

Abstract
A pathological feature of Parkinson's disease is the presence of Lewy bodies within selectively vulnerable neurons. These are ubiquitinated cytoplasmic inclusions containing α-synuclein, an abundant protein normally associated with presynaptic terminals. Point mutations in the α-synuclein gene (A30P and A53T), as well as triplication of the wild-type (WT) locus, have been linked to autosomal dominant Parkinson's. How these alterations might contribute to disease progression is unclear. Using the genetically tractable yeast Saccharomyces cerevisiae as a model system, we find that both the WT and the A53T isoforms of α-synuclein initially localize to the plasma membrane, to which they are delivered via the classical secretory pathway. In contrast, the A30P mutant protein disperses within the cytoplasm and does not associate with the plasma membrane, and its intracellular distribution is unaffected by mutations in the secretory pathway. When their expression is elevated, WT and A53T, but not A30P, are toxic to cells. At moderate levels of expression, WT and A53T induce the cellular stress (heat-shock) response and are toxic to cells bearing mutations in the 20S proteasome. Our results reveal a link between plasma membrane targeting of α-synuclein and its toxicity in yeast and suggest a role for the quality control (QC) system in the cell's effort to deal with this natively unfolded protein.
THE protein α-synuclein (α-Syn) has been implicated in the pathogenesis of Parkinson's disease (PD), a common movement disorder characterized by a loss of dopaminergic neurons in the substantia nigra pars compacta (Kruger et al. 1998; Polymeropoulos 1998; Singleton et al. 2003). A membrane-associated protein, α-Syn colocalizes with synaptic vesicles and may function in their recycling (Cabin et al. 2002). It is also a major component of Lewy bodies (Spillantini et al. 1997), ubiquitinated cytoplasmic protein aggregates that are a characteristic pathological feature of PD. Two missense mutations in the gene encoding α-Syn, A30P and A53T, have been found in cases of autosomal dominant PD (Polymeropoulos et al. 1997; Kruger et al. 1998). All three isoforms—WT, A53T, and A30P—are toxic to dopaminergic neurons when expressed in Drosophila and transgenic mice (Feany and Bender 2000; Kahle et al. 2000; Masliah et al. 2000; van der Putten et al. 2000; Matsuoka et al. 2001). A recently described family with autosomal dominant PD was found to have triplication of the wild-type α-Syn locus (Singleton et al. 2003), suggesting that overexpression of α-Syn may be sufficient to trigger the disease.
Much effort has been directed toward understanding the role of α-Syn in PD progression. In cell line models, the A30P mutant allele of α-Syn was found to compromise proteasome activity and increase sensitivity to mitochondria-dependent apoptosis (Tanaka et al. 2001). Indeed, proteasome inhibition by lactacystin leads to degeneration of dopaminergic cell bodies and accumulation of α-Syn-containing cytoplasmic inclusions (McNaught et al. 2002). Moreover, families with recessive PD have homozygous mutations in the Parkin gene, which encodes an E3 ubiquitin ligase (Kitada et al. 1998). Brains from these individuals contain nonubiquitinated o-glycosylated α-Syn, a substrate for Parkin (Shimura et al. 2001). Lack of functional Parkin may account for the absence of Lewy bodies in these patients. Mitochondrial dysfunction has also been demonstrated in PD (Jenner and Olanow 1998; Betarbet et al. 2002a,b). Mitochondrial toxins and oxidative stress stimulate α-Syn accumulation and selective loss of dopaminergic neurons (Betarbet et al. 2002a).
Key cellular processes, including membrane trafficking, signaling cascades, protein aggregation, and regulated protein turnover are conserved between human and the budding yeast, Saccharomyces cerevisiae. This suggests that fundamental mechanisms may be studied in yeast, a system that can be readily manipulated by genetic methods. Indeed, pathophysiological processes in neurological diseases such as Creutzfeldt-Jacob and Huntington's have been modeled in yeast (Lindquist 1997; Krobitsch and Lindquist 2000). For example, aggregation of the huntingtin protein (Ht) containing expanded polyQ tracts has been observed (Krobitsch and Lindquist 2000), leading to altered gene transcription, including the upregulation of chaperones (Hughes et al. 2001).
Here we have expressed α-Syn in yeast in an attempt to gain insight into both its biology and its pathophysiology. We have found that the WT and A53T isoforms of α-Syn initially localize to the plasma membrane and are delivered there via the secretory pathway. In contrast, the A30P isoform fails to enter the secretory pathway and disperses within the cytoplasm. Consistent with these differences, WT and A53T are toxic when expressed at elevated levels while A30P is not. While our manuscript was in preparation, a similar study was published (Outeiro and Lindquist 2003). Of the overlapping questions addressed in our two studies, results presented here are largely in concordance with those of Outeiro and Lindquist. We also extend their findings in several important ways. First, as mentioned above, we show that both WT and A53T are delivered to the cell's periphery via the secretory pathway, consistent with bona fide plasma membrane targeting and arguing against mere sequestration. Second, we demonstrate that A30P does not enter the secretory pathway, which may account for both its failure to be delivered to the plasma membrane and its failure to elicit toxicity when overexpressed. Third, we show that WT and A53T induce the cellular heat-shock response, whereas A30P does not—paralleling their differences in intracellular localization and toxicity. Finally, we demonstrate that moderate (nonelevated) levels of α-Syn are toxic to cells bearing mutations in the 20S catalytic subunit of the proteasome, consistent with a link between α-Syn expression and proteasome dysfunction.
MATERIALS AND METHODS
Yeast strains, gene constructions, and cultivation conditions:
The yeast strains used in this study are listed in Table 1. Standard rich (YPD) and defined minimal media (SD) were prepared using standard procedures (Rose et al. 1990). Transformations were carried out as described by Elble (1992). The yeast strain HS22, harboring a pre2-1001 mutation, was isolated as a spontaneous extragenic suppressor of the null transcription phenotype of the noninduced hsp82-P2 promoter. It is derived from parental strain EVS1012. Strain G64, bearing a pre1-1001 mutation, was similarly isolated (H. Singh, G. Alba, E. V. Sambuk, S. B. Kremer and D. S. Gross, unpublished data).
TABLE 1.
Yeast strains used in this study
| Strain | Genotype | Source or reference |
|---|---|---|
| Y382 | MATα ade2 ade3 ura3 leu2 trp1 | A. Bender |
| FY23 | MATaura3-52 trp1-d63 leu2-Δ1 | F. Winston |
| MGG38 | FY23; mip1Δ | Mark Goebl |
| EVS1012 | MATα ura3-1 trp1-1 ade− leu2-3,112 his3-11,15 hsp82-P2 hsp82-P2/HIS3::ura3FOAr hsp82-ΔHSE1/lacZ::LEU2 | E. Sambuk and D. S. Gross |
| G64 | EVS1012; pre1-1001 | G. Alba and D. S. Gross |
| HS22 | EVS1012; pre2-1001 | H. Singh and D. S. Gross |
| EVS1013 | MATα ura3-1 trp1-1 ade− leu2-3,112 his3-11,15 hsp82-P2 hsp82-P2/HIS3::ura3FOAr hsp82-P2/lacZ::LEU2 | E. Sambuk and D. S. Gross |
| TW373 | MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0 | Research Genetics |
| YKL148C | TW373; sdh1Δ | Research Genetics |
| YDR529C | TW373; qcr7Δ | Research Genetics |
| YPL078C | TW373; atp4Δ | Research Genetics |
| LRB906 | MATα his3 leu2 ura3-52 | Babu et al. (2002) |
| LRB931 | MATα his3 leu2 ura3-52 sec12 | Babu et al. (2002) |
| LRB932 | MATα his3 leu2 ura3-52 sec4-2 | Babu et al. (2002) |
| LRB933 | MATα his3 leu2 ura3-52 sec14-3 | Babu et al. (2002) |
| LRB934 | MATα his3 leu2 ura3-52 sec9-4 | Babu et al. (2002) |
| LRB937 | MATα his3 leu2 ura3-52 sec23-1 | Babu et al. (2002) |
The vector pEG(KG) (2μ URA3 leu2d) was used for the expression of N-terminal glutathione S-transferase (GST)-fusion proteins (Mitchell et al. 1993). This vector exists at a moderate copy number (∼10 copies/cell) in cells cultivated in medium containing leucine; it is present in higher copy number in cells cultivated in the absence of leucine (Mitchell et al. 1993). Expression of GST-α-Syn fusion genes was under control of the GAL1 promoter. GST-α-Syn constructs were created by cloning PCR-generated DNA fragments into pEG(KG); pUAST-α-Syn, pUAST-A30P, and pUAST-A53T (kindly provided by Nancy Bonini, University of Pennsylvania) were used as templates in the PCRs. Primers annealing to the 5′ end and 3′ end of α-Syn were appended with either an XbaI or an SalI site, respectively. The forward primer was 5′-GGGTCTAGACATGGATGTATTCATGAAAGGAC-3′ and the reverse primer was 5′-TTTGTCGACTTAGGCTTCAGGTTCGTAGTC-3′. For galactose induction of GST-α-Syn fusions, cells were first grown to early logarithmic phase (A600 ∼0.5) in selective medium (lacking either uracil, for moderate expression, or leucine, for high overexpression) containing 2% sucrose. Cells were then harvested, resuspended in the same medium containing 2% galactose, and incubated for an additional 2–12 hr.
