

Abstract
Variation in vein position and wing shape of Drosophila melanogaster depends on many genes. In the following, we report the results of a QTL analysis of wing shape in D. melanogaster. We identified QTL responsible for natural variation for wing shape and analyzed their interactions with developmental genetic signaling pathways important for vein positioning. The QTL analysis indicated that the total number of QTL segregating in this population is likely to be very large. The locations of putative QTL identified in this study were compared to those identified in previous studies and, while there is more correspondence across studies than expected by chance on the third chromosome, the studies appear to have identified different QTL. Using a complementation design, we tested for interactions among these QTL with the Hedgehog and Decapentaplegic signaling pathways, which are important for the development and position of vein pairs L3-L4 and L2-L5. Three QTL showed strong interactions with these two pathways, supporting the hypothesis that these QTL are involved in these pathways. Naturally segregating variation can therefore act through known signaling pathways to produce variation in vein position.
THE relative positions of wing veins of Drosophila melanogaster reflect an interface between genetic and developmental constraints on one hand (Sturtevant and Bier 1995; Bier 2000; de Celis 2003) and the functional constraints of flight performance on the other (Ennos 1988; Daniel and Combes 2002; Combes and Daniel 2003; Wootton et al. 2003). The developmental positioning of veins is controlled by a host of genes, where mutations can produce a dizzying array of variation (Diaz-Benjumea and Garcia-Bellido 1990; Garcia-Bellido and de Celis 1992). However, in natural populations, mutations that cause major rearrangements of wing veins are generally not observed. Presumably, the relative locations of veins and the shape of the wing are important for flight performance or some other functional aspects of the wing and are under stabilizing selection (Brodsky 1994). Mutations that cause vein rearrangements or have large effects on wing shape are therefore likely to have negative effects on fitness and are removed from the population by selection. However, even if such selection is operating, there is considerable genetic variation for subtle changes in the locations of wing veins and wing shape (Cavicchi et al. 1991; Imasheva et al. 1995; Bublii et al. 1996; Whitlock and Fowler 1999; Birdsall et al. 2000; Gilchrist and Partridge 2001; Phillips et al. 2001; Fowler and Whitlock 2002; Whitlock et al. 2002; Mezey and Houle 2005). Such variation is presumably responsible for the responses to artificial selection on wing shape (e.g., Weber 1990) and for the variation among species (e.g., Houle et al. 2003).
Most loci important for segregating variation in vein position and wing shape are unknown, despite the extensive knowledge of the loci involved in wing development. Studies have indicated that the number may be large. For example, Weber (1992) selected two small, adjacent features of the wing in antagonistic directions and found a localized response in the selected region, which was accompanied by small percentage changes in other traits of the wing. Assuming the potential for such fine-scale responses is a property of all aspects of the wing, such genetic control requires a large number of genes. In another study, Weber (1990) antagonistically selected each of several pairs of distances between vein intersections and estimated the effective number of genes responsible for the resulting response as >100. Mezey and Houle (2005) estimated the number of wing shape dimensions in which there was additive genetic variation and found the minimum number was 20 and may well be higher. Since high genetic dimensionality necessarily requires a number of additive gene effects equal to or greater than the number of dimensions, the lower bound is 20 additive genes. These studies point to the same qualitative picture of a large number of genes contributing to genetic variation in wing shape.
One starting point for characterizing the specific loci that are responsible for quantitative variation in wing features is a genomic scan for quantitative trait loci (QTL). The primary goal of such genomic scans is to narrow the field of candidate genes by identifying promising regions where contributing loci may reside (Mackay 2001). For example, scans for QTL affecting Drosophila bristle number have contributed to the identification of several loci that contribute to naturally occurring variation in bristle number (Long et al. 1998, 2000; Lyman et al. 1998; Robin et al. 2002). Thus far, three scans for QTL with effects on wing shape have been performed (Weber et al. 1999, 2001; Zimmerman et al. 2000). These studies have identified a large number of regions where potential QTL may reside and the analysis of Zimmerman et al. (2000) has been followed by an association study of the candidate Egfr (Palsson and Gibson 2004). Zimmerman et al. (2000) analyzed a cross between two lab populations and Weber et al. (1999)(2001) used lines constructed from strains selected for high and low values of a wing shape index (see below).
The goal of this study was to perform a genomic scan for QTL that contribute to variation in wing vein position in a natural population. The scan was performed on recombinant inbred lines (RILs) produced by brother-sister matings from an F2 of two individuals taken from nature (Winters, CA), such that each RIL is fixed for variation that is segregating in the population. In addition to performing a genomic scan for QTL, we also compared the identified genomic locations to those of previous studies (Weber et al. 1999, 2001; Zimmerman et al. 2000) to assess whether common loci are contributing to the variation analyzed in each study. We did this in two ways: (1) by comparing results on a region-by-region basis and (2) by asking whether overall, the regions identified by independent studies correspond to a greater degree than expected at random (Paterson 2002). Given the large number of loci that could contribute to naturally occurring variation and that each study uses different characterizations of wing shape and distinct lines, the a priori expectation for these comparisons is a lack of similarity across studies. However, if the expectation does not hold for a given region, this suggests that some of the same QTL are being identified.
We were also interested in how the identified QTL correspond or interact with pathways known to be important determinants of vein position during wing development. To assess these relationships, we used quantitative complementation testing (Mackay and Fry 1996). In a quantitative complementation test, genotypes at the QTL locus are each crossed to both a mutant and a wild-type genotype at the candidate locus. If there is a significant interaction between genotypes noncomplementation is inferred. This can indicate either that the QTL and candidate are the same locus or that the QTL is a dominant modifier of the candidate locus (Long et al. 1996).
Previous studies have identified interactions between QTL and candidate genes that affect several morphological and behavioral traits (Long et al. 1996; Mackay and Fry 1996; Palsson and Gibson 2000; Fanara et al. 2002; Kopp et al. 2003; Moehring and Mackay 2004). In this study, we extended this framework by testing multiple candidate loci that are in the same wing vein signaling pathways (Held 2002). We not only tested candidates in the region of individual QTL but also tested for interactions of each QTL with all of the candidates in each pathway. If a QTL is able to produce variation by acting through a specific pathway, then the QTL should interact with many of the candidates in the pathway. This strategy can therefore establish whether a QTL can produce variation in vein position by acting through a specific signaling pathway.
