

Abstract
Because some metabolic intermediates are involved in more than one pathway, crosstalk between pathways is crucial to maintaining homeostasis. AMP and histidine biosynthesis pathways are coregulated at the transcriptional level in response to adenine availability. 5′-Phosphoribosyl-4-carboxamide-5-aminoimidazole (AICAR), a metabolic intermediate at the crossroads between these two pathways, is shown here to be critical for activation of the transcriptional response in the absence of adenine. In this study, we show that both AMP and histidine pathways significantly contribute to AICAR synthesis. Furthermore, we show that upregulation of the histidine pathway clearly interferes with regulation of the AMP pathway, thus providing an explanation for the regulatory crosstalk between these pathways. Finally, we revisit the histidine auxotrophy of ade3 or ade16 ade17 mutants. Interestingly, overexpression of PMU1, encoding a potential phosphomutase, partially suppresses the histidine requirement of an ade3 ade16 ade17 triple mutant, most probably by reducing the level of AICAR in this mutant. Together our data clearly establish that AICAR is not just a metabolic intermediate but also acts as a true regulatory molecule.
METABOLIC pathways generate multiple metabolic intermediates, which contribute widely to intracellular chemical diversity. Evolution has taken advantage of this diversity and several of these small molecules have been shown to play a crucial and direct role in regulation of gene expression (Sze et al. 1992; Feller et al. 1994; Flynn and Reece 1999; Loewen et al. 2004).
On the basis of genetics studies in budding yeast, our previous work has revealed that 5′-phosphoribosyl-4-succinocarboxamide-5-aminoimidazole (SAICAR), an intermediate metabolite of purine biosynthesis, is critical for the expression of AMP biosynthesis genes (ADE genes) in the absence of an extracellular purine source (Rébora et al. 2001). We found that mutations affecting the first seven steps of the IMP biosynthesis pathway (Figure 1) result in permanent repression of ADE1-lacZ expression, whereas mutations affecting the last three steps (ade13 or ade16 ade17) result in a constitutive derepression of ADE1 expression. Study of ADE1 expression in an ade2 ade13 double-mutant strain revealed that ade2 is epistatic to ade13, thus confirming the essential role of SAICAR in the activation of ADE gene expression (Rébora et al. 2001). At the molecular level, we have shown that SAICAR activates ADE gene expression by promoting interaction between Bas1p and Bas2p, two transcription factors required for ADE gene expression (Rébora et al. 2001).
Figure 1.—

Schematic of histidine and IMP biosynthesis pathways and reactions supplying 10 formyl-THF for IMP biosynthesis. FGAR, 5′-phosphoribosyl N-formylglycinamide; IMP, inosine 5′-monophosphate; PRPP, 5-phosphoribosyl-1-pyrophosphate; SAMP, adenylosuccinate; XMP, xanthosine 5′-monophosphate. Gene names are italicized. For simplicity, only intermediate metabolites that are cited in the text are indicated.
While we clearly established a role for SAICAR in ADE pathway regulation, the role of 5′-phosphoribosyl-4-carboxamide-5-aminoimidazole (AICAR) in this regulatory process remained obscure. Unlike SAICAR, which is produced only through the IMP pathway, AICAR not only is produced by Ade13p in the IMP pathway, but also is a by-product of histidine biosynthesis (Figure 1). Interestingly, in yeast it has been shown that histidine and AMP biosynthesis pathways are coregulated (Daignan-Fornier and Fink 1992); i.e., expression of HIS1, HIS4, and HIS7 (Figure 1) is activated by Bas1p and Bas2p and repressed by extracellular adenine (Arndt et al. 1987; Springer et al. 1996; Denis et al. 1998). In Aspergillus nidulans, expression of the hisHF gene (the equivalent of the HIS7 gene in Saccharomyces cerevisiae) is also regulated by adenine (Valerius et al. 2001). The physiological raison d'être of such a cross-pathway regulation is still unclear.
Intriguingly, ade3 and ade16 ade17 mutant strains are auxotrophic for both adenine and histidine, a phenotype also reported for Schizosaccharomyces pombe equivalent mutants, ade9 and ade10, respectively (Whitehead et al. 1966). ADE3 encodes a trifunctional enzyme catalyzing three reactions that lead to the synthesis of 10-formyl tetrahydrofolate (THF), a cosubstrate required for the third and the ninth steps of IMP biosynthesis (Figure 1). Since 5,10-methylene THF, which accumulates in the ade3 mutant, inhibits the fourth enzyme of histidine biosynthesis in vitro, it was proposed that this inhibition could lead to histidine auxotrophy in vivo (discussed in Jones and Fink 1982). However, the finding that an ade16 ade17 double mutant is also auxotrophic for histidine (Tibbetts and Appling 2000) does not fit well with this hypothesis, especially since an ade8 mutant, which is affected in the other 10-formyl-THF-consuming step of the IMP pathway, is prototrophic for histidine. We therefore favor the recent hypothesis of Appling and co-workers who propose that the auxotrophy for histidine is likely linked to accumulation of AICAR, rather than to accumulation of 5,10-methylene THF (Tibbetts and Appling 2000).
Finally, accumulation of AICAR also seems to interfere with methionine biosynthesis (Holmes and Appling 2002). Indeed, studies of the FAU1 gene, encoding methenyl THF synthetase, revealed that a triple fau1 ade16 ade17 mutant strain had a severe growth defect that is alleviated by methionine. This phenotype required the three mutations and could be suppressed by additional combined mutations in both ADE2 and HIS4 genes, blocking the pathways responsible for AICAR biosynthesis. Together these data suggest a more general role for AICAR than first thought.
Because AICAR is at the crossroads between several pathways, it is crucial to fully understand its function in a eukaryotic organism. In this article, we show a major role for AICAR in the regulation of ADE gene expression and we identify the pathways responsible for AICAR synthesis. Moreover, we show that deregulation of the histidine pathway can affect the IMP biosynthesis pathway, thus providing an explanation for histidine-purine coregulation. Finally, we have identified a multicopy suppressor of the histidine auxotrophy of an ade3 ade16 ade17 triple mutant and have documented the toxicity of AICAR and SAICAR accumulation in yeast cells.
MATERIALS AND METHODS
Yeast strains and media:
Yeast strains are listed in Table 1. All double-mutant strains constructed in this work were obtained by mating single knockout mutant strains (purchased from EUROSCARF) isogenic to BY4742 (Brachmann et al. 1998). Triple mutants were obtained by mating two isogenic double mutants (except for ade2 ade3 ade13; see below).
TABLE 1.
