

Abstract
The Hedgehog signaling pathway plays an essential role in the pattern formation and development of metazoan animals. Misregulation of Hedgehog signaling has also been associated with the formation of multiple types of cancer. For these reasons, the Hedgehog pathway has attracted considerable interest. Many proteins required in the Hedgehog pathway have been identified, and while much has been learned about their function in signal transduction, it is clear that this complement of proteins does not comprise the full set necessary for Hedgehog signal transduction. Because significant gaps remain in our knowledge of the molecules required for Hedgehog signaling, we performed an enhancer/suppressor screen in Drosophila melanogaster to identify novel components of the pathway. In addition to the isolation of new alleles of the known pathway components patched and smoothened, this screen identified 14 novel complementation groups and a larger number of loci represented by single alleles. These groups include mutations in the genes encoding the translation factors eRF1 and eIF1A and the kinesin-like protein Pavarotti. It also identified mutations in a gene whose product is necessary for the movement of Hedgehog protein through tissues.
THE Hedgehog (Hh) proteins are secreted morphogens that provide positional information during the development of many multi-cellular animals. Hh was originally identified as a segment polarity gene in Drosophila where it is required for the patterning of the embryonic cuticle. It has since been found to be involved in many other developmental processes, including the patterning of adult legs, eyes, and wings. In vertebrates, Hh signaling is known to function in the patterning of many different structures, including the fore brain, neural tube, somites, eye, and limb. Misregulation of Hh signaling has been implicated in basal cell carcinomas, gliomas, and gastric and prostate cancers (for reviews see Ingham and McMahon 2001; Ruiz i Altaba et al. 2002).
One of the defining characteristics of morphogens such as Hh is their ability to elicit different responses in different cells in a concentration-dependent manner (Tabata and Takei 2004). In the Drosophila wing imaginal disc, Hh is produced by cells in the posterior compartment and moves into the anterior compartment, forming a concentration gradient. Anterior cells close to the anterior/posterior (A/P) compartment boundary see high levels of Hh and express both high-threshold genes, such as engrailed (en) and patched (ptc), and low-threshold genes such as decapentaplegic (dpp). Further from the A/P boundary the levels of Hh are insufficient to generate the high-threshold response and only low-threshold genes are expressed (see Brook 2000).
The ability of Hh to induce differential responses can be partially explained by the dual activity of the zinc-finger transcription factor Cubitus interruptus (Ci), the downstream nuclear effector of the Hh pathway (Methot and Basler 1999). In the absence of Hh signaling, the full-length Ci protein (Ci-155) is sequestered in the cytoplasm by a multi-protein complex containing the kinesin-like protein Costal-2 (Cos2), the serine/threonine kinase Fused (Fu), and the novel protein Suppressor of fused [Su(fu)] (Robbins et al. 1997; Sisson et al. 1997; Jia et al. 2003; Lum et al. 2003; Ogden et al. 2003; Ruel et al. 2003). This complex targets Ci for serial phosphorylation by at least three kinases: protein kinase A (PKA), glycogen synthase kinase 3β, and casein kinase 1α (Y. Chen et al. 1998; Price and Kalderon 1999, 2002; Jia et al. 2002). This phosphorylation targets Ci for ubiquitination by the Slimb/SCF complex (Jiang and Struhl 1998; Theodosiou et al. 1998; Ou et al. 2002). Ubiquitinated Ci then undergoes a proteosome-dependent cleavage to generate a smaller (Ci-75) repressor form of the protein that translocates to the nucleus and constitutively represses the expression of Hh target genes (Aza-Blanc et al. 1997).
Smoothened (Smo) is a seven-pass membrane protein that is essential for all Hh signaling (Alcedo et al. 1996; Chen and Struhl 1996; van den Heuvel and Ingham 1996). The activity of Smo is repressed by the Hh-binding, multi-pass transmembrane receptor Patched (Ptc), since, in the absence of Ptc, downstream signaling events are activated by Smo in a ligand-independent manner (Hooper 1994; Chen and Struhl 1996; Quirk et al. 1997). The binding of Hh to Ptc relieves the repression that Ptc normally exerts on Smo. The mechanism by which Ptc inhibits Smo activity is apparently not mediated by sequestration (Denef et al. 2000) and may involve an amplification step (Taipale et al. 2002), but the actual mechanism of repression remains unknown. In unstimulated cells, Smo resides largely in intracellular vesicles, where it binds the Cos2 complex through a direct interaction between its cytoplasmic C-terminal tail and the Cos2 protein (Jia et al. 2003; Lum et al. 2003; Ogden et al. 2003; Ruel et al. 2003; Zhu et al. 2003). Although the nature of Smo activation is not well understood, it is known that in response to Hh, Smo becomes hyperphosphorylated, is stabilized, and translocates to the cell surface (Denef et al. 2000), bringing along with it the Cos2 complex. These events are correlated with the stabilization of full-length Ci-155 and a concomitant loss of repressor Ci-75 (Alcedo et al. 2000; Denef et al. 2000; Lum et al. 2003).
In wing imaginal discs the derepression of Hh target genes caused by the stabilization of Ci-155 is sufficient for the expression of dpp and other low-threshold target genes (Methot and Basler 1999). The expression of high-threshold genes, however, requires the conversion of Ci-155 into a transcriptional activator and its translocation into the nucleus (Methot and Basler 2000). This activation of Ci-155 is Hh dependent and requires Smo protein. The mechanism of Ci-155 activation is poorly understood, but likely involves relieving the repressive effects that Su(fu) has on Ci-155 (Methot and Basler 2000; Wang et al. 2000).
Many gaps remain in our understanding of Hh signaling. Ptc has homology to bacterial proton-driven transmembrane molecular transporters, and it has been proposed that Ptc functions by moving a small molecule that regulates Smo function across the plasma membrane (Taipale et al. 2002). It is not yet known what the small molecule is or how it regulates the activity of Smo, or, for that matter, what actually is Smo activity. Recent reports have connected Smo with downstream components of the pathway by demonstrating a direct interaction between Smo and Cos2 and have shown that Smo phosphorylation and altered subcellular localization are correlated with pathway activation. But it remains unclear how these interactions and activities result in the stabilization of Ci-155 or the conversion of Ci-155 into a potent transcriptional activator.
