

Abstract
We examine the efficiency of different genotyping and phenotyping strategies in inbred line crosses from an information perspective. This provides a mathematical framework for the statistical aspects of QTL experimental design, while guiding our intuition. Our central result is a simple formula that quantifies the fraction of missing information of any genotyping strategy in a backcross. It includes the special case of selectively genotyping only the phenotypic extreme individuals. The formula is a function of the square of the phenotype and the uncertainty in our knowledge of the genotypes at a locus. This result is used to answer a variety of questions. First, we examine the cost-information trade-off varying the density of markers and the proportion of extreme phenotypic individuals genotyped. Then we evaluate the information content of selective phenotyping designs and the impact of measurement error in phenotyping. A simple formula quantifies the information content of any combined phenotyping and genotyping design. We extend our results to cover multigenotype crosses, such as the F2 intercross, and multiple QTL models. We find that when the QTL effect is small, any contrast in a multigenotype cross benefits from selective genotyping in the same manner as in a backcross. The benefit remains in the presence of a second unlinked QTL with small effect (explaining <20% of the variance), but diminishes if the second QTL has a large effect. Software for performing power calculations for backcross and F2 intercross incorporating selective genotyping and marker spacing is available from http://www.biostat.ucsf.edu/sen.
THE goal of a genetic mapping experiment is to detect and localize the genetic elements responsible for the variation in a phenotype of interest. The design of a mapping experiment involves choosing the type of the cross, the parental strains involved, a method for measuring the phenotype, and a genotyping strategy. Traditionally, genotyping and phenotyping strategies have been evaluated in terms of their power to detect a genetic effect. This depends on the size of the genetic effect and the information in the experiment. The experimenter has no control over the former, but phenotyping and genotyping strategies can be designed to extract the most information subject to cost or other constraints. In this article, we consider inbred line crosses from an information perspective.
Selective genotyping (Lebowitz et al. 1987; Lander and Botstein 1989; Darvasi and Soller 1992) is an effective strategy for reducing genotyping costs when there is a single trait of interest. Lander and Botstein (1989) showed that the contribution of an individual to the expected LOD score is approximately proportional to the squared difference of the individual from the overall mean. Darvasi and Soller (1992) showed that by genotyping approximately one-quarter of the individuals in each extreme (half of the total individuals) one retains most of the power as compared to genotyping the entire cross. Darvasi and Soller (1994) considered genotyping strategies from a cost perspective and showed that for lowering total experimental cost it may be optimal to genotype individuals at very wide spacings if the cost of rearing and trait evaluation is low. A selective phenotyping design with a main trait and a correlated trait was considered by Medugorac and Soller (2001) who also analyzed it from a cost-power perspective. Jin et al. (2004) have proposed a selective phenotyping strategy for crosses where phenotyping is more expensive than genotyping, using a criterion that maximizes the genetic diversity of the phenotyped animals. Belknap (1998) considered the problem of the number of replications of a recombinant inbred (RI) line to achieve power comparable to a backcross or F2 cross subject to heritability constraints. All of these design strategies can be considered and unified by considering the information content of the resulting data.
We were motivated to investigate selective genotyping from an information perspective by considering the genotyping strategy employed in Sugiyama et al. (2001). Figure 1 shows the genotype pattern in this cross. First, we note that half of the marker genotypes are missing. The mice with extreme phenotypes were more heavily genotyped than the intermediate ones and some chromosomes were more heavily genotyped than others because an initial genome scan showed indications of QTL on these chromosomes. Finally, some markers were typed only if the flanking markers recombined (see Figure 2). This was done because if two reasonably close markers do not recombine, the genotypes of all loci in that interval are effectively known, but when flanking markers differ, additional genotyping can help to narrow the location of the recombination. Ronin et al. (2003) investigated the properties of a similar genotyping strategy using simulations. Two-stage genotyping strategies have been considered in the context of linkage analysis in human studies by Elston (1994) and for genetic association studies by Satagopan et al. (2002) and Satagopan and Elston (2003). More generally, selective genotyping can be considered to be a special case of outcome-dependent sampling.
Figure 1.—
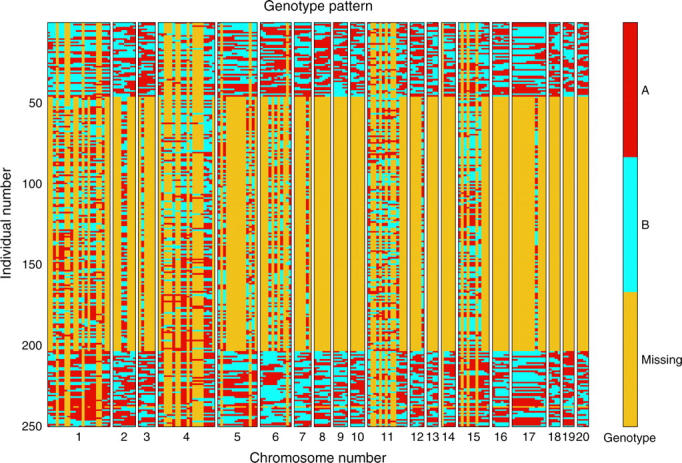
Genotype pattern in the hypertension mouse cross of Sugiyama et al. (2001). Genotypes are colored red if they were from the hypertensive strain (A/J), blue if from the nonhypertensive strain (BL/6), and yellow if missing. Each row represents genotypes from a particular mouse; the mice have been sorted by their blood pressure, so the mouse with the lowest blood pressure appears at the top while the one with the highest blood pressure is at the bottom. The markers are sorted by their position on the genome starting with chromosome 1 through chromosome 20. This figure was generated using Pseudomarker (Sen and Churchill 2001).
Figure 2.—

Close-up of genotype pattern of 50 individuals on chromosome 4 from Sugiyama et al. (2001). Genotypes are denoted by open circles if from the A/J strain and by solid circles if from the BL/6 strain. Each row represents genotypes from a particular mouse. We selected every fifth mouse from the 250 in the cross sorted by blood pressure. The mouse with the lowest blood pressure appears at the bottom while the one with the highest blood pressure is at the top. The marker order and position is shown by centimorgan position. This figure was produced using R/qtl (Broman et al. 2003).
Our goal in this article is to formally investigate the information trade-offs inherent in different genotyping and phenotyping strategies. Although missing-data methods have long been employed to analyze QTL experiments (Lander and Botstein 1989; Xu and Vogl 2000), they have not been employed in their design. We show that the concept of missing information can be used to evaluate genotyping and phenotyping strategies. This approach also provides insight into the bias of the Haley-Knott approximation to LOD scores (Kao 2000). The missing-information perspective provides a unified view of genotyping, noting that information is inversely proportional to the variance of the estimates of genetic model parameters. This suggests answers to the question: “Which individuals and loci are to be genotyped?”
In the next section, we develop the concept of information in a mapping design using the backcross as the example. Next we present our results on the information content of genotyping and phenotyping designs. Mathematical results are detailed in the appendix.
THEORY
Information perspective on QTL mapping:
Let y, g, and m denote the trait, QTL genotype, and observed marker genotypes of a single individual. In a cross with n individuals, we denote them by y = (y1, … , yn), g = (g1, … , gn), and m = (m1, … , mn), respectively.
