regulatory genes also may have played a key role in this regard (Theissen et al. 2000; Theissen 2001). This important role of the SEP subfamily is further supported by the observation that, in Arabidopsis, SEP genes provide the “floral state” upon which the ABC genes can act to promote the formation of all whorls of floral organs (Honma and Goto 2000; Pelaz et al. 2000, 2001a,b; Ditta et al. 2004), in comparison to the more restricted functions of members of the SQUA, DEF/GLO, and AG subfamilies (Ma 1994; Ma and Depamphilis 2000). Furthermore, SEP genes are apparently able to integrate the reproductive meristem determinacy with floral organ identity (Pelaz et al. 2000; Uimari et al. 2004) and may have played a role in the origin of the bisexual flower. The basal placement of SEP homologs from Amborella and Nuphar may represent the most ancestral extant forms of these potentially angiosperm-specific genes, and, as such, studies of their ability to interact with other ABC MADS-box genes may allow for further understanding of the evolution of developmental regulation in flowers. In addition, SEP homologs may be introduced into gymnosperms to test their function in promoting flower formation.
Within the SEP subfamily we see two potential evolutionary novelties: expression throughout the flower (reproductive shoot) and amino acid sequence motifs for transcriptional activation. In addition to expression in the cone, some members of the AGL6 subfamily, notably DAL1 and PrMADS3, are expressed in vegetative shoots (Tandre et al. 1995; Mouradov et al. 1998). The expression of SEP homologs in the floral meristems of angiosperms and that of AGL6 homologs in shoot meristems in gymnosperms suggest an ancestral meristematic function for the common ancestor of the SEP and AGL6 subfamilies. SEP proteins have putative transcriptional activation motifs and some can activate transcription in yeast (Ma et al. 1991; Huang et al. 1995; Immink et al. 2002; Ferrario et al. 2003; Shchennikova et al. 2004). On the basis of functional studies of SEP genes from core eudicots and monocots, it appears that the entire SEP subfamily has a potentially conserved meristematic identity or determinacy function (Bonhomme et al. 1997, 2000; Yu and Goh 2000; Theissen 2001; Lemmetyinen et al. 2004). Although AP1 also has meristem activities (Irish and Sussex 1990; Shannon and Meeks-Wagner 1993), it affects only the identities of the perianth.
An early duplication in the history of the SEP subfamily:
Previous studies have shown that multiple SEP homologs may be found in a single species (Johansen et al. 2002; Becker and Theissen 2003; Litt and Irish 2003; Malcomber and Kellogg 2004). Our results indicate that Amborella, Magnolia, Persea, and Eschscholzia also have at least two SEP homologs. The widespread presence of multiple SEP homologs suggests that these genes resulted from a duplication that occurred either prior to the origin of the angiosperms or very early in their initial diversification. However, it is also possible, although less likely, that these SEP homologs resulted from multiple independent duplication events. The extensive phylogenetic analyses here support an early duplication that occurred before the diversification of all present-day angiosperms. The two clades produced by this duplication event contain either the Arabidopsis AGL2/3/4 genes or the Arabidopsis AGL9 gene. Both the AGL2/3/4 and AGL9 clades have representatives from other eudicots, monocots, magnoliids, and the basal-most angiosperms Amborella and/or Nuphar. Our expanded sampling from phylogenetically critical positions on the angiosperm tree has greatly enhanced the power of phylogenetic analysis to resolve nodes that were previously difficult, supporting the need for extensive and deep sampling.
The occurrence of an early duplication in the SEP subfamily is very similar to preangiosperm duplication events in both the AG and the DEF/GLO subfamilies (Kramer et al. 1998, 2004; Kim et al. 2004) (Figure 8). These parallel duplication events of key regulators of floral form may have allowed functional variations that contributed to the great degree of morphological diversity among early angiosperms. Our analyses indicate that a duplication event occurred in the SEP subfamily sometime after it diverged from the AGL6 and SQUA subfamilies. Because both the SEP and the SQUA subfamilies are apparently absent from gymnosperms, the first duplication in the SEP subfamily likely occurred after the divergence of extant gymnosperms and angiosperms.
Figure 8.—

A hypothetical tree of MADS-box gene subfamilies showing the similarity of shape of the core eudicot clades. At this time there is little evidence to indicate that the AP1/FUL subfamily is more closely related to the AGL6 subfamily than to the SEP subfamily. Note that the arrangement of clades I and II is shown as a hypothetical order and is not meant to suggest that clades assigned to either clade have a similar evolutionary history outside of a duplication event at the base of the core eudicots. Stars represent hypothetical coincidental gene duplication events that may or may not be due to genome duplication events.