Yeast strains used in this study
| Strain | Genotype | Source |
|---|---|---|
| BY4742 | MATα his3-Δ1 leu2-Δ0 lys2-Δ0 ura3-Δ0 | J. Boeke |
| Y1095 | MATα his3-Δ1 leu2-Δ0 lys2-Δ0 ura3-Δ0 ade16::kanMX4 ade17::kanMX4 | Lab collection |
| Y1164 | MATα his3-Δ1 leu2-Δ0 lys2-Δ0 ura3-Δ0 met15-Δ0 ade8::kanMX4 ade16::kanMX4 ade17::kanMX4 | This work |
| Y1166 | MATahis3-Δ1 leu2-Δ0 ura3-Δ0 met15-Δ0 ade3::kanMX4 his1::kanMX4 | This work |
| Y1261 | MATaleu2-3 lys2Δ201 ura3-52 his3Δ200 ade2 ade13-12 | Lab collection |
| Y1551 | MATα his3-Δ1 leu2-Δ0 ura3-Δ0 met15-Δ0 ade3::kanMX4 ade5,7::kanMX4 | This work |
| Y1554 | MATα his3-Δ1 leu2-Δ0 ura3-Δ0 ade3::kanMX4 ade6::kanMX4 | This work |
| Y1657 | MATahis3-Δ1 leu2-Δ0 lys2-Δ0 ura3-Δ0 ade3::kanMX4 his1::kanMX4 ade5,7::kanMX4 | This work |
| Y1661 | MATahis3-Δ1 leu2-Δ0 ura3-Δ0 ade3::kanMX4 his7::kanMX4 | This work |
| Y1975 | MATα ura3 leu2 lys2 his3 | This work |
| Y1976 | MATα ura3 leu2 lys2 his3 ade2 ade13-12 | This work |
| Y1977 | MATaura3 leu2 lys2 his3 ade2 ade13-12 ade3::kanMX4 | This work |
| Y1978 | MATaura3 leu2 lys2 his3 ade13-12 | This work |
| Y1979 | MATα ura3 leu2 lys2 his3 ade2 | This work |
| Y1980 | MATα ura3 leu2 lys2 his3 ade13-12 ade3::kanMX4 | This work |
| Y1981 | MATaura3 leu2 lys2 his3 ade2 ade13-12 ade3::kanMX4 | This work |
| Y1982 | MATaura3 leu2 lys2 his3 | This work |
| Y2030 | MATahis3-Δ1 leu2-Δ0 ura3-Δ0 ade3::kanMX4 met6::kanMX4 | This work |
| Y2031 | MATahis3-Δ1 leu2-Δ0 ura3-Δ0 ade3::kanMX4 fau1::kanMX4 | This work |
| Y2032 | MATahis3-Δ1 leu2-Δ0 ura3-Δ0 ade3::kanMX4 fau1::kanMX4 | This work |
| Y2406 | MATα ura3-Δ0 ade3::kanMX4 ade16::kanMX4 ade17::kanMX4 | Lab collection |
| Y2655 | MATα his3-Δ1 leu2-Δ0 lys2-Δ0 ura3-Δ0 ade3::kanMX4 ade8::kanMX4 his1::kanMX4 | This work |
| PLY122 | MATaleu2-3,112 ura3-52 lys2-Δ201 | P. Ljungdahl |
| 206 | MATaleu2-3,112 ura3-52 lys2-Δ201 ade13-52 | Lab collection |
| 211 | MATaleu2-3,112 ura3-52 lys2-Δ201 ade13-23 | Lab collection |
| 242 | MATaleu2-3,112 ura3-52 lys2-Δ201 ade13-42 | Lab collection |
| Y06591 | MATahis3-Δ1 leu2-Δ0 met15-Δ0 ura3-Δ0 ade3::kanMX4 | EUROSCARF |
| Y10190 | MATα his3-Δ1 leu2-Δ0 lys2-Δ0 ura3-Δ0 his1::kanMX4 | EUROSCARF |
| Y11413 | MATα his3-Δ1 leu2-Δ0 lys2-Δ0 ura3-Δ0 his6::kanMX4 | EUROSCARF |
| Y11583 | MATα his3-Δ1 leu2-Δ0 lys2-Δ0 ura3-Δ0 ade16::kanMX4 | EUROSCARF |
| Y12275 | MATα his3-Δ1 leu2-Δ0 lys2-Δ0 ura3-Δ0 his5::kanMX4 | EUROSCARF |
| Y12669 | MATα his3-Δ1 leu2-Δ0 lys2-Δ0 ura3-Δ0 shm2::kanMX4 | EUROSCARF |
| Y13388 | MATα his3-Δ1 leu2-Δ0 lys2-Δ0 ura3-Δ0 his7::kanMX4 | EUROSCARF |
| Y13403 | MATα his3-Δ1 leu2-Δ0 lys2-Δ0 ura3-Δ0 shm1::kanMX4 | EUROSCARF |
| Y13437 | MATα his3-Δ1 leu2-Δ0 lys2-Δ0 ura3-Δ0 his4::kanMX4 | EUROSCARF |
| Y14244 | MATα his3-Δ1 leu2-Δ0 lys2-Δ0 ura3-Δ0 ade8::kanMX4 | EUROSCARF |
| Y14601 | MATα his3-Δ1 leu2-Δ0 lys2-Δ0 ura3-Δ0 ade5,7::kanMX4 | EUROSCARF |
| Y14691 | MATα his3-Δ1 leu2-Δ0 lys2-Δ0 ura3-Δ0 ade6::kanMX4 | EUROSCARF |
| Y16561 | MATα his3-Δ1 leu2-Δ0 lys2-Δ0 ura3-Δ0 ade17::kanMX4 | EUROSCARF |
| Y16591 | MATα his3-Δ1 leu2-Δ0 lys2-Δ0 ura3-Δ0 ade3::kanMX4 | EUROSCARF |
| Y16719 | MATα his3-Δ1 leu2-Δ0 lys2-Δ0 ura3-Δ0 fau1::kanMX4 | EUROSCARF |
| Y17082 | MATα his3-Δ1 leu2-Δ0 lys2-Δ0 ura3-Δ0 his2::kanMX4 | EUROSCARF |
| 9486-8D | MATα HIS1-17 leu2-3 | G. Fink |
| H552 | MATα gcd2-1 ura3-52 leu2-3,112 | A. Hinnebusch |
| H1453 | MATα GCD2 ura3-52 leu2-3,112 | A. Hinnebusch |
ade3 ade2 ade13 triple mutants were obtained by mating Y1261 (ade2 ade13) and Y16591 (ade3) strains. After sporulation of the resulting diploids, tetrads were dissected and the genotype of the four spores of each tetrad was analyzed. ade3 spores were identified by their geneticine resistance phenotype. To identify ade2 spores, the four spores of each tetrad were mated with a or α ade2 strains. Auxotrophy for adenine and red or white color of the diploid was tested to determine which spores were ade2. To identify ade13 spores, the four spores of each tetrad were mated with a or α ade13 strains. Expression of the ADE1-lacZ fusion was monitored in the resulting diploids by β-galactosidase (β-gal) assays to determine which diploids have a bra phenotype (Guetsova et al. 1997) and thus are homozygous ade13 diploids.
Yeast media (YPD, SC, and SD) were prepared according to Sherman et al. (1986). SD casa medium is SD supplemented with 0.2% (w/v) casaminoacids (DIFCO).
Screening for multicopy suppressors of ade3 ade16 ade17 histidine auxotrophy:
Strain Y2406 was transformed with a multicopy library (kind gift from F. Lacroute). Transformants (∼13,000) were grown and replica plated on medium lacking histidine. Colonies able to grow in the absence of histidine were selected, and plasmid DNA was extracted. Plasmids able to suppress histidine auxotrophy were isolated and the insert boundaries were sequenced.