We develop our ideas in the context of a backcross segregating one QTL. Assume, without loss of generality, that the QTL genotypes can take two values, 0 or 1, and the distribution of the phenotype given the QTL genotypes is Gaussian with unit variance. The conditional mean of the phenotype given the QTL is +δ if gi = 1 and −δ if gi = 0. If we know the QTL genotypes, the LOD score for testing δ = 0 against the alternative that δ ≠ 0 is
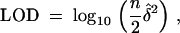 |
where 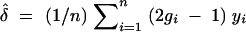 is the maximum-likelihood estimate of δ. Under the null hypothesis, 2 loge(10)LOD has a χ2-distribution with 1 d.f. Under the alternative hypothesis, it has a noncentral χ2-distribution with 1 d.f. and noncentrality parameter nδ2. Thus, the power of the test to detect linkage depends on the sample size n and the square of QTL effect size δ2. More generally, when the QTL genotypes are not known because of incomplete genotyping, the power is a function of Iδ2, where I is the expected Fisher information of the experiment. This follows from the general theory of statistical likelihoods (Cox and Hinkley 1974) as described below. The expected Fisher information depends not only on the sample size, n, but also on the design of the experiment—how we genotype the cross and how accurately we measure the phenotype. Different phenotyping and genotyping strategies will lead to different values of I. Thus, we can compare different strategies by comparing their expected information. In the context of the QTL-mapping problem we may think of information content of an experiment as the “effective sample size.”
is the maximum-likelihood estimate of δ. Under the null hypothesis, 2 loge(10)LOD has a χ2-distribution with 1 d.f. Under the alternative hypothesis, it has a noncentral χ2-distribution with 1 d.f. and noncentrality parameter nδ2. Thus, the power of the test to detect linkage depends on the sample size n and the square of QTL effect size δ2. More generally, when the QTL genotypes are not known because of incomplete genotyping, the power is a function of Iδ2, where I is the expected Fisher information of the experiment. This follows from the general theory of statistical likelihoods (Cox and Hinkley 1974) as described below. The expected Fisher information depends not only on the sample size, n, but also on the design of the experiment—how we genotype the cross and how accurately we measure the phenotype. Different phenotyping and genotyping strategies will lead to different values of I. Thus, we can compare different strategies by comparing their expected information. In the context of the QTL-mapping problem we may think of information content of an experiment as the “effective sample size.”
Power, LOD score, standard errors, and information:
Much of the QTL literature has focused on LOD scores, which are equivalent to a log-likelihood ratio. The Fisher information is the expected curvature of the log-likelihood function. Suppose θ is the parameter of interest, ℓ(θ) is the log-likelihood, and we want to test θ = θ0 against the alternative that θ ≠ θ0. As outlined in the appendix, the log-likelihood ratio for testing this hypothesis is proportional to a noncentral χ2 variable with s d.f. and noncentrality parameter (θ − θ0)TI(θ)(θ − θ0), where I(θ) is the expected Fisher information matrix. Furthermore, the variance of the maximum-likelihood estimate, θ̂, is given by I(θ)−1. Thus, Fisher information is a fundamental quantity that affects both the power of our test and the standard errors of the estimates of QTL effect size. It is therefore the focus of this article.
Before conducting an experiment, we use the expected information from the QTL study design. After conducting the experiment, we can compute the observed information from a design. The observed information is defined as the observed curvature of the log-likelihood function that may vary from sample to sample (Efron and Hinkley 1978). To see this, consider a cartoon example of a very disorganized laboratory that tends to lose a quarter of its data at random (both phenotypes and genotypes). In the backcross scenario considered in the previous section, assume that the lab plans to conduct an experiment with n individuals. After the experiment is performed, we will have data from n* individuals, where n* follows a binomial distribution with parameters n and 3/4. Thus, the observed information from the experiment would be n*, which is the number of individuals for whom we actually have data, and this will vary. On the average, data from 3n/4 individuals are collected, and so the expected Fisher information is 3n/4. For n = 100, the expected information and realized sample size, n*, will vary from ∼60 to ∼90 with a mean of 75.
In a realistic QTL setting, note that at any locus in a short nonrecombinant marker interval, we have complete knowledge of the genotype, whereas in the middle of a recombinant marker interval, we have virtually no information about the genotype. Since the distribution of marker genotypes is random, the information content of a specific marker interval can be known only after conducting the experiment. Therefore, we make a distinction between observed and expected information.
By comparing the observed information to the expected information if all individuals were genotyped, we can quantify the amount of missing information in a realized cross. This can help us decide which individuals should be genotyped or phenotyped more intensely after collecting preliminary data on the cross.
Missing data and information:
A key element in the statistical analysis of QTL data is to adjust for the fact that the genotypes of the individuals in the cross are known only at typed markers. The genotypes at intermediate locations must be inferred from the observed data. In other words, the individual QTL genotypes are “missing data.” Some marker genotypes may also be missing. This may be intentional if we have used a selective genotyping strategy.
Missing-data methods used in QTL analysis include the EM algorithm (Lander and Botstein 1989), Markov chain Monte Carlo (Satagopan et al. 1996), and multiple imputation (Sen and Churchill 2001). In this article, we focus on design (as opposed to the analysis) of QTL experiments. We calculate the observed information content of genotyping strategy relative to a perfect complete-data case using the “missing information principle” (Orchard and Woodbury 1972; McLachlan and Krishnan 1996). This states that the complete-data information (Ic) is the sum of the observed information (Io) and the missing information (Im),
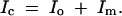 |
This allows us to calculate the amount of missing information due to incomplete genotyping relative to the expected information under complete genotyping. This gives us the expected information from a genotyping strategy. We use this to evaluate competing approaches with different cost profiles.
Likelihood function:
To calculate the observed, missing, and expected information, we need to write down the joint-likelihood function of the observed as well as the missing-data structures. We consider the general case here. Let θ denote the genetic model parameters and λ the QTL locations. When the phenotypes are observed, the likelihood function
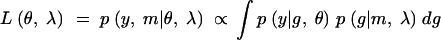 |
has the form of a mixture distribution (see the appendix). This leads us to consider the complete-data likelihood,
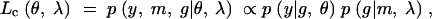 |
which is the likelihood that would apply if the QTL genotypes, g, were actually observed. Using this likelihood function, we can calculate the maximum possible information attainable with complete genotype information.
Note, from the form of the likelihood function, that we assume that the distribution of the phenotypes is independent of the marker genotypes conditional on the QTL genotypes. It is important that the missing-data pattern be “ignorable” (that is, all data that were used to decide that other data would not be collected must be included in the likelihood computation), which would ensure that likelihood-based inference gives asymptotically unbiased estimates of the parameters. This is not guaranteed if the missing-data pattern is “nonignorable” or if nonlikelihood methods are used (Little and Rubin 1987; Schafer 1997). An example of nonignorable missing data would be when selective genotyping of individuals with extreme phenotypes is performed, and the phenotypes of the intermediate individuals are discarded. It is well known that in this case QTL effect estimates are biased. Fortunately, the most common forms of intentional missing data, such as selective genotyping, are ignorable, and hence appropriate likelihood methods will give asymptotically unbiased results.
RESULTS
In this section we first consider genotyping strategies and present a formula for calculating the observed fraction of missing information. This serves as the building block for subsequent sections. We note that the observed fraction of missing information is connected to the bias of the Haley-Knott method for approximating LOD scores. We then calculate the expected information from genotyping strategies when a fraction of the extreme phenotypic individuals are genotyped. Using these results, we analyze the trade-offs between the cost of genotyping and information content. We then analyze the information content of phenotyping designs and consider the situation when a phenotype measurement is replicated for greater accuracy. Next we present a formula for calculating the missing information for a phenotyping design combined with a genotyping design. This is followed by a section where we calculate the expected information under selective genotyping for crosses with more than two genotype classes, such as the F2 intercross. We conclude the section by considering information content in the presence of a second unlinked additive QTL.