Many questions also remain as to how the Hh concentration gradient is formed and shaped. The Hh protein undergoes several processing events, and the mature signaling form contains two different lipid additions. A cholesterol group is added to the C-terminal end of the protein during the autocatalytic cleavage of Hh (Lee et al. 1994; Bumcrot et al. 1995; Porter et al. 1995), and an acyl group is added near the N terminus by the transmembrane acyltransferase Sightless (Chamoun et al. 2001; Lee and Treisman 2001; Micchelli et al. 2002). The expression of the sterol-sensing domain protein Dispatched in Hh-producing cells, but not Hh-receiving cells, is essential for the movement of mature Hh proteins from Hh-producing cells into the Hh-receiving tissue (Burke et al. 1999). In contrast, there is a requirement for proteins involved in glycosaminoglycan biosynthesis (with acetylglucosaminyltransferase activity) in Hh-receiving cells, but not in Hh-producing cells, which has led to the idea that heparin sulfate proteoglycans (HSPGs) are necessary for the movement Hh proteins in receiving tissues (The et al. 1999; Han et al. 2004; Takei et al. 2004). However, the mechanism by which lipid-modified Hh proteins move through tissues, and the role of HSPG in this process, remains unknown.
These unresolved questions about Hh signaling and morphogen movement suggest that additional components to the pathway have yet to be identified. To identify novel proteins required for Hh signaling, we conducted a large-scale genetic screen in Drosophila. In this screen we tested the ability of newly induced mutations to enhance or suppress a partial Hh loss-of-function phenotype generated by the transgenic expression of dominant-negative form of Smo in the developing wing. In addition to new alleles of known components of the Hh pathway, 105 interacting mutations, of which 34 are grouped into 14 novel complementation groups, were identified. The isolation and genetic characterization of these mutations are described here.
MATERIALS AND METHODS
Molecular biology:
The UAS-Smo5A was constructed using standard molecular biology techniques. The Smo coding sequence was mutated by PCR-mediated mutagenesis to generate the mutations TC2237GC, T2287G, AG2368GC, A2501A, and A2539G with the corresponding amino acid changes: S667A S687A, S740A, T755A, and T785A (base-pair and acid numbers according to accession no. NM_078719). The sequence GACTACAAGGACGACGATGACAAG was added immediately before the stop codon, adding a FLAG epiope (DYKDDDDK) to the C-terminal end of the encoded protein. The resulting construct was ligated into the NotI and XhoI sites of the Drosophila cloning and transformation vector pUAST.
Mutagenesis:
Isogenized w1118 males were starved for 8 hr and then added to vials containing filters soaked with 25 mm EMS (ethyl methanesulfonate; Sigma, St. Louis) and 1% sucrose in water. Males were mutagenized overnight, allowed to recover for 24 hr, and then crossed to C756-Gal4,UAS-Smo5A/TM3, and HS-hid virgins. F1 flies with enhancement or suppression of the C756-Gal4,UAS-Smo5A (C765-SmoDN) phenotype were crossed to Bl/CyO;C756-Gal4,UAS-Smo5A/TM6b flies. F2 flies with the same enhancement or suppression of the C765-SmoDN phenotype as the F1 flies were crossed again to Bl/CyO;C756-Gal4,UAS-Smo5A/TM6b. After the enhancement or suppression of the C765-SmoDN phenotype was confirmed again in the F3 generation males and females of genotype mutation/CyO;TM6b were crossed to establish balanced mutant stocks.
The rate of mutagenesis was estimated by dividing the total number of F1 TM3-HS-hid flies by the number of ebony-/TM3-HS-hid flies (i.e., the number of new mutations in the ebony gene). This gave a rate of one new ebony mutation every 1600 F1 genomes, or approximately eight loss-of-function mutations per F1 genome. If one-fourth of the genes in Drosophila are essential (Brizuela et al. 1994), then the mutation rate for the screen is about two lethal mutations per F1 genome.
Fly stocks and genetics:
Fly stocks used included w1118, pavB200, ptcG12, smo3, sitT398, dispS037707, hhAC, ptcG12, smo3, Pka-C101272, ttv00681, Cos2W1, eRF1neo28, eRF1U3, mirrSaiD1, mirrCre2, Ptp69D1, Ptp69D7, Ptp69D18, C765-Gal4, EP(3)0935, EP(3)3121, EP(3)3350, Df(3R)DG4, Df(3R)Cha1a, Df(3L)XS533, Df(3L)iro-2, Df(3L)GN19, Df(3L)GN50 (FlyBase 2003), and pav+t10.5 (pav rescue construct, RC1; Adams et al. 1998). Df(3L)ED4710, Df(3L)ED224, Df(3L)ED225, Df(3L)ED4782, Df(3L)ED4799, Df(3L)ED229, Df(3L)ED4858, Df(3L)ED4861, Df(3L)ED4978, and Df(3L)ED230 (Ryder et al. 2004) and third chromosome deficiencies from the Bloomington Stock Center deficiency kit.
jaft mutant clones were generated by crossing males of genotype w;;FRT80, jaft447/TM6B or w;dpp-LacZ;FRT80, jaft447/TM6B with females of genotype hs-flp;;FRT80,Ubi-GFP. Larvae were heat-shocked for 1 hr at 37° during the first and second instar to induce mitotic recombination. Clones of cells expressing Smo5A were generated by crossing hs-flip; actin > CD2 > Gal4;UAS-GFP flies with w;;UAS-Smo5A flies and heat-shocking the larvae at 37° for 30 min.
To rescue pavarotti (pav) mutants, flies of genotype pav+t10.5/CyO;pav831/TM6b or pav+t10.5/CyO;pav2046/TM6b were crossed to flies of genotype pavB200/TM6b, pav831/TM6b, pav963/TM6b, or pav2046/TM6b.
Dissections and immunohistochemistry:
Wing imaginal discs from climbing, third instar larvae were dissected into cold PBS and then fixed for 20 min at room temperature with 4% formaldehyde in PBS. Discs were then washed three times for 10 min with PBT (PBS with 0.2% Triton X-100), blocked for 45 min with BBT (PBT with 0.1% bovine serum albumin), and incubated overnight at 4° in primary antibodies. Primary antibodies and dilutions used were anti-Dll (1:250; Wu and Cohen 1999), anti-phospho-Mad (1:1000; Persson et al. 1998), anti-Ci (Mab 2A1,1:2; Motzny and Holmgren 1995), mouse anti-Ptc (1:3; Capdevila et al. 1994), and rabbit anti-β-Gal (1:500; Kappel). After four, 30-min washes at room temperature in BBT, discs were incubated for 2 hr in appropriate fluorescent-labeled secondary antibodies diluted 1:200 in BBT. Discs were washed four times with PBT and mounted in 80% glycerol in PBS for analysis with a Leica confocal microscope.
To mount adult wings, flies were incubated in SH buffer (20% glycerol, 80% ethanol) overnight. After a rinse with water, the wings were dissected in water and then mounted in Faure's mounting media.
