

Abstract
We constructed a genetic linkage map between two divergent populations of Mimulus guttatus. We genotyped an F2 mapping population (N = 539) at 154 AFLP, microsatellite, and gene-based markers. A framework map was constructed consisting of 112 marker loci on 14 linkage groups with a total map length of 1518 cM Kosambi. Nearly half of all markers (48%) exhibited significant transmission ratio distortion (α = 0.05). By using a Bayesian multipoint mapping method and visual inspection of significantly distorted markers, we detected 12 transmission ratio distorting loci (TRDL) throughout the genome. The high degree of segregation distortion detected in this intraspecific map indicates substantial genomic divergence that perhaps suggests genomic incompatibilities between these two populations. We compare the pattern of transmission ratio distortion in this map to an interspecific map constructed between M. guttatus and M. nasutus. A similar level of segregation distortion is detected in both maps. Collinear regions between maps are compared to determine if there are shared genetic patterns of non-Mendelian segregation distortion within and among Mimulus species.
POSTZYGOTIC reproductive isolating mechanisms often accumulate gradually in geographically isolated populations over time, eventually yielding distinct species (Mayr 1963). Genetic mapping in hybrid populations permits reconstruction of some of the genetic changes that occur during the process of speciation (Rieseberg et al. 1999). The advent of molecular marker technology has made it possible to construct linkage maps for many wild species to understand the nature of genomic divergence between taxa (Whitkus 1998; Rieseberg et al. 2000) and to study quantitative trait loci (QTL) responsible for divergence in ecologically important traits. By investigating the pattern of segregation of mapped molecular markers among hybrid progeny, one also can identify loci that may act as reproductive barriers, even if they do not contribute to obvious phenotypic differences between the parental taxa. For example, markers that exhibit non-Mendelian segregation ratios in hybrid populations could be linked to genes causing hybrid lethality or sterility or gametophytic competition (Harushima et al. 2001).
Deviations from expected Mendelian segregation ratios (or transmission ratio distortion) in hybrid populations are a common observation that potentially represents some level of reproductive isolation due to chromosomal rearrangements or genic interactions (Rieseberg et al. 1995). Recent evidence shows that distorted markers cluster nonrandomly along linkage maps, suggesting underlying distorting loci causing this pattern (Jiang et al. 2000; Fishman et al. 2001; Schwarz-Sommer et al. 2003; Myburg et al. 2004; Solignac et al. 2004). The degree of transmission ratio distortion (as measured by the overall number of distorted markers) is thought to be positively correlated with the level of genomic divergence between taxa (Palopoli and Wu 1996; Jenczewski et al. 1997; Whitkus 1998; Harushima et al. 2001; Taylor and Ingvarsson 2003). Empirical studies have provided evidence for fewer distorted markers in intraspecific crosses relative to interspecific crosses in agricultural plants (Zamir and Tadmore 1986; Causse et al. 1994; Jenczewski et al. 1997), suggesting a positive correlation between the degree of transmission ratio distortion and the level of genomic divergence. Unfortunately, patterns of distortion have not been compared at both intra- and interspecific levels in a wild system that has not been subjected to artificial selection.
In this article, we examine the pattern of segregation distortion in an interpopulational cross of the wildflower Mimulus guttatus and compare our results to those previously published on an interspecific cross between M. guttatus and M. nasutus (Fishman et al. 2001). Mimulus has been a model plant system for ecological and evolutionary genetics for >50 years (Vickery 1951) and is an ideal group for analyzing levels of genomic divergence and speciation. In particular, the M. guttatus species complex includes numerous highly diverse natural populations and closely related species. Recently, a genetic linkage map was constructed between the largely outcrossing M. guttatus and highly selfing M. nasutus and significant transmission ratio distortion (TRD) was observed at ∼50% of marker loci. Furthermore, the distorted markers clustered nonrandomly on the linkage map and exhibited a strong pattern: 9 of the 11 distorted chromosomal regions had an excess of M. guttatus alleles and a deficit of M. nasutus alleles. This nonrandom pattern was attributed to interactions between the heterospecific genomes, suggesting that substantial genetic divergence has occurred between these two species (Fishman et al. 2001). More recently, detailed genetic experiments designed to elucidate the underlying mechanism of the TRD against the M. nasutus marker alleles on one chromosomal region, linkage group 11 (LG11), have implicated nearly complete female-specific meiotic drive due to interspecific divergence at a single locus (Fishman and Willis 2005).
Here, we construct and analyze a genetic linkage map based on an intraspecific F2 hybrid cross between two phenotypically divergent populations of M. guttatus. This map allows for direct examination of genomic interactions at the level of population differentiation and for a comparison of the distortion found between two M. guttatus populations to that found in the interspecific cross between M. guttatus and M. nasutus. We first ask whether there is evidence of intraspecific transmission ratio distortion in crosses between these wild populations; and, if so, what are the potential causes of the distortion? Second, we ask whether there are similar or different levels of distortion between and within species of Mimulus. Finally, we ask whether there is the potential for a shared genetic basis for distortion between and within species. Many of the molecular markers used in this study were also mapped in the interspecific study of Fishman et al. (2001), facilitating the identification of homologous chromosomal regions and a comparison of regions of distortion between maps. These more detailed comparisons are a first step toward investigating the potential for common genetic factors to cause transmission ratio distortion at multiple levels of divergence in Mimulus.
MATERIALS AND METHODS
Study system:
The M. guttatus species complex (historically Scrophulariaceae, order Lamiales) is highly polymorphic and geographically widespread throughout western North America (Pennell 1951; Vickery 1978; Sweigart and Willis 2003). Populations differ in morphology, mating system, life history strategy, and habitat type. Although widely studied in ecology and evolutionary biology, taxonomic classification of the M. guttatus species complex has been inconsistent. In fact, some authors have subdivided this group into 17 morphologically distinct species (Pennell 1951), while others designate just a few subspecies within the complex (Hitchcock and Cronquist 1973). M. guttatus (2n = 28) is the most common and variable species in the complex.
Populations of M. guttatus can exist as either annuals or perennials, with perennial populations widespread along the Pacific coast. Perennial plants can also be found inland along streams, rivers, and drainage ditches where there is year-round moisture. Annual populations are typically located at inland sites like seepy hillside meadows, rocky cliff faces, or road cuts that have abundant soil moisture in the spring and early summer, but little during the late summer. Plants from these populations are facultative annuals due to seasonally dry environmental conditions, and they can be maintained indefinitely under standard greenhouse conditions. Flower size and vegetative traits can differ dramatically between annual and perennial populations, with annuals typically being smaller than perennials for most size-related traits in the field and in common garden experiments (M. Hall, unpublished data).