Observed fraction of missing information:
As before, assume the conditional distribution of the phenotype given the QTL genotype is Gaussian with unit variance. Conditional on the observed phenotype and marker data, it can be shown (details in the appendix) that the observed fraction of missing information is
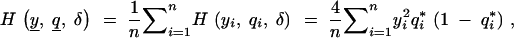 |
1 |
where 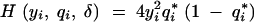 is the fraction of missing information from the ith individual, qi is the prior probability of the QTL genotype given the marker data alone, and
is the fraction of missing information from the ith individual, qi is the prior probability of the QTL genotype given the marker data alone, and 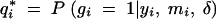 is the posterior probability of the QTL genotype of the ith individual given the observed data. This formula has two uses. Equation 1 can be used to decide which loci will yield the most information from additional genotyping. The missing information is greatest for individuals with extreme phenotypes (the y2 term) and for those with ambiguous genotypes. Thus, it is advantageous to genotype the individuals with extreme phenotypes. On the other hand, if two flanking markers have been typed and are not recombinant, the genotype at the location of interest is effectively known since qi1 − qi ≃ 0, and it will not be worthwhile to genotype intermediate positions. If the flanking markers are recombinant and the putative location is in the middle of the marker interval, qi(1 − qi) ≃ 1/4, it will be worthwhile genotyping an intermediate locus.
is the posterior probability of the QTL genotype of the ith individual given the observed data. This formula has two uses. Equation 1 can be used to decide which loci will yield the most information from additional genotyping. The missing information is greatest for individuals with extreme phenotypes (the y2 term) and for those with ambiguous genotypes. Thus, it is advantageous to genotype the individuals with extreme phenotypes. On the other hand, if two flanking markers have been typed and are not recombinant, the genotype at the location of interest is effectively known since qi1 − qi ≃ 0, and it will not be worthwhile to genotype intermediate positions. If the flanking markers are recombinant and the putative location is in the middle of the marker interval, qi(1 − qi) ≃ 1/4, it will be worthwhile genotyping an intermediate locus.
Missing information and bias of Haley-Knott method:
The Haley-Knott (HK) method (Haley and Knott 1992) is a popular method for approximating LOD scores. Kao (2000) showed that the bias of the HK method is a function of how close the prior genotype probabilities qi were to the posterior genotype probabilities q*i. Further insight into the bias may be obtained by noting that the HK method is equivalent to a single step of the EM algorithm when the starting values of the genotype means correspond to the null hypothesis. Dempster et al. (1977) showed that the EM algorithm is a linear iteration, and its rate of convergence is given by
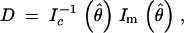 |
where θ̂ is the maximum-likelihood estimate of the parameter of interest, θ, and Ic and Im are the complete and missing information matrices. Therefore the extent of the bias of the HK method depends on the rate of convergence of the EM algorithm. Note the rate of convergence D is just the observed fraction of missing information. Thus, Equation 1 helps us decide when using the HK approximation will have a large bias.
Missing information under selective genotyping:
What is the expected information from a selective genotyping strategy? In this section we consider genotyping strategies where we type an α-fraction of the extreme phenotypic individuals at markers that are spaced d cM apart. The expected information is a function of the effect size, δ, the selection fraction, α, as well as marker spacing, d. To be conservative, we calculate the fraction of missing information in the middle of a marker interval where it is greatest. Let w(α, δ) be the upper α-point of the (marginal) distribution of the phenotype; that is,
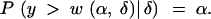 |
We assume that every individual with a phenotype > +w(α/2, δ) or < −w(α/2, δ) is genotyped at markers spaced d cM apart. Now consider a locus in the middle of this marker interval and thus it is a distance d/2 cM away from each of the flanking markers. Let r be the recombination fraction corresponding to d cM and r′ be that corresponding to d/2 cM. Then, if the phenotype |y| ≤ w(α/2, δ), no genotype information is available, and hence q, the prior probability of the QTL genotype is 1/2. If the phenotype |y| > w(α/2, δ), the flanking markers are typed. In this case, with probability r the flanking markers will recombine and q = 1/2. Complementarily, with probability 1 − r, the markers do not recombine, and the prior probability q = r′2/(r′2 + (1 − r′)2) or (1 − r′)2/(r′2 + (1 − r′)2) with equal probability depending on the genotype of the flanking markers. Using these facts, we can use numerical integration to calculate the expected fraction of missing information,
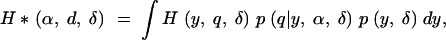 |
2 |
where q is a function of y, α, and δ, since the prior probabilities depend on them. Figure 3 plots the fraction of missing information as a function of the selection fraction (α), the size of the QTL effect (δ) under four scenarios, corresponding to four different marker densities. We note that the fraction of missing information decreases with increasing selection fraction (α) and increasing QTL effect size (δ). More interestingly, irrespective of the QTL effect, less than one-eighth of the information is missing if the selection fraction is ≥50%. This is consistent with the finding of Darvasi and Soller (1992) that little power is lost if a quarter of each extreme is genotyped. However, if the extremes are not densely genotyped, and the distance between markers is 10–20 cM, we may lose between 17 and 25% of the information that would be available if all individuals were genotyped.
Figure 3.—

Fraction of missing information in a backcross as a function of the fraction of the extremes typed (α), the strength of the QTL (δ), and recombination distance flanking markers (θ1, θ2). The proportion of variance explained by the QTL is δ2/(1 + δ2). When there is very little genotype information at a marker (top left, θ1 = θ2 = 0.2), there is very little to be gained by selectively genotyping the faraway markers as it does not add much information. When we have very densely spaced markers (bottom right, θ1 = θ2 = 0.01), the fraction of missing information decreases with increased extreme genotyping. The intermediate cases (top right, θ1 = θ2 = 0.1; bottom left, θ1 = θ2 = 0.05) represent more realistic scenarios. We find that by genotyping 60% of the extremes we lose <10% of the information with markers approximately every 10 cM.
Expected information for small QTL effect:
Missing information is greatest when the QTL effect is small, so we consider the worst-case scenario when δ = 0. In this case it can be shown (see appendix) that the expected information from a cross with n individuals using the selective genotyping strategy described above is
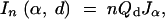 |
where
 |
is the expected information content per observation under dense typing of α-fraction of the extremes,
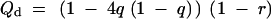 |
3 |
is a deflation factor that depends on the density of markers (and the informativeness of the markers), wα/2 is the upper α/2 point of the standard normal distribution, φ(·) is the density function of the standard normal distribution, and q = r′2/(r′2 + (1 − r′)2) is the probability that the genotype of an individual in the middle of a nonrecombinant marker interval is different from the flanking markers.
Information-cost trade-offs:
Now we evaluate the information content of an experiment by explicitly considering the role of genotyping cost. Let c be the cost of genotyping an individual densely (d ≃ 0) relative to the cost of rearing an individual. Then the ratio of the information and cost of the experiment is
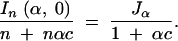 |
The best selective genotyping strategy for a given cost, c, is one that minimizes this ratio as a function of the selection fraction α. Figure 4 shows the optimal selection fraction calculated by numerically maximizing the information-cost ratio as a function of the cost of genotyping an individual, c. Predictably, when the cost of genotyping is low, it pays to genotype a larger fraction of the cross. As costs increase, one should genotype a progressively smaller fraction. Interestingly, when the cost of genotyping is comparable to the cost of rearing (c = 1), then the optimal selection fraction is 43% or just under half the cross. This is roughly consistent with the finding of Darvasi and Soller (1992) who used a different analytic strategy. In practice, we never densely genotype an individual, we just genotype at a set of markers spaced roughly regularly along the genome. We consider the information in the middle of a d-cM marker interval. Then we consider the information-cost ratio, where the total cost of genotyping is a function of the per-marker cost, c, and the genome size, G, in centimorgans. This leads us to the ratio
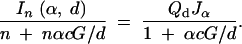 |
4 |
Figure 4.—

Optimal selection fraction (α) as a function of the cost of genotyping an individual completely, c, when the QTL effect, δ, is very small. The unit of cost is relative to the cost of rearing an individual.