The morphogen Hedgehog (Hh), expressed in the posterior portion of the wing disc, is important for the positioning of veins L3-L4; Decapentaplegic (Dpp), which is activated by Hh, is important for positioning veins L2-L5 (de Celis 2003). We performed complementation tests for eight candidates that are members of the Hh or Dpp pathways (Sturtevant et al. 1997; Haerry et al. 1998; Bier 2000; Torres-Vazquez et al. 2000; Held 2002; Fujise et al. 2003; Lunde et al. 2003; Cook et al. 2004): (1) engrailed (en), activates expression of Hedgehog; (2) tout velu (ttv), involved in the movement of Hh; (3) patched (ptc), activated by Hh; L3-L4 placement; (4) decapentaplegic (dpp), production of Dpp; (5) schnurri (shn), activation and repression of Dpp target genes; (6) saxophone (sax), type I receptor, mediates downstream response to Dpp; (7) punt (put), type II receptor, mediates downstream response to Dpp; and (8) spalt-major (salm), activated by Dpp, L2 forms anterior to the domain of salm expression. We also tested two additional candidate loci: (9) rhomboid (rho), promotes production of vein material; and (10) crossveinless c (cv-c), crossvein development, interacts with rho.
MATERIALS AND METHODS
RILs and molecular markers:
The RILs and associated marker data have been described previously (Kopp et al. 2003) so only a brief summary is provided here. The RILs were derived from two flies collected from a natural population (Winters, CA). The F2 genomes derived from the parental cross were isogenized by 25 generations of full-sib inbreeding. Since the original flies used to produce the RILs were not inbred, there are four original parental haplotypes. This means that each locus of the original parents may have been heterozygous and there are therefore up to four different alleles at each locus in the RILs. Transposable elements of the roo family were used as markers for these lines (see Charlesworth and Langley 1989 for review). Five individuals of each line were genotyped for the presence of elements by in situ hybridization to polytene salivary gland chromosomes. Markers were designated to be present if detected in all larvae, absent if detected in no larvae, and segregating otherwise. Segregating markers were ignored in the QTL analysis. Parental chromosomes were homosequential with the exception of an inversion on the right arm of CHR3 from approximately 89EF to 96A.
Linkage groups and recombination rates:
The inbreeding design used to produce the RILs differs from the designs typically used because of the presence of four original parental haplotypes for each chromosome (typically there are two). Software for analyzing this specific design is not currently available. However, the two-allele RIL design analyzed in QTLCartographer (Wang et al. 2003) applies when analyzing markers present in a single parental haplotype vs. the other haplotypes. For example, for a given parental linkage group (LG; i.e., markers linked in an original parental haplotype), the markers are coded as one type if they were linked in a parental haplotype and all markers not in this parental haplotype are coded as the second type. This avoids the problem of uncertainty as to which chromosomes are involved in any given recombination event. By analyzing each LG vs. the others, a recombination event is counted when there is recombination between the focal LG and any of the other three LGs. When analyzing a given focal LG, the hypothesis being assessed is whether there is an allele at a location along the LG with an effect that differs from the effect of a weighted sum of (up to three) alleles on the other LGs.
The original parental haplotypes were estimated using correlations among markers in the RIL as described in Kopp et al. (2003). Cases where a marker was present on more than one parental haplotype make estimating recombination rates difficult because it is uncertain as to whether markers out of phase reflect a recombination event or the original parental configuration. These markers were therefore dropped from the analysis. The LGs and associated markers used in this study are presented in Figure 1. Note that the fourth chromosome is not considered, and there are only two linkage groups for the X chromosome (XCHR) and three for the third chromosome (CHR3). Only a few markers were present on the fourth chromosome and third XCHR LG so these were excluded from the analysis. LGs 2 and 3 of CHR3 had most markers in common so these were considered as a single LG 2–3 (dropping all markers not shared in common). In total, the analysis used 117 markers. For the LGs of the XCHR, recombination rates were estimated using r = 3/(8/R − 12), using r = 1/(4/R − 6) for LG 2–3 of CHR3 and using r = 1/(3/R − 6) for the rest, where R is the proportion of RILs for which recombination occurred between adjacent markers in the LG and any of the other LGs (Haldane and Waddington 1931; see appendix).
Figure 1.—

Linkage groups (LGs) reflecting the marker associations in parental haplotypes for the (a) XCHR, (b) CHR2, and (c) CHR3. Distances between markers reflect estimated recombination distances. Note that there are two XCHR LGs because the third had only three markers and there are only three CHR3 groups because two (LG2–3) had most markers in common.
Wing measurement:
The left wings of males from a total of 131 RILs were measured. For each RIL, five females (paired with four to five males) were allowed to lay eggs in two replicate vials on a cornmeal, sucrose, brewer's yeast medium at 25°. Only wings of males were measured. The final analysis included a total of 3204 measured wings with an average of 22.2 individuals per line (minimum, 5; maximum, 42). Variation in number measured per line was due to deaths or damage to wings before vials were chosen for measurement and extra measurements for some lines. All measurements were carried out using WingMachine, an automated image analysis system, the details of which are described elsewhere (Houle et al. 2003).
Morphometrics:
The 12 landmarks of wings (Figure 2) were subjected to a morphometric alignment using a Procrustes generalized least-squares superimposition (Rohlf and Slice 1990) and scaled by centroid size using the tpsRegr program (Rohlf 1998). For each wing, the x- and y-coordinates of the displacement of each landmark from the centroid were calculated. This resulted in 24 coordinate traits summarizing wing shape, although there are only 20 d.f. because the superimposition and scaling resulted in a loss of 4 d.f. (Rohlf and Slice 1990; Mezey and Houle 2004). These data capture all variation for any function of the coordinate traits. In the case of wings where it is not explicitly clear how vein placement relates to flight and other functional aspects, this quantification is particularly useful, since any functional aspect of the wing that depends on relative location of these landmarks can be modeled. Since functional wing traits have not yet been clearly defined, we analyze the first seven principal components [PC1–PC7] of the variance-covariance matrix calculated using RIL means. These PCs in this case are the same as relative warps (Rohlf 1999). We use these first seven PCs because they account for the bulk (93%) of the total variation among the lines (Table 1). For each PC, an ANOVA was performed and there was significant among-RIL variation for each (P < 0.01). No significant effect of the inversion was found for any of the PCs (ANOVA).
Figure 2.—

Wing landmarks used in the analysis.
TABLE 1.