For this analysis, we focus on two populations of M. guttatus that have a high degree of divergence in overall size, habitat, and life history. The well-studied IM population consists of small-flowered, diminutive annuals that live on Iron Mountain, in Oregon's western Cascades (Willis 1993). These plants are predominantly outcrossing (Willis 1993; Sweigart et al. 1999) and have a short period of growth and reproduction, with flowering occurring over a 3- to 5-week period in June through early July. The montane environment experiences fluctuations in temperature and precipitation ranging from below freezing and >2 m of snow in the winter to well above 40° with little or no rainfall in the late summer months. The DUN population consists of large-flowered perennial plants with larger, nearly succulent leaves that inhabit the temperate environment of Oregon's coastal sand dunes south of Florence in the Oregon Dunes National Recreation Area. At this site, temperatures vary <20° from summer to winter, and there is continual moisture available to plants from heavy rain (1930 mm average annual precipitation) and coastal fog. DUN plants typically flower from early June through October or November.
Generation of F2 mapping population:
We generated an F2 mapping population from IM and DUN parents as part of a larger QTL mapping experiment designed to investigate the genetic basis for quantitative trait differences between these populations of M. guttatus. The IM parent was a highly fertile inbred line (IM62) derived from the Iron Mountain site. This parental line is the same parental line used to construct the previous interspecific map (Fishman et al. 2001). Two separate wild-collected plants (DUN1 and DUN2) were used as parents from the DUN perennial population. Each of the DUN parents was reciprocally crossed to IM62 to produce four classes of F1 individuals, and one plant from each class was selected at random to produce the F2 generation. One F1 plant (IM62 maternal parent, DUN1 paternal parent) was reciprocally crossed to another F1 plant (DUN2 maternal parent, IM62 paternal parent) to produce two classes of F2 seeds. The other two F1 plants were also reciprocally crossed to each other to produce two other classes of F2 seeds, for a grand total of four classes of F2 seeds. Each F2 individual therefore has a nuclear genome derived from contributions of three individuals (IM62, DUN1, and DUN2) and a cytoplasmic genome derived from either the DUN or IM population. Note that this crossing design enforces outbreeding with respect to alleles derived from the DUN population, but allows for homozygosity of alleles from the highly viable and fertile inbred IM62 line, thereby reducing the potential for transmission ratio distortion in F2 progeny to be caused by inbreeding depression. All seeds used in the common garden experiment described below were the same age: the F1 plants and the parental plants were recreated by selfing IM62 and reciprocally crossing DUN1 and DUN2 at the same time as the creation of the F2 lines.
In June 2000, we grew 100 IM62 plants, 50 each of the DUN1 × DUN2 plants and their reciprocal crosses, and 200 F1 plants along with the F2 mapping population (N = 600 total, with each of the four F2 classes equally represented) in individual pots in a common garden experiment at the University of Oregon Department of Biology greenhouse. The plants were grown in 4-inch pots filled with sand over a thin layer of hemlock bark on the bottom, to prevent sand from escaping the pot. A thin layer of organic potting mix (Black Gold potting soil; Sun Gro Horticulture, Bellevue, WA) was sprinkled on top to prevent dessication of seeds. To ensure the presence of seedlings in each pot, we planted five seeds of the same class per pot on June 12, 2000, and pots were placed in flats in a fully randomized design in the greenhouse during the long days when flowering begins for each of the native populations. Plants were watered as needed two to three times daily and left unfertilized. Seedlings were thinned to the centermost individual after germination, 2 weeks after planting.
Tissue collection and DNA extraction:
A total of four corollas from each F2 individual and each of the three parents were collected into separate 1.5-ml Eppendorf tubes, immediately placed on dry ice, and stored at −80°. Genomic DNA was isolated from the corollas using a modified hexadecyl trimethyl-ammonium bromide chloroform extraction protocol (Lin and Ritland 1996; Kelly and Willis 1998). DNA concentration was quantified with a Hoechst fluorometer. Of the 600 F2 lines, we collected corolla tissue from 539 individuals for genotyping. The remaining 61 individuals were not genotyped due to a variety of factors: failed DNA extractions, mortality before tissue collection occurred, or insufficient quantity of floral tissue for DNA extraction.
Molecular marker analyses:
We used three different types of PCR-based molecular genetic markers, including amplified fragment length polymorphisms (AFLPs), microsatellites, and gene-based markers, for genotyping in this hybrid mapping population. The AFLPs were scored using standard protocols (Vos et al. 1995; Remington et al. 1998; Fishman et al. 2001) with modifications for high throughput and low DNA content per reaction. The procedure followed that of Fishman et al. (2001) in using a standard restriction digest-ligation step, followed by preamplifications and then final selective amplifications. Each selective primer combination was visualized with the Li-Cor automated sequencing system. Polymorphic fragments were scored visually on TIFF image files using RFLPSCAN 3.0 (Scanalytics). See Fishman et al. (2001) for details on primer combinations and scoring procedure. We used standard EcoRI (E) and TaqI (T) primers with single selective nucleotides for preamplifications. For final amplification, we used different combinations of three E primers with three selective nucleotides (E + ACG, E + ACC, and E + AGG) and the three T + 1 primers. In total, we used eight different primer combinations and nomenclature follows that described in Fishman et al. (2001). Only fragments that were consistently present in both DUN parents and absent in IM62 (or, conversely, absent in both DUN parents and present in IM62) were scored for this analysis. Most AFLP markers were scored as dominant markers. A small number of AFLP fragments clearly segregated as alternative alleles at a single locus and were scored as codominant markers. We used eight primer combinations to produce a total of 126 polymorphic markers, 3 of which were codominant. Scored fragments ranged in length from 55 to 518 bp.
Microsatellite and several gene-based markers have previously been developed for genetic mapping in M. guttatus and its close relative M. nasutus (Kelly and Willis 1998; Fishman et al. 2001). We tested all of the codominant markers originally used in the interspecific map (Fishman et al. 2001) for polymorphism in this cross and used all of these that consistently segregated alternative alleles among the F2's for genotyping. The primers, GenBank accession numbers, PCR conditions, and names of these markers are given in Fishman et al. (2001). We tested 32 different markers in total. We identified 11 informative microsatellite loci and three informative gene-based markers. We made a few minor changes in our PCR and genotyping protocol, where the forward primers were 5′ labeled with fluorescent dyes for detection on an ABI 3700 genetic analyzer. The PCR products were run on the ABI 3700, fragments were detected using GeneScan 3.5.1, and their sizes were determined using Genotyper 3.6 (Applied Biosystems, Foster City, CA).