In Figure 5 we plot this ratio for a genome size of 1450 cM (corresponding to the laboratory mouse) as a function of the selection fraction, α, and marker spacing, d, for four different marker genotyping costs, c, expressed in units of the cost of rearing a single individual. When the cost of genotyping a single marker is comparable to rearing an individual, the optimal strategy is to genotype a small fraction (∼6%) of the extremes at a wide spacing (∼46 cM or a recombination fraction of 30%). As the cost of genotyping decreases, the optimal strategy is to type more individuals more densely. When the genotyping cost is one-tenth of the cost of rearing, one should genotype ∼23% of the cross at ∼36 cM (recombination fraction 26%). When the genotyping cost is one-hundredth of the cost of rearing, one should genotype ∼49% of the cross at ∼21 cM (recombination fraction 17%). When the genotyping cost is one-thousandth of the cost of rearing, one should genotype ∼71% of the cross at ∼9 cM (recombination fraction 8%). These conclusions are broadly consistent with the findings of Darvasi and Soller (1994), who considered marker spacing strategies (without selective genotyping). For well-characterized model organisms such as the mouse, the cost of genotyping is a tiny fraction of the cost of rearing and phenotyping. For those organisms, genotyping the whole cross every 10 cM is reasonable. For organisms such as some plants without well-developed markers, the cost of genotyping a marker is comparable to raising an individual and in those cases it suffices to genotype a small fraction of the cross at a few, sparse sets of markers. The exact trade-offs depend on the particulars of the mapping problem, and we provide software (see below) to make the calculations for different scenarios. To obtain the optimal selection fraction subject to a given marker spacing, we can minimize the information-cost ratio above as a function of α given d and this is the solution of the equation
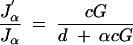 |
5 |
(see appendix for proof), where 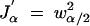 is the derivative of Jα with respect to α. In Figure 6 we show the optimum selection fraction as a function of marker spacing and cost of genotyping a single marker for the laboratory mouse.
is the derivative of Jα with respect to α. In Figure 6 we show the optimum selection fraction as a function of marker spacing and cost of genotyping a single marker for the laboratory mouse.
Figure 5.—

The information per unit cost ratio plotted as a function of the selection fraction, α, and average spacing between markers, d. We calculate the information in the middle of the marker interval. The information per unit cost numbers are normalized to the maximum possible information per unit cost. This way we can see what ranges of the selection fraction and spacings give near-optimum returns. Each number corresponds to a the cost, c, of genotyping a single marker relative to that of rearing an individual. The genome size is assumed to be 14.5 morgans (similar to that of the mouse). The top left corner corresponds to c = 1, the top right corner to c = 0.1, the bottom left corner to c = 0.01, and the bottom right corner to c = 0.001.
Figure 6.—

The optimal selection fraction (α) plotted as a function of marker spacing d for four cost scenarios. The lines are for when the cost of genotyping a single marker (c) is expressed in the units of the cost of rearing.
Selective phenotyping strategies:
If a phenotype is observed noisily, then although the noisy version is observed, the “true” phenotype remains unobserved or missing. For example, we may have to measure blood pressure several times, to achieve an accurate phenotyping of an individual mouse, or we may have to phenotype multiple individuals from a recombinant inbred line. Another example of selective phenotyping would be when a suite of related phenotypes are of interest (such as measuring body weight weekly), but we phenotype selectively (weigh the heaviest and lightest animals at birth, every week, but everyone else every 4 weeks). Yet another class of selective phenotyping strategies was considered by Jin et al. (2004). In their approach, which is based on an individual's genotype, some individuals are phenotyped accurately or not at all.
When the phenotypes are not directly observed, but are observed with error through z, the surrogate phenotype, the likelihood function has to be modified accordingly. We assume that the surrogate phenotype depends on the true phenotype through the parameter ρ, which gives the likelihood function
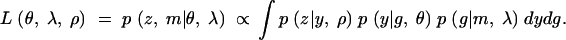 |
In this case, the complete-data likelihood would treat the phenotype as well as the QTL genotypes as missing data and will be
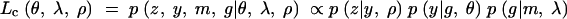 |
We also assume that the surrogate phenotype is independent of the marker data (and the QTL genotypes) given the phenotypes.
The rationale behind selective phenotyping is the same as that of selective genotyping: we want to maximize information while controlling cost. Suppose our true phenotype, y, is not completely observed and instead we observe a noisy version, z. Assume that zi = yi + ηi, where ηi is the independent random measurement error with mean zero and variance τ2i. When the phenotype is noiselessly observed τi = 0. The correlation between zi and yi is 1 + τ2i−1.
Consider the case when the QTL genotype is completely observed. Then the information from each individual is proportional to the inverse of the variance of the ith observation, 1 + τ2i. Thus, the information from the whole experiment is
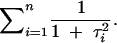 |
It is worthwhile considering the special case, when an investigator has the choice of either replicating the measurement or measuring multiple individuals. Let the measurement error variance be τ2, so that if a measurement is replicated t times, the measurement error variance 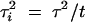 . Thus the information content of an experiment with n individuals, when the phenotype measurement is replicated t times, is
. Thus the information content of an experiment with n individuals, when the phenotype measurement is replicated t times, is
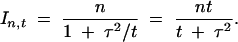 |
Now suppose, without loss of generality, that the cost of raising an individual is unity and the cost of phenotyping is c. Then the cost of the experiment is
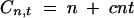 |
and the information-cost ratio of this strategy is
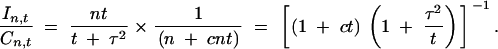 |
6 |
The maximum of the information-cost ratio as a function of t depends on the ratio τ2/c. In Figure 7 we show the optimal replication number t as a function of the phenotyping variance-cost (τ2/c) ratio. It can be shown (see appendix) that the optimal replication number is
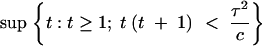 |
7 |
Figure 7.—

Optimal replication number (m) as a function of the variance-cost ratio (τ2/c). Here τ2 is the ratio of the variance of the measurement instrument to the environmental variance in the phenotype, and c is the ratio of the cost of phenotyping to the cost of raising an individual. We can see that when the variance of the phenotyping instrument is low relative to the cost of phenotyping, there is no point in replicating (m = 1). It makes more sense to replicate the measurement if the cost of phenotyping relative to raising an individual is low or if the phenotyping variance is high relative to the environmental variance.
Selective phenotyping and genotyping:
Consider selective genotyping and phenotyping together. The fraction of missing information is
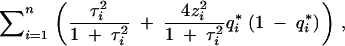 |
8 |
where 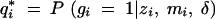 is the posterior probability of the QTL genotype given the observed data. This formula allows us to evaluate any genotyping and phenotyping strategy. The main message is that it is most profitable to phenotype and genotype the extreme phenotypic individuals carefully, because they contribute the most information.
is the posterior probability of the QTL genotype given the observed data. This formula allows us to evaluate any genotyping and phenotyping strategy. The main message is that it is most profitable to phenotype and genotype the extreme phenotypic individuals carefully, because they contribute the most information.
Multigenotype crosses:
A backcross population can be parameterized using a single parameter; this simplifies the analysis of information. In this section we present the generalizations to multigenotype crosses such as the F2. In this case, information is a matrix, and therefore to compare different scenarios we have to obtain one-dimensional summaries. The two most common summaries correspond to D-optimality and c-optimality criteria (Cox and Reid 2000). If I is the expected information matrix from an experiment, for D-optimality, one compares the determinant, det(I), from different experiments. This corresponds to comparing the volume of the confidence ellipsoid of the parameter estimates. For c-optimality with the contrast vector u, one compares (inverse of) the asymptotic variance of the contrast, uTI−1u. This is equivalent to comparing the width of the confidence intervals for the contrast.