Principal component loadings
| Proportion of variation: 0.347: | 0.246: | 0.103: | 0.101: | 0.062: | 0.047: | 0.026: | ||
|---|---|---|---|---|---|---|---|---|
| Loadings: PC1 | PC2 | PC3 | PC4 | PC5 | PC6 | PC7 | ||
| x.1 | 0.58 | 0.48 | −0.17 | −0.27 | ||||
| y.1 | 0.48 | 0.22 | 0.18 | |||||
| x.2 | −0.39 | 0.38 | ||||||
| y.2 | −0.38 | 0.24 | ||||||
| x.3 | −0.16 | −0.42 | −0.16 | |||||
| y.3 | −0.18 | −0.31 | −0.23 | |||||
| x.4 | −0.44 | 0.2 | −0.40 | 0.52 | ||||
| y.4 | 0.41 | −0.22 | 0.45 | −0.18 | −0.32 | |||
| x.5 | −0.25 | 0.47 | ||||||
| y.5 | −0.2 | 0.16 | −0.18 | |||||
| x.6 | −0.16 | 0.21 | −0.17 | 0.15 | ||||
| y.6 | −0.16 | −0.21 | −0.16 | |||||
| x.7 | 0.51 | 0.3 | −0.15 | −0.32 | −0.16 | |||
| y.7 | 0.33 | 0.21 | 0.42 | −0.41 | ||||
| x.8 | 0.49 | −0.18 | 0.16 | 0.15 | 0.61 | |||
| y.8 | 0.21 | 0.28 | 0.16 | |||||
| x.9 | −0.34 | 0.17 | −0.33 | −0.26 | ||||
| y.9 | ||||||||
| x.10 | −0.38 | 0.22 | −0.35 | −0.27 | ||||
| y.10 | ||||||||
| x.11 | 0.2 | |||||||
| y.11 | ||||||||
| x.12 | −0.15 | 0.21 | ||||||
| y.12 | −0.16 | |||||||
Loadings ≥|0.15| are displayed; loadings ≥|0.25| are in italics.
QTL analysis:
PC1–PC7 were analyzed using composite interval mapping (CIM) using Windows Version 2.0 QTL Cartographer (Wang et al. 2003). Separate analyses were performed for each LG. This approach has two consequences. First, the results for multiple LGs of a chromosome are not expected to be independent. Second, separate analysis of each LG is not expected to produce highly reliable estimates of QTL effect since not all markers are included in any given analysis. We used CIM model 6 of QTL Cartographer, using five background parameters chosen by forward stepwise regression and a 10-cM window size. Analyses using different parameter combinations including larger window sizes (up to 50 cM) and different numbers of background markers did not appear to have a major impact on the locations of peaks, although the number of markers had an effect on resolution of the peaks as expected (results not presented). Significance was assessed by 1000 random permutations for each LG. While this approach accounts for the multiple-comparisons problem for a given LG, there are still multiple comparisons for the different LGs and multiple traits, a total of (9 LG) × (7 traits) = 63 comparisons. With this many comparisons, a Bonferroni adjustment of the significance value would be far too conservative. What is more, many of these tests will be highly correlated, so it is unclear how to best adjust for multiple comparisons. As a compromise between accounting for number of false positives and power to detect true differences, we settled on a significance threshold of α = 0.01, which was used to assess locations across all analyses. Dominance effects cannot be estimated when using RILs.
Each peak in each analysis is not necessarily expected to reflect a distinct QTL, since analysis of different LGs should identify the same QTL. Likewise, if a QTL has a pleiotropic effect, the same QTL could produce significant results for different PCs. We used two strategies to assess whether different peaks reflect the same QTL. First, for a given LG, if there were significant peaks for different PCs between the same two markers of the LG, these were considered to reflect a single putative QTL with a pleiotropic effect. Second, if peaks were found for the same PC for overlapping marker regions in distinct LGs, these were also considered to reflect a single putative QTL. In Table 2, we report each case of a significant peak for all analyses of PC1–PC7 as well as the set of QTL that we consider to be distinct by these criteria.
TABLE 2.
QTL identified in RILs
| Peak/QTL | Trait | Chr./LG | Position in LG | LG interval (QTL interval) |
% variation | Candidate locus/marker tested |
|---|---|---|---|---|---|---|
| P1/Q1 | PC2 | X/1 | 0.023 | 3A–4B | 18.8 | |
| P2/Q2 | PC6 | X/2 | 0.078 | 4B–5A | 13.7 | |
| P3/Q3 | PC7 | X/1 | 0.571 | 11F–12C | 13.8 | |
| P4/Q4 | PC7 | X/1 | 0.969 | 17E–19AB | 23.0 | |
| P5/Q5 | PC1 | 2/2 | 0.027 | 22A–23F24A (22C–24CD) | 16.0 | dpp/22F |
| P6/Q5 | PC3 | 2/2 | 0.095 | 23F24A–30D (22C–24CD) | 23.8 | |
| P7/Q5 | PC6 | 2/3 | 0.02 | 22B–24CD (22C–24CD) | 27.5 | |
| P8/Q5 | PC6 | 2/1 | 0.028 | 22C–23F34A (22C–24CD) | 13.5 | |
| P9/Q6 | PC7 | 2/2 | 0.28 | 31F–36F | 20.5 | salm/32F |
| P10/Q7 | PC4 | 2/2 | 0.347 | 37C–49F (41F–42B) | 21.4 | |
| P11/Q7 | PC5 | 2/2 | 0.347 | 37C–49F (41F–42B) | 23.7 | |
| P12/Q7 | PC1 | 2/3 | 0.471 | 41F–42B (41F–42B) | 55.2 | |
| P13/Q8 | PC1 | 2/3 | 0.712 | 42B–47E | 20.9 | sax/43E, ptc/44C, shn/47D |
| P14/Q9 | PC7 | 2/3 | 1.000 | 47E–48A | 35.0 | en/48A |
| P15/Q10 | PC1 | 2/2 | 0.559 | 51A–54A | 16.0 | ttv/51A |
| P16/Q11 | PC1 | 2/2 | 0.648 | 54C–55C | 24.4 | |
| P17/Q11 | PC7 | 2/2 | 0.674 | 54C–55C | 23.8 | |
| P18/Q12 | PC1 | 2/2 | 0.807 | 55C–57B (55D–57B) | 13.7 | |
| P19/Q12 | PC1 | 2/1 | 0.917 | 55D–58D (55D–57B) | 12.5 | |
| P20/Q12 | PC2 | 2/1 | 0.917 | 55D–58D (55D–57B) | 17.6 | |
| P21/Q12 | PC3 | 2/2 | 0.754 | 55C–57B (55D–57B) | 18.3 | |
| P22/Q13 | PC4 | 3/1 | 0.045 | 61C–64D (61C–63F) | 10.8 | rho/62A |
| P23/Q14 | PC6 | 3/4 | 0.117 | 65E–73D (66B–67D) | 48.8 | |
| P24/Q15 | PC4 | 3/1 | 0.207 | 69A–70C | 25.7 | |
| P25/Q16 | PC2 | 3/4 | 0.309 | 77E–83F84A (79A–79C) | 19.8 | |
| P26/Q16 | PC4 | 3/1 | 0.312 | 79A–79C (79A–79C) | 58.2 | |
| P27/Q17 | PC1 | 3/1 | 0.565 | 86D–87A | 61.9 | |
| P28/Q17 | PC5 | 3/1 | 0.544 | 86D–87A | 19.4 | |
| P29/Q18 | PC3 | 3/1 | 0.855 | 87E–87F | 37.9 | |
| P30/Q19 | PC4 | 3/1 | 0.889 | 87F–89AB (88C–89AB) | 43.7 | cv-c/88C, put/88C |
| P31/Q20 | PC3 | 3/23 | 0.975 | 89EF–90EF (89EF–90EF) | 16.1 | |
| P32/Q20 | PC5 | 3/1 | 0.906 | 89AB–94D (89EF–90EF) | 24.9 | |
| P33/Q21 | PC2 | 3/4 | 1.000 | 99F–100B | 14.7 | |
| P34/Q21 | PC3 | 3/4 | 0.929 | 99F–100B | 53.0 |
The position in the LG is the relative position of the peak in a LG scaled to unit length. LG interval lists the flanking markers of the peak in the LG where the peak occurred and the QTL interval is the inferred location of the QTL using the criteria described in the text. Effects are in standard deviation units of variation along a PC always coding the focal LG the same way.