Functional implications of the persistence of multiple copies of SEP homologs:
The redundancy caused by gene duplication may have provided the SEP subfamily with a plethora of genes among which sub- or neofunctionalization can occur. In addition to the preangiosperm duplication event described above, our analyses revealed a number of additional gene duplication events in eudicots and monocots of both the AGL2/3/4 and the AGL9 lineages. Similar duplication events have been identified in each of the SQUA/AP1, DEF/GLO, and AG subfamilies (Kramer et al. 1998, 2004; Litt and Irish 2003; Kim et al. 2004; Stellari et al. 2004). More notably, there have been three distinct core eudicot duplications within the SQUA/AP1 lineage (Litt and Irish 2003), similar to that observed in the AGL2/3/4 clade. The duplicate gene pairs in the core eudicots may have resulted from a genome-wide duplication in the ancestor of the eudicots that was proposed on the basis of genomic sequence analysis (Ku et al. 2000; Bowers et al. 2003); alternatively, separate gene duplication events may have occurred in the common ancestor of the extant eudicots.
Our analysis strongly supports the hypothesis that the SEP subfamily has been present in the angiosperms since before the diversification of the extant basal-most angiosperms Amborella and the Nymphaeales. The long-term coexistence of functional duplicated copies suggests that subfunctionalization and/or neofunctionalization (Force et al. 1999; Lynch and Force 2000) may have occurred among paralogs in the SEP subfamily. For example, in Arabidopsis, the SEP1/2 (AGL2/4) genes are expressed in all four whorls, whereas SEP3 (AGL9) is expressed in the inner three whorls but not in the outermost whorl (sepals) and SEP4 is expressed throughout the early floral meristem (Flanagan and Ma 1994; Savidge et al. 1995; Mandel and Yanofsky 1998; Ditta et al. 2004). Nonetheless, AGL2 and AGL9 paralogs have considerable functional overlap and redundancy. The Arabidopsis SEP1, -2, -3, and -4 genes are largely functionally redundant in controlling the identity of floral organs and floral meristem determinacy (Pelaz et al. 2000; Ditta et al. 2004). Only the quadruple mutant lacking all four genes exhibits a complete loss of floral organ identity. Therefore, it is likely that the sequences of the encoded proteins do not differ sufficiently to cause changes in the biochemical functions, but the expression patterns and perhaps some interaction partners of these genes can change, thereby resulting in functional changes.
The expression of SEP homologs in core eudicots and monocots is generally similar to that of SEP1/2 and SEP3, although expression varies in early developing sepals. The expression results presented here support the idea that SEP homologs in a basal eudicot and a basal-most angiosperm also have similar expression patterns. On the basis of these studies, it appears that the entire SEP subfamily has a potentially conserved function in controlling the identity of all floral organs; this may have been lost in specific lineages, such as the carpel-specific DEF49 (Davies et al. 1996) in the AGL2 clade or the stamen-specific GRCD1 in the AGL9 clade (Kotilainen et al. 2000). Additionally, it seems that the SEP subfamily has a conserved expression in the floral meristem and ovules. It is possible that the meristematic function(s) are redundant with other closely related families such as AP1 (Irish and Sussex 1990; Shannon and Meeks-Wagner 1993).
Duplicate gene copies may have been retained within the genomes of the earliest angiosperms as a result of possibly flexible interactions among A-, B-, C-, and E-function MADS-box proteins that selected against their loss (Kim et al. 2004). Because these genes encode components of multimeric complexes, new gene copies could perhaps have been more easily assimilated than if they functioned singly. Studies of DEF/GLO homologs from monocots demonstrate greater flexibility in the number of protein-protein interactions than are possible in Arabidopsis (Hsu and Yang 2002; Winter et al. 2002; Kanno et al. 2003). Such flexibility has also been proposed for DEF/GLO homologs in Amborella and might be due to specific properties of the C-terminal regions (Kim et al. 2004), which have been shown or are thought to be involved in protein-protein interactions among MADS-box proteins (Egea-Cortines et al. 1999; Ferrario et al. 2003; Lamb and Irish 2003). The relative similarity of the C-terminal regions of the Amborella SEP homologs is similar to those observed in DEF/GLO homologs (Kim et al. 2004; Stellari et al. 2004). Our study demonstrates that the C-terminal regions of SEP homologs from the basal-most angiosperms are structurally very similar. This suggests that paralogs may have retained greater similarity in function than those from the monocots and eudicots and, therefore, they may be less specialized and, hypothetically, more flexible in their ability to interact with other MADS-box proteins. These duplicate gene copies may then have been under sufficiently relaxed selection to allow for the diversification of function through sub- and/or neofunctionalization due to changes in both the coding and the regulatory regions of these genes, similar to that observed in the AG subfamily (L. M. Zahn, J. H. Leebens-Mack, C. W. dePamphilis and H. Ma, unpublished data). This duplication and subsequent diversification of genes in these lineages may have provided the raw material that has allowed for the diversification of the floral forms observed today (Irish 2003; Kramer et al. 2003).