RESULTS
Smo5A acts as a dominant negative:
Previous work has demonstrated that Smo becomes phosphorylated in response to Hh and that this phosphoylation is correlated with Hh signal transduction (Denef et al. 2000). A search of the primary structure of the Smo with PROSITE identified five putative PKA phosphorylation sites in the C-terminal, cytoplasmic tail (red boxes in Figure 1A). To test if these sites are important for Smo function, transgenic flies were generated to express a mutant protein in which the target serines and threonines have been replaced with alanines (UAS-Smo5A; see materials and methods). Clones of cells in the wing imaginal disc expressing the mutant Smo5A protein and GFP failed to express the Hh target Ptc (arrows in Figure 1, B and C), suggesting that Smo5A protein has dominant-negative activity.
Figure 1.—
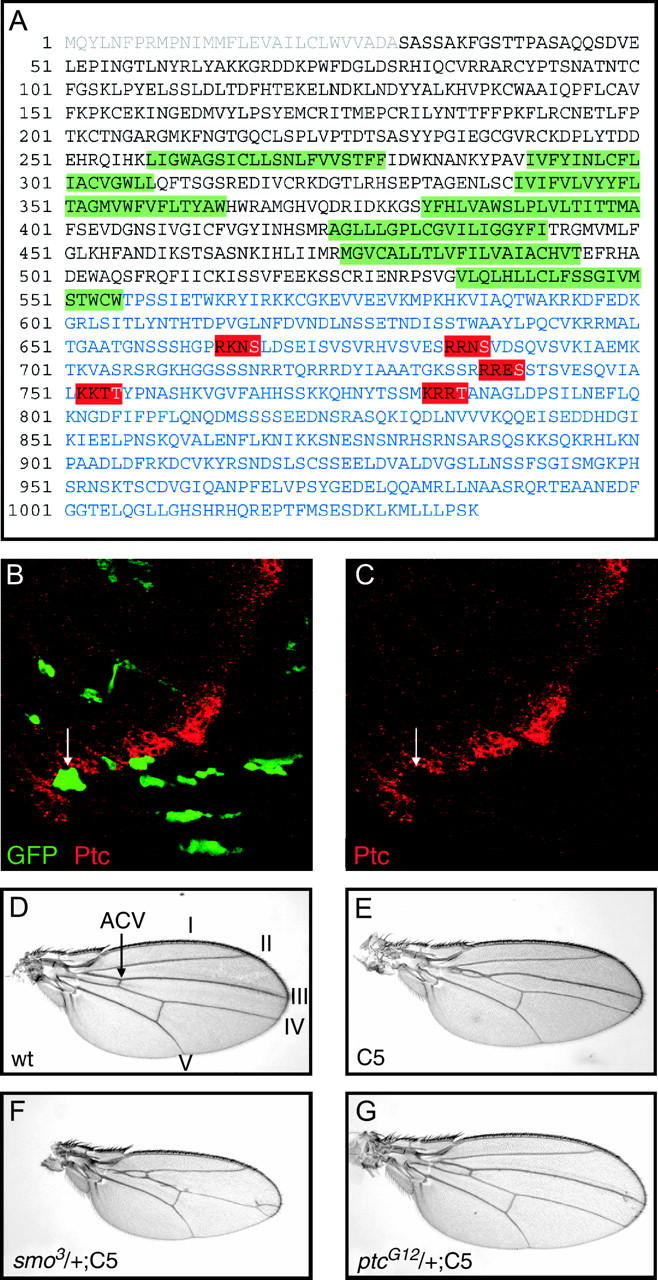
Smo5A is a dominant negative. (A) The primary sequence of smoothened, signal sequence (gray), transmembrane domains (green) C-terminal cytoplasmic tail (blue), and five putative PKA phosphorylation sites (red) that were mutated to alanines are highlighted. Expression of Smo5A in flip-out clones (green GFP-expressing cells in B) represses the expression of Patched expression (red; B and C). Expression of Smo5A in the wing with the C765-Gal4 (E) driver causes a reduction of the central portion of the wing between longitudinal veins III and IV and fusion of these veins proximal to the anterior cross-vein (compare to wild type; D). These phenotypes are strongly enhanced in flies heterozygous for smo (F) and are completely suppressed by removing one copy of ptc. Longitudinal veins I–V and the anterior cross-vein (ACV) are indicated in D.
Expression of UAS-Smo5A throughout the developing wing imaginal disc with the C765-Gal4 driver caused a reduction in the distance between veins III and IV and a partial fusion of these veins proximal to the anterior cross-vein (compare Figure 1E with 1D). Hh signaling directly specifies the vein III–IV intervein region, and the UAS-Smo5A,C765-Gal4 phenotype (hereafter referred to as the C765-SmoDN phenotype) is consistent with a partial loss of Hh signaling during wing development. Indeed, reducing the dosage of Smo by one-half resulted in strong enhancement in the C765-SmoDN phenotype (Figure 1F). Conversely, reducing repression of the pathway by removal of one copy of ptc resulted in nearly normal wings (Figure 1G).
A modifier screen for novel components of the Hedgehog pathway:
The expression of UAS-Smo5A with C765-Gal4 produces a mild Hh loss-of-function phenotype, primarily in the wing of flies that are otherwise viable and fertile. The C765-SmoDN phenotype is visible in adult flies under the dissecting microscope, very consistent from individual to individual, and readily modified by reducing the dosage of genes encoding components of the Hh pathway. Furthermore, the C765-SmoDN phenotype is not significantly modified in flies heterozygous for components of other signaling pathways necessary for wing development (e.g., Wg or Dpp; data not shown). Because of these characteristics, we used the C765-SmoDN phenotype as sensitized background in a F1 enhancer/suppressor screen to identify novel components of the Hh pathway.
In the screen isogenic w1118 males were mutagenized with EMS and crossed to females carrying the UAS-Smo5A and C765-Gal4 transgenes (the crossing scheme is summarized in Figure 2). The progeny of these flies were scored for enhancement or suppression of the C765-SmoDN phenotype. F1 flies with modifying mutations were crossed back to flies with UAS-Smo5A and C765-Gal4 transgenes and second and third chromosome balancers. Male progeny of backcrosses that confirmed the original modification were again backcrossed. This second backcross permitted the establishment of balanced mutant stocks, mapped the interacting mutation to the second or third chromosome, and confirmed the modification of the C765-SmoDN phenotype in a third generation.
Figure 2.—

Crossing scheme for the screen. EMS-mutagenized males were crossed to females with the C765-Gal4,UAS-Smo5A tester chromosome (C5). The F1 progeny with enhancement or suppression of the C5 phenotype were crossed to the backcross strain (containing the C5 chromosome, the second chromosome dominant marker Bristle, and second and third chromosome balancers). F2 flies that showed penetrant modification of the C5 phenotype were again crossed to the backcross strain to generate a balanced mutant stock and to map the modifying mutation to the second or third chromosome.
A total of ∼90,000 genomes were screened, and 2558 F1 flies were selected as having potentially interesting mutations. After the two backcrosses, balanced stocks had been established for 107 mutants that showed a consistent and penetrant modification of the C765-SmoDN phenotype.