Expression of α-Syn derivatives N-terminally fused to GFP was also regulated by the GAL1 promoter; fusion genes were cloned into a pRS314 (TRP1 CEN6)-based vector, pHY314GFP (Dixon et al. 2003). GFP-α-Syn constructs were created by cloning PCR-generated DNA fragments using the α-Syn pUAST constructs as templates as described above. Primers annealing to the 5′ end and 3′ end of α-Syn were designed to incorporate SpeI and SacI sites, respectively, using forward primer 5′-GGGACTAGTATGGATGTATTCATGAAAGGAC-3′ and reverse primer 5′-TTTGAGCTCTTAGGCTTCAGGTTCGTAGTC-3′. The PCR products were cleaved with the appropriate enzymes and ligated into pHY314GFP. Y382 cells transformed with GFP-α-Syn expression vectors were grown to midlog phase in medium containing 2% sucrose lacking tryptophan, harvested, resuspended in medium containing 2% galactose lacking tryptophan, and grown for a further 2–12 hr. For experiments conducted with LRB906 and its isogenic sec4, sec9, sec12, sec14, and sec23 derivatives, GFP-α-Syn fusion genes were subcloned into pRS316 (URA3, CEN6).
DNA sequencing:
To confirm the fidelity of the PCR and cloning steps, all GFP- and GST-α-Syn fusion constructs were sequenced. Primers complementary to both Watson and Crick strands were used (Arizona State University sequencing facility).
Localization studies:
For differential interference contrast and fluorescence microscopy, cells were visualized with an Olympus UPLan F1 objective. Images were recorded with a Photometrics Cool Snap HQ CCD camera (KAF1400 Kodak chip) with Scanalytics IPLab Spectrum (version 3.6) software (Fairfax, VA). The GFP signal was visualized with an S65T filter. Samples were removed at specific time points (Figures 1–4; Tables 2 and 3) following galactose induction and several microscopic fields were captured for each sample. In the majority of experiments (Figures 1–3; Table 2), cells were grown to midlog phase in 2% sucrose and then induced as described above by the addition of 2% galactose for the times indicated (see figure and table legends). In the experiments of Figure 4 and Table 3, cultures were pregrown in 2% raffinose and then induced through addition of galactose to a final concentration of 2%.
Figure 1.—

α-Syn initially localizes to the yeast plasma membrane prior to dispersing within the cytoplasm. Y382 cells expressing GFP-α-Syn (WT) were grown to midlog phase in −Trp medium containing 2% sucrose, harvested, and resuspended in −Trp medium containing 2% galactose. Fluorescence micrographs were taken at the indicated times following resuspension.
Figure 4.—


Intracellular localization of A53T, but not that of A30P, is disrupted in sec mutants. (A) Localization of GFP and the indicated GFP-α-Syn fusion proteins in the sec9 mutant LRB934 at either 23° or 37° was determined by fluorescence and differential interference microscopy. Cultures were pregrown in 2% raffinose −Ura medium to early log, and then galactose was added to a final concentration of 2% simultaneous with temperature upshift (37° samples). Cells were harvested and photographed 2 hr later. (B) As in A, except the sec14 mutant LRB933 was employed.
TABLE 2.
Time course of intracellular localization of WT, A53T, and A30P α-Syn
| α-Syn isoform |
Time (hr) | Plasma membrane (%) |
Cytoplasm (%) | Nucleus (%) |
|---|---|---|---|---|
| WT | 0 | 0 | 0 | 0 |
| 2 | 209 (100) | 0 | 0 | |
| 4 | 90 (38) | 148 (62) | 0 | |
| 8 | 4 (18) | 230 (82) | 0 | |
| 12 | 11 (4) | 263 (96) | 0 | |
| A30P | 0 | 0 | 0 | 0 |
| 2 | 0 | 206 (93) | 15 (7) | |
| 4 | 0 | 269 (81) | 64 (19) | |
| 8 | 0 | 251 (82) | 55 (18) | |
| 12 | 0 | 164 (77) | 49 (23) | |
| A53T | 0 | 0 | 0 | 0 |
| 2 | 20 (8) | 238 (92) | 0 | |
| 4 | 38 (15) | 217 (85) | 0 | |
| 8 | 228 (59) | 157 (41) | 0 | |
| 12 | 131 (34) | 250 (66) | 0 |
Y382 cultures were grown to midlog phase in −Trp medium containing 2% sucrose, harvested, and resuspended in −Trp medium containing 2% galactose to induce expression of the indicated GFP fusion proteins. At various times thereafter, an aliquot of cells was removed and scored for subcellular localization of GFP fluorescence. Cells that exhibited clear enhancement of peripheral fluorescence were scored as localizing to the plasma membrane; otherwise, they were scored as localizing to the cytoplasm if diffusely fluorescent or to the nucleus if they exhibited enhanced nuclear fluorescence. For each time point, multiple random fields were counted.
TABLE 3.
Intracellular localization of WT, A53T, and A30P α-Syn inSEC+,sec9, andsec14 cells
| α-Syn isoform |
Plasma membrane |
Plasma membrane aggregates |
Cytoplasm | Cytoplasmic aggregates |
|---|---|---|---|---|
| SEC+ | ||||
| GFP | ||||
| 23° | 0 | 0 | 80 | 0 |
| 37° | 13 | 0 | 174 | 0 |
| WT | ||||
| 23° | 36 | 0 | 33 (9) | 0 |
| 37° | 135 | 0 | 25 | 0 |
| A53T | ||||
| 23° | 22 | 0 | 8 | 0 |
| 37° | 72 | 0 | 9 (1) | 0 |
| A30P | ||||
| 23° | 1 | 0 | 99 (2) | 0 |
| 37° | 1 | 0 | 160 (1) | 0 |
| sec9 | ||||
| GFP | ||||
| 23° | 0 | 0 | 107 (2) | 0 |
| 37° | 0 | 0 | 96 (15) | 0 |
| WT | ||||
| 23° | 27 | 0 | 27 (7) | 1 |
| 37° | 18 | 16 | 31 (9) | 0 |
| A53T | ||||
| 23° | 71 | 5 | 47 (8) | 0 |
| 37° | 16 | 9 | 23 (10) | 0 |
| A30P | ||||
| 23° | 0 | 0 | 49 (6) | 0 |
| 37° | 0 | 0 | 40 (3) | 3 |
| sec14 | ||||
| GFP | ||||
| 23° | 0 | 0 | 36 | 0 |
| 37° | 0 | 0 | 30 (9) | 0 |
| WT | ||||
| 23° | 39 | 1 | 9 (2) | 0 |
| 37° | 9 | 8 | 10 | 3 |
| A53T | ||||
| 23° | 114 | 0 | 10 (1) | 0 |
| 37° | 10 | 15 | 15 | 14 |
| A30P | ||||
| 23° | 0 | 0 | 32 (9) | 1 |
| 37° | 0 | 0 | 39 (6) | 0 |
Yeast strains LRB906 (SEC+), LRB934 (sec9), and LRB933 (sec14), expressing GFP alone or GFP-α-Syn fusion proteins as indicated, were pregrown at 23° in 2% raffinose to early log phase. Galactose was then added at a final concentration of 2%, and cells were cultivated for an additional 2 hr at either 23° or 37°. Plasma membrane, uniform enhancement of fluorescence at cell periphery; plasma membrane aggregates, enhancement of peripheral fluorescence characterized by the presence of punctate foci; cytoplasm, fluorescence restricted to cytosol and internal organelles (those cells exhibiting enhanced nuclear fluorescence are indicated in parentheses); cytoplasmic aggregates, fluorescence restricted to the cytoplasm with one or more punctate foci.