Implications of conservation in evolutionary paths of interacting MADS-box genes:
Multiple phylogenetic studies of MADS-box gene subfamilies have demonstrated a consistent pattern of gene duplication among the eudicots (Soltis et al. 2005). Studies in Arabidopsis and Lycopersicon (Solanum; Spooner et al. 1993) suggest that many core eudicots have retained at least three copies of each of the DEF/GLO, AG, and SEP lineages and that more basal taxa have maintained at least two copies. Although members of different clades may have distinct gene functions, genes within a single subfamily often retain similar functional capacity (Litt and Irish 2003; Kramer et al. 2004; L. M. Zahn, J. H. Leebens-Mack, C. W. dePamphilis and H. Ma, unpublished data). However, there also appears to be significant variation in expression and, most likely, function among some closely related homologs (Malcomber and Kellogg 2004). It is worth noting that the duplication in the DEF/GLO subfamily resulted in the DEF and GLO clades that represent genes that have nonequivalent functions. In the AG subfamily, while there is some functional redundancy among AG, SHATTERPROOF1 and -2, and SEEDSTICK (formerly AGL11) in Arabidopsis, there is also evidence for considerable functional divergence among orthologs and paralogs (Kramer et al. 2004; L. M. Zahn, J. H. Leebens-Mack, C. W. dePamphilis and H. Ma, unpublished data). It is possible that the function of the members of the SEP subfamily may have remained more conservative than that of either the DEF/GLO or the AG subfamilies. This greater conservation of function in SEP homologs may be due to a greater functional constraint, in part because these genes are often involved in the development of multiple floral organs, unlike the requirement of B- and C-function genes for two adjacent whorls.
Recent yeast two-hybrid experiments have demonstrated that SEP proteins have conserved interactions with proteins of the SQUA, DEF/GLO, and AG subfamilies (Davies et al. 1996; Fan et al. 1997; Honma and Goto 2001; Pelaz et al. 2001a; Favaro et al. 2003; Immink et al. 2003). Such interactions have contributed to the quartet model (Theissen 2001; Theissen and Saedler 2001), which suggests that these genes must evolve in concert to maintain their ancestral functions. Therefore, on the basis of their similar phylogenies (Figure 8), it is logical to hypothesize that the SQUA, DEF/GLO, AG, and SEP subfamilies have evolved in a concerted manner. However, studies involving genes from the SEP subfamily indicate that interactions of members of one subfamily are not specific, as both AGL9 and AGL2/3/4 proteins interact with proteins of the AG and AGL11 clades in the AG subfamily. In addition, SEP proteins have demonstrated interactions with other MADS-box proteins outside these subfamilies (i.e., with members of the AGL20 and AGL6 subfamilies) (Ferrario et al. 2003). Therefore, patterns of protein-protein interactions are not directly correlated with the phylogenetic topologies of these subfamilies. In other words, there is no evidence either for specific protein-protein interaction driving the divergence of genes or for specific patterns of gene trees providing the basis for specific protein interaction.
Acknowledgments
We thank Yi Hu for help with construction of the cDNA libraries, John Carlson, Bill Farmerie, Marlin Druckenmiller, and Jennifer Arrington for DNA sequencing, Yoshita Oza and Michael Kosco for help with RNA in situ hybridization experiments, and Anthony Omeis for plant care. We are grateful for helpful comments and discussion of the manuscript from Jill Ricker, Barbara Bliss, Liying Cui, John Carlson, and anonymous reviewers. This work was funded by a National Science Foundation Plant Genome Grant for the Floral Genome Project DBI-0115684.