Assume that there are K genotypes possible at a given locus and let q be the probability distribution of the genotypes at an unlinked locus. For the backcross, K = 2, and q = (1/2, 1/2). For the intercross, K = 3 and q = (1/4, 1/2, 1/4). In general, the QTL genotypes, g, can take K values 1, … , K. We assume that the distribution of the phenotype given the QTL genotype g = k is Gaussian with mean μk and unit variance. We calculate the information under the worst-case scenario when all the QTL genotype means are equal and when we genotype densely an α-fraction of the extreme phenotypic individuals. It is shown in the appendix that for the backcross the expected value of the information matrix is
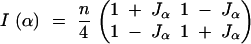 |
Since the determinant of this matrix is equal to n2Jα/4, using the D-optimality criterion, we get the same conclusions as we did with the scalar parameterization of the problem in previous sections. The inverse of the information matrix is
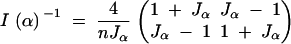 |
Therefore the variance of the contrast of interest, u = (+1, −1), is 4/(nJα). Since this is inversely proportional to Jα, we get the same conclusions with c-optimality criteria as with the the D-optimality criterion. For the F2, the expected information matrix is
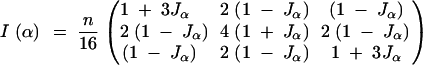 |
(see appendix). The determinant of this matrix is
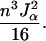 |
The inverse of the variance of any contrast u = (u1, u2, u3) is
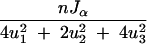 |
and hence proportional to Jα. Thus, judged by c-optimality criteria, the information content of an F2 cross changes with the selection fraction in a similar manner as a backcross. For a multigenotype cross (such as a four-way cross), the expected information matrix is
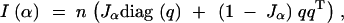 |
with determinant
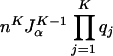 |
9 |
and inverse
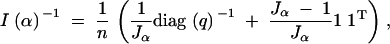 |
which implies that the inverse of the variance of any contrast u is
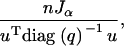 |
10 |
which is proportional to Jα. Thus our results for the backcross can be interpreted very generally in the context of c-optimality.
Multiple-QTL models:
Thus far, we have developed our ideas in the context of single-QTL models. For complex traits, it is generally understood that many QTL contribute to the trait. In this section, we investigate the usefulness of selective genotyping in the context of multiple-QTL models. If the effect of each QTL is small, then we can use the results of the previous section on multigenotype crosses to conclude that any contrast between QTL genotype combinations benefits from selective genotyping, in the same way as in a backcross. In particular, linked and epistatic QTL also benefit from selective genotyping.
When one or more QTL have strong effects, it is not obvious that selective genotyping is still beneficial for detection of the smaller QTL. Consider two additive unlinked QTL in a backcross following the model for the phenotype of the ith individual,
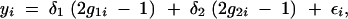 |
where εi is the Gaussian residual error with zero mean and unit variance, and gji is the QTL genotype of the ith individual for the jth QTL taking value either 0 or 1 with equal probability, j = 1, 2. The least favorable condition for detecting a QTL is when its effect is small, so we consider the case when δ1 = 0, while varying the effect of the second QTL, δ2 = β, for various values of β. For an ungenotyped individual the missing information matrix for (δ1, δ2) = (0, β) is shown in the appendix to be equal to
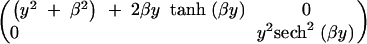 |
Note that the missing information content for δ2 is the same as that in Equation 1 for an ungenotyped individual (when the prior probabilities of the QTL genotypes are equal to one-half). This is consistent with intuition that the information for the second QTL should be the same as that in a single-QTL model since the first QTL has a negligible effect. Using this result we can calculate the expected information under selective genotyping (where an extreme phenotypic individual is completely genotyped and the other not at all). From Figure 8 we can judge the impact of the selection fraction in the presence of a linked additive QTL of varying effect size. When the other QTL has small effect, the fraction of missing information with a selection fraction of 50% is ∼10% as in Figure 3, bottom right. The loss of information due to selective genotyping with a fixed selection fraction increases with the strength of the other QTL. However, the loss of information is modest if the portion of variance explained by the second QTL is <20% (β = 1/2).
Figure 8.—

Fraction of missing information for a QTL with small effect under selective genotyping, as a function of the selection fraction (α), and for the effect size of a second unlinked additive QTL (β). The difference between the genotype means is 2β, and the proportion of variance explained by the second QTL is β2/(1 + β2). The solid lines correspond to the limiting cases of (i) when the second QTL also has a negligible effect (β = 0) and (ii) when the second QTL has a really large, obvious effect (β = ∞). The dashed lines correspond to intermediate cases of β = 0.5 (variance explained 20%), β = 1 (variance explained 50%), and β = 1.5 (variance explained 69%).
In the limiting case, when the strength of the second QTL is really big (β = ∞), the information from the experiment is ∼(n/2)J2α (see appendix). It is easier to understand the result by considering the case when α = 1/2. In this situation, by genotyping half of the extreme phenotypic individuals, we get only half of the information relative to complete genotyping. This result may appear surprising at first. Since the second QTL has a huge effect, we essentially know its QTL genotypes, and we can get the residuals adjusting for its effect. Half of the individuals with negative residuals are those whose overall phenotype was in the lower quartile. Similarly, half of the individuals with positive residuals are those whose overall phenotype was in the upper quartile. In other words, the distribution of the residuals of the genotyped population is the same as that of the ungenotyped population, and in terms of the residual phenotype the genotyped population was unselected. Since half the individuals were genotyped, the loss of information is 50%, and selective genotyping on the overall phenotype is the same as random selection.
DISCUSSION
The information perspective provides useful insight into phenotyping and genotyping designs. Most information is provided by extreme phenotypic individuals. It is most important to phenotype and genotype them well. Indeed, this is the rationale behind case-control designs. In specific scenarios, we can use simple formulas to explicitly calculate the trade-offs between cost and information. Our conclusions are consistent with previous work on selective genotyping. In particular, we show that genotyping 25% of either extreme phenotypic individual gives most of the information in the data when we are genotyping densely. When individuals are not densely typed, the amount of information lost depends additionally on the marker density. It is preferable to type markers ∼20 cM apart (or closer) unless the cost of genotyping approaches the cost of rearing.
In this article we have focused on the backcross for simplicity. However, as shown in Multigenotype crosses, the results for the backcross generalize to c-optimality. Specifically, when the QTL effect is small, the dependence of the variance of any contrast in any cross on the densely genotyped selection fraction is the same. When a cross is not densely genotyped, the information will have to be discounted by a deflation factor that depends on the informativeness of neighboring markers. In the backcross, it is given by (3), but in general it will depend not only on the cross, but also on the nature of the markers (for example, in an F2, whether the markers are dominant or codominant).
Our results for multigenotype crosses indicate that the information trade-offs in inbred line crosses are also relevant for other settings such as human association studies. In an association study, the different haplotypes segregating in the population may be considered as different alleles. Therefore, if we are interested only in linear contrasts between the haplotypes, we get the same information trade-offs with the selection fraction as in a backcross. These results were derived assuming that the genetic effect is very small, which is realistic for studies of most complex traits. When the genetic effect is substantial, the information will depend on the selection fraction in a more complex manner, but the information expressions for the small genetic effect may be considered as lower bounds. More generally, our technique of calculating the expected information of an experiment may be relevant to outcome-dependent sampling where the correlation structure between predictors is known.