Figure 3.—

A53T, like WT, principally localizes to the plasma membrane, while A30P remains in the cytoplasm. (A) Y382 cells expressing WT, A30P, and A53T α-Syn were grown to midlog phase, galactose induced, and photographed as in Figure 1. (B) As in A; images were taken after two doublings (∼4 hr). White arrow, α-Syn localization at the bud neck; black arrows, α-Syn localization within the nucleus.
Protein preparation and immunoblotting:
Cells bearing GST-α-Syn expression vectors were grown to midlog phase in medium lacking leucine and containing 2% sucrose. They were then harvested and resuspended in medium lacking leucine and containing 2% galactose and grown for a further 3 hr. Polyclonal anti-GST antibodies (Abs) were obtained from Sigma; polyclonal anti-Cdc34 Abs were provided by Mark Goebl (Indiana University). Yeast lysate preparations and immunoblot analysis were carried out as described previously (Dixon et al. 2003).
Fractionation of whole-cell lysates:
Y382 cultures expressing GST, GST-α-Syn(WT), and GST-α-Syn(A30P) were grown at 30° to midlog phase in −Leu 2% sucrose medium, harvested, and then resuspended in −Leu 2% galactose for 4 hr. Cells were lysed and 0.5 mg of each extract was fractionated on a Sephacryl S-300HR column (Amersham Biosciences) that had been previously equilibrated in 150 mm NaCl, 50 mm Tris-HCl (pH 7.5), 0.5 mm EDTA. Protein from 1-ml fractions was precipitated with 10% trichloroacetic acid. The precipitate was washed with acetone, resolubilized in 20 μl of SDS-polyacrylamide gel electrophoresis loading buffer, and subjected to immunoblot analysis.
Expression assays:
The yeast strain EVS1013, bearing an integrated stress-responsive hsp82-lacZ reporter gene, was transformed with each of the four GST-containing expression vectors (WT, A30P, A53T, or GST alone). Transformants were grown overnight in 2% galactose −Ura medium at 30° to midlog phase (A600 = 0.3–0.7) and then split into two aliquots, control and heat shock. For heat shock, cells were incubated 45 min at 39° followed by a 30-min recovery at 30°. β-Galactosidase activity was determined as previously described by Erkine and Gross (2003). The reporter gene bears the promoter and 5′ coding region of a highly inducible hsp82 mutant termed P2 (McDaniel et al. 1989). In this construct, hsp82 sequence extends from −806 to +308 (relative to the AUG) and is fused in frame to the lacZ ORF at codon 8. Two-tailed t-tests were conducted to test the statistical significance of differences in β-galactosidase activity exhibited by the GST, WT, A30P, and A53T transformed strains. t-values were calculated using the extended t-test (Norman and Streiner 2000). Confidence levels are reported as P-values, defined as the probability that no significant difference exists between the two groups of data being compared.
RESULTS
α-Syn utilizes the secretory pathway to localize to the yeast plasma membrane:
α-Syn is an amphipathic protein that principally associates with phospholipid membranes and presynaptic vesicles in neurons (George et al. 1995; Irizarry et al. 1996; McLean et al. 2000). To investigate whether α-Syn likewise localizes to membranous structures in yeast, we constructed a strain expressing a GFP-α-Syn fusion under control of the galactose-inducible GAL1 promoter and monitored its localization by fluorescence microscopy. Consistent with its membrane-binding behavior in human cells, α-Syn initially localizes to the plasma membrane in yeast strain Y382 (Figure 1, 2–4 hr after galactose induction). At later times, however, α-Syn accumulates in the cytoplasm (6 hr), eventually being recruited away from the plasma membrane (8 hr; see also Figure 3A). A similar pattern of localization was observed in a genetically unrelated strain, FY23 (data not shown).
The secretory pathway has been implicated in delivering proteins to the plasma membrane and is a highly regulated and evolutionarily conserved process (Ferro-Novick and Jahn 1994; Rothman 1994). As α-Syn has been detected in the rough endoplasmic reticulum, small vesicles, and the Golgi apparatus in neurons, it has been suggested that α-Syn trafficking may employ this pathway (Gosavi et al. 2002; Mori et al. 2002). To test whether the secretory pathway is involved in delivering α-Syn to the plasma membrane in yeast and to distinguish between sequestration and bona fide plasma membrane targeting, we monitored the localization of GFP-α-Syn in each of five conditional secretory pathway defective yeast strains (Novick et al. 1980). When shifted to the nonpermissive temperature, these mutants block secretory traffic at a specific step while allowing protein synthesis to continue, resulting in the accumulation of protein at the affected internal compartment. The sec mutants are defective in carrying out discrete steps in the secretory pathway, including ER to Golgi transport (sec12 and sec23) (Barlowe et al. 1994), intra-Golgi trafficking (sec14) (Bankaitis et al. 1989), secretory vesicle docking at the plasma membrane (sec4) (Brennwald et al. 1994), and secretory vesicle fusion with the plasma membrane (sec9) (Lehman et al. 1999).
To confirm that the secretory pathway had been compromised in each sec mutant, we monitored the localization of Yck2, a protein that employs the classical secretory pathway for its delivery to the plasma membrane (Babu et al. 2002). Yeast cells expressing GFP-tagged Yck2 or α-Syn (each regulated by the GAL1 promoter) were induced by the addition of galactose and either maintained at the permissive temperature (23°) or shifted to the nonpermissive temperature (37°) for 90 min. At the permissive temperature, both Yck2 and α-Syn localized to the plasma membrane in all strains tested (Figure 2). Interestingly, in SEC+ strains, α-Syn frequently accumulated at the bud neck, a site of new membrane synthesis [see Figures 1 (2 and 3 hr), 2 (SEC+), and 3B (white arrow)]. In the sec4 and sec12 mutants, α-Syn formed distinctive, plasma membrane-associated punctate structures, suggesting that even at the permissive temperature, the proteins encoded by SEC4 and SEC12 are not fully functional. When the cells were shifted to the nonpermissive temperature, plasma membrane localization was blocked (Figure 2; Table 2). This block in membrane trafficking was specific to the sec mutants and is not a property of α-Syn, since its localization was unaffected by elevated temperature in the wild-type (SEC+) strain (Figure 2). Together, these observations indicate that α-Syn is delivered to the plasma membrane by the classical secretory pathway.
Figure 2.—
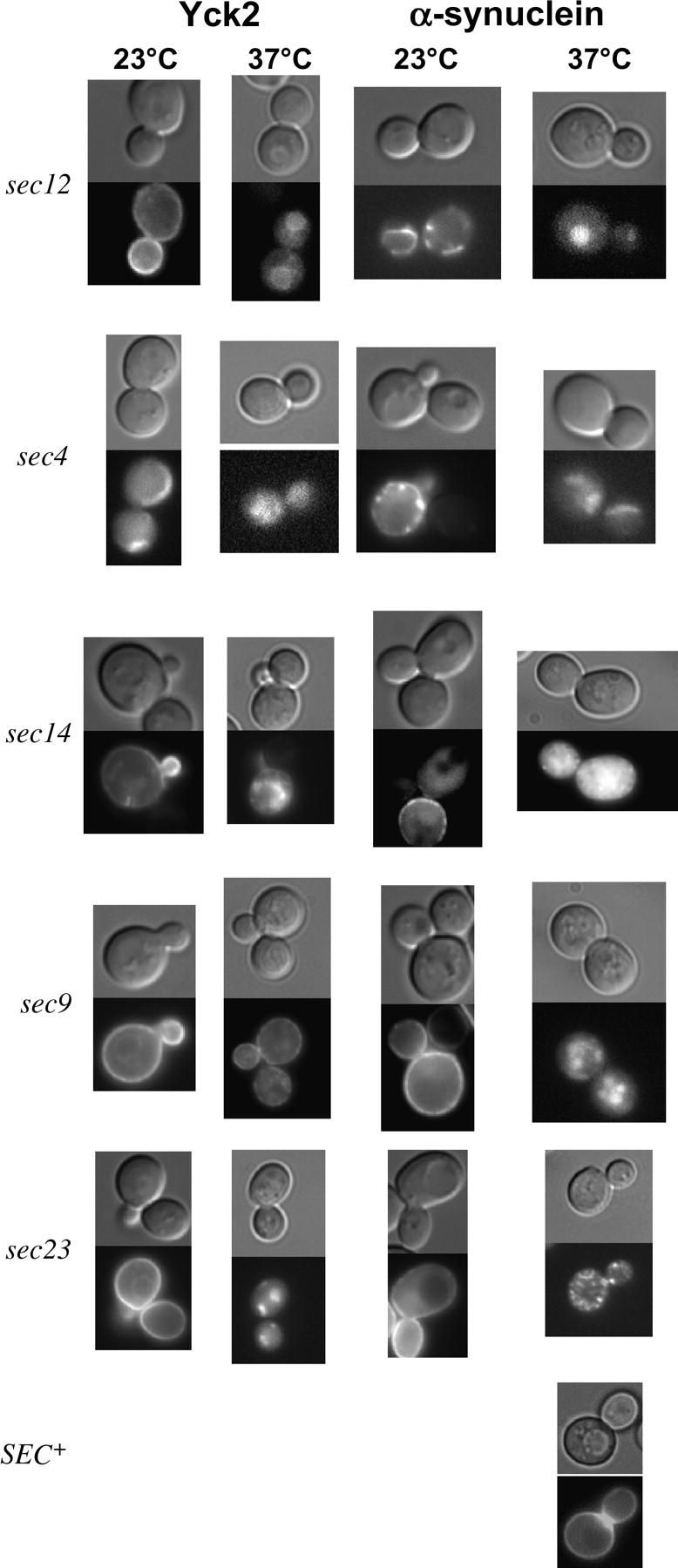
α-Syn targeting to the plasma membrane is blocked in sec mutants. Isogenic strains bearing the indicated sec mutations and expressing either GFP-Yck2 or GFP-α-Syn (WT) were maintained at the permissive temperature (23°) or shifted to the nonpermissive temperature (37°) for 90 min simultaneous with the addition of 2% galactose (cells were pregrown at 23° in 2% sucrose). The isogenic SEC+ strain is depicted at the bottom. Each composite consists of a differential interference contrast (DIC) image and corresponding fluorescence (GFP) micrograph.