Plasmids:
P1933 (Rébora et al. 2001) is a URA3 centromeric plasmid expressing ADE4 under control of a tetracycline regulatable promoter (Gari et al. 1997). P2122 is a tet-ADE4 LEU2 centromeric plasmid obtained by swapping the P1933 marker with a fragment of the pUL9 plasmid (Cross 1997). P2191 is a centromeric LEU2 plasmid expressing the HIS3 gene, which was used to complement the his3 mutation in all strains in which the His− phenotype was tested. P2818 is a PMU1 multicopy plasmid. P2819 is a derivative of P2818 obtained by digestion at the single BstEII restriction site, Klenow filling, and religation; this treatment introduces a frameshift in the PMU1 open reading frame. P2821 is a URA3 centromeric plasmid expressing PMU1 under control of a tetracycline regulatable promoter (Gari et al. 1997).
LacZ fusions and β-gal assays:
The lacZ fusions used in this study have been previously described (Daignan-Fornier and Fink 1992; Guetsova et al. 1997). P115 is a plasmid carrying an ADE1-lacZ fusion in the 2μ URA3 vector YEp356R (Myers et al. 1986). P473 is a plasmid carrying an ADE1-lacZ fusion in a 2μ LEU2 vector YEp367 (Myers et al. 1986). β-Gal assays were performed as described by Kippert (1995) on cells grown for 6 hr in the presence or in the absence of adenine. β-Gal units are defined as (OD420 × 1000)/[OD600 × t(min) × vol(ml)]. In each experiment, at least two independent β-gal assays were performed, and each assay was done on three independent transformants. The variation between assays in each experiment was <20%.
Adenylosuccinate lyase assay:
Adenylosuccinate lyase activity was measured as previously described (Guetsova et al. 1997).
Northern blot:
Northern blot experiments were made as previously described (Pinson et al. 2001).
RESULTS
Both AICAR and SAICAR can activate expression of the ADE genes:
As mentioned in the Introduction, SAICAR accumulation is clearly correlated with the activation of ADE gene expression. However, the role of AICAR in the regulation of the ADE genes remains to be elucidated. Indeed, AICAR accumulation in the ade16 ade17 double mutant could lead to activation of the ADE genes either directly or indirectly through “retro-synthesis” of SAICAR or feedback inhibition of Ade13p. To discriminate between these possibilities, we used the cells to synthesize AICAR from the histidine pathway. In an ade8 ade16 ade17 triple mutant, ADE1-lacZ expression was only slightly lower than that in the ade16 ade17 double mutant (Figure 2), indicating that AICAR provided by the histidine pathway appeared sufficient to activate ADE gene expression. Expression of ADE1-lacZ was also high in an ade3 mutant that is unable to synthesize 10-formyl-THF, a cosubstrate used in the reactions catalyzed by Ade8p and Ade16p Ade17p (Figure 2). Therefore, since the triple ade8 ade16 ade17 mutant phenocopies the single ade3 mutant, the ade3 mutant was used in the rest of the study.
Figure 2.—

Effect of a mutation blocking IMP biosynthesis pathway on accumulation of AICAR. Yeast strains transformed with a plasmid carrying the ADE1-lacZ fusion (P115) were grown for 6 hr in SD casa medium containing adenine at either low (0.025 mm low ade) or high concentration (0.3 mm, high ade). β-Galactosidase activity was then measured as described in materials and methods. Strains were BY4742 (wild type), Y14244 (ade8), Y1095 (ade16 ade17), Y1164 (ade8 ade16 ade17), and Y16591 (ade3).
We then monitored expression of ADE1-lacZ in an ade2 ade3 ade13 triple-mutant strain. In such a strain, AICAR synthesized from the histidine pathway can accumulate (see previous section), while SAICAR cannot be produced through either normal synthesis or “retro-synthesis” from AICAR, because of the ade2 and ade13 mutations, respectively (Figure 1). An ade2 ade13 double mutant was mated to an ade3 mutant and expression of ADE1-lacZ was measured in the parental strains (Figure 3A) and in the meiotic progeny (Figure 3B). Expression of ADE1-lacZ was high in the ade3 ade13 and ade2 ade3 double mutants as well as in the two triple ade2 ade3 ade13 mutant spores that can accumulate AICAR but not SAICAR (Figure 3B). Consistently, Northern blot analyses revealed that expression of another ADE gene, ADE17, was high in the ade2 ade3 ade13 mutant (Figure 3C). These two results establish that AICAR, like SAICAR, can directly activate expression of the ADE genes.
Figure 3.—

Expression of ADE1 and ADE17 in ade3 ade2 ade13 triple-mutant strains. Strains Y16591 (ade3) and Y1261 (ade2 ade13) were mated and after sporulation of the resulting diploid, tetrads were isolated. (A and B) The parental strains and the spores of two tetrads were transformed with a plasmid carrying the ADE1-lacZ fusion (P115) and grown for 6 hr in SD casa medium containing adenine at either low (0.025 mm) or high (0.3 mm) concentration. β-Gal activity was then measured as described in materials and methods. Strains were (A) parental strains Y1261 (ade2 ade13) and Y16591 (ade3); and (B) meiotic progeny strains Y1975 (wild type), Y1976 (ade2 ade3), Y1977 (ade2 ade3 ade13), Y1978 (ade13), Y1979 (ade2), Y1980 (ade3 ade13), Y1981 (ade2 ade3 ade13), and Y1982 (wild type). (C) Northern blot analysis of ADE17 gene expression in the ade2 ade3 ade13 mutant strain. Strains Y1975 (wild type), Y1261 (ade2 ade13), Y1980 (ade3 ade13), Y1976 (ade2 ade3), and Y1977 (ade2 ade3 ade13) were grown to an OD600 of 0.5 in SD casa medium with or without adenine as indicated. Extraction of RNA and hybridization were performed as described in materials and methods. Radioactivity was detected using a Phosphorimager and signal was quantified using the ImageQuant software. The quantification is presented as the ADE17/ACT1 ratio, which was arbitrarily set up as “1” in the wild-type “+ade” control lane.
Genetic analysis reveals the role of histidine and IMP pathways in AICAR synthesis:
As shown in the previous section, in an ade8 ade16 ade17 or in an ade3 mutant, a significant amount of AICAR can be provided through an alternative pathway, most probably the histidine pathway.
To further evaluate the cellular supply routes for AICAR, we constructed ade3 his1 and ade3 his7 double-mutant strains in which both the IMP and histidine pathways are blocked upstream of AICAR synthesis (Figure 1). To our surprise, ADE1-lacZ was still expressed in ade3 his1 and ade3 his7 (Figure 4A) mutant strains, although at a lower level than in an ade3 single mutant. This result indicated that AICAR produced through histidine biosynthesis indeed affected expression of the ADE1-lacZ fusion, but, intriguingly, it also suggested that AICAR can be produced even when both the histidine and the IMP pathways are blocked. However, there is no direct evidence that the ade3 mutation leads to a total block of the Ade8p catalyzed step. We therefore combined ade3 with various IMP pathway mutants. Clearly, expression of the ADE1-lacZ fusion was lower in the ade3 ade5,7 and the ade3 ade6 (Figure 4B) double-mutant strains than in the ade3 mutant strain (Figure 4B). This result strongly argued for residual AICAR synthesis in the ade3 mutant. This was confirmed by the fact that in an ade3 ade5,7 his1 triple mutant, when both the histidine and the IMP pathways are blocked, the expression of ADE1-lacZ was almost undetectable (Figure 4C). Furthermore, similar results were obtained when expression of the ADE17 gene was assayed by Northern blot in the different mutant strains (Figure 4D). Importantly, we found that ADE1-lacZ expression in low-adenine medium was the same in ade3 ade5,7 his1 and ade3 ade8 his1 triple mutants (data not shown), thus establishing that the leakage observed in the ade3 mutant is due to residual Ade8p activity in the absence of 10-formyl-THF synthesized by Ade3p. Therefore, our results show that both the histidine and the IMP pathways significantly contribute to AICAR accumulation in yeast.