We also tested 25 additional gene-based markers for polymorphism in our cross. These markers were developed as part of a larger collaborative project and will be described in detail elsewhere. Briefly, these new markers were designed so that primer pairs flanked introns in nuclear genes. Any intron length polymorphisms would therefore be revealed as PCR-product length polymorphisms. These markers were developed first by sequencing random clones from a cDNA library constructed from RNA isolated from IM62 floral bud tissue. The resulting expressed sequence tags were then assembled into contigs, and the contigs were searched against the Arabidopsis protein database with BLASTX for contigs with high similarity to a small number of Arabidopsis proteins. Putative intron positions in the subset of selected IM62 contigs were determined using the Arabidopsis annotations with alignments of the contigs and the Arabidopsis proteins, and primers flanking the introns were designed. We initially tested 25 of these new markers, named with a prefix of MgSTS (for M. guttatus sequence-tagged site), and determined which were polymorphic for PCR product size in our cross. Ten of the MgSTS markers were informative, and we genotyped them in all of the F2 individuals using the same PCR conditions as in the microsatellite reactions, except that we used a standard annealing temperature of 52° and 31 cycles for all markers. All forward MgSTS primers were 5′ labeled with a fluorescent dye for detection on the ABI 3700. All markers that were mapped are included, along with forward and reverse primers, in Table 1. Individual F2 genotypes were analyzed and scored in the same manner as the microsatellites. All of the microsatellite and gene-based markers produced fragments inherited in a codominant manner.
TABLE 1.
Names and primers for mappedM. guttatus sequence-tagged site (MgSTS) markers
| Marker name | Forward primer (5′–3′) | Reverse primer (5′–3′) |
|---|---|---|
| MgSTS18 | GGTTGGCCAAGTATCGATTT | AGGCAAACCCACATAGCATC |
| MgSTS19 | ATTTGCCGTTCCACAATCTC | AGTTCCATTCGACCGATACG |
| MgSTS25 | AATGGAGATGTGGGCAAGAT | AATTGCGGGAACAGCATTAG |
| MgSTS35 | AAAATCGGGGAGAATTTTGG | CACACGTGGCTGGATTACAC |
| MgSTS38 | ATGAGCATGGCATCGACATA | GTCTCACCGTGTCGGATTTT |
| MgSTS41 | GGTAGCGGAATTCATCCTCA | GCAGAGCTTTCACCACCTTC |
| MgSTS43 | CCGGGAAACGATAGAACAAA | CAAGGGAGTTCCCTGCAATA |
| MgSTS54 | TCAAATTCGATGTGGGATCA | AAACCCGACTGCTGCTAATG |
| MgSTS56 | GGACTGATGCCAAACCCTAA | AATCTGCCTTCCCAAAAGGT |
| MgSTS87 | CTTCGACGATGCAGAGAGTG | ACATAAGCCCTCCTCGTGAA |
Linkage map construction:
Using the molecular markers genotyped for 539 F2 individuals, we constructed a genetic linkage map using MAPMAKER 3.0 (Lander et al. 1987; Lincoln et al. 1992), using the same methodology as that of Fishman et al. (2001). All of the distances between markers were estimated using the Kosambi mapping function (Kosambi 1944). The error detection data and the table of two-point distances were used to identify unreliable markers. We repeatedly tried placing unlinked markers and eliminated unreliable markers until we reached a consistent linear order for each group that included a subset of the most reliable markers. A small subset of markers was removed from the mapping data set due to the possibility of marker alleles that were not identical by descent. These markers were easily distinguished by unequal representations of particular marker genotypes in two of four F2 classes relative to reciprocal pairs.
Genome length and map coverage:
The total genome length was estimated in several different ways. First, we calculated s, the average framework marker spacing, by dividing the combined total length of all linkage groups by the number of intervals. Then the genome length L was estimated using various methods. First, we added 2s to the length of each linkage group to account for chromosome ends beyond the terminal markers. Second, we used method 4 of Chakravarti et al. (1991) to calculate the length of each linkage group. This method multiplies the length of each linkage group by (m + 1)/(m − 1), where m is the number of framework markers on each group. We also estimated the map coverage c. The proportion c of the genome that is within distance d cM of a marker, assuming random distribution of markers, was estimated using c = 1 − e−2dn/L, where n is the number of markers and L is the estimated genome length.
Segregation distortion analysis:
We tested each marker for significant deviations from expected Mendelian genotype frequencies (χ2 with 1 d.f. for dominant markers, 2 d.f. for codominant markers, α = 0.05; Sokal and Rohlf 1995). It is possible for distorted genotypic ratios to appear by chance alone or as a result of linkage to a locus important for hybrid fitness. Therefore, we attempted to correct for multiple tests using a more conservative threshold of significance (α = 0.001), as well. To examine the pattern of distortion across the framework map, we also calculated the deviation of the parental homozygote frequency from the Mendelian expectation of 0.25 at each locus.
We used the Bayesian multipoint mapping method developed by Vogl and Xu (2000) to estimate the location and effects of transmission ratio distorting loci (TRDL). This method assumes that different TRDL act independently. We used the program ANITA (supplied by C. Vogl) to analyze our genotypic data with 10,000 iterations per linkage group. We ran the program with the maximum number of TRDL set to one and two for all linkage groups. The position of each TRDL was estimated as the mode of the posterior distribution of detected TRDL. Genotype and allele frequencies were calculated from the mean of the 100 nearest values to the mode of the posterior distribution. Also included is the highest posterior density interval at 95% for each of the TRDL detected, as estimated from the posterior distribution.
RESULTS
Linkage map construction:
We were able to genotype the F2 population at a total of 154 markers. Although there were some missing data due to occasional PCR failure, a large proportion of the individuals were genotyped per marker (mean = 493, SD = 54). We initially evaluated linkage by grouping linked markers on the basis of linkage criteria between a pair of markers with a minimum LOD of 6 and a maximum distance of 40 cM. Fourteen groups of linked markers were obtained using these criteria, and only 3 of the 154 markers were found to be unlinked to any linkage group.
We then constructed a linkage map based on a subset of markers that displayed the most reliable placement on their linkage groups. To arrive at this framework linkage map, we subjected each linkage group to exhaustive grouping, ordering and comparing of different marker combinations. We excluded some markers from the final framework map due to unreliable or nonlinear placement based on comparisons between two-point and multipoint analyses. One marker on LG11, AAT356, behaved inconsistently with respect to flanking markers. The three markers appeared to be equally linked to each other, generating an apparent triangulation of linkage among the markers. Ordinarily we would have excluded this marker from the framework map, but because this codominant marker is tightly linked to a TRDL in the interspecific map (Fishman et al. 2001), we decided to present maps of LG11 with and without that marker. The linkage group with AAT356 excluded is referred to as LG11a, and the framework linkage map containing LG11a is referred to as map A.