The balanced mutations were tested for complementation with genes for known components of the Hh pathway, including sitT398, dispS037707, hhAC, ptcG12, smo3, Pka-C101272, ttv00681, and Cos2W1. New alleles of ptc (ptc1232) and smo (smo848) were isolated, demonstrating that the screening strategy was effective in finding mutations in genes required for Hh signaling.
The remaining mutations were tested for cross-complementation with the other mutations that mapped to the same chromosome. These complementation crosses placed 34 of the mutations into 14 different lethal complementation groups, 2 on the second chromosome and 12 on the third chromosome (Table 1). Remarkably, one mutant fly contained mutations for two separate complementation groups (group B-left and group B-right). These mutations are separable by recombination, each enhances the C765-SmoDN phenotype, and each complements members of one group and fails to complement members of the other. The remaining 71 mutations represent single hits.
TABLE 1.
Screen summary
| Cross | No. of flies |
|---|---|
| F1 flies screened | ∼90,000 |
| F1 enhancer/suppressor selected | 2558 |
| Balanced mutant stocks | 107 |
| Complementation groups | No. of alleles |
| smoothened (chromosome 2) | 1 |
| patched | 1 |
| Group N | 2 |
| Group O | 2 |
| Group Aa (chromosome 3) | 3 |
| Group B-lefta | 2 |
| Group B-righta | 2 |
| Group C (jaft) | 4 |
| Group D (pavarotti)b | 3 |
| Group E | 2 |
| Group F (mirror) | 2 |
| Group Gc | 3 |
| Group I (eIF1A)a | 2 |
| Group J (eRF1)a | 3 |
| Group Ka | 2 |
| Group Ma | 2 |
| Single hits | 71 |
No clones of mutant cells were recovered in adults or third instar wing discs, possibly cell lethal.
Required for cytokinesis, clones of mutant cells were not recovered in adults or third instar wing discs.
Clones of mutant cells were not recovered in adults or third instar wing discs, but persist long enough to occasionally induce phenotypes in wings and eyes (see text).
We next sought to identify the genes disrupted by the mutations. Mapping mutations is greatly simplified when there are multiple alleles, and most of our complementation groups are on the third chromosome. We have therefore concentrated our efforts on mapping and characterizing these groups.
The 12 lethal complementation groups on the third chromosome were mapped with several techniques, including standard meiotic recombination onto a chromosome with multiple recessive markers (rucuca), complementation crosses with deficiencies, P-element-mediated meiotic recombination mapping (Zhai et al. 2003), and P-element-mediated male recombination mapping (B. Chen et al. 1998). The mapping results for the third chromosome groups are summarized in Figure 3 and, for several groups, discussed in detail in the following sections. The interaction between each of the groups and C765-Gal4,UAS-Smo5A is shown in Figure 4.
Figure 3.—

Summary of mapping complementation groups on the third chromosome. The cytological map positions or intervals in the third chromosome complementation groups are indicated by bars and open boxes, respectively.
Figure 4.—
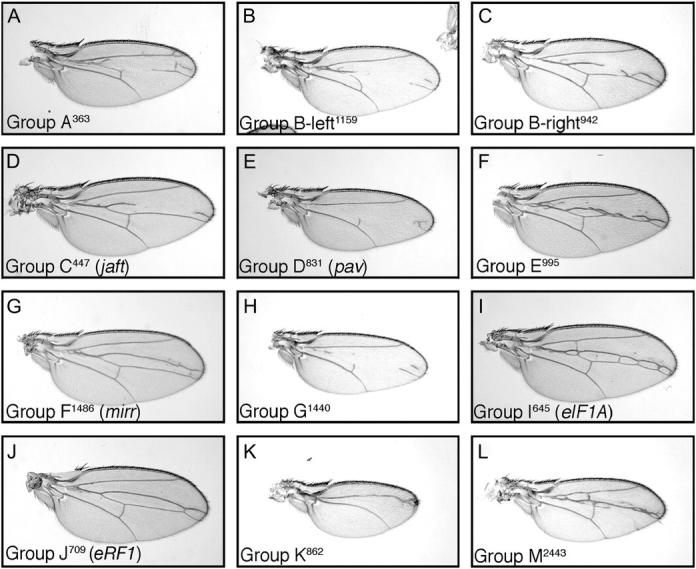
Modification of C765-Gal4,UAS-Smo5A by third chromosome complementation groups. The 12 complementation groups on the third chromosome enhance the C5 phenotype (see Figure 1E for comparison). The group and mutant number are indicated.
To better study the function of the complementation groups in Hh signaling, individual mutations were recombined onto an FRT chromosome, and clones of mutant cells were generated by somatic recombination (Harrison and Perrimon 1993). Both alleles of group E are heterozygous fertile; however, recombinants of either allele on the FRT80 chromosome appear to be male sterile. It has, therefore, not been possible to generate FRT stocks for group E. No mutant clones were recovered for 8 of the 12 groups (groups A, B-left, B-right, D, I, J, K, and M; see Table 1) even though wild-type twin spots, with two copies of the clonal marker, were present in adult eyes. This suggests that the mutations in these groups are cell lethal.
While clones of cells that are mutant for group G do not seem to persist in the adult, these cells can survive long enough to occasionally have dramatic effects on wing and eye development. Some flies in which clones have been induced have scars in the eye/missing photoreceptors, rough eyes, wing blisters, wing margin notching, and altered wing blade morphology (data not shown). No obvious phenotypes were observed in legs, halteres, antennae, thorax, or abdomen. However, other flies with distinct twin spots in the eye appeared morphologically normal.
Translation factors eIF1A and eRF1:
Group I (comprising two members: I654 and I2232) was mapped by meiotic recombination between the rucuca markers stripe and ebony. Complementation crosses with deficiencies in the region demonstrated that both group I alleles failed to complement Df(3R)Cha7 (90F01-F04;91F05), but complemented two overlapping deficiencies: Df(3R)DG4 (90D02-04;90F03-06) and Df(3R)Cha1a (91A02-B03; 91F13-92A01). This defined an interval for the group I region of ∼425 kb between the genes stripe and fruitless. Testing for complementation with lethal P insertions in the region revealed that the P insertions EP(3)0935 and EP(3)3350 failed to complement both alleles of group I. Both P insertions are located in the gene encoding the translation initiation factor eIF-1A (Lasko 2000; Pena-Rangel et al. 2002), suggesting that group I is allelic with eIF-1A.