References
- Albert, V. A., D. E. Soltis, J. E. Carlson, W. G. Farmerie, P. K. Wall et al., 2005. Floral gene resources from basal angiosperms for comparative genomics research. BMC Plant Biol. (in press). [DOI] [PMC free article] [PubMed]
- Aoki, S., K. Uehara, M. Imafuku, M. Hasebe and M. Ito, 2004. Phylogeny and divergence of basal angiosperms inferred from APETALA3- and PISTILLATA-like MADS-box genes. J. Plant Res. 117: 229–244. [DOI] [PubMed] [Google Scholar]
- Angiosperm Phylogeny Group, 2003. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG II. Bot. J. Linn. Soc. 141: 399–436. [Google Scholar]
- Barkman, T. J., G. Chenery, J. R. McNeal, J. Lyons-Weiler, W. J. Ellisens et al., 2000. Independent and combined analyses of sequences from all three genomic compartments converge on the root of flowering plant phylogeny. Proc. Natl. Acad. Sci. USA 97: 13166–13171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker, A., and G. Theissen, 2003. The major clades of MADS-box genes and their role in the development and evolution of flowering plants. Mol. Phylogenet. Evol. 29: 464–489. [DOI] [PubMed] [Google Scholar]
- Bonhomme, F., H. Sommer, G. Bernier and A. Jacqmard, 1997. Characterization of SaMADS D from Sinapis alba suggests a dual function of the gene: in inflorescence development and floral organogenesis. Plant Mol. Biol. 34: 573–582. [DOI] [PubMed] [Google Scholar]
- Bonhomme, F., B. Kurz, S. Melzer, G. Bernier and A. Jacqmard, 2000. Cytokinin and gibberellin activate SaMADS A, a gene apparently involved in regulation of the floral transition in Sinapis alba. Plant J. 24: 103–111. [DOI] [PubMed] [Google Scholar]
- Bowers, J. E., B. A. Chapman, J. Rong and A. H. Paterson, 2003. Unravelling angiosperm genome evolution by phylogenetic analysis of chromosomal duplication events. Nature 422: 433–438. [DOI] [PubMed] [Google Scholar]
- Buzgo, M., D. E. Soltis, P. S. Soltis and H. Ma, 2004. Towards a comprehensive integration or morphological and genetic studies of floral development. Trends Plant Sci. 9: 164–173. [DOI] [PubMed] [Google Scholar]
- Coen, E. S., and E. M. Meyerowitz, 1991. The war of the whorls: genetic interactions controlling flower development. Nature 353: 31–37. [DOI] [PubMed] [Google Scholar]
- Davies, B., and Z. Schwarz-Sommer, 1994. Control of floral organ identity by homeotic MADS-box transcription factors. Results Probl. Cell Differ. 20: 235–258. [DOI] [PubMed] [Google Scholar]
- Davies, B., M. Egea-Cortines, E. De Andrade Silva, H. Saedler and H. Sommer, 1996. Multiple interactions amongst floral homeotic MADS box proteins. EMBO J. 15: 4330–4343. [PMC free article] [PubMed] [Google Scholar]
- De Bodt, S., J. Raes, Y. Van De Peer and G. Theissen, 2003. And then there were many: MADS goes genomic. Trends Plant Sci. 8: 475–483. [DOI] [PubMed] [Google Scholar]
- Ditta, G., A. Pinyopich, P. Robles, S. Pelaz and M. F. Yanofsky, 2004. The SEP4 gene of Arabidopsis thaliana functions in floral organ and meristem identity. Curr. Biol. 14: 1935–1940. [DOI] [PubMed] [Google Scholar]
- Edgar, R. C., 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32: 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egea-Cortines, M., H. Saedler and H. Sommer, 1999. Ternary complex formation between the MADS-box proteins SQUAMOSA, DEFICIENS and GLOBOSA is involved in the control of floral architecture in Antirrhinum majus. EMBO J. 18: 5370–5379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ermolaeva, M. D., M. Wu, J. A. Eisen and S. L. Salzberg, 2003. The age of the Arabidopsis thaliana genome duplication. Plant Mol. Biol. 51: 859–866. [DOI] [PubMed] [Google Scholar]
- Fan, H. Y., Y. Hu, M. Tudor and H. Ma, 1997. Specific interactions between the K domains of AG and AGLs, members of the MADS domain family of DNA binding proteins. Plant J. 12: 999–1010. [DOI] [PubMed] [Google Scholar]
- Favaro, R., R. G. Immink, V. Ferioli, B. Bernasconi, M. Byzova et al., 2002. Ovule-specific MADS-box proteins have conserved protein-protein interactions in monocot and dicot plants. Mol. Genet. Genomics 268: 152–159. [DOI] [PubMed] [Google Scholar]