The final framework map (Figure 1) includes a total of 112 markers (111 for map A). Codominant markers make up 24% of the markers on the framework map. These codominant markers as well as the large size of the mapping population allowed us to construct a single map that included both classes of dominant AFLP markers. The two classes of dominant AFLPs on the map are not equally abundant (30 and 70% of the markers had DUN and IM homozygotes, respectively), although their frequencies do not differ substantially from the distribution of total AFLP marker genotypes (40 and 60%). Because some of the DUN homozygote AFLP markers were eliminated from the data set due to the chance of alleles that were not identical by descent, this deviation from 50% for each marker class is not surprising.
Figure 1.—
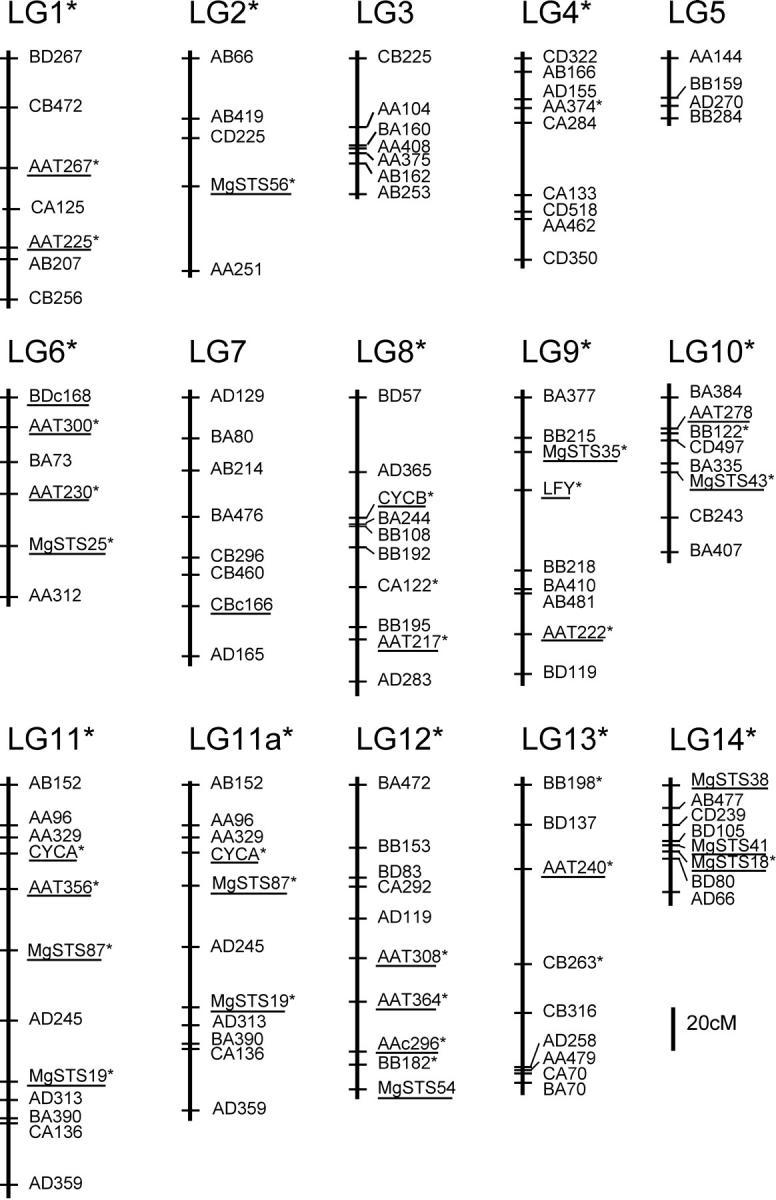
Framework linkage map of M. guttatus (IM) × M. guttatus (DUN) F2 hybrid population. The names of all codominant markers are underlined. All markers and linkage groups that are shared with the interspecific M. guttatus (IM) × M. nasutus map are marked with an asterisk (*).
The 14 linkage groups correspond to the haploid chromosome number for M. guttatus. A total of 27 (both dominant and codominant) markers are common to both this map and the M. guttatus × M. nasutus map (Fishman et al. 2001; L. Fishman and J. H. Willis, unpublished results for the new gene-based markers) and they include 10 microsatellite loci, 10 gene-based markers, one codominant AFLP, and six dominant AFLP markers. These markers map to 11 of the 14 linkage groups. The marker order is preserved between maps, making it possible to compare collinear regions between an intra- and interspecific cross. The 11 linkage groups with common markers were assigned the same numbers as the linkage groups from Fishman et al. (2001). Seven of the 8 linkage groups with at least 2 shared markers maintain collinearity, although intramarker distances vary slightly. The other linkage group (LG11) has a very minor difference between maps, as more recent unpublished mapping results show very tight linkage of several codominant markers. These markers are more spread out with a slightly different ordering in the intraspecific map relative to the very tightly linked markers in the interspecific map (Figure 3). Three other linkage groups share a single marker (LG2, LG4, and LG14), several of which have been added to the interspecific map since publication (L. Fishman and J. H. Willis, unpublished data).
Figure 3.—

Comparative map of TRDL between and within species of M. guttatus. The linkage group is indicated above both intraspecific [g × g (M. guttatus × M. guttatus)] and interspecific [g × n (M. guttatus × M. nasutus)] maps. Hatch marks indicate marker placement. Only terminal markers and communal markers are labeled on each map, with dashed lines connecting communal markers. Linkage groups with a single communal markers are matched up arbitrarily; note the orientation could be rotated. Due to placement of additional codominant markers to the previously published interspecific map (Fishman et al. 2001), several AFLP markers were ousted to maintain basic mapping criteria. The following changes were made to the g × n map: LG2, addition of MgSTS56, slight separation of BA172 and CB280; LG6, addition of MgSTS25; LG9, addition of MgSTS35 and removal of BC108, slight separation of CA261 and CB115; LG10, addition of MgSTS43 and removal of AA153c and AA100; LG11, addition of MgSTS19 and MgSTS87, removal of BA387, change in map order for CYCA, AAT356, BD100, BB124, BA196, slight separation of BB124 and BA196; and LG14, addition of MgSTS18. Arrows point to locations of TRDL or detected regions of distortion. Solid arrows represent markers distorted toward excess IM (M. guttatus) alleles, open arrows represent markers distorted toward either excess DUN or M. nasutus alleles, stippled arrow represents distortion with an excess of heterozygotes, and shaded arrow represents distortion with a deficiency of heterozygotes.