Comparison of putative QTL location among studies:
Results from Zimmerman et al. (2000), Weber et al. (1999)(2001), and this study that have the following aspects in common are compared (other results are not considered): (1) males, (2) high marker coverage (analyses in Zimmerman et al. 2000 using W6 and W29 lines are ignored), (3) CHR2 and CHR3, and (4) flies raised at 25°–26°. The differences among the studies are compared in Table 3.
TABLE 3.
Wing shape QTL studies
| Study
|
|||
|---|---|---|---|
| Weber et al. (2001)/Weber et al. (1999) | Zimmerman et al. (2000) | This study | |
| Strains/design | Recombinant chromosomes from high-/low-index F lines (low background) | RILs derived from Oregon R × Russian 2b |
RILs derived from two wild-type parents (Winters, CA) |
| No. possible alleles per locus | 2 | 2 | 4 |
| Marker coverage per centimorgan, CHR2/CHR3 |
2.3/1.8 | 4.1/2.6 | 2.4/1.7 |
| Map distances | Genes close to markers | RIL markers | RIL markers |
| Estimated using traits | F-index (ratio of widths) | Landmarks | Landmarks |
| Analysis method | MIM | CIM | CIM |
Average distance between markers assumes 110 cM for CHR2 and 111 cM for CHR3. The average distance between markers for Zimmerman et al. (2000) includes all markers listed in Nuzhdin et al. (1997). MIM, multiple-interval mapping; CIM, composite-interval mapping.
Each study used a different set of markers so it is not possible to directly align the mappings. We therefore localized each reported QTL to a pair of flanking markers used in the study where the QTL was identified and determined how many of these regions overlapped among studies (summarized in Figure 4). Using the locations of putative QTL, we used a correspondence test to assess the hypothesis that clustering of putative QTL locations was not greater than expected at random. This was done separately for CHR2 and CHR3. The test statistic for comparing the locations of QTL found for a chromosome was the number of QTL regions that overlapped a QTL region in at least one other study. The null distribution of this statistic was produced by 1000 randomizations of the locations of the QTL for a chromosome in each study. Each randomization preserved the same number of QTL with nonoverlapping intervals, spanning the same number of markers; i.e., each randomization would look like Figure 4, a or b, with the horizontal lines reflecting QTL shuffled at random. The test is based on the binning approach described in Paterson (2002), but should be more powerful, since it takes the structure of the individual study markers into account. We used this approach to compare pairs of studies and all three studies together for each chromosome.
Figure 4.—


Comparison among QTL studies of wing shape for (a) CHR2 and (b) CHR3. Abbreviations are M (this study), Z (Zimmerman et al. 2000), W99 (Weber et al. 1999), and W01 (Weber et al. 2001). The horizontal bars indicate the markers spanning the inferred locations of putative QTL identified in a study (see text). The labels on the horizontal bars for M correspond to QTL numbers in Table 1 and also indicate the PCs they affect, the labels for Z correspond to traits used in Zimmerman et al. (2000)(see their Table 3), and the labels for W99 and W01 correspond to QTL numbers in Weber et al. (1999)(their Table 5) and Weber et al. (2001)(their Table 3), respectively. Vertical lines indicate overlap of locations identified among studies. Triangles indicate the locations of the centromeres. The part of CHR2 indicated by the hatched rectangle was not analyzed in Zimmerman et al. (2001) and was not used in the test of correspondence.
Complementation tests of candidate loci:
We used complementation tests to analyze 10 candidate loci involved in wing vein development that fall within identified QTL regions that affect PC1, PC4, and PC7 (Table 2). These PCs were chosen because they account for variation in vein pairs L3-L4 and L2-L5 (see results) and because good candidates were located in the regions of the QTL that affect these PCs. Suitable markers (eight total) were chosen within seven QTL regions and a complementation test was carried out between these markers and candidate loci. To account for effects of genetic background, we use the approach of Kopp et al. (2003) and randomize backgrounds by testing 12 RIL for QTL vs. two wild-type backgrounds and two mutant backgrounds. The wild-type backgrounds were two isofemale lines produced from the same population used to produce the RILs (Winters, CA). Mutant stocks containing the following loss-of-function and amorphic/hypomorphic alleles were tested: dpp[d6, d12], salm[1, 03602], sax[4, KG07525], ptc[S2, tuf-1], shn[1, 04738], en[1, Xho25], ttv[00681b, KG05875], rho[ve-1, 7M43], put[135, 10460], and cv-c[1] (Bloomington Stock Center nos. 256, 429, 472, 628, 1471, 2062, 2070, 3008, 3100, 3274, 3527, 5404, 5801, 6332, 10949, 11386, 11745, 11340, 14130, and 14920). An average of 8 ± 4 wings were measured for males of each cross, a total of 1838 wings. We tested for noncomplementation between a marker in each QTL region and an associated candidate locus for the trait affected by the QTL. Additionally, we tested for noncomplementation for all other combinations of QTL and candidates for each of these traits, resulting in (10 candidates) × (8 markers) × (3 PCs) = 240 total tests for noncomplementation. ANOVAs were implemented in the SAS Proc Mixed procedure (SAS Version 8.01; SAS Institute 2002), treating marker and cross (wild-type and mutant stock) as fixed effects, with RIL and wild-type/mutant stocks as random effects nested within marker and cross, respectively.
RESULTS
Variation in wing shape:
The first two PCs account for >50% of variation (Table 1). PC1 has large loadings on landmarks 4, 7, and 8. This is consistent with our observation that larger wings tended to move the distal crossvein toward the wing margin and curl the distal part of vein L2 proximally (hence moving landmark 4) and vice versa for smaller wings. PC2, PC3, PC6, and PC7 had large loadings on these landmarks plus landmarks 1, 9, and 10. Thus, much of the variation in wing shape among these lines appears to involve movement of the relative locations of the crossveins and the relative distances between veins L2 and L5. PC4 and PC5 have large loadings on landmarks 2 and 3. These PCs appear to account for variation in the distance between veins L3 and L4.