- Favaro, R., A. Pinyopich, R. Battaglia, M. Kooiker, L. Borghi et al., 2003. MADS-box protein complexes control carpel and ovule development in Arabidopsis. Plant Cell 15: 2603–2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein, J., 1985. Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39: 783–791. [DOI] [PubMed] [Google Scholar]
- Ferrario, S., R. G. Immink, A. Shchennikova, J. Busscher-Lange and G. C. Angenent, 2003. The MADS box gene FBP2 is required for SEPALLATA function in petunia. Plant Cell 15: 914–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanagan, C. A., and H. Ma, 1994. Spatially and temporally regulated expression of the MADS-box gene AGL2 in wild-type and mutant Arabidopsis flowers. Plant Mol. Biol. 26: 581–595. [DOI] [PubMed] [Google Scholar]
- Force, A., M. Lynch, F. B. Pickett, A. Amores, Y. L. Yan et al., 1999. Preservation of duplicate genes by complementary, degenerative mutations. Genetics 151: 1531–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gascuel, O., 1997. BIONJ: an improved version of the NJ algorithm based on a simple model of sequence data. Mol. Biol. Evol. 14: 685–695. [DOI] [PubMed] [Google Scholar]
- Graham, S. W., and R. G. Olmstead, 2000. Utility of 17 chloroplast genes for inferring the phylogeny of the basal angiosperms. Am. J. Bot. 87: 1712–1730. [PubMed] [Google Scholar]
- Guindon, S., and O. Gascuel, 2003. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52: 696–704. [DOI] [PubMed] [Google Scholar]
- Honma, T., and K. Goto, 2000. The Arabidopsis floral homeotic gene PISTILLATA is regulated by discrete cis-elements responsive to induction and maintenance signals. Development 127: 2021–2030. [DOI] [PubMed] [Google Scholar]
- Honma, T., and K. Goto, 2001. Complexes of MADS-box proteins are sufficient to convert leaves into floral organs. Nature 409: 525–529. [DOI] [PubMed] [Google Scholar]
- Hsu, H. F., and C. H. Yang, 2002. An orchid (Oncidium Gower Ramsey) AP3-like MADS gene regulates floral formation and initiation. Plant Cell Physiol. 43: 1198–1209. [DOI] [PubMed] [Google Scholar]
- Huang, H., M. Tudor, C. A. Weiss, Y. Hu and H. Ma, 1995. The Arabidopsis MADS-box gene AGL3 is widely expressed and encodes a sequence-specific DNA-binding protein. Plant Mol. Biol. 28: 549–567. [DOI] [PubMed] [Google Scholar]
- Immink, R. G., T. W. Gadella, Jr., S. Ferrario, M. Busscher and G. C. Angenent, 2002. Analysis of MADS box protein-protein interactions in living plant cells. Proc. Natl. Acad. Sci. USA 99: 2416–2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Immink, R. G., S. Ferrario, J. Busscher-Lange, M. Kooiker, M. Busscher et al., 2003. Analysis of the petunia MADS-box transcription factor family. Mol. Genet. Genomics 268: 598–606. [DOI] [PubMed] [Google Scholar]
- Irish, V. F., 2003. The evolution of floral homeotic gene function. BioEssays 25: 637–646. [DOI] [PubMed] [Google Scholar]
- Irish, V. F., and I. M. Sussex, 1990. Function of the apetala-1 gene during Arabidopsis floral development. Plant Cell 2: 741–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack, T., 2004. Molecular and genetic mechanisms of floral control. Plant Cell 16: S1–S17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen, B., L. B. Pedersen, M. Skipper and S. Frederiksen, 2002. MADS-box gene evolution-structure and transcription patterns. Mol. Phylogenet. Evol. 23: 458–480. [DOI] [PubMed] [Google Scholar]
- Kanno, A., H. Saeki, T. Kameya, H. Saedler and G. Theissen, 2003. Heterotopic expression of class B floral homeotic genes supports a modified ABC model for tulip (Tulipa gesneriana). Plant Mol. Biol. 52: 831–841. [DOI] [PubMed] [Google Scholar]
- Kim, S., M.-J. Yoo, V. A. Albert, J. S. Farris, P. S. Soltis et al., 2004. Phylogeny and diversification of B-function MADS-box genes in angiosperms: evolutionary and functional implications of a 260-million-year-old duplication. Am. J. Bot. 91: 2102–2118. [DOI] [PubMed] [Google Scholar]
- Kim, S., J. Koh, H. Ma, Y. Hu, P. K. Endress et al., 2005. Sequence and expression studies of A-, B-, and E-class MADS-box genes in Eupomatia (Eupomatiaceae): support for the bracteate origin of the calyptra. Int. J. Plant Sci. 166: 185–198. [Google Scholar]