Map length and genome coverage:
The total map length spans 1517.8 cM Kosambi (1481.7 cM Kosambi for map A). We used several approaches to estimate genome length L, all of which indicate that the framework map provides fairly complete coverage of the M. guttatus genome. Using the first method, which assumes a random distribution of markers across the genome, we added twice the average interval length (s = 15.49 and 15.28 cM for map A) to each linkage group to account for chromosome ends beyond the terminal markers. The estimated genome length using this method is 1951.46 and 1909.4 cM for map A. Using method 4 of Chakravarti et al. (1991), we estimated a nearly identical genome length of 1952.56 cM (1912.77 cM for map A) by including only the markers placed on the linkage groups. When using markers that were linked but excluded from the framework map, we estimated a slightly smaller length of 1843.53 cM for both maps. All of these numbers are very close to (although somewhat smaller than) the genome length estimated for the M. guttatus × M. nasutus genome, which was 2092 cM, using method 1. Using the different methods of genome length estimates, we estimated that 68–70% of the genome is within 10 cM of a linked marker, and 89–91% is within 20 cM of a linked marker. These estimates of genome coverage are slightly lower than that for the M. guttatus × M. nasutus map.
Intraspecific segregation distortion:
Genotyping of all 154 markers revealed substantial non-Mendelian inheritance (48% at α = 0.05, 29% at α = 0.001) across all markers. Of the 112 markers that were included on the framework map, 47 and 27% (α = 0.05 and α = 0.001, respectively) showed significant distortion from Mendelian expectations. For the mapped markers, a larger percentage of the codominant markers (74% at α = 0.05, 52% at α = 0.001) were distorted when compared to dominant markers (39% at α = 0.05, 19% at α = 0.001). This may be due to the fact that codominant markers have more complete genetic information and therefore have greater power to detect distortion. The distorted loci were equally split between deviation toward the IM or the DUN genotype (49% in either direction), 3 codominant markers showed heterozygote excess, and 6 codominant markers showed a deficiency of heterozygotes.
Each of the markers was analyzed within the context of its position along the linkage groups relative to other markers (Figure 2), allowing us to visually examine genomic regions of distortion in either direction. Many of the distorted markers are clustered in one direction or another along the linkage groups. We identified five distorted regions with two or more adjacent loci that were significantly distorted in the same direction, all on different linkage groups. Three of these regions display an excess of DUN alleles (or a deficiency in IM alleles) and are located on LG2, LG8, and LG14, whereas two display an excess of IM alleles (or a deficiency of DUN alleles). These are located on LG9 and LG10 (Figure 2).
Figure 2.—

Transmission ratio distortion across the M. guttatus (IM) × M. guttatus (DUN) framework map. The dot and the plus symbol represent the two homozygous parental genotypes (DUN/DUN and IM/IM, respectively) at marker loci on each of the 14 linkage groups. The vertical position of each symbol shows the magnitude and direction of the deviation of genotype frequencies from the Mendelian expectation (0.25). The biases were graphed directly. DUN homozygote deviations (DUN/DUN) were graphed as positive values [deviation = f(DUN/DUN) − 0.25], and IM homozygotes (IM/IM) were graphed as negative values [deviation = −(f(IM/IM) − 0.25)]. The shaded peaks show the frequency distributions of the location of TRDL as estimated by the Bayesian mapping method of Vogl and Xu (2000). The average frequency of detecting a TRDL is indicated next to each peak. Each frequency distribution is scaled to the maximum frequency per group for visualization purposes.
The results from the multipoint Bayesian method developed by Vogl and Xu (2000) corresponded fairly well with the visual analysis of detection and placement of TRDL, as described above. The results with the maximum number of TRDL set to one fit the data most reliably, with the exception of LG8, where the maximum number of TRDL of two was used. A single TRDL was detected on 12 of the 14 linkage groups a majority of the time (>50%) after 10,000 iterations. The estimated position of each TRDL and its estimated allele and genotype frequencies based on the posterior frequency distribution are reported in Table 2. Five regions demonstrated distortion toward excess of IM homozygotes (or deficiency of DUN homozygotes) and are located on LG3, LG6, LG8, LG9, and LG10, whereas four regions displayed the reciprocal (located on LG2, LG5, LG8, and LG14, see Table 2; Figure 2). The remaining three TRDL do not result in greater frequencies of either the IM or DUN alleles. In two of these cases, there is apparently an excess of heterozygotes over that expected with Mendelian ratios (LG7 and LG11), and in one case there is an apparent deficiency of heterozygotes (LG12). Using a more stringent criterion for significance (frequency of iterations in which a TRDL was detected >75%), TRDL are detected on 8 of the 14 linkage groups. Four have distortion toward the IM allele, 3 have excesses of the DUN allele, and 2 have a pattern with no strong allele. The positions of the distorted regions we detected visually were the same as those detected using the Bayesian method, although the Bayesian method detected additional TRDL.
TABLE 2.
Summary of TRDL positions and their associated genotype and allele frequencies
| DUN homozygote
|
Heterozygote
|
IM homozygote
|
Allele frequency |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Position (cM) |
Frequency | HPDIa | Frequency | HPDIa | Frequency | HPDIa | DUN | IM | |
| LG2 | 77.5 | 0.36 | 0.17–0.61 | 0.48 | 0.23–0.78 | 0.15 | 0.015–0.30 | 0.60 | 0.40 |
| LG3 | 46.4 | 0.21 | 0.013–0.52 | 0.45 | 0.097–0.77 | 0.35 | 0.21–0.68 | 0.43 | 0.57 |
| LG5 | 26.4 | 0.39 | 0.10–0.72 | 0.45 | 0.068–0.87 | 0.16 | 0.0095–0.49 | 0.62 | 0.38 |
| LG6 | 70.7 | 0.27 | 0.15–0.40 | 0.41 | 0.27–0.54 | 0.33 | 0.11–0.45 | 0.47 | 0.53 |
| LG7 | 125.2 | 0.20 | 0.12–0.42 | 0.60 | 0.37–0.73 | 0.19 | 0.062–0.28 | 0.51 | 0.49 |
| LG8-1 | 51.0 | 0.29 | 0.056–0.56 | 0.36 | 0.11–0.62 | 0.35 | 0.13–0.61 | 0.47 | 0.53 |
| LG8-2 | 94.7 | 0.26 | 0.055–0.51 | 0.59 | 0.37–0.81 | 0.15 | 0.052–0.41 | 0.56 | 0.44 |
| LG9 | 61.2 | 0.25 | 0.15–0.36 | 0.32 | 0.17–0.43 | 0.43 | 0.060–0.58 | 0.41 | 0.59 |
| LG10 | 0.3 | 0.15 | 0.070–0.23 | 0.62 | 0.46–0.80 | 0.23 | 0.068–0.38 | 0.46 | 0.54 |
| LG11 | 66.5 | 0.18 | 0.087–0.29 | 0.66 | 0.55–0.79 | 0.16 | 0.082–0.35 | 0.51 | 0.49 |
| LG11a | 53.0 | 0.19 | 0.082–0.29 | 0.66 | 0.54–0.79 | 0.16 | 0.080–0.37 | 0.51 | 0.49 |
| LG12 | 98.6 | 0.30 | 0.041–0.46 | 0.38 | 0.21–0.76 | 0.32 | 0.14–0.45 | 0.49 | 0.51 |
| LG14 | 0.3 | 0.44 | 0.30–0.56 | 0.38 | 0.27–0.60 | 0.18 | 0.058–0.30 | 0.63 | 0.37 |
Map position of TRDL detected by the Bayesian mapping methodology is shown (Vogl and Xu 2000). Genotype and allele frequencies were estimated from 100 values nearest to the peak (position) of the posterior distributions of individual TRDL.