A53T but not A30P localizes to the plasma membrane in yeast:
It has been reported that the α-Syn mutant A30P fails to bind to membranes in either HeLa cells or primary neuronal cultures, whereas WT and A53T do so readily (Jensen et al. 1998; Cole et al. 2002). To determine whether either A30P or A53T localizes to the plasma membrane in yeast, strains expressing galactose-inducible, GFP-tagged versions of A30P and A53T were constructed. Localization was monitored in a time course experiment and compared to WT α-Syn. Similar to WT, A53T targeted to the plasma membrane (Figure 3). In certain strain backgrounds (Figure 3, Table 2), although not in others (Table 3), A53T targeting occurred with delayed kinetics and reduced efficiency. In contrast, the A30P mutant failed to target to the plasma membrane at any time point and in any strain background and instead displayed a tendency to accumulate in the nucleus (Figure 3B, black arrows; Tables 2 and 3). Therefore, as in human cells, A53T exhibits prominent membrane association, whereas A30P does not.
Plasma membrane localization of A53T is disrupted by mutations in the sec pathway:
To strengthen the notion that A53T localizes to the plasma membrane, we investigated whether its intracellular localization is affected in sec mutants. We reasoned that if A53T is targeted to the plasma membrane, it should exhibit a sensitivity to defects in the sec pathway similar to that of WT. In contrast, A30P should not be sensitive to sec mutations, given that it fails to localize to the plasma membrane in a SEC+ strain. As predicted, the intracellular localization of A53T is altered in each of the four sec mutants analyzed—sec4, sec9, sec12, and sec14 (Figure 4; Table 3; data not shown). In the case of sec9, sec12, and sec14, the peripheral localization of A53T is disrupted at the nonpermissive temperature; in the case of sec4, sec9, and sec14, A53T is additionally (or instead) prone to aggregation. By contrast, the intracellular distribution of A30P is unaffected by the sec mutations. Together, these data suggest that A53T, like WT, enters the secretory pathway, whereas A30P does not.
The WT and A53T isoforms of α-Syn are toxic to yeast, while A30P is not:
Because transgenic overproduction of α-Syn results in dopaminergic cell death in both mice and Drosophila (Feany and Bender 2000; Kahle et al. 2000; Masliah et al. 2000; van der Putten et al. 2000; Matsuoka et al. 2001), we tested whether its overexpression likewise caused cell death in yeast. To achieve overproduction, we expressed GST-α-Syn fusions from a plasmid whose copy number can be increased by altering the composition of the growth medium (see materials and methods). As shown by the spot dilution assays of Figure 5A, WT and A53T are toxic when expressed at higher levels (SGal −Leu medium), whereas A30P is not. Importantly, as shown by immunoblot analysis, this difference in toxicity is not due to differences in expression (Figure 5B). Overexpression of WT and A53T, but not of A30P, is toxic in a variety of genetic backgrounds, including Y382, TW373, and FY23 (Figure 5A; data not shown). Interestingly, the difference in α-Syn expression in cells grown on SGal −Leu vs. those grown on SGal −Ura is just 30% (3-hr galactose induction; data not shown), demonstrating that a small increase in the level of either WT or A53T is sufficient to elicit toxicity.
Figure 5.—
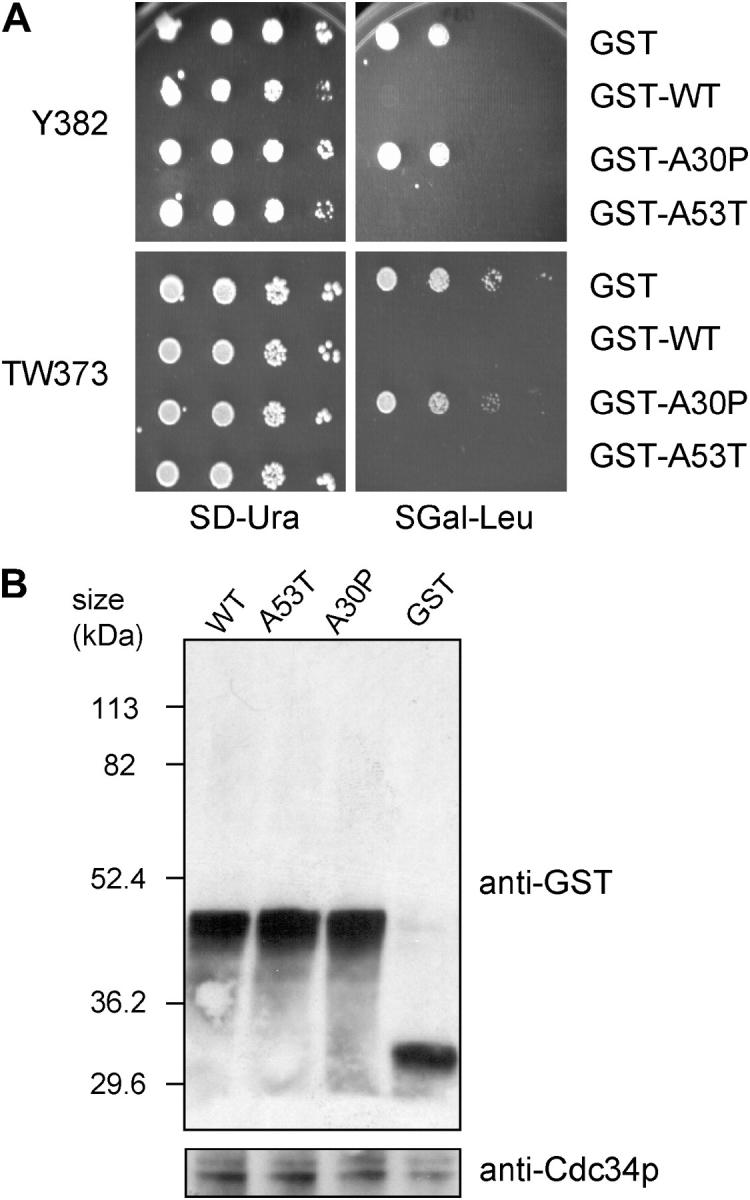
Overproduced WT and A53T are toxic in yeast, while A30P is not. (A) Y382 or TW373 cells carrying the indicated GST fusions were grown to stationary phase. Tenfold serial dilutions were spotted onto synthetic medium lacking uracil and containing 2% glucose (dextrose) (SD −Ura) or synthetic medium lacking leucine and containing 2% galactose (SGal −Leu) and incubated at 30° for 3 days. (B) Y382 cultures expressing the indicated GST fusions were grown to midlog phase in −Leu medium and shifted to galactose for 3 hr to induce expression of the indicated fusion proteins. Lysates were prepared and subjected to immunoblot analysis using an anti-GST antibody. The blot was then reprobed with an anti-Cdc34 antibody as a loading control.