- Kotilainen, M., P. Elomaa, A. Uimari, V. A. Albert, D. Yu et al., 2000. GRCD1, an AGL2-like MADS box gene, participates in the C function during stamen development in Gerbera hybrida. Plant Cell 12: 1893–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer, E. M., R. L. Dorit and V. F. Irish, 1998. Molecular evolution of genes controlling petal and stamen development: duplication and divergence within the APETALA3 and PISTILLATA MADS-box gene lineages. Genetics 149: 765–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer, E. M., V. S. Di Stilio and P. M. Schluter, 2003. Complex patterns of gene duplication in the APETALA3 and PISTILLATA lineages of the Ranunculaceae. Int. J. Plant Sci. 164: 1–11. [Google Scholar]
- Kramer, E. M., M. A. Jaramillo and V. S. Di Stilio, 2004. Patterns of gene duplication and functional evolution during the diversification of the AGAMOUS subfamily of MADS box genes in angiosperms. Genetics 166: 1011–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku, H. M., T. Vision, J. Liu and S. D. Tanksley, 2000. Comparing sequenced segments of the tomato and Arabidopsis genomes: large-scale duplication followed by selective gene loss creates a network of synteny. Proc. Natl. Acad. Sci. USA 97: 9121–9126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb, R. S., and V. F. Irish, 2003. Functional divergence within the APETALA3/PISTILLATA floral homeotic gene lineages. Proc. Natl. Acad. Sci. USA 100: 6558–6563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawton-Rauh, A. L., E. R. Alvarez-Buylla and M. D. Purugganan, 2000. Molecular evolution of flower development. Trends Ecol. Evol. 15: 144–149. [DOI] [PubMed] [Google Scholar]
- Lemmetyinen, J., M. Hassinen, A. Elo, I. Porali, K. Keinonen et al., 2004. Functional characterization of SEPALLATA3 and AGAMOUS orthologues in silver birch. Physiol. Plant 121: 149–162. [DOI] [PubMed] [Google Scholar]
- Litt, A., and V. F. Irish, 2003. Duplication and diversification in the APETALA1/FRUITFULL floral homeotic gene lineage: implications for the evolution of floral development. Genetics 165: 821–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch, M., and A. Force, 2000. The probability of duplicate gene preservation by subfunctionalization. Genetics 154: 459–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, H., 1994. The unfolding drama of flower development: recent results from genetic and molecular analyses. Genes Dev. 8: 745–756. [DOI] [PubMed] [Google Scholar]
- Ma, H., and C. Depamphilis, 2000. The ABCs of floral evolution. Cell 101: 5–8. [DOI] [PubMed] [Google Scholar]
- Ma, H., M. F. Yanofsky and E. M. Meyerowitz, 1991. AGL1–AGL6, an Arabidopsis gene family with similarity to floral homeotic and transcription factor genes. Genes Dev. 5: 484–495. [DOI] [PubMed] [Google Scholar]
- Maddison, W. P., and D. R. Maddison, 1989. Interactive analysis of phylogeny and character evolution using the computer program MacClade. Folia Primatol. 53: 190–202. [DOI] [PubMed] [Google Scholar]
- Malcomber, S. T., and E. A. Kellogg, 2004. Heterogeneous expression patterns and separate roles of the SEPALLATA gene LEAFY HULL STERILE1 in grasses. Plant Cell 16: 1692–1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandel, M. A., and M. F. Yanofsky, 1998. The Arabidopsis AGL9 MADS box gene is expressed in young flower primordia. Sex. Plant Reprod. 11: 22–28. [Google Scholar]
- Martinez-Castilla, L. P., and E. R. Alvarez-Buylla, 2003. Adaptive evolution in the Arabidopsis MADS-box gene family inferred from its complete resolved phylogeny. Proc. Natl. Acad. Sci. USA 100: 13407–13412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews, S., and M. J. Donoghue, 1999. The root of angiosperm phylogeny inferred from duplicate phytochrome genes. Science 286: 947–950. [DOI] [PubMed] [Google Scholar]