Wing shape QTL:
The analysis identified a total of four XCHR, eight CHR2, and nine CHR3 distinct QTL. These QTL explained from 48.6% (PC5) to >100% (PC1, PC3, PC4, and PC7) when summing effects across LGs and chromosomes. The latter result must reflect overestimation of QTL effects. These percentages are similar to those found in Zimmerman et al. (2000) (10–70%) and Weber et al. (1999)(2001) (94.7 and 95.1%). Pleiotropic effects or closely linked QTL may also result in the same QTL signal producing significant peaks across more than one PC. There were a total of eight putative QTL with pleiotropic effects on two to three PCs.
Qualitatively, the likelihood profiles for different LGs of the same chromosome were similar (Figure 3). The profiles are not expected to mirror each other exactly, since the power of tests differs among LGs, a consequence of each LG incorporating different numbers of markers and testing vs. a composite value of other LGs. Different LGs tended to have peaks in the same general areas, although the peaks were not necessarily significant. These nonsignificant peaks were used to localize the QTL as described above.
Figure 3.—

Likelihood-ratio profiles (y-axis) vs. the linkage groups (x-axis) of the (a) XCHR, (b) CHR2, and (c) CHR3. Significant peaks are labeled as P#/Q#, where P# is the peak number and Q# reflects distinct putative QTL (Table 2). Note there were no significant peaks for LG of CHR2 so this LG is not shown.
Comparison of QTL location across studies:
The locations of QTL identified in Weber et al. (1999)(2001), Zimmerman et al. (2000), and this study localized to flanking markers used in each study are presented in Figure 4. Comparing region by region, there is only one location at the end of 3R where all studies identified a QTL: 99A–99F. Between any two pairs of studies, there are many locations in common. Overall on CHR2 there are seven regions that are unique to specific study. The picture for CHR3 is quite different. Almost all QTL overlap with QTL in another study.
The test of correspondence also produces a different picture for CHR2 and CHR3 (Table 4). For CHR2, none of the pairwise tests or the test considering all three studies produced a significant result. There is therefore no evidence that regions are clustering more than expected at random on CHR2. For CHR3, none of the pairwise comparisons produced a significant result. In contrast to the pairwise tests, the P-value for a test among all studies for CHR3 was significant even after a Bonferroni correction for all [(2 chromosomes) × (4 tests) = 8] tests (α = 0.00625). The QTL therefore cluster on CHR3 more than expected at random.
TABLE 4.
Results of study correspondence tests
| Chr. 2
|
Chr. 3
|
|||
|---|---|---|---|---|
| Study | This study | Weber et al. (2001) | This study | Weber et al. (1999) |
| Zimmerman et al. (2000) | 3/P < 0.32 | 2/P < 0.55 | 11/P < 0.2 | 14/P < 0.03 |
| This study | 9/P < 0.27 | 10/P < 0.013 | ||
| All | 13/P < 0.2 | 26/P < 0.0045* | ||
The number before the slash is the number of identified regions that overlap with at least one other region in another study and the number after the slash is the P-value, where * reflects a significant result.
Complementation tests:
A total of 240 tests for noncomplementation were performed. Accounting for multiple tests with a Bonferroni adjustment is likely to be far too conservative with this many tests. We therefore use an estimate of the proportion of the significant tests that are expected to be false positives (Manly et al. 2004). At a significance level of α = 0.05, 36 tests are significant, indicating noncomplementation. This produces an expected false positive rate of 33%, such that two-thirds of the tests at this significance level are expected to correctly reject the null hypothesis. This approach does not indicate which of the tests are false positives, but does indicate that we have found far more significant tests than expected at random when performing this many tests. Additionally, this estimate of false positives assumes tests are independent, which is clearly not the case for the tests considered here. A false positive rate of 33% should therefore be viewed as very conservative and is probably much lower. We present all of the significant tests at α = 0.05 in Table 5.
TABLE 5.
Results of complementation tests
| QTL | QTL-PC | Marker | Candidate | PC-tested | P-value |
|---|---|---|---|---|---|
| Q5 | 1, 3, 6 | 22F | en | 1 | 0.0079 |
| Q5 | 1, 3, 6 | 22F | ttv | 1 | 0.0029 |
| Q8 | 1 | 43E | dpp | 7 | 0.028 |
| Q8 | 1 | 43E | salm | 7 | 0.025 |
| Q8 | 1 | 43E | salm | 4 | 0.04 |
| Q8 | 1 | 43E | sax | 7 | 0.049 |
| Q8 | 1 | 43E | ptc | 7 | 0.036 |
| Q8 | 1 | 43E | ptc | 4 | 0.044 |
| Q8 | 1 | 43E | shn | 4 | 0.027 |
| Q8 | 1 | 43E | shn | 7 | 0.04 |
| Q8 | 1 | 43E | shn | 1 | 0.047 |
| Q8 | 1 | 43E | en | 7 | 0.022 |
| Q8 | 1 | 43E | en | 4 | 0.025 |
| Q8 | 1 | 43E | put | 7 | 0.034 |
| Q8 | 1 | 44C | ttv | 1 | 0.028 |
| Q9 | 7 | 48A | dpp | 1 | 0.018 |
| Q9 | 7 | 48A | salm | 7 | 0.03 |
| Q9 | 7 | 48A | ptc | 1 | 0.00027 |
| Q9 | 7 | 48A | ptc | 7 | 0.029 |
| Q9 | 7 | 48A | shn | 7 | 0.03 |
| Q9 | 7 | 48A | shn | 4 | 0.039 |
| Q9 | 7 | 48A | en | 7 | 0.023 |
| Q9 | 7 | 48A | en | 4 | 0.03 |
| Q9 | 7 | 48A | ttv | 1 | 0.02 |
| Q9 | 7 | 48A | put | 7 | 0.036 |
| Q9 | 7 | 48A | put | 4 | 0.046 |
| Q9 | 7 | 48A | cv-c | 1 | 0.00057 |
| Q19 | 4 | 88C | salm | 7 | 0.029 |
| Q19 | 4 | 88C | salm | 4 | 0.044 |
| Q19 | 4 | 88C | sax | 7 | 0.045 |
| Q19 | 4 | 88C | ptc | 4 | 0.046 |
| Q19 | 4 | 88C | shn | 1 | 0.00062 |
| Q19 | 4 | 88C | en | 1 | 0.024 |
| Q19 | 4 | 88C | en | 4 | 0.048 |
| Q19 | 4 | 88C | ttv | 1 | 0.003 |
| Q19 | 4 | 88C | put | 4 | 0.045 |
The tests indicate that QTL Q8, Q9, and Q19 interact with almost all of the candidate loci tested. These interaction effects are picked up on all three of the PCs tested, even though these QTL were found to affect only a single PC each; i.e., epistatic effects were found for traits that the QTL were not expected to affect. With the exceptions of Q9 (en) and Q19 (put), none of the QTL interacted with candidates for the expected PC, when the QTL and candidate were located in the same chromosomal region. This indicates that none of the QTL are likely to be any of the candidates tested. rho was not involved in any significant tests.