For each TRDL, the highest posterior density interval (HPDI) at 95% is presented.
By combining the results of the TRDL mapping with our count of regions with multiple adjacent distorted markers, we estimated a minimum number of distorted loci causing unequal transmission of parental alleles. The TRDL mapping identified 12 loci that substantially altered parental allele and/or genotype frequencies in the F2 cross, using a cutoff of detecting a TRDL at ≥50% of the iterations. In total, 12 TRDL were detected in this intraspecific cross, and they show no real bias favoring DUN or IM alleles on a genome-wide scale. Five of the 12 TRDL showed excesses of IM allele frequencies (located on LG3, LG6, LG8, LG9, and LG10), whereas four regions demonstrated excesses of DUN alleles (on LG2, LG5, LG8, and LG14). The remaining TRDL displayed either heterozygote excesses (2 TRDL) or deficiency (1 TRDL) relative to the two equally frequent homozygote classes. This estimate of the number of TRDL is likely to be an underestimate of the true number of distorting regions because we allowed a maximum of only 1 or 2 loci per linkage group and because it is difficult to detect TRDL in regions with low marker density or no codominant markers.
Intra- vs. interspecific segregation distortion:
Because of the collinearity between portions of this map and the interspecific M. guttatus × M. nasutus map, we can compare many of the linkage groups and their patterns of distortion. Results from the interspecific map detected an equivalent number of TRDL (11–12) to that in the intraspecific map, but the pattern of distortion differed. Nine of the 11 TRDL detected had an excess of M. guttatus (IM) homozygotes (or a deficiency of M. nasutus homozygotes), whereas our map had unbiased directional distortion.
Of the 11 collinear regions of linkage groups, some display no significant distortion in either cross, while others display distortion in one or both of the crosses. One linkage group (LG4) contains common undistorted regions between the two maps (Figure 3). Four linkage groups display distortion in just one map, either in the intraspecific map but not the interspecific map (LG8 and LG9) or in the opposite pattern (LG1 and LG13). The remaining 6 collinear linkage groups have distortion on both maps, two of which have distorted regions in generally the same direction (LG6 and LG10), toward excessive IM homozygotes (or deficiency of the alternative homozygote). One linkage group (LG14) has distorted regions in the same direction, but toward excessive DUN or M. nasutus homozygotes (or deficiency of IM homozygotes). On LG2, although there is distortion on both maps, it is in different directions on each map (Figure 3). Three of these distorted regions (LG11, LG12, and LG14) potentially map to the same regions on both maps, although with different effects (Figure 3).
DISCUSSION
Intraspecific segregation distortion:
We found a high degree of transmission ratio distortion in a cross between populations of M. guttatus, suggesting that these two populations have undergone substantial genetic divergence. The level of distortion in this study (48% of all markers at α = 0.05) is much greater than that reported for other intraspecific crosses (13–18% at α = 0.05; Zamir and Tadmore 1986; Jenczewski et al. 1997; Lu et al. 2002), perhaps reflecting greater intraspecific differentiation in M. guttatus or differences in experimental details or analysis. We found that many of the distorted markers cluster in regions nonrandomly, implying the existence of underlying loci generating this pattern of transmission ratio distortion. There are several potential explanations for this pattern. One potential source of transmission ratio bias is inbreeding depression. Our crossing design makes it unlikely that inbreeding depression is a source of the observed transmission ratio distortion. The IM parent was from a highly inbred line with normal fitness, and the use of two separate DUN parents provides assurance that any single recessive deleterious allele in either DUN parent was not in a homozygous state in the F2 progeny. It is conceivable that both DUN parents were carriers for the same rare recessive deleterious alleles at exactly the same loci, but this seems unlikely and should not account for the overall pattern of distortion across the genome.
Many other potential genetic mechanisms of transmission ratio distortion involve interactions between divergent genomes. These interactions, which may arise at several stages of the life cycle, can bias the genotype frequencies ultimately observed in the F2 hybrids: meiotic drive may distort allele frequencies among the viable F1 gametophytes, gametophytic competition or pollen-pistil interactions may also distort allele frequencies among the gametes that achieve fertilization, or differential viability of genotypic classes in the F2 zygotes may cause TRD observed in the F2 adults.
Unfortunately our results from this single mapping population do not allow us to discriminate among the various potential causes of TRD, particularly since our markers and crossing designs do not allow us to separately follow the maternal and paternal inheritance of particular genomic regions. It is conceivable that one or more of the regions exhibit distortion as a result of meiotic drive acting in one or both sexes, a possibility that is particularly intriguing given the recent finding of female-specific meiotic drive in the M. guttatus × M. nasutus cross (Fishman and Willis 2005). Competition among F1 gametophytes, such as pollen grains competing for access to ovules, is a plausible explanation of at least some of the distortion if competitive ability is caused by allelic variation of genes expressed in haploids. Differential pollen tube growth and pollen-pistil interactions can act to prevent hybrid formation in many intra- and interspecific crosses (Rieseberg and Carney 1998). The substantial differences in style length between these two populations of M. guttatus may provide an arena for pollen competition. Experiments involving application of mixed pollen to M. guttatus and M. nasutus demonstrate that M. guttatus pollen tubes outgrow M. nasutus pollen tubes on the longer M. guttatus style (Kiang and Hamrick 1978; Diaz and Macnair 1999). DUN pistils are almost twice as long on average as IM pistils in a common garden, with F1 styles being roughly intermediate in length (M. Hall, unpublished data). If DUN pollen is competitively superior to IM pollen on long styles due to haploid gene expression, then segregation of pollen growth alleles in the F1 pollen may be a cause of the distortion observed on any of the four TRDL that exhibit an excess of DUN alleles. Finally, differential zygotic survival among the F2 zygotes could explain any of the 12 TRDL. Further experiments, such as the reciprocal backcrosses and introgression studies recently reported for the M. guttatus × M. nasutus cross (Fishman and Willis 2005), are needed to further investigate the mechanisms of the distortion reported here.