Figure 4.—
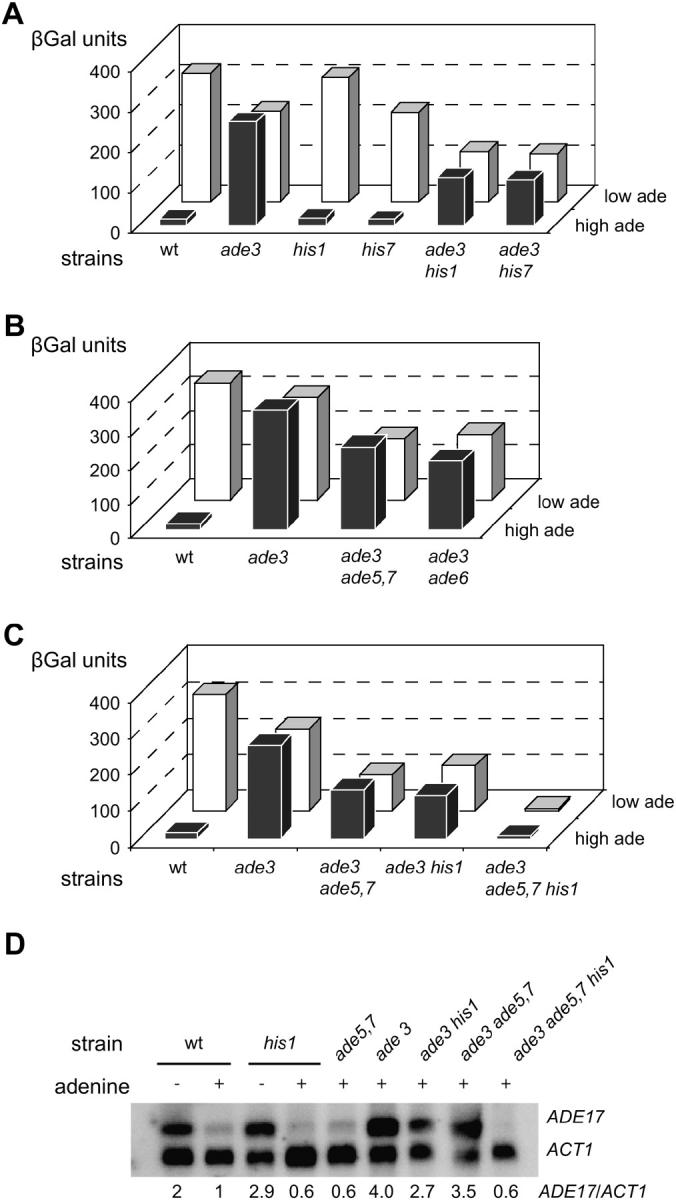
Effect of mutations blocking both the IMP and the histidine biosynthesis pathways from accumulating AICAR. (A–C). Yeast strains transformed with a plasmid carrying the ADE1-lacZ fusion (P115) were grown for 6 hr in SD casa medium containing adenine at either low (0.025 mm) or high (0.3 mm) concentration. β-Gal activity was then measured as described in materials and methods. Strains were: (A) BY4742 (wild type), Y16591 (ade3), Y10190 (his1), Y13388 (his7) Y1166 (ade3 his1), and Y1661 (ade3 his7); (B) BY4742 (wild type), Y16591 (ade3), Y1551 (ade3 ade5,7), and Y1554 (ade3 ade6); and (C) BY4742 (wild type), Y16591 (ade3), Y1166 (ade3 his1), Y1551 (ade3 ade5,7), and Y1657 (ade3 ade5,7 his1). (D) Northern blot analysis of ADE17 gene expression in the ade2 ade3 ade13 mutant strain. Strains BY4742 (wild type), Y10190 (his1), Y14601 (ade5,7), Y16591 (ade3), Y1166 (ade3 his1), Y1551 (ade3 ade5,7), and Y1657 (ade3 ade5,7 his1) were grown to an OD600 of 0.5 in SD casa medium with or without adenine as indicated. Extraction of RNA and hybridization were performed as described in materials and methods. Radioactivity was detected using a phosphorimager and signal was quantified using the ImageQuant software. The quantification is presented as the ADE17/ACT1 ratio, which was arbitrarily set up as “1” in the wild-type “+ade” control lane.
Deregulation of the histidine biosynthesis pathway impairs regulation of AMP biosynthesis:
Since AICAR contributes to expression of ADE genes and can be provided through the histidine biosynthesis pathway, we evaluated the role of the histidine biosynthesis pathway in the regulation of AMP biosynthesis. This was done by using mutants either decreasing or increasing the flux in the histidine pathway.
Deletion of any of the HIS genes had no effect on the expression of the ADE1-lacZ fusion (data not shown). This result indicates that AICAR synthesis from the histidine biosynthesis pathway is not required for full induction of the AMP biosynthesis pathway in the absence of adenine.
To stimulate the histidine pathway, we used a gcd2 mutant known to deregulate HIS gene transcription (Hinnebusch 1988). GCD2 encodes a protein that negatively regulates synthesis of Gcn4p, a major transcription activator of HIS genes (Hinnebusch 1988). Expression of ADE1-lacZ was measured in gcd2 and GCD2 isogenic strains in the presence or absence of adenine. The ratio of ADE1-lacZ activity under derepression (no adenine) and repression (with adenine) defines the adenine repression factor. This repression factor was significantly lower in the gcd2 mutant (7.8 ± 0.8) than in the wild-type strain (15.1 ± 1.7). As expected, this difference was abolished when the same strains were grown in the absence of histidine. This result indicates that the activation of the histidine pathway in the presence of adenine results in partial derepression of the ADE1-lacZ expression. This was confirmed by monitoring ADE1-lacZ expression in the HIS1-17 dominant mutant that is not feedback inhibited by histidine (Rasse-Messenguy and Fink 1973) and consequently overproduces histidine, which is excreted in the medium. The HIS1-17 mutant was mated to a wild-type strain and, after sporulation and tetrad dissection, the ADE1-lacZ plasmid was introduced into 88 spores (22 tetrads). Expression of the fusion measured in the presence or absence of adenine revealed a clear cosegregation between the HIS1-17 allele (monitored by excretion of histidine) and a lower factor of repression by adenine: the repression factor in wild-type strains was 16.0 ± 5.0, while it was 5.2 ± 2.5 in HIS1-17 mutant strains. These results establish that, in the presence of adenine, a constitutively active histidine biosynthesis pathway can lead to a partial deregulation of AMP biosynthesis. In both gcd2 and HIS1-17, expression of the fusion was affected only in high-adenine medium while expression levels in low-adenine medium were similar.