Our results for the backcross are also applicable to recombinant inbred lines. Modifications are necessary for map expansion on RI lines and in the cost functions. In a recombinant inbred line, one may be limited by the number of lines one can raise, whereas in a backcross one is limited to a single replication of a phenotype measurement, which entails killing the animal. Also, typically, in a set of RI lines there is essentially no cost of genotyping; the only cost is in phenotyping. Jin et al. (2004) considered the selective phenotyping problem by choosing individuals to phenotype who were as “dissimilar” as possible. This may be interpreted as them trying to choose a design matrix as “large” as possible and hence increasing the information content of the experiment. For example, note that the determinant of the information matrix in a multigenotype cross, as given by (9), depends not only on the selection fraction through Jα, but also on the product of the allele frequencies. Thus, in an F2, if we can undersample the heterozygotes so that all three genotypes at a locus are equally frequent, we will get more information for the same number of individuals phenotyped and genotyped at that locus.
The results of this article have been developed in the context of phenotypes that have a Gaussian distribution conditional on the QTL genotype. If this assumption is grossly violated, we may need to modify our selective genotyping criteria. For example, for a phenotype with a long tail, it may be more efficient to oversample individuals in the long tail. An example of this setting would be when we have survival phenotypes. When there are many traits to be analyzed, knowledge of the correlation structure between the phenotypes may be necessary to employ selective phenotyping and genotyping.
If a cross were selectively genotyped and the phenotypes of the ungenotyped individuals are discarded, the statistical analysis has to proceed with care. If we proceed with an analysis as if the discarded phenotypes were never collected, the effect estimates are biased. The LOD scores are biased (inflated or deflated) relative to a fully genotyped population. However, if we proceed with a likelihood that accounts for the ascertainment, the effect estimates are unbiased, and the LOD scores are deflated relative to a fully genotyped population. If two or more linked QTL are present, then recombination fraction estimates from the selectively genotyped individuals may be biased. For example, if the two QTL are linked in coupling, the recombination fractions are biased downward; if the QTL are linked in repulsion, recombination fractions are biased upward (Lin and Ritland 1996). Unlinked loci may appear linked in the selected population. For example, if two unlinked, additive loci both have similar effects on the phenotype, then individuals with the most extreme phenotypes will have similar genotypes for both loci. In other words, the selection of individuals based on their phenotype will introduce linkage disequilibrium between the unlinked loci. In general, if the data used to make the selective genotyping decisions are not observed (violating the missing at random condition), parameter estimates may be biased.
If the QTL effects are small, the benefit derived from selective genotyping if multiple QTL are segregating is the same as that if a single QTL is segregating. However, the benefit is diminished if some QTL have large effect. In the context of human association and linkage studies, Allison et al. (1998) came to a similar conclusion by examining power using simulations and analytic calculations. Although our results quantify the information content when two unlinked additive QTL are segregating, our approach can be extended to cover linked and epistatic QTL. If the QTL effects are small, selective genotyping does not adversely affect detection of epistasis; it is still beneficial. However, it is unclear to what degree the benefit remains in the presence of some QTL main effects or epistatic effects of moderate size or when QTL are linked. This needs to be explored further.
Most of this article is concerned with the efficiency of a genotyping design. We note that there may be a concern for robustness of a genotyping design. From an efficiency perspective, it might seem that there is never a good reason to type more than half of the individuals. From a robustness perspective (such as for checking recombination fractions,or for segregation distortion), it may be desirable to genotype all individuals (or a subsample of the intermediate individuals) at a few markers on each chromosome.
Our information analysis considers information for the detection of linkage. When linkage has been established, one is interested in localizing the QTL. In this setting, different notions of information should be considered (Darvasi 1997). We also considered cost functions that are linear in the sample size and the number of markers. In practice, there may be economies of scale in which case the cost function will be a concave function. Our optimality results warrant modification in those settings.
The design of QTL experiments involves balancing many competing biological, practical, and statistical priorities. The information perspective provides the experimenter with conceptual, as well as quantitative, tools to address the statistical aspects of experimental design. The essence of this point of view is captured in the formula for the fraction of missing information presented in Equation 1.
Software for performing power calculations, for generating the figures in this article, and for symbolic computation used for some proofs is available from http://www.biostat.ucsf.edu/sen/. The software for performing power and minimum detectable effect size calculations for backcross and F2 intercross populations accounts for marker spacing as well as selective genotyping of extremes. It is packaged as R/qtlDesign, an add-on package to the R programming language (http://www.r-project.org). Programs for numerical computation and for generating the figures in this article were also written in R. Proofs that used symbolic computation were performed using Maxima (http://maxima.sourceforge.net). Both R and Maxima are freely available under the GNU General Public License.
Acknowledgments
We thank B. Paigen and F. Sugiyama for permission to use the hypertension data. We are grateful for the comments of two anonymous referees and the associate editor; they prompted the work on multiple-QTL models. We thank Chuck McCulloch, Mark Segal, and Brian Yandell for helpful discussions. Inspiration for symbolic computation came from Jamie Stafford and Karl Broman. Support for this work was provided by National Institutes of Health grants GM60457 (J.M.S.), CA098438 (J.M.S.), and GM070683 (G.A.C.).
APPENDIX
Likelihood: The likelihood function is
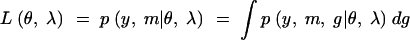 |
A1 |
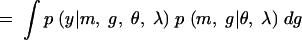 |
A2 |
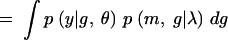 |
A3 |
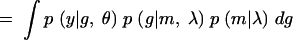 |
A4 |
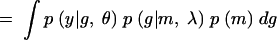 |
A5 |
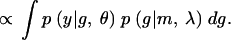 |
A6 |
In Equation A1 we introduce and integrate over the unobserved QTL genotypes, g. Next we condition over m and g to get (A2). Since the phenotype depends on the markers only through the QTL genotypes, p(y|m, g, θ, λ) = p(y|g, θ). Furthermore, the joint distribution of the marker and QTL genotypes does not depend on the genetic model parameters θ, which gives us (A3). Conditioning on the markers gives us (A4). If we assume no segregation distortion or marker-assisted selection, then the marginal distribution of the markers does not depend on the QTL location, and so p(m|λ) = p(m), which gives us (A5). In other words, the likelihood function has the form of a mixture distribution with the probability of the QTL genotypes given the marker information as the mixing probabilities. Sen and Churchill (2001) consider the Bayesian analog of this likelihood function.
Formula for fraction of missing information:
Since the phenotype given the QTL genotypes is normally distributed with variance 1, and means + δ for gi= 1 and − δ for gi= 0,
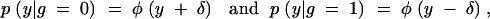 |
where φ(·) is the standard normal density function.
In our context, the missing data are the unobserved QTL genotypes and the observed data consist of the marker genotypes and the phenotypes. The parameter of interest is δ. Thus the distribution of the missing data conditional on the observed data is
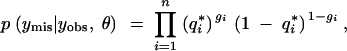 |
where 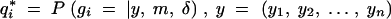 (the observed phenotypes), and m = (m1, m2, … , mn) (the observed marker genotype data).
(the observed phenotypes), and m = (m1, m2, … , mn) (the observed marker genotype data).
Let qi= P(gi= 1|m), that is, the probability of the QTL genotype given the marker data only (not including the phenotype information). Then by the Bayes theorem and using the functional form of the standard normal density function it is easy to see that
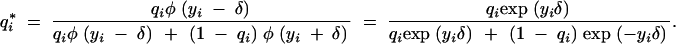 |
A7 |
Let
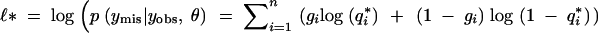 |
Then,
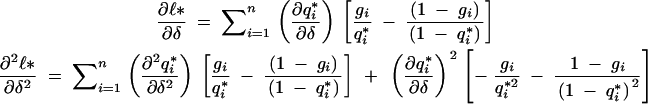 |
Hence,
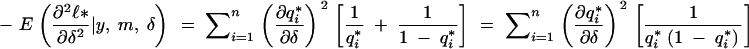 |
A8 |
Using (A7) and differentiating,
 |
A9 |
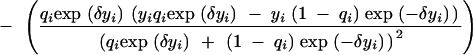 |
A10 |
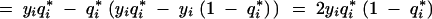 |
A11 |
(A9) follows from the rules of differentiation. Using the definition of q*i as in (A7), we get (A10). And algebraic simplification results in (A11).