Group J (comprising three members: J709, J2604, and J2633) mapped between the markers scarlet and curled by meiotic recombination. Group J members failed to complement Df(3L)rdgC-co2 (77A01;77D01) but complemented the overlapping deficiencies Df(3L)XS533 (76B04;77B) and Df(3L)ri-79c (77B-C;77F-78A). This placed group J in the interval between 77B and 77C. Complementation crosses with lethal P insertions in the interval identified one insertion that failed to complement group J. This P element, EP(3)3121, is inserted in the gene encoding the elongation release factor eRF1(Abdelilah-Seyfried et al. 2001), suggesting that the group J alleles are mutations in this gene. Indeed, both a second P insertion in the eRF1 locus (eRF1neo28) and an EMS allele (eRF1U3) failed to complement the group J mutations.
The Iroquois complex gene mirror is mutated in group F:
Two lethal enhancer mutations (F1486 and F1825) compose group F. These mutations mapped between the recessive markers roughoid (61F) and thread (72C) by meiotic recombination. Testing for complementation with deficiencies in the region identified three overlapping deficiencies that failed to complement both group F mutations: Df(3L)eygC1 (69A4-5:69D4-6), Df(3L)BSC10 (69D4-5; 69F5-7), and Df(3L)iro-2 (69B1-5;69D1-6). This suggests that the gene mutated in group F is in an interval between 69D4 and 69D6. We tested for complementation with mutations of known genes in the region and found that both alleles of group F failed to complement mutations in the mirror gene (mirrCre2 and mirrSaiD1) (McNeill et al. 1997; Kehl et al. 1998).
The group F mutations also failed to complement Ptp69D1. However, this allele of Protein tyrosine phosphatase 69D was generated by the local transposition of the mirrCre2 P insertion into the Ptp69D locus, followed imprecise excision (Desai et al. 1996). mirrCre2 fails to complement the lethality of Ptp69D1, whereas it is complemented by mirrSaiD1, suggesting that a lethal mutation in mirr is retained in the Ptp69D1 stock. It is therefore most likely that the group F mutations fail to complement the Ptp69D1 stock because of the second lethal mutation in mirr and not the mutation in Ptp69D and thus the group F mutations are new alleles of mirr.
Group D are mutations in the gene encoding the kinesin-like protein Pavarotti:
The three members of group D compose a single lethal complementation group, but, interestingly, D831 and D2046 were identified as enhancers whereas D963 was identified as a suppressor. Group D mapped between roughoid and hairy by meiotic recombination and deficiencies in this area were tested for complementation. Df(3L)GN24 (63F06-07;64C13-15) and Df(3L)GN50 (63E01-02;64B17) failed to complement the group D mutations, whereas the overlapping deficiencies Df(3L)ZN47 (64C; 65C) and Df(3L)GN19 (63F04-07;64B09-11) complemented the group D mutations. This defined an interval of ∼895 kb between the genes ImpL2 [uncovered by Df(3L)GN19] (Garbe et al. 1993) and Srp54K [uncovered by Df(3L)ZN47] (FlyBase 2003) that contains group D.
To further refine the position of the group D mutations, a mapping technique of measuring the recombination distance between the mutations and the molecularly defined P-element insertions was used (Zhai et al. 2003). Of the 20,412 recombination events scored, only 3 occurred between group D and the EP(3)1135 insertion. This places group D at a distance of 0.015 cM from this P insertion (see Figure 5A for details). Lethal mutations and insertions in nearby genes were tested for complementation. All three group D members failed to complement a mutation in pavarotti (pavB200), suggesting that the group D mutants are alleles of the pav gene. To confirm this, we tested whether the lethality of the group D mutations could be rescued by the pav rescue construct RC1 (Adams et al. 1998). A single copy of the RC1 transgene rescued to viability D831 and D2046 when trans-heterozygous with pavB200. D963 was also rescued by RC1, when homozygous or in trans with the other group D alleles. However, the RC1 transgene did not rescue the lethality of either D831 or D2046 when homozygous or the D831/D2046 trans-heterozygous combination. pavB200 enhances the C765-SmoDN phenotype but not as strongly as D831 or D2046 (compare Figure 5E with B and C). This suggests that pavB200 may be hypomorphic and that D831 or D2046 are stronger, possibly null alleles. This could explain the failure of the RC1 construct to rescue the homozygous lethality of D831 or D2046.
Figure 5.—

Group D mutations are alleles of pavarotti. (A) The cytological positions, and the distance in centimorgans from group D, of the insertions used for P-element-mediated meiotic recombination mapping are indicated on the top line. The region around EP(3)1135, which is only 0.015 cM from group D, is shown in the bottom line. pav is ∼10 kb from the EP(3)1135 insertion site. Mutations in pav modify the C5 phenotype. Two alleles of pav identified in the screen, pav831 and pav2046, enhance the C5 phenotype (B and D, respectively) whereas the third allele, pav963, suppresses the C5 phenotype (C). The previously described pavB200 allele also enhances the C5 phenotype (E).
Group C disrupts a novel gene required for Hh movement:
With four members (C447, C477, C789, and C2075), group C is the largest complementation group identified in the screen. It has been mapped by meiotic recombination between FRT80B and scarlet (73A) on chromosome 3L. All of the deficiencies in the third chromosome deficiency kit (as provided by the Bloomington Stock Center) that map to this region complement group C. The following DrosDel (Ryder et al. 2004) deletions—Df(3L)ED4710 (74D1;75B11), Df(3L)ED224 (75B2;75C6), Df(3L)ED225 (75C1;77E6), Df(3L)ED4782 (75F2;76A1), Df(3L)ED4799 (76A1;76B3) Df(3L)ED229 (76A1;76E1), Df(3L)ED4858 (76D3;77C1), Df(3L)ED4861 (76F1;77E6), Df(3L)ED4978 (78D5;79A2), and Df(3L)ED230 (79C2;80A4)—also complemented the group C mutation.
In wing imaginal discs, Hh is expressed in the posterior compartment and the expression of Ci, an essential downstream effector, is restricted to the anterior compartment. Hh protein produced by posterior compartment cells moves into the anterior compartment, creating an activity gradient that extends into the anterior compartment from the A/P compartment boundary. In the absence of Hh, the full-length Ci protein (Ci-155) is degraded into a smaller, repressor form (Ci-75). The activation of Hh signaling inhibits this degradation and induces the stabilization and accumulation of Ci-155 in anterior cells along the A/P compartment boundary (Brook 2000). The upregulation Ci-155 can be detected with the rat monoclonal anti-2A1 (see Figure 6, A, C, D, and F) whose epitope is cleaved from the Ci-75 protein (Motzny and Holmgren 1995).