Three genomic regions show strong transmission ratio distortion of diploid genotype frequencies without attendant distortion of allele frequencies. Two of these TRDL (on LG7 and LG11) exhibit an excess of heterozygotes while one (on LG12) exhibits a heterozygote deficiency (Table 2). These patterns may be due either to selection acting on a single locus or to selection acting on multiple linked loci. Distinguishing between true overdominance (or underdominance) and pseudo-overdominance (or pseudo-underdominance) as well as the potential mechanisms of selection will require more detailed crossing studies and fine-scale linkage analysis.
Intra- vs. interspecific segregation distortion:
A goal of this study was to compare the magnitude and patterns of TRD observed in this intraspecific cross to that documented in the interspecific M. guttatus × M. nasutus cross (Fishman et al. 2001). Previous work suggests that the degree of distortion between parental genotypes is correlated with degree of divergence, with greater numbers of genomic regions showing significant TRD in interspecific crosses than in intraspecific crosses (Jenczewski et al. 1997; Whitkus 1998). We therefore were surprised by our results showing that the total proportion of distorted markers does not differ between the intraspecific and interspecific studies (48 vs. 49% at α = 0.05 and 29 vs. 31% at α = 0.001, respectively). It is not yet clear why the intraspecific distortion was so prevalent and it may be the result of several factors. For example, it may be that the two populations of M. guttatus studied here may be more divergent than populations within other species, and indeed M. guttatus is well known to harbor tremendous phenotypic (e.g., Vickery 1978) and molecular genetic (Sweigart and Willis 2003) diversity. Methodological differences between our study and other mapping studies in sample size, number or type of genetic markers, or statistical methods may also have resulted in differences in the power to detect TRDL.
When we examined the genomic locations of the distorted markers in this study, we identified a total of 12 distorted chromosomal regions. A minimum of 11–12 TRDL were identified in the interspecific M. guttatus × M. nasutus cross (Fishman et al. 2001). While the number of TRDL is similar in both mapping populations, the pattern of allele frequency distortion at the TRDL differed strikingly. In the interspecific study, a preponderance of the TRDL (9 of 11) exhibited a higher frequency of M. guttatus (IM62) genotypes and/or alleles compared to Mendelian expectations. In contrast, there is no obvious tendency for markers or TRDL to show distortion toward an excess of IM alleles or DUN alleles in the current intraspecific study.
Twenty-seven of the markers mapped in this study were also mapped in the interspecific M. guttatus × M. nasutus study (Fishman et al. 2001). To the extent that these shared markers identify homologous regions of linkage groups, we may be able to go beyond a basic comparison of the prevalence and direction of transmission ratio distortion at the intra- and interspecific levels. The shared markers map to 11 linkage groups, and 8 of these genomic regions contain 2 or more markers. For these 11 linkage group regions that are apparently homologous to those in the interspecific study, we can classify them according to whether they show similar or different patterns of TRD in the two studies. Only 1 of the 11 linkage groups (LG4) shows no TRD in either cross. Four other regions show significant TRD in one cross but not in the other: portions of LG8 and LG9 are distorted in the intraspecific cross only, whereas portions of LG1 and LG13 are distorted only in the interspecific cross.
Six of the apparently homologous linkage groups show TRD in both crosses. A single region of LG14 is biased against the IM alleles in both crosses and may be in the same genomic region (Figure 3). However, because there is only a single shared marker between maps, we cannot orient the linkage groups with respect to each other to distinguish for certain whether or not these distorted regions are likely to be the same. Two of the linkage groups (LG6 and LG10) contain regions with distortion toward an excess of IM homozygotes (or deficiency of either DUN or M. nasutus homozygotes), but these are clearly not in the same genomic regions (Figure 3). Finally, two collinear linkage groups (LG11 and LG12) have distorted regions on both maps in approximately the same location, but the pattern of distortion is inconsistent between maps. On LG12, there are reduced numbers of heterozygote genotypes (but no strong allele) on the intraspecific map, whereas there are excessive IM alleles on the interspecific map. There are clearly different types of genetic interactions at this region between maps. On LG11, there is a shared distorted region where there is apparent heterosis in the intraspecific map and excess of IM alleles in the interspecific map. This collinear region is of particular interest because in the interspecific cross it is tightly linked to a locus that causes essentially complete meiotic drive of the IM allele over the M. nasutus alleles in female meioses (Fishman and Willis 2005). It is possible that a similar but less extreme pattern of meiotic drive exists in the intraspecific cross, although in the absence of additional crosses alternative causes of distortion cannot be ruled out. Although marker AAT356 (excluded in LG11a) follows the interspecific pattern (excess of IM homozygotes and deficit of DUN homozygotes), both flanking markers (MgSTS87 and CYCA) show a different pattern. These markers have a deficit of DUN homozygotes relative to Mendelian expectation, and they also have an excess of heterozygotes rather than an excess of IM homozygotes.
Ultimately, a more detailed and complete comparison of transmission ratio distortion in the two crosses will require the addition of many more codominant markers to each of the genetic maps. In addition, extensive crossing experiments and fine-scale mapping will be needed to elucidate the genetic mechanisms underlying the patterns of distortion in these intraspecific and interspecific crosses (Fishman and Willis 2005). A clearer understanding of the mechanisms causing the observed transmission ratio distortion will provide richer insight into the nature of genetic divergence between populations and species. Such an understanding will also help generate hypotheses about the evolutionary dynamics of such loci within and between populations and the potential of these genomic regions to act as barriers to gene flow between populations and closely related species.
Acknowledgments
We are grateful to A. Kelly and J. Aagaard for marker development and to C. Vogl for sharing a TRDL mapping program. Early drafts of this article benefited from comments by A. Case, A. Sweigart, A. Cooley, Y. W. Lee, and S. McDaniel. We also thank L. Fishman, M. Rausher, and W. Morris for helpful discussions of this material and L. Fishman for providing additional marker data that enabled map comparisons. This material is based upon work supported by the National Science Foundation under grant nos. 9727578, 0075704, 010577, and 0328636 and by Sigma Xi GIAR.
References
- Causse, M. A., T. M. Fulton, Y. G. Cho, S. N. Ahn, J. Chunwongse et al., 1994. Saturated molecular map of the rice genome based on an interspecific backcross population. Genetics 138: 1251–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarti, A., L. K. Lasher and J. E. Reefer, 1991. A maximum likelihood method for estimating genome length using genetic linkage data. Genetics 128: 175–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz, A., and M. R. MacNair, 1999. Pollen tube competition as a mechanism of prezygotic isolation between Mimulus nasutus and its presumed progenitor M. guttatus. New Phytol. 144: 471–478. [DOI] [PubMed] [Google Scholar]
- Fishman, L., and J. H. Willis, 2005. A novel meiotic drive locus completely distorts segregation in Mimulus (monkeyflower) hybrids. Genetics 169: 347–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishman, L., A. Kelly, E. Morgan and J. H. Willis, 2001. A genetic map in the Mimulus guttatus species complex reveals transmission ratio distortion due to heterospecific interactions. Genetics 159: 1701–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harushima, Y., M. Nakagahra, M. Yano, T. Sasaki and N. Kurata, 2001. A genome-wide survey of reproductive barriers in an intraspecific hybrid. Genetics 159: 883–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitchcock, C. L., and A. Cronquist, 1973 Flora of the Pacific Northwest. University of Washington Press, Seattle.