Auxotrophy for histidine of ade3 and ade16 ade17 mutant strains cannot be simply suppressed by overexpression of HIS genes or by mutations in the folate interconversion pathway:
Intriguingly, both ade3 and ade16 ade17 mutant strains are auxotrophic for histidine. Because these two strains are blocked in the ninth step of IMP biosynthesis, we thought that this auxotrophy was likely to be due to an accumulation of AICAR. A simple hypothesis to account for this phenotype is that AICAR inhibits one of the histidine biosynthesis pathway enzymes. We reasoned that, conversely, overexpression of the inhibited enzyme might partially overcome this inhibition and result in bradytrophy for histidine. We therefore individually expressed the HIS genes in the ade3 and ade16 ade17 mutant strains and monitored growth of these strains in the presence or absence of histidine. None of these constructs was sufficient to restore histidine prototrophy while all the constructs complemented the cognate his mutation (data not shown). Since we did not succeed in constructing a plasmid overexpressing HIS1, we took advantage of the dominant HIS1-17 mutant, which leads to overproduction and excretion of histidine (Rasse-Messenguy and Fink 1973). The ade3 HIS1-17 double mutant was still auxotrophic for histidine and could no longer excrete histidine, indicating that the HIS pathway is still blocked in this mutant (data not shown). Importantly, our results in the previous sections clearly show that, in an ade3 ade5,7 double mutant, AICAR can be synthesized only from the histidine pathway, and in such a mutant, ADE1-lacZ expression was still activated. This result indicates that the early steps of histidine biosynthesis required for AICAR production are still functional in the ade3 ade5,7 double mutant and therefore are unlikely targets for inhibition by AICAR. The inhibition by AICAR is thus rather likely to affect one (or several) of the last four steps of the pathway. The last step, catalyzed by histidinol dehydrogenase, allows synthesis of histidine from histidinol. Histidinol cannot be taken up by wild-type yeast cells, but a HOL1-1,101 dominant allele of the HOL1 gene was shown to allow utilization of histidinol as a histidine source (Gaber et al. 1990). Introduction of the HOL1-1,101 allele carried on a plasmid in the ade3 and ade16 ade17 strains allowed growth on histidinol, showing that histidinol dehydrogenase is not inhibited in these strains (data not shown). Therefore, the inhibition of AICAR should affect one of the three remaining late steps catalyzed by His3p, His5p, and His2p (Figure 1). However, overexpression of these three genes in the same strain was not sufficient to suppress the auxotrophy for histidine (data not shown).
Since accumulation of AICAR was shown to interfere with folate and methionine biosynthesis (Holmes and Appling 2002), we thought that the AICAR effect on the histidine pathway could indirectly be due to accumulation of an intermediate in the methionine or folate pathways. We therefore tried to suppress the His− phenotype of ade3 by combining ade3 with mutations of these two pathways (Figure 5).
Figure 5.—

Schematic of connections among histidine, purine, folate, and methionine metabolism in yeast. Gene names are italicized. CH + THF, methenyl-tetrahydrofolate; 5-CHO-THF, 5-formyl-tetrahydrofolate; CH2-THF, methylene-tetrahydrofolate; CH3-THF, methyl-tetrahydrofolate.
The ade3 fau1 double mutants had a severe growth defect even on rich medium (Figure 6A) and were auxotrophic for methionine (Figure 6B) as reported for the triple ade16 ade17 fau1 mutant (Holmes and Appling 2002). Therefore, this result further shows that ade3 phenocopies an ade16 ade17 mutant. We observed that the ade3 fau1 methionine auxotrophy was much tighter at 30° than at 25° (Figure 6B).
Figure 6.—
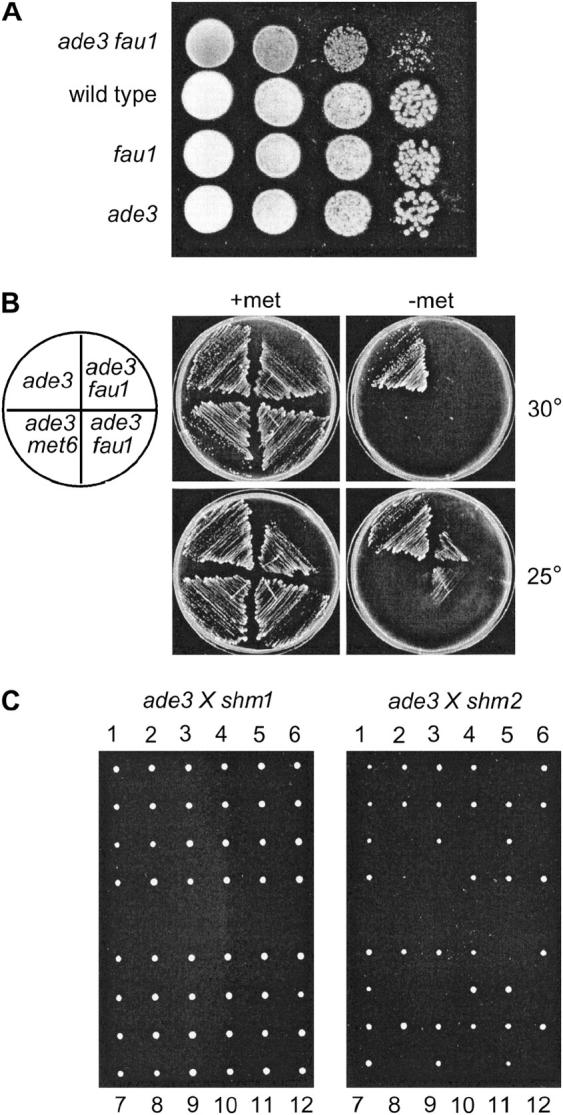
Phenotypes of double mutants combining ade3 mutations with mutations in folate metabolism. (A) Growth of the four spores of a tetratype obtained by mating Y06591 (ade3) and Y16179 (fau1) strains. A serial dilution of the spores was dropped onto YPD medium, and growth of cells was observed after 2 days at 30°. (B) Growth of the ade3 fau1 double mutant in the presence or the absence of methionine. Strains Y16591 (ade3), Y2030 (ade3 met6), Y2031 (ade3 fau1), and Y2032 (ade3 fau1) were streaked onto SD medium with (+met) or without (−met) methionine, and growth of the strains was observed after 3 days at 25° or 30°, as indicated. (C) The Y06591 (ade3) strain was mated with either Y13403 (shm1) or Y12669 (shm2) strains, and after sporulation of the resulting diploids, 12 tetrads (shown as vertical alignments of four spores) were dissected onto YPD medium supplemented with adenine (0.3 mm). Growth of spores was observed after 3 days at 30°.
Combining ade3 with mtd1, met6, met12, or met13 did not result in any synthetic phenotype (data not shown). However, we found a clear synthetic lethality between ade3 and shm2 as suggested by multiple incomplete tetrads (Figure 6C) and confirmed by further genetics analysis (data not shown). Such a synthetic lethality had been previously reported by others (Nigavekar and Cannon 2002) although ade3 shm2 double mutants appear viable in other backgrounds (McNeil et al. 1996). Finally, ade3 shm1 double mutants were perfectly viable in our background (Figure 6C) in contradiction with previously published results (Koren et al. 2003). Importantly, none of these double mutants were prototrophic for histidine (data not shown).