Figure 6.—
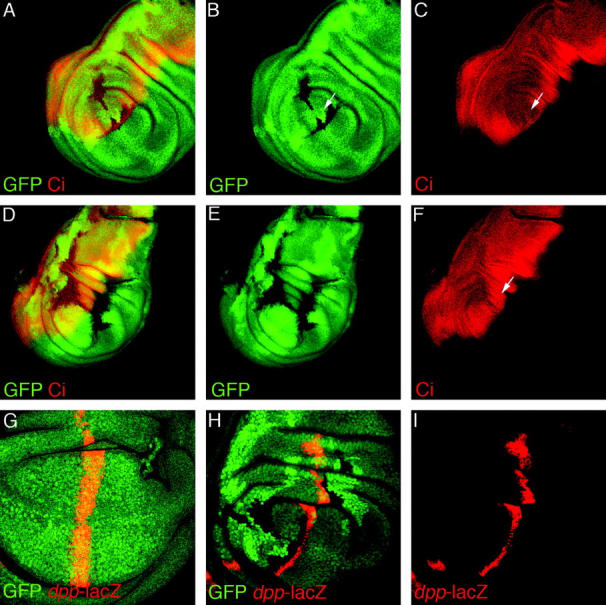
Group C is required for Hedgehog signaling. Confocal images of third instar wing imaginal discs containing clones of cells mutant for group C (loss of green GFP expression in A, B, D, E, and H) and stained for full-length Ci protein (red in A, C, D, and F) or anti-β-Gal (red in G–I) to show expression of dpp-LacZ. In wild-type tissue, Ci-155 is stabilized by Hh signaling, and this results in a broad band of more heavily stained cells on the posterior side of the A/P boundary. Clones of cells mutant for group C fail to upregulate Ci-155 (A and C) except, notably, those cells that are immediately adjacent to the Hh source. Also, wild-type cells on the opposite side of the clone from the source of Hh nonautonomously fail to upregulate Ci-155 (arrow in C). Ci-155 is upregulated normally in anterior cells (arrow in F) when these cells are adjacent to a large, posterior clone at the A/P boundary (loss of green GFP expression), suggesting that group C in not required in Hh-producing cells. In wild-type wing imaginal discs, dpp-lacZ is expressed in a stripe along the posterior side of the A/P boundary (red in G). In clones of cells for group C (loss of green GFP expression in H), dpp-lacZ expression (red in H and I) was lost, except in a single row of cells immediately adjacent to the A/P boundary and the Hh source.
Clones of cells in the anterior compartment that are mutant for group C (absence of green GFP clonal marker in Figure 6, A, B, D, and E) fail to upregulate full-length Ci (red in Figure 6, A, C, D, and F), suggesting that the group C gene product is essential for Hh signaling. Interestingly, Ci is normally upregulated in anterior cells opposite large clones of cells in the posterior compartment that abuts the A/P boundary (Figure 6, D–F). Therefore, group C is dispensable in Hh-producing cells for proper Hh signaling in wing discs.
Although most cells in anterior compartment clones fail to upregulate Ci-155, mutant cells adjacent to the clone border closest to the source of Hh do accumulate full-length Ci (Figure 6, A–C). This nonautonomous effect can be seen more easily by examining the expression of dpp, a transcriptional target of Hh signaling, using a dpp-lacZ transgene. In wild-type discs dpp-lacZ is expressed in a stripe ∼10 cells wide along the anterior side of the A/P compartment boundary (red in Figure 6G). This expression of dpp-lacZ is lost in clones of cells mutant for group C, except for a single row of mutant cells along the border of the clone closest to the Hh source (Figure 6, H and I). Furthermore, wild-type cells on the opposite side, or “downstream,” of the Hh source also fail to upregulate Ci-155 (arrow in Figure 6, B and C). Thus, cells that are mutant for group C are competent to respond to, and transduce, the Hh signal, but do not permit the movement of Hh protein. Therefore, the Hh response is absent from cells in the interior of the clones and in wild-type cells downstream of clones because these cells never see the Hh signal.
The phenotypes described above are nearly identical to those caused by clones of cells mutant for the tout-velu genes [tout-velu (ttv), brother of tout-velu (botv), and sister of tout-velu (sotv); Han et al. 2004; Takei et al. 2004]. We have therefore named the group C gene jaft, for just another tout-velu. ttv genes encode acetylglucosaminyltransferases and are required for HSPG biosynthesis. HSPGs are thought to be required for the movement of Hh, as well as the movement of other signaling molecules such as Wg and Dpp (Han et al. 2004; Takei et al. 2004).
Clones of cells in wing discs that are mutant for ttv, botv, or sotv severely disrupt Dpp signaling and have more subtle effects on Wg signaling. We tested whether clones of jaft would induce similar disruptions to these pathways. Dpp is expressed in a stripe of cells along the anterior side of the A/P boundary. The secreted Dpp protein moves away from these producing cells to form a gradient in both the anterior and posterior compartments (Lecuit et al. 1996; Nellen et al. 1996; Entchev et al. 2000; Teleman and Cohen 2000). The activity of the Dpp pathway can be detected using a phospho-specific antibody against the Mothers against dpp protein (Mad), a downstream component that becomes phosphorylated in response to Dpp signaling (Persson et al. 1998). Phosphorylated Mad (P-Mad) is undetectable in jaft mutant clones (arrows in Figure 7, B and C) except in a single row of cells closest to the source of Dpp (red cells indicated with arrows in Figure 7A).
Figure 7.—
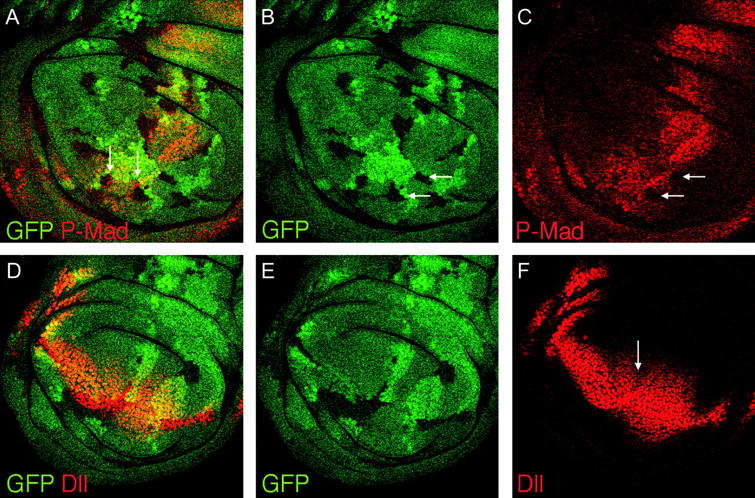
Wg and Dpp signaling are disrupted in jaft mutants. Dpp is expressed along the A/P compartment boundary and signals in a graded manner in both anterior and posterior compartments. Pathway activation can be detected with antibodies against the phosphorylated form of the downstream component Mad (P-Mad, red in A and C). In clones of cells mutant for jaft (absence of green GFP expression in A and B), there is no detectable phosphorylated Mad (arrows in B and C). Wg secreted from the D/V margin activated the graded expression of the Dll target gene in both dorsal and ventral compartments (red in D and F). In clones of cells mutant for jaft (absence of green GFP expression in D and E), only those cells closest to the Wg source express Dll, and in jaft mutant clones, Dll expression ends in a sharp border rather than the graded expression in wild-type tissue.