Thus, using (A8),
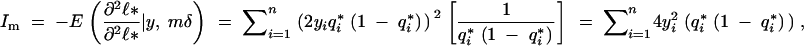 |
which establishes (1).
Optimal selection fraction and marker spacing:
In this section we consider selecting the optimal selection fraction and marker spacing when the QTL effect is small. We consider the most conservative limiting scenario when δ = 0 for which we can derive formulas. The expected information when the selection fraction is α is
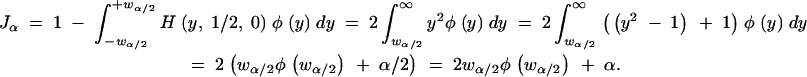 |
The first line follows from Equation 2, noting that there is no information loss for the extreme individuals who are genotyped. Individuals with phenotype between −wα/2 and +wα/2 are not genotyped, and hence the prior probability of their genotype is 1/2. The second line follows from the definition of the function H and algebraic simplification. The final line follows from integration, noting that ∫(y2 − 1)φ(y) = −yφ(y). When the location of the QTL is in the middle of a marker interval that is of length d cM, the expected information is
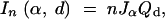 |
where Qd = (1 − 4q(1 − q))(1 − r), r is the recombination fraction corresponding to the genetic distance d, and q is the conditional probability that the QTL has the same genotype as its flanking marker genotypes given that the flanking markers are not recombinant. Assuming the Haldane map function, we would have
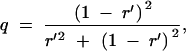 |
where r′ is the recombination fraction corresponding to a genetic distance of d/2. To see this, note that only nonrecombinant individuals contribute information. The contribution from the nonrecombinant intervals is 1 − 4q(1 − q) times the contribution of a completely genotyped location.
The information-cost ratio given a marker spacing d is given by Equation 4. Note that it has the form
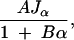 |
for constants A = Qd and B = cG/d when d is fixed. Differentiating with respect to α we get that the maximum must satisfy
 |
where J′α is the derivative of Jα with respect to α. Since the denominators are nonzero, we get
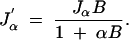 |
Finally, note that
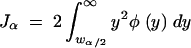 |
and therefore using Leibniz's theorem for differentiation of an integral
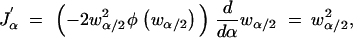 |
since
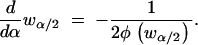 |
Optimal replication number:
From Equation 6 we get the information-cost ratio as a function of t. It is sufficient to minimize its reciprocal as a function of t, At = (1 + ct)(1 + (τ2/t)):
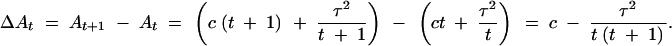 |
Hence, At is minimum for the largest t such that the difference above is positive. This establishes the optimal replication number (7).
Information in multigenotype crosses:
In this section we calculate the information content of multigenotype crosses under selective genotyping. We calculate the observed information matrix using the missing information principle. The complete-information matrix is calculated as the conditional expectation given the observed data of the curvature of the complete-data log-likelihood; the missing information matrix is calculated as the conditional dispersion given the observed data of the score function of the missing-data log-likelihood (Louis 1982; McLachlan and Krishnan 1996). The complete-data log-likelihood is a Gaussian log-likelihood
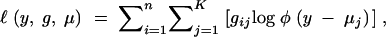 |
where gij is the indicator if gi= j. Hence the complete-information matrix is
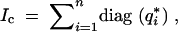 |
where q*i denotes the posterior probabilities of the K genotypes for the ith individual given the marker and phenotype data. For the F2 this reduces to
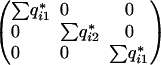 |
The diagonal entries in this matrix are the number of individuals from each genotype category given the observed data. The distribution of the missing data (the QTL genotypes) given the observed data is multinomial and therefore the missing-data log-likelihood is
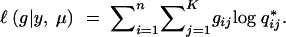 |
This leads to the conditional score function,
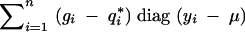 |
It follows that the variance of the conditional score function is
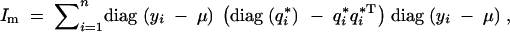 |
which is
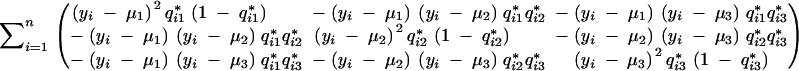 |
for F2's. As with the backcross, we consider selective genotyping an α-fraction of the extreme phenotypic individuals, when the phenotype means in all QTL genotype classes are approximately equal. Additionally, assume that when we genotype, we genotype densely. In this special case, the posterior distribution of the QTL genotypes for ungenotyped individuals is the same as their prior distribution. Also, since the QTL effect is negligible, all genotypes will be equally represented in each phenotype class. Therefore, the complete-information matrix, in expectation over all realizations of the data, is
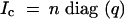 |
and for the F2 case is
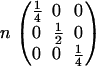 |
For the missing-information matrix, note that the only contributions come from individuals who are not genotyped. For those individuals, the contribution is proportional to y2 multiplied by the variance matrix of the QTL genotypes in the cross. For the F2's this is
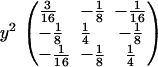 |
Therefore, when we are genotyping individuals only a fraction α of the extreme phenotypic individuals, i.e., those exceeding wα/2 in absolute value, the expected value over all realizations of the data of the missing-information matrix becomes
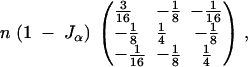 |
where Jα is the expectation of a squared normal variable, truncated above wα/2 in absolute value. For more general crosses, the information matrix is
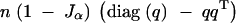 |
Hence the expected value (under all realizations of the data) of the observed information matrix is
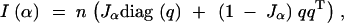 |
which is, for F2's,
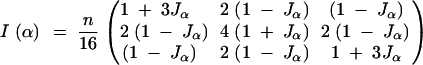 |
Algebraic computation reveals that
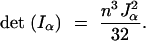 |
The variance of a contrast, u = (u1, u2, u3), is then
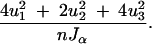 |
The inverse of the information matrix is
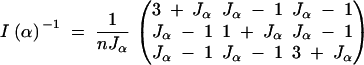 |
For the more general multigenotype case, the determinant of the information matrix is
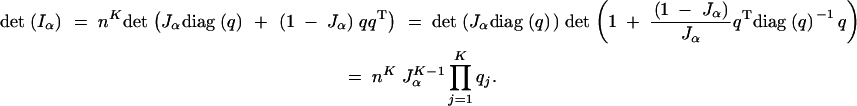 |
The second line follows from noting that
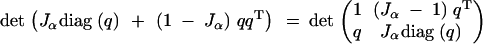 |
The inverse of the information matrix is
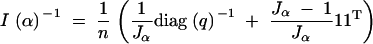 |
Verify by multiplication.