Both WT and A30P form heterogeneous-sized soluble aggregates under conditions of overproduction:
To investigate whether the difference in toxicity elicited by WT vs. A30P α-Syn stems from a difference in their tendency to aggregate, we isolated whole-cell extracts (WCEs) from Y382 cells expressing GST fusions of WT and A30P, as well as GST alone. Cells were galactose induced for 4 hr, and then WCEs were isolated and fractionated on a Sephacryl S-300HR column. GST-containing proteins were detected by immunoblotting. As shown in Figure 6, soluble complexes of both WT and A30P eluted in all but the lowest molecular weight fractions. This behavior is in marked contrast to GST alone, whose elution peaked sharply at ∼25 kDa, as expected. It is also atypical of GST fusion proteins expressed in yeast (Dixon et al. 2003). The presence of soluble GST-α-Syn aggregates likely reflects self-aggregation and is similar to what has been observed for recombinant WT and A30P α-synucleins incubated in solution at physiological ionic strength (Hoyer et al. 2004). Importantly, this analysis does not rule out formation of large, insoluble aggregates. Under analogous conditions of elevated expression, such aggregates, in the form of cytoplasmic inclusions, have been detected for WT (and A53T) α-Syn but not for A30P (Outeiro and Lindquist 2003).
Figure 6.—

WT and A30P form heterogeneous-sized aggregates under conditions of overproduction. WCEs isolated from Y382 cells (pregrown in 2% sucrose −Leu medium; galactose induced for 4 hr) were fractionated on a Sephacryl S-300HR column. Protein from 1-ml fractions was precipitated with 10% trichloroacetic acid. The precipitate was washed with acetone, resolubilized in 20 μl of SDS-polyacrylamide gel electrophoresis loading buffer, and subjected to immunoblot analysis. The column was calibrated using blue dextran (2 MDa), thyroglobulin (669 kDa), apoferritin (440 kDa), catalase (232 kDa), aldolase (158 kDa), ovalbumin (44 kDa), chymotrypsinogen A (25 kDa), and myoglobin (17 kDa).
Proteasomal mutations enhance α-Syn-induced toxicity:
Given the suggestive link between α-Syn accumulation, Lewy body formation, and impaired proteasomal function in neuronal cells (Lindersson et al. 2004), we asked whether moderate levels of α-Syn might affect the viability of cells bearing a pre2 mutation in the 20S proteasome. pre2 mutants exhibit defects in chymotrypsin-like proteolysis, stress response, and ubiquitin-mediated protein degradation (Heinemeyer et al. 1991, 1993). Moderate expression of GST-α-Syn fusions was induced by growth on medium containing 2% galactose. While expression of each α-Syn isoform had little effect on the growth of PRE+ cells, that of A53T significantly impaired growth of the pre2 strain (Figure 7, A and B, GAL medium; compare A53T-expressing cells with those expressing GST alone or those transformed with an empty vector). WT and A30P also appeared to exacerbate the pre2 slow growth phenotype, but their effect was less pronounced. When pre2 cells were grown under conditions that repress expression of α-Syn (DEX), growth of the α-Syn transformants was comparable to that of the negative controls transformed with either GST or pRS316. As similar results were obtained with a second 20S proteasomal mutant (pre1; data not shown), these results are consistent with the notion that α-Syn, particularly its A53T isoform, is synthetically toxic in combination with mutations in the 20S proteasome.
Figure 7.—


α-Syn expression impairs growth of a pre2 proteasomal mutant. (A) PRE+ and pre2-1001 strains (EVS1013 and HS22, respectively) were transformed with GST-α-Syn expression vectors and streaked onto synthetic medium lacking uracil. Transformants were grown under either repressing (2% dextrose; DEX) or inducing (2% galactose; GAL) conditions. Plates were incubated at 30° for either 2.5 or 3 days, respectively (in the case of PRE+) and for 6.5 or 9 days (in the case of pre2-1001). Vector was pRS316. (B) The isogenic strains EVS1012 and HS22 were transformed with GST-α-Syn expression vectors as above and grown in liquid SD −Ura to stationary phase. Fivefold serial dilutions were spotted onto −Ura medium containing either 2% dextrose (DEX) or 2% galactose + 2% raffinose (GAL). Plates were incubated at 30° either for 2 or 3 days (PRE+) or for 5 or 8 days (pre2).
Expression of α-Syn induces the heat-shock response:
Given the genetic interaction between α-Syn and the 20S proteasome suggested above, we wished to know whether α-Syn affected other cellular quality control (QC) systems. This might be expected, given that α-Syn has been linked to oxidative stress in neurons (Jenner and Olanow 1998; Betarbet et al. 2002a). Moreover, chaperones diminish the aggregation of abnormal proteins by interacting with folding intermediates and off-pathway folding products (Gething 1997), and their increased expression might be expected to reduce the cytotoxicity of α-Syn, itself a natively unfolded protein (Syme et al. 2002). We therefore tested the possibility that α-Syn induces the heat-shock transcriptional response. To conduct this test, we employed a strain bearing an integrated, stress-responsive reporter gene, hsp82-lacZ. As illustrated in Figure 8A, hsp82-lacZ is strongly induced (>20-fold) by an acute heat shock (30° to 39° shift for 45 min). It is also induced, albeit to a lesser degree, by oxidative stress (exposure to hydrogen peroxide), consistent with the relative responsiveness of HSP genes to thermal and oxidative stresses (Raitt et al. 2000).
Figure 8.—
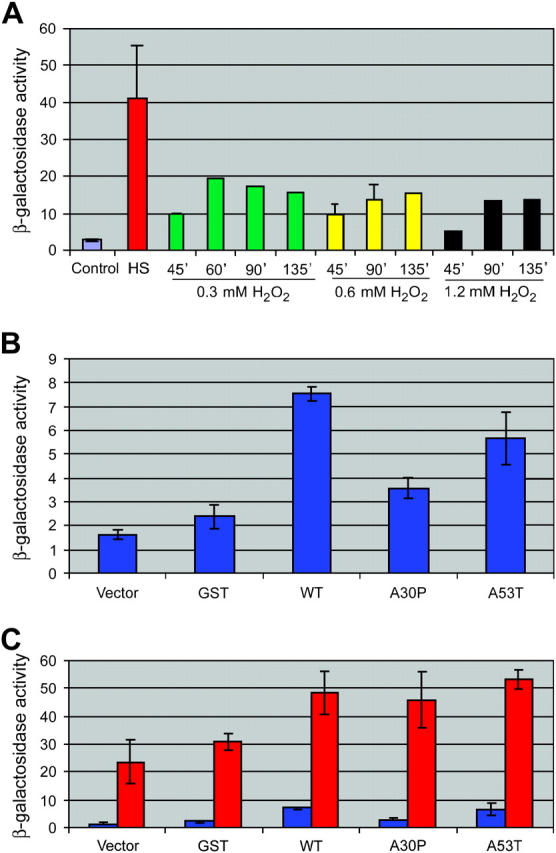
α-Syn induces the heat-shock response. (A) Induction of hsp82-lacZ in response to thermal and oxidative stress. Strain EVS1013 was maintained at 30°, subjected to a heat shock (HS), or exposed to 0.3, 0.6, or 1.2 mm hydroperoxide for the times indicated. Cells were harvested and lysed, and extracts were assayed for β-galactosidase activity (expressed in Miller units). For control, heat-shocked, and 0.6 mm hydroperoxide-treated cultures, the means of three to six independent assays ±SEM are depicted. (B) Induction of hsp82-lacZ in response to moderate overexpression of α-Syn. EVS1013 cells, transformed with the plasmids indicated, were cultivated overnight in synthetic medium lacking uracil and containing 2% galactose and then processed as above (n = 8 for GST, n = 10 for WT, and n = 11 for the others). (C) Induction of hsp82-lacZ in α-Syn-expressing cells either subjected to an acute heat shock (red) or maintained at 30° (blue) (n = 3). The data presented in B and C represent replicate assays of the same transformants. Independent transformants gave essentially identical results.