Wg is expressed along the dorsal/ventral (D/V) boundary and forms a concentration gradient in both dorsal and ventral compartments where it activates the expression of target genes such as Distal-less (Dll; Zecca et al. 1996; Neumann and Cohen 1997; Strigini and Cohen 2000). Clones of cells mutant for jaft cause a reduction in the expression of Dll, especially in wild-type cells downstream from the clone (arrow in Figure 7F). Large clones of mutant cells reduce the range of Dll expression and often have a more sharply defined border between expressing and nonexpressing cells than does wild-type tissue (compare the broad, graded expressing of Dll in the posterior/ventral region with the narrower, defined expression in the clone in the anterior/ventral region in Figure 7, D–F). The disruption of signaling caused by jaft mutant clones is nearly identical to those reported for clones of cells mutant for ttv, botv, and sotv, suggesting that jaft may likewise encode a protein required for HSPG biosynthesis.
DISCUSSION
We set out to identify novel components of the Hh-signaling pathway by screening newly induced mutations for the ability to enhance or suppress a partial Hh loss-of-function phenotype in the Drosophila wing. In addition to new alleles of known components of the Hh pathway, ptc1232 and smo848, 105 novel interacting mutations were isolated. Of these mutations, 36 can be grouped into 14 lethal complementation groups.
Although numerous screens have been conducted to identify Hh-signaling components, we isolated mutations in a large number of genes not identified in these previous screens. Interestingly, it was not possible to recover clones of mutant cells in either adult or larval imaginal discs for at least eight of our complementation groups, suggesting that these mutations are cell lethal. The pleiotropic effects of these mutations would likely prevent the isolation of these genes in genetic screens, except in an enhancer/suppressor screen such as the one presented here. Therefore, the genes identified in this screen may be important, novel mediators of Hh signaling. However, the cell lethality of many of the mutations makes it difficult to confirm or further examine the role of the encoded proteins in Hh signaling.
Two of the cell-lethal complementation groups are mutations in genes encoding proteins essential for translation. The group I mutations, eIF1A645 and eIF1A2232, are allelic with the gene encoding the eukaryotic initiation factor 1A (eIF1A), which is required for the stable association of the 40S complex with the 5′ cap (Pestova et al. 2001). The three group J mutations, eRF1709, eRF12604, and eRF12633, are new alleles of the eukaryotic release factor 1 (eRF1) gene. eRF1 is necessary for recognition of the stop codon and for the termination of protein synthesis (Kisselev and Buckingham 2000).
It is possible that reducing the dosage of eIF1A or eRF1 diminishes the capacity of a cell to translate proteins. Because Hh signaling requires new translation of Smo protein (Alcedo et al. 2000; Denef et al. 2000) and the downstream effects of Hh likely require the synthesis of new proteins, a generalized reduction in protein translation could cause an enhancement of the C765-SmoDN phenotype. However, the Drosophila Minute (M) mutations are generally thought to be mutations in genes encoding ribosomal proteins, and flies heterozygous for M mutations have reduced protein synthesis. These flies present stereotypical, haplo-insufficient phenotypes, including delayed development, small body size, and small, thin bristles (Lambertsson 1998). None of the 107 mutants isolated in this screen display these phenotypes. Thus, M mutations were apparently not selected as enhancers of the C765-SmoDN phenotype, whereas two alleles of eIF1A and three alleles of eRF1 were isolated. This suggests that there is a specific interaction between these genes and Hh signaling and raises the possibility that Hh signaling may directly promote protein translation.
Although Hh signaling is best known for its role in pattern formation, it has also been shown to promote growth during normal development (Marti and Bovolenta 2002; Ruiz i Altaba et al. 2002) and in tumorigenesis (Berman et al. 2003; Thayer et al. 2003; Watkins et al. 2003). In wing imaginal discs, Hh signaling is necessary for the growth of the central, vein III–IV intervein region (Mullor et al. 1997; Strigini and Cohen 1997). It has been reported that this growth is mediated by the helix-loop-helix transcription factor encoded by the Hh target gene knot (Crozatier et al. 2002), but Hh may also promote growth by increasing translation of new proteins through the upregulation eIF1A and eRF1. eIF1A has been shown to be upregulated in response to hyperactivation of JAK/STAT signaling (Myrick and Dearolf 2000); however, it is not known if increasing levels of eIF1A promote protein translation. The potential regulation of eIF1A and eRF1 expression by Hh signaling and its possible role in tissue growth await further experimentation.
Two new alleles of mirror, mirr1486, and mirr1825, were identified as enhancers of the C765-SmoDN phenotype. mirr is a member of the Iroquois complex and encodes a homeodomain transcription factor (McNeill et al. 1997; Kehl et al. 1998). The other members of the Iroquois complex, araucan and caupolican, are transcriptional targets of Hh signaling in the wing imaginal disc and are necessary for the patterning of the veins and sensory organs (Gomez-Skarmeta and Modolell 1996). The isolation of mirr mutations suggests that mirr may also be a target of Hh signaling. However, in third instar discs, expression of mirr was not detected in the central portion of the wing pouch (Kehl et al. 1998), the region of the wing disc where the genetic interaction with Hh was observed. It is possible that mirr is expressed there, but below detectable levels. Alternatively, mirr may be expressed in this central region of the wing pouch during earlier stages of development and disruption of this earlier expression may result in the enhancement of the C765-SmoDN phenotype.
The mutations in group D, pav831, pav963, and pav2046, are new alleles of pav, a gene encoding a kinesin-like protein. Pav has been shown to be required for the organization of central spindle and the contractile ring, and cytokinesis fails in cells lacking Pav (Adams et al. 1998). Although the role of pav in cytokenesis has been studied in some detail no function for Pav in Hh signaling has yet been demonstrated, and pav was not identified in previous screens for Hh pathway components. However, cells lacking Pav are unable to complete cytokinesis and the resulting multi-nucleated cells are cleared by apoptosis. This nearly cell-lethal phenotype would likely mask any role of Pav in Hh signaling and prevent its isolation in genetic screens other than a modifier screen such as this.
In response to Hh signaling, Ptc moves from the cell surface to intracellular vesicles, and Smo moves from intracellular vesicles in the cytoplasm to the plasma membrane. This movement of Smo to the cell surface is correlated with signal transduction, and the movement of both Smo and Ptc requires the actin cytoskeleton and the microtubule network (Jia et al. 2003; Lum et al. 2003; Zhu et al. 2003). It is possible that Pav mediates the movement of Smo, Ptc, or both, in response to Hh signaling. Alternatively, Pav may function to move or maintain Ci-155 in the nucleus of Hh-responding cells, as Pav is primarily nuclear localized during interphase. However, expression of a mutant Pav protein that is unable to localize to the nucleus is able to rescue pav mutants to adulthood (Minestrini et al. 2003), suggesting that Pav nuclear localization is not essential for cytokinesis or Hh signaling.