- Jenczewski, E., M. Gherardi, I. Bonnin, J.-M. Prosperi, I. Olivieri et al., 1997. Insight on segregation distortions in two intraspecific crosses between annual species of Medicago (Leguminosae). Theor. Appl. Genet. 94: 682–691. [Google Scholar]
- Jiang, C.-X., P. W. Chee, X. Draye, P. L. Morrell, C. W. Smith et al., 2000. Multilocus interactions restrict gene introgression in interspecific populations of polyploid Gossypium (Cotton). Evolution 54: 798–814. [DOI] [PubMed] [Google Scholar]
- Kelly, A., and J. Willis, 1998. Polymorphic microsatellite loci in Mimulus guttatus and related species. Mol. Ecol. 7: 769–774. [Google Scholar]
- Kiang, Y. T., and J. L. Hamrick, 1978. Reproductive isolation in the Mimulus guttatus-M. nasutus complex. Am. Midl. Nat. 100: 269–276. [Google Scholar]
- Kosambi, D. D., 1944. The estimation of map distances from recombination values. Annu. Eugen. 12: 172–175. [Google Scholar]
- Lander, E. S., P. Greene, J. Abrahamson, A. Barlow, M. J. Daly et al., 1987. MAPMAKER: an interactive computer package for constructing primary genetic linkage maps of experimental and natural populations. Genomics 1: 174–181. [DOI] [PubMed] [Google Scholar]
- Lin, J.-Z., and K. Ritland, 1996. Construction of a genetic linkage map in the wild plant Mimulus using RAPD and isozyme markers. Genome 39: 63–70. [DOI] [PubMed] [Google Scholar]
- Lincoln, S. E., M. J. Daly and E. S. Lander, 1992 Mapping genes controlling quantitative traits with MAPMAKER/QTL 1.1. Whitehead Institute Technical Report, Whitehead Institute, Cambridge, MA.
- Lu, H., J. Romero-Severson and R. Bernardo, 2002. Chromosomal regions associated with segregation distortion in maize. Theor. Appl. Genet. 105: 622–628. [DOI] [PubMed] [Google Scholar]
- Mayr, E., 1963 Animal Species and Evolution. Harvard University Press, Cambridge, MA.
- Myburg, A. A., C. Vogl, A. R. Griffen, R. R. Sederoff and R. W. Whetten, 2004. Genetics of postzygotic isolation in Eucalyptus: whole-genome analysis of barriers to introgression in a wide interspecific cross of Eucalyptus grandis and E. globulus. Genetics 166: 1405–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palopoli, M. F., and C.-I Wu, 1996. Rapid evolution of a coadapted gene complex: evidence from the Segregation Distorter (SD) system of meiotic drive in Drosophila melanogaster. Genetics 143: 1675–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennell, F. W., 1951 Illustrated Flora of the Pacific States. Stanford University Press, Stanford, CA.
- Remington, D. L., R. W. Whetten, B. H. Liu and D. M. O'Malley, 1998. Construction of an AFLP genetic map with nearly complete genome coverage in Pinus taeda. Theor. Appl. Genet. 98: 1279–1292. [DOI] [PubMed] [Google Scholar]
- Rieseberg, L. H., and S. E. Carney, 1998. Plant hybridization. New Phytol. 140: 599–624. [DOI] [PubMed] [Google Scholar]
- Rieseberg, L. H., C. R. Linder and G. J. Seiler, 1995. Chromosomal and genic barriers to introgression in Helianthus. Genetics 141: 1163–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieseberg, L. H., J. Whitton and K. Gardner, 1999. Hybrid zones and the genetic architecture of a barrier to gene flow between two sunflower species. Genetics 152: 713–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieseberg, L. H., S. J. E. Baird and K. A. Gardner, 2000. Hybridization, introgression, and linkage evolution. Plant Mol. Biol. 42: 205–224. [PubMed] [Google Scholar]
- Schwarz-Sommer, Z., E. de Andrade Silva, R. Berndtgen, W. E. Lonnig, A. Muller et al., 2003. A linkage map of an F2 hybrid population of Antirrhinum majus and A. molle. Genetics 163: 699–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokal, R. R., and F. J. Rohlf, 1995 Biometry: The Principles and Practice of Statistics in Biological Research. W. H. Freeman, New York.
- Solignac, M., D. Vautrin, E. Baudry, F. Mougel, A. Loiseau et al., 2004. A microsatellite-based linkage map of the honeybee, Apis mellifera L. Genetics 167: 253–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweigart, A. L., and J. H. Willis, 2003. Patterns of nucleotide diversity in two species of Mimulus are affected by mating system and asymmetric introgression. Evolution 57: 2490–2506. [DOI] [PubMed] [Google Scholar]
- Sweigart, A. L., K. Karoly, A. Jones and J. H. Willis, 1999. The distribution of individual inbreeding coefficients and pairwise relatedness in a population of Mimulus guttatus. Heredity 83: 625–632. [DOI] [PubMed] [Google Scholar]
- Taylor, D. R., and P. K. Ingvarsson, 2003. Common features of segregation distortion in plants and animals. Genetica 117: 27–35. [DOI] [PubMed] [Google Scholar]
- Vickery, R. K., 1951. Genetic differences between the races and species of Mimulus. Carnegie Inst. Wash. Publ. 50: 118–119. [Google Scholar]
- Vickery, R. K., 1978. Case studies in the evolution of species complexes in Mimulus. Evol. Biol. 11: 405–507. [Google Scholar]
- Vogl, C., and S. Xu, 2000. Multipoint mapping of viability and segregation distorting loci using molecular markers. Genetics 155: 1439–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos, P., R. Hogers and M. Bleeker, 1995. AFLP: a new technique for DNA fingerprinting. Nucleic Acids Res. 23: 4407–4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitkus, R., 1998. Genetics of adaptive radiation in Hawaiian and Cook Islands species of Tetramolopium (Asteraceae). II. Genetic linkage map and its implicatons for interspecific breeding barriers. Genetics 150: 1209–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis, J. H., 1993. Effects of different levels of inbreeding on fitness components in Mimulus guttatus. Evolution 47: 864–876. [DOI] [PubMed] [Google Scholar]
- Zamir, D., and Y. Tadmore, 1986. Unequal segregation of nuclear genes in plants. Bot. Gaz. 147: 355–358. [Google Scholar]