Moderate expression of α-Syn was likewise stressful, as both WT and A53T induced hsp82-lacZ transcription to a significantly greater degree (approximately threefold) than did GST alone (P < 0.02; two-tailed t-test) (Figure 8B). A30P expression was far less stressful. Statistical comparison of WT- vs. A53T-expressing cells suggests that there is no significant difference between them, whereas a significant difference exists between WT and A30P (P < 0.001). Importantly, by immunoblot analysis, GST, WT, A30P, and A53T were uniformly expressed in these transformants (data not shown), so differences in hsp82-lacZ responsiveness cannot be attributed to different levels of α-Syn expression. Interestingly, α-Syn-expressing cells show robust hsp82-lacZ transcription following an acute heat-shock, which exceeds that seen in non α-Syn-expressing cells (Figure 8C). Stress elicited by α-Syn may therefore be additive (or synergistic) with thermal stress. We conclude that accumulation of WT and A53T α-Syn per se triggers the heat-shock response, consistent with their ability to induce toxicity at elevated expression levels. (Note that we consider the terms “stress” and “toxicity” to have distinct meanings. A “stressful” agent is one that activates the heat-shock response, but does not, by itself, impair growth rate. A “toxic” agent, on the other hand, impairs cell growth. Thus, moderate levels of α-Syn are stressful, whereas elevated levels are toxic.)
DISCUSSION
α-Syn is delivered to the plasma membrane via the classical secretory pathway:
α-Syn has been reported to bind membranes in a selective manner both in vivo and in vitro (reviewed in Lucking and Brice 2000). Consistent with this, we have found that when expressed in S. cerevisiae, α-Syn initially localizes to the plasma membrane. Two lines of evidence suggest that α-Syn binds to the yeast plasma membrane and is not simply sequestered at the cell's periphery. First, similar to its behavior in mammalian brain cells (Maroteaux et al. 1988), we found that α-Syn initially accumulates at sites of new membrane formation (bud sites). Second, targeting of α-Syn is disrupted in sec mutants defective for discrete steps in the secretory pathway, including ER vesicle budding, ER-derived vesicle fusion at the Golgi, intra-Golgi trafficking, secretory vesicle docking, and secretory vesicle fusion to the plasma membrane. These disruptions closely parallel those of Yck2, a protein known to associate with the yeast plasma membrane (Babu et al. 2002), and are reminiscent of findings in mammalian cells that implicate ER to Golgi signaling in α-Syn trafficking (Nishimura et al. 1999). It is thus likely that α-Syn is delivered to the plasma membrane through its association with vesicular intermediates of the classical secretory pathway. Our data do not distinguish whether the protein itself is sorted into vesicles or associates with other cytosolic proteolipid components.
We have further investigated the localization of two α-Syn point mutants associated with autosomal dominant PD, A30P and A53T. We find that like WT, A53T is also delivered to the yeast plasma membrane via the classical secretory pathway. In contrast, A30P remains within the cytoplasm and does not enter the sec pathway. Therefore, consistent with its behavior in mammalian cells (Jensen et al. 1998; Jo et al. 2002; Bussell and Eliezer 2003), the A30P mutation appears to abolish the protein's ability to target to specific membranes in S. cerevisiae. Importantly, we failed to observe aggregate formation with either WT or A53T (up to 12 hr postinduction) in SEC+ strains of varied genetic backgrounds. It is worth noting that others report seeing aggregation of WT and A53T, but only when the GAL1-regulated GFP fusion genes are integrated and present in two copies. Similar to our findings, α-Syn fusions expressed from single copy genes fail to aggregate, and those expressed from 2μ plasmids do so very infrequently (Outeiro and Lindquist 2003). Therefore, by increasing the intracellular concentration of WT or A53T just twofold, their tendency to aggregate is markedly enhanced.
WT and A53T α-Syn are stressful to yeast when expressed at moderate levels and are toxic when overexpressed:
Given that α-Syn is a natively unfolded protein in aqueous solution (Weinreb et al. 1996; Kim 1997), it has been suggested that a diminished capacity of the cell's QC system may contribute to age-dependent, α-Syn-associated neurodegeneration (Taylor et al. 2002; Berke and Paulson 2003). To test whether the QC system plays a role in permitting yeast to cope with ectopically expressed α-Syn, we investigated the role of two QC systems, the cellular stress response and the 20S proteasome, in maintaining viability. We found that, when expressed at nonelevated levels (on SGal −Ura medium), both WT and A53T induced the cellular stress (heat-shock) response. Nonetheless, cells grew at a rate indistinguishable from that of cells expressing GST, suggesting that when expressed at these levels, WT and A53T are adequately handled by the cellular QC system. However, coupled with mutations in the 20S proteasome, moderate levels of A53T are toxic. And when their expression is elevated, both WT and A53T are toxic, even in the absence of a proteasomal mutation. Interestingly, prior exposure to a brief 42° heat shock protects cells from the toxic effects of subsequently overexpressing either WT or A53T α-Syn (S. N. Witt and T. R. Flower, personal communication), implicating a role for heat-shock protein chaperones in ameliorating α-Syn-induced toxicity.
Our data are compatible with either of two (not mutually exclusive) possibilities. First, a dysfunctional proteasome could allow α-Syn to accumulate to sufficiently high levels such that it could enter cellular compartments from which it would be otherwise excluded. However, this is unlikely, since we have found no evidence that WT, A53T, or A30P α-Syn accumulates to elevated levels in the pre2 mutant (data not shown). Second, it is possible that both 20S subunit mutations and α-Syn diminish the activity of the proteasome, so that when present together, proteasome activity is drastically impaired, resulting in cell death. Consistent with this, α-Syn filaments and oligomers have been shown to inhibit proteasomal function by markedly reducing its chymotrypsin-like activity (Lindersson et al. 2004). Thus, the accentuated slow growth (or lethal) phenotype conferred by moderate expression of α-Syn (particularly A53T) in the pre2 and pre1 mutants may stem from a further loss of proteasomal function. Also consistent with this, GFPu, an unstable GFP derivative that acts as a reporter for general proteasome activity (Bence et al. 2001), was found to specifically accumulate in yeast cells expressing high levels of any one of the three α-Syn isoforms (Outeiro and Lindquist 2003).
A potential concern is that the presence of the GST tag may materially affect the behavior of the α-Syn derivatives studied here. However, WT- and A53T-specific toxicity has also been seen in an unrelated yeast strain using untagged derivatives (Outeiro and Lindquist 2003). So too has α-Syn-induced impairment of proteasomal activity, as discussed above. Moreover, GFP-tagged α-synucleins have been previously shown to behave similarly to their untagged counterparts (Outeiro and Lindquist 2003). Thus, it is likely that α-Syn fusions behave similarly to their untagged counterparts under the conditions that we have employed, although we cannot discount the possibility that the mechanism of toxicity operative here differs from that employed by the native proteins.
Insights into the mechanism of α-Syn-mediated toxicity:
Models for explaining α-Syn toxicity generally invoke its nucleated polymerization as central to the toxic process. Indeed, dopaminergic neurons in PD brains are characterized by the presence of Lewy bodies, which are aggregates of α-Syn and other proteins. Supporting this view, when expressed at elevated levels in S. cerevisiae, WT and A53T form cytoplasmic inclusions and are toxic to yeast, while under identical conditions of expression, A30P neither forms aggregates nor is toxic (Outeiro and Lindquist 2003). The absence of A30P toxicity in yeast may relate to its inability to associate with the plasma membrane and, perhaps, other membranes. This in turn could be a consequence of proline, an α-helix breaker, blocking the unfolded to folded transition in α-Syn thought to be crucial for α-Syn interaction with phospholipid membranes (Chandra et al. 2003). Indeed, the A30P, but not the A53T, mutation decreases the affinity of α-Syn for lipid surfaces (Bussell and Eliezer 2004). By contrast, A30P toxicity in mammalian cells may stem from a loss of function, leading to mistargeting (and possible sequestration of WT α-Syn in inappropriate intracellular compartments). Note that our experiments do not address whether insoluble aggregates or cytoplasmic inclusions form as a consequence of elevated expression of either WT or A53T, as we have monitored subcellular localization only under conditions of moderate expression.