DISCUSSION
A striking result of the genomic scan is the number of distinct QTL that were identified. Even using the conservative criteria that each peak is reflective of a single true QTL and that many of these peaks reflect the same QTL (identified on different LGs or using different traits), the analysis indicates at least 21 distinct QTL. From the perspective that many loci may be able to affect vein positioning, this may not be surprising. However, the variation surveyed is that present in just two individuals sampled from a natural population. The total number of QTL segregating for wing variation in the population is likely to be far greater due to several factors. First, low-frequency alleles will often not segregate in such a small sample. Second, the sampled individuals were related as shown by the shared haplotype for part of CHR3. Third, the analysis is unlikely to identify QTL unless they have large effects. Fourth, the fairly small number of markers makes it likely that some of the regions with QTL actually reflect the effects of more than one locus. This result implies a large mutational target for alleles with effects on vein position. The subtle differences in wing shape that have evolved in Drosophila (Houle et al. 2003) could therefore involve substitutions at a large number of loci.
Our results are qualitatively the same as previous QTL studies of wing shape (Weber et al. 1999, 2001; Zimmerman et al. 2000). A large number of distinct genomic regions are identified, the QTL appear to have pleiotropic effects, and the amount of variation in traits accounted for is high. The proportion of variation explained must be an overestimate, due to the Beavis (1998) effect (Bost et al. 2001). Interestingly, both Zimmerman et al. (2000) and this study identified very few locations on the XCHR. This may be a function of the number of loci on the XCHR (Noor et al. 2001).
The studies do differ in the locations identified as containing QTL (Figure 4). While many regions overlapped between pairs of studies, there is only one region (98A–99F) found in common among Weber et al. (1999)(2001), Zimmerman et al. (2000), and this study. At first glance, it therefore appears that these studies are identifying different QTL. However, there is evidence for more clustering of QTL than expected at random on CHR3, which is surprising given that the studies analyze different lines and different traits. Several scenarios could produce this pattern: First, these QTL could reflect alleles that are identical by descent. Second, these QTL may reflect variation at the same loci, but different alleles. This could occur if these loci are particularly prone to be involved in local adaptation or are more mutable than others. Third, concordance may reflect regions that have concentrations of loci that affect wing shape, without the same loci being involved. This would be intensified in regions with low recombination (Noor et al. 2001).
While the analysis of correspondence suggests the possibility that some of the same loci on CHR3 could be contributing to the variation analyzed in the different studies, the more likely explanation for the general lack of correspondence is that different loci are being identified in each study. This result is consistent with a highly complex genetic basis for quantitative variation in wing shape. Allelic variants at a very large number of loci seem to be able to produce a variety of effects on wing shape (Weber 1992; Mezey and Houle 2004). One possible explanation for this is that there are many developmental genetic pathways where genetic variation can affect wing shape. If no pathways dominate the production of variation in wing shape, we might not expect to find that the majority of QTL interact with specific pathways since there are many alternative ways in which allelic variation can introduce variation. However, the complementation tests revealed that three of the seven QTL tested (Q8, Q9, and Q19) had significant interactions with almost all of the candidate loci in the Hh and Dpp pathways (Table 5). The fact that almost half of the tested QTL can produce variation by acting through the Hh and Dpp pathway argues for these pathways playing an important role in the production of quantitative variation in wing vein position.
It is notable that the candidate locus rho was not associated with any of the significant tests and cv-c was associated only with one. While the developmental roles of the other candidates have been directly connected to positioning of veins, rho and cv-c have not. rho promotes production of vein material and mutations of cv-c cause the partial or complete absence of the crossveins (Diaz-Benjumea and Garcia-Bellido 1990; Held 2002). These genes are also developmentally downstream from the other candidates. We have shown that the Dpp and Hh pathways are implicated in the quantitative aspects of vein position; perhaps it is also the case that variation at loci involved in later stages of wing development or production of vein material will not produce variation in vein position.
The complementation tests also demonstrate another aspect wing quantitative genetics: the multivariate effects of epistasis. QTL Q8, Q9, and Q19, which had epistatic interactions with most of the candidate loci, were each defined by effects on a single PC. However, noncomplementation of each of these QTL was detected on all three PCs (1, 4, and 7), not just on the PC affected by the QTL. One explanation for this result is that each of these QTL do affect all three PCs and these effects were simply not detected in the QTL analysis. However, if this were the case, we might not expect to find the noncomplementation pattern of Q8, where the epistatic effects almost all involve PC4 and PC7, while the QTL was found to affect PC1. Another explanation that seems more likely is that the presence of different alleles at the candidate loci can not only change the trait-specific effects of a QTL, but also alter which traits the QTL affects. This means not only the magnitude but also the type of effect on the wing produced by a QTL can be substantially altered by the effects of other loci.
The overall picture that has emerged from this study is that the quantitative aspects of wing vein placement are complex. Both a large number of potential QTL and the possibility of epistatic effects cause substantial rearrangements of QTL effects. However, despite the diversity in the effects of QTL, it appears that many QTL may be acting through the Hh and Dpp signaling pathways to produce variation in wing vein position. This implies that even when quantitative variation has an extremely complex genetic basis, it may be possible to understand the genetics of this variation by analyzing a few important pathways. An approach that combines quantitative genetic analysis and analysis of expression of genes important in the developmental position of veins seems the most promising for understanding the genetics and evolution of vein position and wing shape.
Acknowledgments
We thank the following people for their contribution to this research and comments on the manuscript: L. Harshman, F. James, L. Moyle, I. Dworkin, and an anonymous reviewer. We also thank K. van der Linde, J. Birdsley J. Moss, J. Polland, A. Allen, A. J. Steel, and A. Brock for assisting with wing measurement. This research was supported by National Science Foundation grant NSF-0129219 and National Institutes of Health grants 1R01GM61773-01 and R24GM65513-01.