- Mouradov, A., T. V. Glassick, B. A. Hamdorf, L. C. Murphy, S. S. Marla et al., 1998. Family of MADS-Box genes expressed early in male and female reproductive structures of monterey pine. Plant Physiol. 117: 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam, J., C. W. Depamphilis, H. Ma and M. Nei, 2003. Antiquity and evolution of the MADS-box gene family controlling flower development in plants. Mol. Biol. Evol. 20: 1435–1447. [DOI] [PubMed] [Google Scholar]
- Nam, J., J. Kim, S. Lee, G. An, H. Ma et al., 2004. Type I MADS-box genes have experienced faster birth-and-death evolution than type II MADS-box genes in angiosperms. Proc. Natl. Acad. Sci. USA 101: 1910–1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholas, K. B., H. B. Nicholas, Jr, and D. W. Deerfield, II, 1997. GeneDoc: analysis and visualization of genetic variation. EMBNEW.NEWS 4: 14. [Google Scholar]
- Parenicova, L., S. De Folter, M. Kieffer, D. S. Horner, C. Favalli et al., 2003. Molecular and phylogenetic analyses of the complete MADS-box transcription factor family in Arabidopsis: new openings to the MADS world. Plant Cell 15: 1538–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelaz, S., G. S. Ditta, E. Baumann, E. Wisman and M. F. Yanofsky, 2000. B and C floral organ identity functions require SEPALLATA MADS-box genes. Nature 405: 200–203. [DOI] [PubMed] [Google Scholar]
- Pelaz, S., C. Gustafson-Brown, S. E. Kohalmi, W. L. Crosby and M. F. Yanofsky, 2001. a APETALA1 and SEPALLATA3 interact to promote flower development. Plant J. 26: 385–394. [DOI] [PubMed] [Google Scholar]
- Pelaz, S., R. Tapia-Lopez, E. R. Alvarez-Buylla and M. F. Yanofsky, 2001. b Conversion of leaves into petals in Arabidopsis. Curr. Biol. 11: 182–184. [DOI] [PubMed] [Google Scholar]
- Posada, D., and K. A. Crandall, 1998. MODELTEST: testing the model of DNA substitution. Bioinformatics 14: 817–818. [DOI] [PubMed] [Google Scholar]
- Purugganan, M. D., 1998. The molecular evolution of development. BioEssays 20: 700–711. [DOI] [PubMed] [Google Scholar]
- Qiu, Y. L., J. Lee, F. Bernasconi-Quadroni, D. E. Soltis, P. S. Soltis et al., 1999. The earliest angiosperms: evidence from mitochondrial, plastid and nuclear genomes. Nature 402: 404–407. [DOI] [PubMed] [Google Scholar]
- Riechmann, J. L., and E. M. Meyerowitz, 1997. Determination of floral organ identity by Arabidopsis MADS domain homeotic proteins AP1, AP3, PI, and AG is independent of their DNA-binding specificity. Mol. Biol. Cell 8: 1243–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savidge, B., S. D. Rounsley and M. F. Yanofsky, 1995. Temporal relationship between the transcription of two Arabidopsis MADS box genes and the floral organ identity genes. Plant Cell 7: 721–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon, S., and D. R. Meeks-Wagner, 1993. Genetic interactions that regulate inflorescence development in Arabidopsis. Plant Cell 5: 639–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shchennikova, A. V., O. A. Shulga, R. Immink, K. G. Skryabin and G. C. Angenent, 2004. Identification and characterization of four chrysanthemum MADS-box genes, belonging to the APETALA1/FRUITFULL and SEPALLATA3 subfamilies. Plant Physiol. 134: 1632–1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth, D. R., J. L. Bowman and E. M. Meyerowitz, 1990. Early flower development in Arabidopsis. Plant Cell 2: 755–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soltis, P. S., D. E. Soltis and M. W. Chase, 1999. Angiosperm phylogeny inferred from multiple genes as a tool for comparative biology. Nature 402: 402–404. [DOI] [PubMed] [Google Scholar]
- Soltis, D. E., P. S. Soltis, M. W. Chase, M. E. Mort, D. C. Albach et al., 2000. Angiosperm phylogeny inferred from 18S rDNA, rbcL, and atpB sequences. Bot. J. Linn. Soc. 133: 381–461. [Google Scholar]
- Soltis, D. E., P. S. Soltis, V. A. Albert, D. G. Oppenheimer, C. W. Depamphilis et al., 2002. Missing links: the genetic architecture of flowers and floral diversification. Trends Plant Sci. 7: 22–31. [DOI] [PubMed] [Google Scholar]
- Soltis, D. E., V. A. Albert, S. Kim, M.-J. Yoo, P. S. Soltis et al., 2005 Evolution of the flower, pp. 165–200 in Diversity and Evolution of Plants, edited by R. Henry. CABI Publishers, Wallingford, UK.