Therefore, while our data do not provide a definitive explanation for the underlying cause of α-Syn-induced toxicity, they do indicate that α-Syn's association with membranes, the plasma membrane in particular, positively correlates with its toxicity. As α-Syn undergoes an unfolded to folded transition upon binding to negatively charged phospholipid membranes (Chandra et al. 2003), it is possible that α-Syn-induced toxicity arises from correctly folded WT and A53T polypeptides that have entered the secretory pathway and targeted the plasma membrane. Binding of α-Syn to cellular membranes might influence its self-assembly, promoting formation of soluble α-Syn protofibrils or insoluble α-Syn filaments (Rochet et al. 2004) (note that the latter would not have been detectable in the WCE fractionation of Figure 6). As discussed above, such α-Syn complexes can impair proteasome function (Lindersson et al. 2004) and thus, at least in theory, could account for the isoform-specific toxicity that we and others see. Future experiments will be necessary to test this and other possibilities.
Acknowledgments
We thank Lee Ellen Brunson and Jorge Herrera-Diaz for technical assistance; Lucy Robinson for helpful discussions; and Nancy Bonini, Lucy Robinson, and Mark Goebl for their generous gifts of plasmids, yeast strains, and antibodies. This work was supported by a grant from the National Institutes of Health (GM-45842) to D.S.G. and by grants from the National Science Foundation (MCB 0090403) and the Parkinson's Disease Resource of Northwest Louisiana to N.M.
References
- Babu, P., J. D. Bryan, H. R. Panek, S. L. Jordan, B. M. Forbrich et al., 2002. Plasma membrane localization of the Yck2p yeast casein kinase 1 isoform requires the C-terminal extension and secretory pathway function. J. Cell Sci. 115: 4957–4968. [DOI] [PubMed] [Google Scholar]
- Bankaitis, V. A., D. E. Malehorn, S. D. Emr and R. Greene, 1989. The Saccharomyces cerevisiae SEC14 gene encodes a cytosolic factor that is required for transport of secretory proteins from the yeast Golgi complex. J. Cell Biol. 108: 1271–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlowe, C., L. Orci, T. Yeung, M. Hosobuchi, S. Hamamoto et al., 1994. COPII: a membrane coat formed by Sec proteins that drive vesicle budding from the endoplasmic reticulum. Cell 77: 895–907. [DOI] [PubMed] [Google Scholar]
- Bence, N. F., R. M. Sampat and R. R. Kopito, 2001. Impairment of the ubiquitin-proteasome system by protein aggregation. Science 292: 1552–1555. [DOI] [PubMed] [Google Scholar]
- Berke, S. J., and H. L. Paulson, 2003. Protein aggregation and the ubiquitin proteasome pathway: gaining the UPPer hand on neurodegeneration. Curr. Opin. Genet. Dev. 13: 253–261. [DOI] [PubMed] [Google Scholar]
- Betarbet, R., T. B. Sherer, D. A. Di Monte and J. T. Greenamyre, 2002. a Mechanistic approaches to Parkinson's disease pathogenesis. Brain Pathol. 12: 499–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betarbet, R., T. B. Sherer and J. T. Greenamyre, 2002. b Animal models of Parkinson's disease. BioEssays 24: 308–318. [DOI] [PubMed] [Google Scholar]
- Brennwald, P., B. Kearns, K. Champion, S. Keranen, V. Bankaitis et al., 1994. Sec9 is a SNAP-25-like component of a yeast SNARE complex that may be the effector of Sec4 function in exocytosis. Cell 79: 245–258. [DOI] [PubMed] [Google Scholar]
- Bussell, R., Jr., and D. Eliezer, 2003. A structural and functional role for 11-mer repeats in alpha-synuclein and other exchangeable lipid binding proteins. J. Mol. Biol. 329: 763–778. [DOI] [PubMed] [Google Scholar]
- Bussell, R., Jr., and D. Eliezer, 2004. Effects of Parkinson's disease-linked mutations on the structure of lipid-associated alpha-synuclein. Biochemistry 43: 4810–4818. [DOI] [PubMed] [Google Scholar]
- Cabin, D. E., K. Shimazu, D. Murphy, N. B. Cole, W. Gottschalk et al., 2002. Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking alpha-synuclein. J. Neurosci. 22: 8797–8807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra, S., X. Chen, J. Rizo, R. Jahn and T. C. Sudhof, 2003. A broken alpha-helix in folded alpha-Synuclein. J. Biol. Chem. 278: 15313–15318. [DOI] [PubMed] [Google Scholar]
- Cole, N. B., D. D. Murphy, T. Grider, S. Rueter, D. Brasaemle et al., 2002. Lipid droplet binding and oligomerization properties of the Parkinson's disease protein alpha-synuclein. J. Biol. Chem. 277: 6344–6352. [DOI] [PubMed] [Google Scholar]
- Dixon, C., L. E. Brunson, M. M. Roy, D. Smothers, M. G. Sehorn et al., 2003. Overproduction of polypeptides corresponding to the amino terminus of the F-box proteins Cdc4p and Met30p inhibits ubiquitin ligase activities of their SCF complexes. Eukaryot. Cell 2: 123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elble, R., 1992. A simple and efficient procedure for transformation of yeasts. Biotechniques 13: 18–20. [PubMed] [Google Scholar]
- Erkine, A. M., and D. S. Gross, 2003. Dynamic chromatin alterations triggered by natural and synthetic activation domains. J. Biol. Chem. 278: 7755–7764. [DOI] [PubMed] [Google Scholar]
- Feany, M. B., and W. W. Bender, 2000. A Drosophila model of Parkinson's disease. Nature 404: 394–398. [DOI] [PubMed] [Google Scholar]
- Ferro-Novick, S., and R. Jahn, 1994. Vesicle fusion from yeast to man. Nature 370: 191–193. [DOI] [PubMed] [Google Scholar]
- George, J. M., H. Jin, W. S. Woods and D. F. Clayton, 1995. Characterization of a novel protein regulated during the critical period for song learning in the zebra finch. Neuron 15: 361–372. [DOI] [PubMed] [Google Scholar]
- Gething, M. J., 1997. Protein folding. The difference with prokaryotes. Nature 388: 329, 331. [DOI] [PubMed] [Google Scholar]
- Gosavi, N., H. J. Lee, J. S. Lee, S. Patel and S. J. Lee, 2002. Golgi fragmentation occurs in the cells with prefibrillar alpha-synuclein aggregates and precedes the formation of fibrillar inclusion. J. Biol. Chem. 277: 48984–48992. [DOI] [PubMed] [Google Scholar]
- Heinemeyer, W., J. A. Kleinschmidt, J. Saidowsky, C. Escher and D. H. Wolf, 1991. Proteinase yscE, the yeast proteasome/multicatalytic-multifunctional proteinase: mutants unravel its function in stress induced proteolysis and uncover its necessity for cell survival. EMBO J. 10: 555–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinemeyer, W., A. Gruhler, V. Mohrle, Y. Mahe and D. H. Wolf, 1993. PRE2, highly homologous to the human major histocompatibility complex-linked RING10 gene, codes for a yeast proteasome subunit necessary for chrymotryptic activity and degradation of ubiquitinated proteins. J. Biol. Chem. 268: 5115–5120. [PubMed] [Google Scholar]
- Hoyer, W., D. Cherny, V. Subramaniam and T. M. Jovin, 2004. Impact of the acidic C-terminal region comprising amino acids 109–140 on alpha-synuclein aggregation in vitro. Biochemistry 43: 16233–16242. [DOI] [PubMed] [Google Scholar]
- Hughes, R. E., R. S. Lo, C. Davis, A. D. Strand, C. L. Neal et al., 2001. Altered transcription in yeast expressing expanded polyglutamine. Proc. Natl. Acad. Sci. USA 98: 13201–13206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry, M. C., T. W. Kim, M. McNamara, R. E. Tanzi, J. M. George et al., 1996. Characterization of the precursor protein of the non-A beta component of senile plaques (NACP) in the human central nervous system. J. Neuropathol. Exp. Neurol. 55: 889–895. [DOI] [PubMed] [Google Scholar]
- Jenner, P., and C. W. Olanow, 1998. Understanding cell death in Parkinson's disease. Ann. Neurol. 44: S72–S84. [DOI] [PubMed] [Google Scholar]
- Jensen, P. H., M. S. Nielsen, R. Jakes, C. G. Dotti and M. Goedert, 1998. Binding of alpha-synuclein to brain vesicles is abolished by familial Parkinson's disease mutation. J. Biol. Chem. 273: 26292–26294. [DOI] [PubMed] [Google Scholar]