Finally, since AICAR is thought to affect the interaction between the transcription factors Bas1p and Bas2p, we wondered whether the histidine auxotrophy would persist in the absence of these proteins. Indeed, both ade3 bas1 and ade3 bas2 double-mutant strains were auxotrophic for histidine as was the single ade3 mutant (data not shown). Therefore, the His− phenotype, which is likely to result from AICAR accumulation, is independent of the effect of AICAR on Bas1p or Bas2p.
Overexpression of PMU1 partially bypasses histidine auxotrophy of an ade3 ade16 ade17 mutant by detoxifying AICAR:
A search for multicopy suppressors of histidine auxotrophy of an ade3 ade16 ade17 triple mutant allowed us to identify five plasmids carrying a 3-kb DNA fragment from chromosome XI that could clearly restore histidine prototrophy to the triple mutant (Figure 7A). Subcloning experiments revealed that PMU1, encoding a potential phosphomutase, was responsible for the suppression (Figure 7B). A simple hypothesis would be that overexpression of PMU1 bypasses histidine auxotrophy by replacing the impaired activity in the late histidine pathway. Clearly, overexpression of PMU1 in his2, his3, or his5 mutants could not suppress histidine auxotrophy (data not shown). We therefore hypothesized that PMU1 directly affects AICAR, which is thought to be responsible for the inhibition of a late step of histidine biosynthesis. Indeed, because it encodes a potential phosphomutase, PMU1 could simply transform AICAR into another derivative with lower inhibitory ability. To test this hypothesis, we took advantage of the fact that overproduction of AICAR is toxic for yeast cells. The IMP biosynthesis pathway was made constitutively active by overexpressing the ADE4 gene (Rébora et al. 2001) in various ade mutant strains. The overexpression of ADE4 strongly affected growth of an ade16 ade17 double-mutant strain whereas it had no effect on the ade16 or ade17 single-mutant strains (Figure 7C). This result strongly suggests that AICAR overproduction is toxic for yeast cells. Consistently, overexpression of ADE4 had no toxic effect on ade3 or ade8 ade16 ade17 mutant strains (Figure 7C), which are blocked upstream in the pathway and are therefore insensitive to overexpression of ADE4. Further, concomitant overexpression of ADE4 and PMU1 resulted in improved growth (Figure 7D), indicating that PMU1 could partially bypass the negative effect of AICAR accumulation. We therefore believe that PMU1 suppresses histidine auxotrophy by somehow detoxifying AICAR.
Figure 7.—

Histidine auxotrophy of an ade3 ade16 ade17 triple mutant is suppressed by overexpression of PMU1. (A) A serial dilution of Y2406 transformed with P2818 (PMU1 2μ) or by the control vector was dropped onto SC medium with or without histidine as indicated. Pictures were taken after 3 days at 30° for the +histidine plate and 7 days for the −histidine plate. (B) Interruption of the PMU1 reading frame (P2819) abolishes suppression while overexpression of PMU1 alone (tet-PMU1) allows suppression. Suppression, indicated on the right, was monitored after transformation in the Y2406 strain. (C) Effect of accumulation of AICAR on cell growth. Strains were transformed with either a control plasmid (pCM189, vector) or a plasmid expressing the tet-ADE4 fusion (P1933, tet-ADE4). A serial dilution of the different transformants was dropped onto SD casa medium supplemented with adenine and tryptophan, and growth of cells was observed after 3 days at 30°. The following strains were used: Y1095 (ade16 ade17), BY4742 (wild type), Y11583 (ade16), Y16561 (ade17), Y16591 (ade3), and Y1164 (ade3 ade16 ade17). (D) Effect of overexpression of PMU1 on AICAR toxicity. A serial dilution of the Y1095 (ade16 ade17) strain transformed with tet-ADE4 (P2122) and either PMU1 overexpressing plasmids or the control vector was dropped onto SC medium with or without tetracycline as indicated. Growth of cells was observed after 3 days at 30°. (E) Toxicity of SAICAR accumulated in ade13 mutant strains. The following strains were used: PLY122 (wild type), 206 (ade13-52), 211 (ade13-23), and 242 (ade13-42). Residual adenylosuccinate lyase activity was measured in the four strains as described in materials and methods and indicated as a percentage of wild-type activity. Growth of cells was observed after 3 days at 30°.
Importantly, accumulation of SAICAR also appears toxic since we found that overexpression of ADE4 strongly affected growth of various ade13 mutant strains (Figure 7E). Consistently, the growth defect was strongly correlated with the strength of the ade13 allele, estimated by direct measurement of residual adenylosuccinate lyase activity in the mutant strains (Figure 7E). Thus toxicity appears to be correlated with the amount of SAICAR accumulated. However, it should be noted that while both AICAR and SAICAR accumulation appear toxic for yeast cells, only AICAR accumulation is associated with histidine auxotrophy since ade13 mutants are prototrophic for histidine biosynthesis.
DISCUSSION
Both AICAR and SAICAR activate expression of ADE genes:
In this article, we establish that AICAR can activate expression of ADE genes, as reported previously for another purine metabolic intermediate, SAICAR (Rébora et al. 2001). AICAR is produced through the IMP biosynthesis pathway, but also by His7p during histidine biosynthesis. However, mutations in the first steps of IMP synthesis, upstream of AICAR synthesis, result in complete adenine auxotrophy, indicating that AICAR synthesis from the histidine pathway cannot provide sufficient synthesis of purine nucleotides for cell growth in the absence of extracellular adenine. This is not surprising since AICAR provided by the histidine pathway is produced at the cost of one ATP molecule (Figure 1) and is then recycled through the AMP biosynthesis pathway. Therefore, AICAR produced during histidine biosynthesis does not contribute to net AMP de novo synthesis.
However, we observed that AICAR produced during histidine biosynthesis can affect the regulation of AMP biosynthesis in the presence of adenine under certain circumstances. Indeed, while a block in the histidine pathway had no effect on ADE1-lacZ expression, either transcriptional or enzymatic deregulation of histidine biosynthesis resulted in a partial derepression of ADE1-lacZ. This result clearly justifies the coregulation of HIS1, HIS4, and HIS7 genes with ADE genes. Indeed, in the presence of extracellular adenine, expression of ADE genes should be turned off and the level of AICAR and SAICAR (which are required as coactivators of ADE gene transcription) should be kept as low as possible. The observed repression of early steps of histidine biosynthesis by adenine is most probably necessary to avoid unwanted expression of ADE genes due to the synthesis of AICAR by the histidine pathway.
Additionally, the histidine pathway could affect IMP biosynthesis regulation by depleting the adenylate pool. Indeed, such an ATP depletion due to deregulation of histidine synthesis has been extensively documented in bacteria and it has been shown that hisG deregulated mutants lead to adenine auxotrophy in Escherichia coli and Salmonella (Shedlovsky and Magasanik 1962a,b; Johnston and Roth 1979; Galloway and Taylor 1980). No such phenotype was associated with the HIS1-17 yeast mutation (K. Rébora and B. Daignan-Fornier, unpublished observations), which suggests that the respective flux in the IMP and histidine pathways differs in yeast and bacteria. Furthermore, while similar ATP depletion was observed in both ade3 and ade3 ade5,7 his1 mutants grown in low-adenine medium (down to 10% of wild-type level; B. Laloo and B. Daignan-Fornier, unpublished observations), expression of ADE1-lacZ was high in the former and low in the latter. These results further confirm that AICAR synthesis is critical for transcriptional activation of ADE genes in the absence of adenine. However, it should be kept in mind that, in the presence of adenine, ATP has an important inhibitory effect on the first enzyme of the pathway, Ade4p (Rébora et al. 2001).