- Spooner, D. M., G. J. Anderson and R. K. Jansen, 1993. Chloroplast DNA evidence for the interrelationships of tomatoes, potatoes, and pepinos (Solanaceae). Am. J. Bot. 80: 676–688. [Google Scholar]
- Stellari, G. M., M. A. Jaramillo and E. M. Kramer, 2004. Evolution of the APETALA3 and PISTILLATA lineages of MADS-box-containing genes in the basal angiosperms. Mol. Biol. Evol. 21: 506–519. [DOI] [PubMed] [Google Scholar]
- Sung, S. K., G. H. Yu, J. Nam, D. H. Jeong and G. An, 2000. Developmentally regulated expression of two MADS-box genes, MdMADS3 and MdMADS4, in the morphogenesis of flower buds and fruits in apple. Planta 210: 519–528. [DOI] [PubMed] [Google Scholar]
- Swofford, D. L., 1993. PAUP: a computer program for phylogenetic inference using maximum parsimony. J. Gen. Physiol. 102: A9. [Google Scholar]
- Tandre, K., V. A. Albert, A. Sundas and P. Engstrom, 1995. Conifer homologues to genes that control floral development in angiosperms. Plant Mol. Biol. 27: 69–78. [DOI] [PubMed] [Google Scholar]
- Theissen, G., 2001. Development of floral organ identity: stories from the MADS house. Curr. Opin. Plant Biol. 4: 75–85. [DOI] [PubMed] [Google Scholar]
- Theissen, G., and H. Saedler, 2001. Plant biology. Floral quartets. Nature 409: 469–471. [DOI] [PubMed] [Google Scholar]
- Theissen, G., J. T. Kim and H. Saedler, 1996. Classification and phylogeny of the MADS-box multigene family suggest defined roles of MADS-box gene subfamilies in the morphological evolution of eukaryotes. J. Mol. Evol. 43: 484–516. [DOI] [PubMed] [Google Scholar]
- Theissen, G., A. Becker, A. Di Rosa, A. Kanno, J. T. Kim et al., 2000. A short history of MADS-box genes in plants. Plant Mol. Biol. 42: 115–149. [PubMed] [Google Scholar]
- Uimari, A., M. Kotilainen, P. Elomaa, D. Yu, V. A. Albert et al., 2004. Integration of reproductive meristem fates by a SEPALLATA-like MADS-box gene. Proc. Natl. Acad. Sci. USA 101: 15817–15822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenbussche, M., G. Theissen, Y. Van De Peer and T. Gerats, 2003. a Structural diversification and neo-functionalization during floral MADS-box gene evolution by C-terminal frameshift mutations. Nucleic Acids Res. 31: 4401–4409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenbussche, M., J. Zethof, E. Souer, R. Koes, G. B. Tornielli et al., 2003. b Toward the analysis of the petunia MADS box gene family by reverse and forward transposon insertion mutagenesis approaches: B, C, and D floral organ identity functions require SEPALLATA-like MADS box genes in petunia. Plant Cell 15: 2680–2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigel, D., and E. M. Meyerowitz, 1994. The ABCs of floral homeotic genes. Cell 78: 203–209. [DOI] [PubMed] [Google Scholar]
- Winter, K. U., C. Weiser, K. Kaufmann, A. Bohne, C. Kirchner et al., 2002. Evolution of class B floral homeotic proteins: obligate heterodimerization originated from homodimerization. Mol. Biol. Evol. 19: 587–596. [DOI] [PubMed] [Google Scholar]
- Yang, Y., L. Fanning and T. Jack, 2003. The K domain mediates heterodimerization of the Arabidopsis floral organ identity proteins, APETALA3 and PISTILLATA. Plant J. 33: 47–59. [DOI] [PubMed] [Google Scholar]
- Yang, Z., 1994. Maximum likelihood phylogenetic estimation from DNA sequences with variable rates over sites: approximate methods. J. Mol. Evol. 39: 306–314. [DOI] [PubMed] [Google Scholar]
- Yu, H., and C. J. Goh, 2000. Identification and characterization of three orchid MADS-box genes of the AP1/AGL9 subfamily during floral transition. Plant Physiol. 123: 1325–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanis, M. J., D. E. Soltis, P. S. Soltis, S. Mathews and M. J. Donoghue, 2002. The root of the angiosperms revisited. Proc. Natl. Acad. Sci. USA 99: 6848–6853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, L. Q., S. K. Pond and B. S. Gaut, 2001. A survey of the molecular evolutionary dynamics of twenty-five multigene families from four grass taxa. J. Mol. Evol. 52: 144–156. [DOI] [PubMed] [Google Scholar]
- Zhao, D., Q. Yu, C. Chen and H. Ma, 2001 Genetic control of reproductive meristems, pp. 89–142 in Meristematic Tissues in Plant Growth and Development, edited by M. T. McManus and B. Veit. Sheffield Academic Press, Sheffield, UK.