- Jo, E., N. Fuller, R. P. Rand, P. St. George-Hyslop and P. E. Fraser, 2002. Defective membrane interactions of familial Parkinson's disease mutant A30P alpha-synuclein. J. Mol. Biol. 315: 799–807. [DOI] [PubMed] [Google Scholar]
- Kahle, P. J., M. Neumann, L. Ozmen, V. Muller, H. Jacobsen et al., 2000. Subcellular localization of wild-type and Parkinson's disease-associated mutant alpha-synuclein in human and transgenic mouse brain. J. Neurosci. 20: 6365–6373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J., 1997. Evidence that the precursor protein of non-A beta component of Alzheimer's disease amyloid (NACP) has an extended structure primarily composed of random-coil. Mol. Cells 7: 78–83. [PubMed] [Google Scholar]
- Kitada, T., S. Asakawa, N. Hattori, H. Matsumine, Y. Yamamura et al., 1998. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392: 605–608. [DOI] [PubMed] [Google Scholar]
- Krobitsch, S., and S. Lindquist, 2000. Aggregation of huntingtin in yeast varies with the length of the polyglutamine expansion and the expression of chaperone proteins. Proc. Natl. Acad. Sci. USA 97: 1589–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruger, R., W. Kuhn, T. Muller, D. Woitalla, M. Graeber et al., 1998. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat. Genet. 18: 106–108. [DOI] [PubMed] [Google Scholar]
- Lehman, K., G. Rossi, J. E. Adamo and P. Brennwald, 1999. Yeast homologues of tomosyn and lethal giant larvae function in exocytosis and are associated with the plasma membrane SNARE, Sec9. J. Cell Biol. 146: 125–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindersson, E., R. Beedholm, P. Hojrup, T. Moos, W. Gai et al., 2004. Proteasomal inhibition by alpha-synuclein filaments and oligomers. J. Biol. Chem. 279: 12924–12934. [DOI] [PubMed] [Google Scholar]
- Lindquist, S., 1997. Mad cows meet psi-chotic yeast: the expansion of the prion hypothesis. Cell 89: 495–498. [DOI] [PubMed] [Google Scholar]
- Lucking, C. B., and A. Brice, 2000. Alpha-synuclein and Parkinson's disease. Cell. Mol. Life Sci. 57: 1894–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroteaux, L., J. T. Campanelli and R. H. Scheller, 1988. Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci. 8: 2804–2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah, E., E. Rockenstein, I. Veinbergs, M. Mallory, M. Hashimoto et al., 2000. Dopaminergic loss and inclusion body formation in alpha-synuclein mice: implications for neurodegenerative disorders. Science 287: 1265–1269. [DOI] [PubMed] [Google Scholar]
- Matsuoka, Y., M. Vila, S. Lincoln, A. McCormack, M. Picciano et al., 2001. Lack of nigral pathology in transgenic mice expressing human alpha-synuclein driven by the tyrosine hydroxylase promoter. Neurobiol. Dis. 8: 535–539. [DOI] [PubMed] [Google Scholar]
- McDaniel, D., A. J. Caplan, M. S. Lee, C. C. Adams, B. R. Fishel et al., 1989. Basal-level expression of the yeast HSP82 gene requires a heat shock regulatory element. Mol. Cell. Biol. 9: 4789–4798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean, P. J., H. Kawamata, S. Ribich and B. T. Hyman, 2000. Membrane association and protein conformation of alpha-synuclein in intact neurons. Effect of Parkinson's disease-linked mutations. J. Biol. Chem. 275: 8812–8816. [DOI] [PubMed] [Google Scholar]
- McNaught, K. S., L. M. Bjorklund, R. Belizaire, O. Isacson, P. Jenner et al., 2002. Proteasome inhibition causes nigral degeneration with inclusion bodies in rats. Neuroreport 13: 1437–1441. [DOI] [PubMed] [Google Scholar]
- Mitchell, D. A., T. K. Marshall and R. J. Deschenes, 1993. Vectors for the inducible overexpression of glutathione S-transferase fusion proteins in yeast. Yeast 9: 715–722. [DOI] [PubMed] [Google Scholar]
- Mori, F., C. Inenaga, M. Yoshimoto, H. Umezu, R. Tanaka et al., 2002. Alpha-synuclein immunoreactivity in normal and neoplastic Schwann cells. Acta Neuropathol. 103: 145–151. [DOI] [PubMed] [Google Scholar]
- Nishimura, N., S. Bannykh, S. Slabough, J. Matteson, Y. Altschuler et al., 1999. A di-acidic (DXE) code directs concentration of cargo during export from the endoplasmic reticulum. J. Biol. Chem. 274: 15937–15946. [DOI] [PubMed] [Google Scholar]
- Norman, G. R., and D. L. Streiner, 2000 Biostatistics: The Bare Essentials. B. C. Decker, London.
- Novick, P., C. Field and R. Schekman, 1980. Identification of 23 complementation groups required for post-translational events in the yeast secretory pathway. Cell 21: 205–215. [DOI] [PubMed] [Google Scholar]
- Outeiro, T. F., and S. Lindquist, 2003. Yeast cells provide insight into alpha-synuclein biology and pathobiology. Science 302: 1772–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymeropoulos, M. H., 1998. Autosomal dominant Parkinson's disease. J. Neurol. 245: 1–3. [DOI] [PubMed] [Google Scholar]
- Polymeropoulos, M. H., C. Lavedan, E. Leroy, S. E. Ide, A. Dehejia et al., 1997. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science 276: 2045–2047. [DOI] [PubMed] [Google Scholar]
- Raitt, D. C., A. L. Johnson, A. M. Erkine, K. Makino, B. Morgan et al., 2000. The Skn7 response regulator of Saccharomyces cerevisiae interacts with Hsf1 in vivo and is required for the induction of heat shock genes by oxidative stress. Mol. Biol. Cell 11: 2335–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochet, J. C., T. F. Outeiro, K. A. Conway, T. T. Ding, M. J. Volles et al., 2004. Interactions among alpha-synuclein, dopamine, and biomembranes: some clues for understanding neurodegeneration in Parkinson's disease. J. Mol. Neurosci. 23: 23–34. [DOI] [PubMed] [Google Scholar]
- Rose, M. D., F. Winston and P. Hieter, 1990 A Laboratory Course. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Rothman, J. E., 1994. Mechanisms of intracellular protein transport. Nature 372: 55–63. [DOI] [PubMed] [Google Scholar]
- Shimura, H., M. G. Schlossmacher, N. Hattori, M. P. Frosch, A. Trockenbacher et al., 2001. Ubiquitination of a new form of alpha-synuclein by parkin from human brain: implications for Parkinson's disease. Science 293: 263–269. [DOI] [PubMed] [Google Scholar]
- Singleton, A. B., M. Farrer, J. Johnson, A. Singleton, S. Hague et al., 2003. alpha-Synuclein locus triplication causes Parkinson's disease. Science 302: 841. [DOI] [PubMed] [Google Scholar]
- Spillantini, M. G., M. L. Schmidt, V. M. Lee, J. Q. Trojanowski, R. Jakes et al., 1997. Alpha-synuclein in Lewy bodies. Nature 388: 839–840. [DOI] [PubMed] [Google Scholar]
- Syme, C. D., E. W. Blanch, C. Holt, R. Jakes, M. Goedert et al., 2002. A Raman optical activity study of rheomorphism in caseins, synucleins and tau. New insight into the structure and behaviour of natively unfolded proteins. Eur. J. Biochem. 269: 148–156. [DOI] [PubMed] [Google Scholar]
- Tanaka, Y., S. Engelender, S. Igarashi, R. K. Rao, T. Wanner et al., 2001. Inducible expression of mutant alpha-synuclein decreases proteasome activity and increases sensitivity to mitochondria-dependent apoptosis. Hum. Mol. Genet. 10: 919–926. [DOI] [PubMed] [Google Scholar]
- Taylor, J. P., J. Hardy and K. H. Fischbeck, 2002. Toxic proteins in neurodegenerative disease. Science 296: 1991–1995. [DOI] [PubMed] [Google Scholar]
- van der Putten, H., K. H. Wiederhold, A. Probst, S. Barbieri, C. Mistl et al., 2000. Neuropathology in mice expressing human alpha-synuclein. J. Neurosci. 20: 6021–6029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinreb, P. H., W. Zhen, A. W. Poon, K. A. Conway and P. T. Lansbury, Jr., 1996. NACP, a protein implicated in Alzheimer's disease and learning, is natively unfolded. Biochemistry 35: 13709–13715. [DOI] [PubMed] [Google Scholar]