APPENDIX
The formulas for estimating recombination rates between markers for RILs produced by brother-sister mating from an original mating of inbred parents, i.e., two possible alleles at each locus, are derived in Haldane and Waddington (1931) for autosomal linked markers, r = 1/(4/R − 6), and for X-linked markers, r = 3/(8/R − 12), where r is the recombination rate (Lynch and Walsh 1998) and R is the proportion of RILs for which recombination occurred between adjacent markers. In the current treatment, estimators of recombination rate for a case where there are two parental haplotypes in the proportion 1:3 is needed. This estimator is derived following the instructions in Haldane and Waddington (1931)(pp. 370 and 371). In their notation, this is the case where the initial population of zygotes is 1AABB:3aabb. Assume zygote pairing at random for one generation producing the following mating proportions: 1 of AABBxAABB, 3 of AABBxaabb, 3 of aabbxAABB and 9 of aabbxaabb. After this one generation, add this to a population that has zygotes in the proportion 3AABB:1aabb and has produced the mirror image of matings (the population is now symmetrical). Of 32 typical zygote pairings in this mixed population, there are 10 of each of the two types of Cn and 6 of each of the types of En (Haldane and Waddington 1931, p. 364). Note that there are none of any of the other types. After, dividing by 16 to produce a total of two pairings, from Haldane and Waddington (1931)(p. 365),
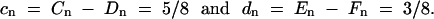 |
Substituting these into Equation 2.4 (Haldane and Waddington 1931, p. 366) produces
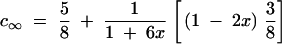 |
Using the relationship y = 1/2(1 − c∞) (Haldane and Waddington 1931, p. 366),
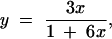 |
where y = R is the proportion of crossover zygotes and x = r is the recombination rate. Solving for r produces r = 1/(3/R − 6).
References
- Beavis, W. D., 1998 QTL analyses: power, precision and accuracy, pp. 145–162 in Molecular Dissection of Complex Traits, edited by A. H. Patterson. CRC Press, Boca Raton, FL/New York.
- Bier, E., 2000. Drawing lines in the Drosophila wing: initiation of wing vein development. Curr. Opin. Genet. Dev. 10: 393–398. [DOI] [PubMed] [Google Scholar]
- Birdsall, K., E. Zimmerman, K. Teeter and G. Gibson, 2000. Genetic variation for the positioning of wing veins in Drosophila melanogaster. Evol. Dev. 2: 16–24. [DOI] [PubMed] [Google Scholar]
- Bost, B., D. de Vienne, F. Hospital, L. Moreau and C. Dillmann, 2001. Genetic and nongenetic bases for the L-shaped distribution of quantitative trait loci effects. Genetics 157: 1773–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodsky, A. K., 1994 The Evolution of Insect Flight. Oxford University Press, New York.
- Bublii, O. A., A. G. Imasheva and O. E. Lazebny, 1996. Variation of a set of wing characters in natural populations of Drosophila melanogaster. Genetica 32: 1513–1520. [PubMed] [Google Scholar]
- Cavicchi, S., G. Giorgi, V. Natali and D. Guerra, 1991. Temperature related divergence in experimental populations of Drosophila melanogaster. 4. Fourier and centroid analysis of wing shape and relation between shape variation and fitness. J. Evol. Biol. 4: 141–159. [Google Scholar]
- Charlesworth, B., and C. H. Langley, 1989. The population genetics of Drosophila transposable elements. Annu. Rev. Genet. 23: 251–287. [DOI] [PubMed] [Google Scholar]
- Combes, S. A., and T. L. Daniel, 2003. Flexural stiffness in insect wings. I. Scaling and the influence of wing venation. J. Exp. Biol. 206: 2979. [DOI] [PubMed] [Google Scholar]
- Cook, O., B. Biehs and E. Bier, 2004. brinker and optomotor-blind act coordinately to initiate development of the L5 wing vein primordium in Drosophila. Development 131: 2113–2124. [DOI] [PubMed] [Google Scholar]
- Daniel, T. L., and S. A. Combes, 2002. Flexible wings and fins: bending by inertial or fluid-dynamic forces. Integr. Comp. Biol. 45: 1044–1049. [DOI] [PubMed] [Google Scholar]
- de Celis, J. F., 2003. Pattern formation in the Drosophila wing: the development of the veins. BioEssays 25: 443–451. [DOI] [PubMed] [Google Scholar]
- Diaz-Benjumea, F. J., and A. Garcia-Bellido, 1990. Genetic analysis of the wing vein pattern of Drosophila. Roux's Arch. Dev. Biol. 198: 336–354. [DOI] [PubMed] [Google Scholar]
- Ennos, A. R., 1988. The importance of torsion in the design of insect wings. J. Exp. Biol. 140: 137–160. [Google Scholar]
- Fanara, J. J., K. O. Robinson, S. M. Rollmann, R. R. H. Anholt and T. F. C. Mackay, 2002. Vanaso is a candidate quantitative trait gene for Drosophila olfactory behavior. Genetics 162: 1321–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler, K., and M. C. Whitlock, 2002. Environmental stress, inbreeding, and the nature of phenotypic and genetic variance in Drosophila melanogaster. Proc. R. Soc. Lond. Ser. B 269: 677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujise, M., S. Takeo, K. Kamimura, T. Matsuo, T. Aigaki et al., 2003. Dally regulates Dpp morphogen gradient formation in the Drosophila wing. Development 130: 1515–1522. [DOI] [PubMed] [Google Scholar]
- Garcia-Bellido, A., and J. F. de Celis, 1992. Developmental genetics of the venation pattern of Drosophila. Annu. Rev. Genet. 26: 277–304. [DOI] [PubMed] [Google Scholar]
- Gilchrist, A. S., and L. Partridge, 2001. The contrasting genetic architecture of wing size and shape in Drosophila melanogaster. Heredity 86: 144–152. [DOI] [PubMed] [Google Scholar]
- Haerry, T. E., O. Khalsa, M. B. O'Connor and K. A. Wharton, 1998. Synergistic signaling by two BMP ligands through SAX and TKV receptors controls wing growth and patterning in Drosophila. Development 125: 3977–3987. [DOI] [PubMed] [Google Scholar]
- Haldane, J. B. S., and C. H. Waddington, 1931. Inbreeding and linkage. Genetics 16: 357–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Held, L. I., 2002 Imaginal Discs: The Genetic and Cellular Logic of Pattern Formation. Cambridge University Press, Cambridge, UK/London/New York.