What is the role of AICAR accumulation in producing auxotrophy for histidine of the ade3 and ade16 ade17 mutant strains?
ade3 and ade16 ade17 mutant strains are the only mutants of the AMP biosynthesis pathway that are auxotrophic for both adenine and histidine. Since these mutants cannot metabolize AICAR and are therefore likely to accumulate this metabolite, it was proposed by Appling and co-workers that AICAR accumulation was responsible for histidine auxotrophy, possibly by inhibiting an enzyme of the histidine pathway (Tibbetts and Appling 2000). However, we found that neither a constitutive HIS1 dominant allele nor the overexpression of any of the HIS2–HIS7 genes is able to suppress the requirement for histidine of an ade3 or ade16 ade17 mutant strain. Together these results suggest that the histidine auxotrophy of ade3 and ade16 ade17 mutants might not be due simply to an inhibition by AICAR of one of the steps of the histidine biosynthesis pathway, but rather takes place via a more complex mechanism. However, we cannot rule out that overexpression of the HIS genes under control of the tet repressible promoter might not be sufficient to relieve AICAR inhibition. Finally, while a link among AICAR, methionine, and folate metabolism has previously been described (Holmes and Appling 2002), we could not find any mutant in these pathways that, when combined to ade3, alleviated the requirement of this mutant for histidine.
Importantly, we found that overexpression of PMU1 can detoxify AICAR and suppress histidine auxotrophy of the ade3 ade16 ade17 triple mutant. This result further correlates AICAR accumulation and histidine auxotrophy in these mutants. The mechanism by which Pmu1p acts on AICAR is not clear, but since PMU1 encodes a potential phosphomutase, it presumably could directly modify AICAR, which is a monophosphate nucleotide derivative. Consistently, PMU1 was previously isolated as a multicopy suppressor of the temperature sensitivity of a tps2 mutant lacking trehalose-phosphatase activity and shown to reduce the accumulation of trehalose-6-phosphate in this mutant (Elliott et al. 1996).
Toxicity of AICAR and SAICAR accumulation:
In the course of this work we found that accumulation of SAICAR and AICAR can be toxic for yeast. The toxicity is detected only when the pathway is made constitutive by overexpressing Ade4p, the first enzyme of the pathway. This effect is clearly due to overproduction of AICAR (or SAICAR) from the IMP de novo pathway since mutations upstream in the pathway totally abolish the toxicity. In vitro AICAR can activate mammalian AMP-activated protein kinase (AMPK) (Sullivan et al. 1994; Corton et al. 1995; Stefanelli et al. 1998), and therefore AICAR toxicity could at least partly result from incorrect regulation of AMPK due to accumulation of the metabolite. However, the effect of AICAR on Snf1p, the yeast equivalent of AMPK, has not yet been documented.
Interestingly, adenylosuccinate lyase (ASL, the equivalent of yeast Ade13p; see Figure 1) deficiency in humans is characterized by accumulation in body fluids of SAICAriboside and succinyladenosine, the two dephosphorylated derivatives of SAICAR and adenylosuccinate, which are the substrates of ASL (Jaeken and Van den Berghe 1984). ASL deficiency leads to varying degrees of neurological abnormalities, i.e., mental retardation and autistic features. Several mutations that lead to ASL deficiency have been reported. The mutated enzymes showed different levels of residual enzymatic activity that are correlated to different degrees of mental retardation (Race et al. 2000). The link between SAICAriboside and succinyladenosine accumulation and neurological abnormalities is not understood yet and future studies on SAICAR toxicity could provide important clues. Strikingly, accumulation of AICAR derivatives due to a mutation in AICAR transformylase (the equivalent of yeast Ade16p Ade17p activity; see Figure 1) was recently found to be associated with severe neurological defects (Marie et al. 2004). Therefore, accumulation of AICAR and SAICAR is clearly deleterious for humans. In the future, yeast genetics could certainly prove useful in investigating the molecular mechanisms leading to this toxicity.
Our results, together with previous data in yeast and other organisms, strongly suggest that AICAR and SAICAR interfere with histidine and methionine synthesis pathways and other cellular processes. Clearly, these two intermediary metabolites are not just inert molecules “waiting” for further metabolizing to IMP but are involved in regulatory crosstalks. Because their concentration reflects the flux in the AMP biosynthesis pathway, these small molecules are informative and could significantly contribute to the regulation of cellular homeostasis.
Acknowledgments
We are grateful to G. Fink, R. Gaber, F. Lacroute, and A. Hinnebusch for providing biological materials. We thank C. Saint-Marc for characterization of several double mutants, C. Desmoucelles for construction of the ade8 ade16 ade17 triple mutant, A. Devin for advice, and I. Sagot for critical reading of the manuscript. This work was supported by grants from Conseil Régional d'Aquitaine, Université Bordeaux 2, and CNRS (UMR5095). K.R. was supported by Ministère de la Recherche and Fondation pour la Recherche Médicale training fellowships.
References
- Arndt, K. T., C. Styles and G. R. Fink, 1987. Multiple global regulators control HIS4 transcription in yeast. Science 237: 874–880. [DOI] [PubMed] [Google Scholar]
- Brachmann, C. B., A. Davies, G. J. Cost, E. Caputo, J. Li et al., 1998. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 14: 115–132. [DOI] [PubMed] [Google Scholar]
- Corton, J. M., J. G. Gillespie, S. A. Hawley and D. G. Hardie, 1995. 5-Aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur. J. Biochem. 229: 558–565. [DOI] [PubMed] [Google Scholar]
- Cross, F. R., 1997. ‘Marker swap’ plasmids: convenient tools for budding yeast molecular genetics. Yeast 13: 647–653. [DOI] [PubMed] [Google Scholar]
- Daignan-Fornier, B., and G. R. Fink, 1992. Coregulation of purine and histidine biosynthesis by the transcriptional activators BAS1 and BAS2. Proc. Natl. Acad. Sci. USA 89: 6746–6750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denis, V., H. Boucherie, C. Monribot and B. Daignan-Fornier, 1998. Role of the myb-like protein bas1p in Saccharomyces cerevisiae: a proteome analysis. Mol. Microbiol. 30: 557–566. [DOI] [PubMed] [Google Scholar]