Information in the presence of an unlinked QTL:
Let gjki be the indicator that g1i= j and g2i= k, j, k = 0, 1. The complete-data log-likelihood is
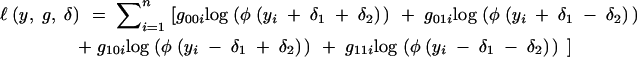 |
This gives the complete-information matrix,
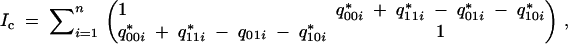 |
where q*jki is the posterior expectation of gjki given the phenotype data. When δ1 = 0 it reduces to the sum of identity matrices. The missing-information matrix is the second derivative of the missing-data (QTL genotypes) likelihood. Since the two loci are unlinked, the prior distributions of the QTL genotypes of the two loci are independent. The posterior distributions given the phenotype are found by the Bayes theorem, and the missing information matrix can be calculated using symbolic computation (see code at http://www.biostat.ucsf.edu/sen/). When (δ1, δ2) = (0, β) it reduces to
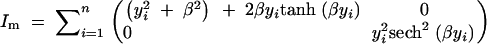 |
We can calculate the expected value of the information matrix by numerical integration. The special cases of β = 0 and β = ∞ deserve special mention. Note that the missing information for δ2 is y2i sech2(βyi), which is the same as H(yi, 1/2, β). For β = 0 the observed information matrix reduces to
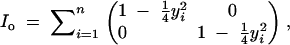 |
whose expected value, using the definition of Jα, is
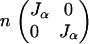 |
For large β it is easy to see that the expected information for δ2 is approximately equal to n. Using the definition of tanh(x) = (exp(−x) − exp(x))/(exp(−x) + exp(x)), we can see that for large β, the missing information for δ1 for the ith observation is equal to
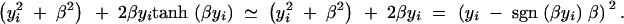 |
Therefore the expected information per observation for δ1 with a selection fraction of α is approximately
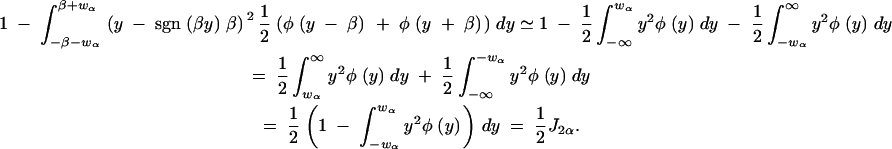 |
The first step follows, noting that the upper α/2 point of the marginal distribution of the phenotype for large β is wα. The second step breaks the integral into sums and then uses the fact that β is large. The final step follows from the definition of Jα.
References
- Allison, D. B., M. Heo, N. J. Schork, E. L. Wong and R. C. Elston, 1998. Extreme selection strategies in gene mapping studies of oligogenic quantitative traits do not always increase power. Hum. Hered. 48: 97–107. [DOI] [PubMed] [Google Scholar]
- Belknap, J. K., 1998. Effect of within-strain sample size on QTL detection and mapping using recombinant inbred mouse strains. Behav. Genet. 28: 29–38. [DOI] [PubMed] [Google Scholar]
- Broman, K., H. Wu, S. Sen and G. Churchill, 2003. R/qtl: Qtl mapping in experimental crosses. Bioinformatics 19: 889–890. [DOI] [PubMed] [Google Scholar]
- Cox, D., and D. Hinkley, 1974 Theoretical Statistics. Chapman & Hall, London.
- Cox, D., and N. Reid, 2000 The Theory of the Design of Experiments. Chapman & Hall/CRC Press, London/Boca Raton, FL.
- Darvasi, A., 1997. The effect of selective genotyping on QTL mapping accuracy. Mamm. Genome 1: 67–68. [DOI] [PubMed] [Google Scholar]
- Darvasi, A., and M. Soller, 1992. Selective genotyping for determination of linkage between a marker locus and a quantitative trait locus. Theor. Appl. Genet. 85: 353–359. [DOI] [PubMed] [Google Scholar]
- Darvasi, A., and M. Soller, 1994. Optimum spacing of genetic markers for determining linkage between marker loci and quantitative trait loci. Theor. Appl. Genet. 89: 351–357. [DOI] [PubMed] [Google Scholar]
- Dempster, A., N. Laird and D. Rubin, 1977. Maximum likelihood from incomplete data via the EM algorithm. J. R. Stat. Soc. Ser. B 39: 1–22. [Google Scholar]
- Efron, B., and D. Hinkley, 1978. Assessing the accuracy of the maximum likelihood estimator: observed versus expected Fisher information. Biometrika 65: 457–482. [Google Scholar]
- Elston, R., 1994 P values, power, and pitfalls in the linkage analysis of psychiatric disorders, pp. 3–21 in Genetic Approaches to Mental Disorders: Proceedings of the Annual Meeting of the American Psychopathological Association, edited by E. Gershon and C. Cloninger. American Psychiatric Press, Washington, DC.
- Haley, C., and S. Knott, 1992. A simple regression method for mapping quantitative trait loci in line crosses using flanking markers. Heredity 69: 315–324. [DOI] [PubMed] [Google Scholar]
- Jin, C., H. Lan, A. D. Attie, G. A. Churchill and B. S. Yandell, 2004. Selective phenotyping for increased efficiency in genetic mapping studies. Genetics 168: 2285–2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao, C.-H., 2000. On the difference between maximum likelihood and regression interval mapping in the analysis of quantitative trait loci. Genetics 156: 855–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander, E. S., and D. Botstein, 1989. Mapping Mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics 121: 185–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebowitz, R., M. Soller and J. Beckmann, 1987. Trait-based analyses for the detection of linkage between marker loci and quantitative trait loci in crosses between inbred lines. Theor. Appl. Genet. 73: 556–562. [DOI] [PubMed] [Google Scholar]
- Lin, J. Z., and K. Ritland, 1996. The effects of selective genotyping on estimates of proportion of recombination between linked quantitative trait loci. Theor. Appl. Genet. 93: 1261–1266. [DOI] [PubMed] [Google Scholar]
- Little, R. J., and D. B. Rubin, 1987 Statistical Analysis With Missing Data. John Wiley & Sons, New York.
- Louis, T., 1982. Finding the observed information matrix when using the EM algorithm. J. R. Stat. Soc. Ser. B 44: 226–233. [Google Scholar]
- McLachlan, G. J., and T. Krishnan, 1996 The EM Algorithm and Extensions. John Wiley & Sons, New York.
- Medugorac, I., and M. Soller, 2001. Selective genotyping with a main trait and a correlated trait. J. Anim. Breed. Genet. 118: 285–295. [Google Scholar]
- Orchard, T., and M. Woodbury, 1972 A missing information principle: theory and applications, pp. 697–715 in Proceedings of the 6th Berkeley Symposium on Mathematical Statistics, Vol. 1, edited by L. M. LeCam, J. Neyman and E. L. Scott. University of California Press, Berkeley, CA.
- Ronin, Y., A. Korol, M. Schtemberg, E. Nevo and M. Soller, 2003. High-resolution mapping of quantitative trait loci by selective recombinant genotyping. Genetics 164: 1657–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satagopan, J., B. Yandell, M. Newton and T. Osborn, 1996. A Bayesian approach to detect quantitative trait loci using Markov chain Monte Carlo. Genetics 144: 805–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satagopan, J. M., and R. C. Elston, 2003. Optimal two-stage genotyping in population-based association studies. Genet. Epidemiol. 25: 149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satagopan, J. M., D. A. Verbel, E. S. Venkatraman, K. E. Offit and C. B. Begg, 2002. Two-stage designs for gene-disease association studies. Biometrics 58: 163–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer, J., 1997 Analysis of Incomplete Multivariate Data. Chapman & Hall, London/New York.
- Sen, S., and G. A. Churchill, 2001. A statistical framework for quantitative trait mapping. Genetics 159: 371–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama, F., G. A. Churchill, D. C. Higgins, C. Johns, K. P. Makaritsis et al., 2001. Concordance of murine quantitative trait loci for salt-induced hypertension with rat and human loci. Genomics 71: 70–77. [DOI] [PubMed] [Google Scholar]
- Xu, S., and C. Vogl, 2000. Maximum likelihood analysis of quantitative trait loci under selective genotyping. Heredity 84: 525–537. [DOI] [PubMed] [Google Scholar]