- Houle, D., J. G. Mezey, P. Galpern and A. Carter, 2003. Automated measurement of Drosophila wings. BMC Evol. Biol. 3: 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imasheva, D. G., O. A. Bulbli, O. E. Lazebny and L. A. Zhivotovsky, 1995. Geographic differentiation in wing shape in Drosophila melanogaster. Genetica 96: 303–306. [DOI] [PubMed] [Google Scholar]
- Kopp, A., R. M. Graze, S. Z. Xu, S. B. Carroll and S. V. Nuzhdin, 2003. Quantitative trait loci responsible for variation in sexually dimorphic traits in Drosophila melanogaster. Genetics 163: 771–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long, A. D., S. L. Mullaney, T. F. C. Mackay and C. H. Langley, 1996. Genetic interactions between naturally occurring alleles at quantitative trait loci and mutant alleles at candidate loci affecting bristle number in Drosophila melanogaster. Genetics 144: 1497–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long, A. D., R. F. Lyman, C. H. Langley and T. F. C. Mackay, 1998. Two sites in the Delta gene region contribute to naturally occurring variation in bristle number in Drosophila melanogaster. Genetics 149: 999–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long, A. D., R. F. Lyman, A. H. Morgan, C. H. Langley and T. F. C. Mackay, 2000. Both naturally occurring insertions of transposable elements and intermediate frequency polymorphisms at the achaete-scute complex are associated with variation in bristle number in Drosophila melanogaster. Genetics 154: 1255–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunde, K., J. L. Trimble, A. Guichard, K. A. Guss, U. Nauber et al., 2003. Activation of the knirps locus links patterning to morphogenesis of the second wing vein in Drosophila. Development 130: 235–248. [DOI] [PubMed] [Google Scholar]
- Lyman, R. F., and T. F. C. Mackay, 1998. Candidate quantitative trait loci and naturally occurring phenotypic variation for bristle number in Drosophila melanogaster: the Delta-Hairless gene region. Genetics 149: 983–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch, M, and B. Walsh, 1998 Genetics and Analysis of Quantitative Traits. Sinauer, Sunderland, MA.
- Mackay, T. F. C., 2001. The genetic architecture of quantitative traits. Annu. Rev. Genet. 35: 303–339. [DOI] [PubMed] [Google Scholar]
- Mackay, T. F. C., and J. D. Fry, 1996. Polygenic mutation in Drosophila melanogaster: genetic interactions between selection lines and candidate quantitative trait loci. Genetics 144: 671–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manly, K. F., D. Nettleton and J. T. Gene Hwang, 2004. Genomics, prior probability and statistical tests of multiple hypotheses. Genome Res. 14: 997–1001. [DOI] [PubMed] [Google Scholar]
- Mezey, J. G., and D. Houle, 2005. Absolute constraints on wing shape of Drosophila melanogaster. Evolution (in press). [PubMed]
- Moehring, A. J., and T. F. C. Mackay, 2004. The quantitative genetic basis of male mating behavior in Drosophila melanogaster. Genetics 167: 1249–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noor, M. A. F., A. L. Cunningham and J. C. Larkin, 2001. Consequences of recombination rate variation on quantitative trait locus mapping studies: simulations based on the Drosophila melanogaster genome. Genetics 159: 581–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuzhdin, S. V., E. G. Pasyukova, C. L. Dilda, Z-B. Zeng and T. F. C. Mackay, 1997. Sex-specific quantitative trait loci affecting longevity in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 94: 9734–9739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palsson, A., and G. Gibson, 2000. Quantitative developmental genetic analysis reveals that the ancestral dipteran wing vein prepattern is conserved in Drosophila melanogaster. Dev. Genes Evol. 210: 617–622. [DOI] [PubMed] [Google Scholar]
- Palsson, A., and G. Gibson, 2004. Association between nucleotide variation in Egfr and wing shape in Drosophila melanogaster. Genetics 167: 1187–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson, A. H., 2002. What has QTL mapping taught us about plant domestication? New Phytol. 154: 591–608. [DOI] [PubMed] [Google Scholar]
- Phillips, P. C., M. C. Whitlock and K. Fowler, 2001. Inbreeding changes the shape of the genetic covariance matrix in Drosophila melanogaster. Genetics 158: 1137–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robin, C., R. F. Lyman, A. D. Long, C. H. Langley and T. F. C. Mackay, 2002. hairy: a quantitative trait locus for Drosophila sensory bristle number. Genetics 162: 155–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohlf, F. J., 1998 tpsDig: Shape Regression. V.1.17. State University of New York, Stony Brook, NY (http://life.bio.sunysb.edu/morph/).
- Rohlf, F. J., 1999. Shape statistics: procrustes superimpositions and tangent spaces. J. Classif. 16: 197–223. [Google Scholar]
- Rohlf, F. J., and D. Slice, 1990. Extensions of the procrustes method for the optimal superimposition of landmarks. Syst. Zool. 39: 40–59. [Google Scholar]
- SAS Institute, 2002 The SAS System for Windows, Release 8.01. SAS Institute, Cary, NC.
- Sturtevant, M. A., and E. Bier, 1995. Analysis of the genetic hierarchy guiding wing vein development in Drosophila. Development 121: 785–801. [DOI] [PubMed] [Google Scholar]
- Sturtevant, M. A., B. Biehs, E. Marin and E. Bier, 1997. The spalt gene links the A/P compartment boundary to a linear adult structure in the Drosophila wing. Development 124: 21–32. [DOI] [PubMed] [Google Scholar]
- Torres-Vazquez, J., R. Warrior and K. Arora, 2000. schnurri is required for dpp-dependent patterning of the Drosophila wing. Dev. Biol. 227: 388–402. [DOI] [PubMed] [Google Scholar]
- Wang, S., C. Basten and Z-B. Zeng, 2003 Window QTL Cartographer, V2.0. North Carolina State University, Raleigh, NC (http://statgen.ncsu.edu/qtlcart/WQTLCart.htm).
- Weber, K. E., 1990. Selection on wing allometry in Drosophila melanogaster. Genetics 126: 975–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber, K. E., 1992. How small are the smallest selectable domains of form? Genetics 130: 345–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber, K., S. Eisman, L. Morey, A. Patty, J. Sparks et al., 1999. An analysis of polygenes affecting wing shape on chromosome 3 in Drosophila melanogaster. Genetics 153: 773–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber, K., R. Eisman, S. Higgins, L. Morey, A. Patty et al., 2001. An analysis of polygenes affecting wing shape on chromosome 2 in Drosophila melanogaster. Genetics 159: 1045–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitlock, M. C., and K. Fowler, 1999. The changes in genetic and environmental variance with inbreeding in Drosophila melanogaster. Genetics 152: 345–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitlock, M. C., P. C. Phillips and K. Fowler, 2002. Persistence of changes in the genetic covariance matrix after a bottleneck. Evolution 56: 1968–1975. [DOI] [PubMed] [Google Scholar]
- Wootton, R. J., R. C. Herbert, P. G. Young and K. E. Evans, 2003. Approaches to the structural modeling of insect wings. Philos. Trans. R. Soc. Lond. B 358: 1577–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman, E., A. Palsson and G. Gibson, 2000. Quantitative trait loci affecting components of wing shape in Drosophila melanogaster. Genetics 155: 671–683. [DOI] [PMC free article] [PubMed] [Google Scholar]