- Elliott, B., R. S. Haltiwanger and B. Futcher, 1996. Synergy between trehalose and Hsp104 for thermotolerance in Saccharomyces cerevisiae. Genetics 144: 923–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feller, A., E. Dubois, F. Ramos and A. Pierard, 1994. Repression of the genes for lysine biosynthesis in Saccharomyces cerevisiae is caused by limitation of Lys14-dependent transcriptional activation. Mol. Cell. Biol. 14: 6411–6418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn, P. J., and R. J. Reece, 1999. Activation of transcription by metabolic intermediates of the pyrimidine biosynthetic pathway. Mol. Cell. Biol. 19: 882–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaber, R. F., M. C. Kielland-Brandt and G. R. Fink, 1990. HOL1 mutations confer novel ion transport in Saccharomyces cerevisiae. Mol. Cell. Biol. 10: 643–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galloway, R. J., and B. L. Taylor, 1980. Histidine starvation and adenosine 5′-triphosphate depletion in chemotaxis of Salmonella typhimurium. J. Bacteriol. 144: 1068–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gari, E., L. Piedrafita, M. Aldea and E. Herrero, 1997. A set of vectors with a tetracycline-regulatable promoter system for modulated gene expression in Saccharomyces cerevisiae. Yeast 13: 837–848. [DOI] [PubMed] [Google Scholar]
- Guetsova, M. L., K. Lecoq and B. Daignan-Fornier, 1997. The isolation and characterization of Saccharomyces cerevisiae mutants that constitutively express purine biosynthetic genes. Genetics 147: 383–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinnebusch, A. G., 1988. Mechanisms of gene regulation in the general control of amino acid biosynthesis in Saccharomyces cerevisiae. Microbiol. Rev. 52: 248–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes, W. B., and D. R. Appling, 2002. Cloning and characterization of methenyltetrahydrofolate synthetase from Saccharomyces cerevisiae. J. Biol. Chem. 277: 20205–20213. [DOI] [PubMed] [Google Scholar]
- Jaeken, J., and G. Van den Berghe, 1984. An infantile autistic syndrome characterised by the presence of succinylpurines in body fluids. Lancet 2: 1058–1061. [PubMed] [Google Scholar]
- Johnston, H. M., and J. R. Roth, 1979. Histidine mutants requiring adenine: selection of mutants with reduced hisG expression in Salmonella typhimurium. Genetics 92: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, E. W., and G. R. Fink, 1982 Regulation of amino acid and nucleotide biosynthesis in yeast, pp. 181–299 in The Molecular Biology of the Yeast Saccharomyces: Metabolism and Gene Expression, edited by J. N. Strathern, E. W. Jones and J. R. Broach. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Kippert, F., 1995. A rapid permeabilization procedure for accurate quantitative determination of beta-galactosidase activity in yeast cells. FEMS Microbiol. Lett. 128: 201–206. [DOI] [PubMed] [Google Scholar]
- Koren, A., S. Ben-Aroya, R. Steinlauf and M. Kupiec, 2003. Pitfalls of the synthetic lethality screen in Saccharomyces cerevisiae: an improved design. Curr. Genet. 43: 62–69. [DOI] [PubMed] [Google Scholar]
- Loewen, C. J., M. L. Gaspar, S. A. Jesch, C. Delon, N. T. Ktistakis et al., 2004. Phospholipid metabolism regulated by a transcription factor sensing phosphatidic acid. Science 304: 1644–1647. [DOI] [PubMed] [Google Scholar]
- Marie, S., B. Heron, P. Bitoun, T. Timmerman, G. Van Den Berghe et al., 2004. AICA-ribosiduria: a novel, neurologically devastating inborn error of purine biosynthesis caused by mutation of ATIC. Am. J. Hum. Genet. 74: 1276–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeil, J. B., A. L. Bognar and R. E. Pearlman, 1996. In vivo analysis of folate coenzymes and their compartmentation in Saccharomyces cerevisiae. Genetics 142: 371–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers, A. M., A. Tzagoloff, D. M. Kinney and C. J. Lusty, 1986. Yeast shuttle and integrative vectors with multiple cloning sites suitable for construction of lacZ fusions. Gene 45: 299–310. [DOI] [PubMed] [Google Scholar]
- Nigavekar, S. S., and J. F. Cannon, 2002. Characterization of genes that are synthetically lethal with ade3 or leu2 in Saccharomyces cerevisiae. Yeast 19: 115–122. [DOI] [PubMed] [Google Scholar]
- Pinson, B., E. M. Brendeford, O. S. Gabrielsen and B. Daignan-Fornier, 2001. Highly conserved features of DNA binding between two divergent members of the Myb family of transcription factors. Nucleic Acids Res. 29: 527–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Race, V., S. Marie, M. F. Vincent and G. Van den Berghe, 2000. Clinical, biochemical and molecular genetic correlations in adenylosuccinate lyase deficiency. Hum. Mol. Genet. 9: 2159–2165. [DOI] [PubMed] [Google Scholar]
- Rasse-Messenguy, F., and G. R. Fink, 1973. Feedback-resistant mutants of histidine biosynthesis in yeast. Basic Life Sci. 2: 85–95. [DOI] [PubMed] [Google Scholar]
- Rébora, K., C. Desmoucelles, F. Borne, B. Pinson and B. Daignan-Fornier, 2001. Yeast AMP pathway genes respond to adenine through regulated synthesis of a metabolic intermediate. Mol. Cell. Biol. 21: 7901–7912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shedlovsky, A. E., and B. Magasanik, 1962. a A defect in histidine biosynthesis causing an adenine deficiency. J. Biol. Chem. 237: 3725–3730. [PubMed] [Google Scholar]
- Shedlovsky, A. E., and B. Magasanik, 1962. b The enzymatic basis of an adenine-histidine relationship in Escherichia coli. J. Biol. Chem. 237: 3731–3736. [PubMed] [Google Scholar]
- Sherman, F., G. R. Fink and J. B. Hicks, 1986 Methods in Yeast Genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Springer, C., M. Kunzler, T. Balmelli and G. H. Braus, 1996. Amino acid and adenine cross-pathway regulation act through the same 5′-TGACTC-3′ motif in the yeast HIS7 promoter. J. Biol. Chem. 271: 29637–29643. [DOI] [PubMed] [Google Scholar]
- Stefanelli, C., I. Stanic, F. Bonavita, F. Flamigni, C. Pignatti et al., 1998. Inhibition of glucocorticoid-induced apoptosis with 5-aminoimidazole-4-carboxamide ribonucleoside, a cell-permeable activator of AMP-activated protein kinase. Biochem. Biophys. Res. Commun. 243: 821–826. [DOI] [PubMed] [Google Scholar]
- Sullivan, J. E., F. Carey, D. Carling and R. K. Beri, 1994. Characterisation of 5′-AMP-activated protein kinase in human liver using specific peptide substrates and the effects of 5′-AMP analogues on enzyme activity. Biochem. Biophys. Res. Commun. 200: 1551–1556. [DOI] [PubMed] [Google Scholar]
- Sze, J. Y., M. Woontner, J. A. Jaehning and G. B. Kohlhaw, 1992. In vitro transcriptional activation by a metabolic intermediate: activation by Leu3 depends on alpha-isopropylmalate. Science 258: 1143–1145. [DOI] [PubMed] [Google Scholar]
- Tibbetts, A. S., and D. R. Appling, 2000. Characterization of two 5-aminoimidazole-4-carboxamide ribonucleotide transformylase/inosine monophosphate cyclohydrolase isozymes from Saccharomyces cerevisiae. J. Biol. Chem. 275: 20920–20927. [DOI] [PubMed] [Google Scholar]
- Valerius, O., O. Draht, E. Kubler, K. Adler, B. Hoffmann et al., 2001. Regulation of hisHF transcription of Aspergillus nidulans by adenine and amino acid limitation. Fungal Genet. Biol. 32: 21–31. [DOI] [PubMed] [Google Scholar]
- Whitehead, E., M. Nagy and H. Heslot, 1966. Interactions entre la biosynthèse de purines nucléotides et celle de l'histidine chez le Schizosaccharomyces pombe. C. R. Acad. Sci. Paris 263: 819. [Google Scholar]