The four members of group C, jaft447, jaft477, jaft789, and jaft2075, are mutations in a novel gene we have named jaft. Clones of cells mutant for jaft in the wing imaginal disc are defective in Hh signaling. The phenotypes caused by clones of mutant cells indicate that, in the absence of jaft, cells can still produce the Hh signal and can respond to the signal if they detect the Hh protein. However, the Hh protein is unable to move through the jaft mutant tissue, causing a loss of Hh signaling in the interior of mutant clones and in wild-type cells “downstream” of the Hh source. The movement of Dpp and Wg proteins is similarly disrupted in these clones, although Wg movement is disrupted to a much lesser extent than either Hh or Dpp.
These phenotypes are identical to those caused by clones of cells mutant for tout-velu, brother of tout-velu, or sister of tout-velu. ttv, botv, and sotv encode proteins with acetylglucosaminyltransferase activity and are required for the biosynthesis of HSPGs (Han et al. 2004; Takei et al. 2004). It is the phenotypes induced by mutations in these genes that have demonstrated the requirement for HSPGs for the movement of Hh and other morphogens. But what HSPGs do that facilitates the movement of signaling molecules is not yet known.
Because mutations of jaft induce phenotypes that are identical to those caused by mutations of the tout-velu-like genes, it is likely that the jaft gene product is also required for HSPG biosynthesis. Mapping of jaft mutations has placed the gene between 73A and 80B on chromosome 3L. None of the genes that reside in this interval encode obvious HSPG biosynthetic proteins. Therefore, jaft may encode a protein that plays a regulatory or modulatory role in HSPG biosynthesis, and the cloning and further characterization of jaft should provide novel insight into the role of HSPGs in morphogen movement.
Acknowledgments
The generation of the UAS-Smo5A transgenic flies and the original observation that the Smo5A protein had dominant-negative activity are the work of Natalie Denef. We thank Natalie and acknowledge her contribution here. We also thank David Glover and Martin Kerr for fly stocks and reagents; Jan Schulte-Holthausen for technical assistance; and David Hipfner, William Norton, and Barry Thompson for thoughtful comments on the manuscript. This work was supported by a National Institutes of Health Individual Postdoctoral Fellowship (GM-64220) to R.T.C. and by the European Molecular Biology Laboratory.
References
- Abdelilah-Seyfried, S., Y. M. Chan, C. Zeng, N. J. Justice, S. Younger-Shepherd et al., 2001. A gain-of-function screen for genes that affect the development of the Drosophila adult external sensory organ. Genetics 157: 455–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams, R. R., A. A. Tavares, A. Salzberg, H. J. Bellen and D. M. Glover, 1998. pavarotti encodes a kinesin-like protein required to organize the central spindle and contractile ring for cytokinesis. Genes Dev. 12: 1483–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcedo, J., M. Ayzenzon, T. Von Ohlen, M. Noll and J. E. Hooper, 1996. The Drosophila smoothened gene encodes a seven-pass membrane protein, a putative receptor for the hedgehog signal. Cell 86: 221–232. [DOI] [PubMed] [Google Scholar]
- Alcedo, J., Y. Zou and M. Noll, 2000. Posttranscriptional regulation of smoothened is part of a self-correcting mechanism in the Hedgehog signaling system. Mol. Cell 6: 457–465. [DOI] [PubMed] [Google Scholar]
- Aza-Blanc, P., F. A. Ramirez-Weber, M. P. Laget, C. Schwartz and T. B. Kornberg, 1997. Proteolysis that is inhibited by hedgehog targets Cubitus interruptus protein to the nucleus and converts it to a repressor. Cell 89: 1043–1053. [DOI] [PubMed] [Google Scholar]
- Berman, D. M., S. S. Karhadkar, A. Maitra, R. Montes De Oca, M. R. Gerstenblith et al., 2003. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature 425: 846–851. [DOI] [PubMed] [Google Scholar]
- Brizuela, B. J., L. Elfring, J. Ballard, J. W. Tamkun and J. A. Kennison, 1994. Genetic analysis of the brahma gene of Drosophila melanogaster and polytene chromosome subdivisions 72AB. Genetics 137: 803–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brook, W. J., 2000. Hedgehog signaling and the axial patterning of Drosophila wings. Biochem. Cell Biol. 78: 585–591. [DOI] [PubMed] [Google Scholar]
- Bumcrot, D. A., R. Takada and A. P. McMahon, 1995. Proteolytic processing yields two secreted forms of sonic hedgehog. Mol. Cell. Biol. 15: 2294–2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke, R., D. Nellen, M. Bellotto, E. Hafen, K. A. Senti et al., 1999. Dispatched, a novel sterol-sensing domain protein dedicated to the release of cholesterol-modified hedgehog from signaling cells. Cell 99: 803–815. [DOI] [PubMed] [Google Scholar]
- Capdevila, J., M. P. Estrada, E. Sanchez-Herrero and I. Guerrero, 1994. The Drosophila segment polarity gene patched interacts with decapentaplegic in wing development. EMBO J. 13: 71–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamoun, Z., R. K. Mann, D. Nellen, D. P. von Kessler, M. Bellotto et al., 2001. Skinny hedgehog, an acyltransferase required for palmitoylation and activity of the hedgehog signal. Science 293: 2080–2084. [DOI] [PubMed] [Google Scholar]
- Chen, B., T. Chu, E. Harms, J. P. Gergen and S. Strickland, 1998. Mapping of Drosophila mutations using site-specific male recombination. Genetics 149: 157–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Y., and G. Struhl, 1996. Dual roles for patched in sequestering and transducing Hedgehog. Cell 87: 553–563. [DOI] [PubMed] [Google Scholar]
- Chen, Y., N. Gallaher, R. H. Goodman and S. M. Smolik, 1998. Protein kinase A directly regulates the activity and proteolysis of cubitus interruptus. Proc. Natl. Acad. Sci. USA 95: 2349–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crozatier, M., B. Glise and A. Vincent, 2002. Connecting Hh, Dpp and EGF signalling in patterning of the Drosophila wing: the pivotal role of collier/knot in the AP organiser. Development 129: 4261–4269. [DOI] [PubMed] [Google Scholar]
- Denef, N., D. Neubuser, L. Perez and S. M. Cohen, 2000. Hedgehog induces opposite changes in turnover and subcellular localization of patched and smoothened. Cell 102: 521–531. [DOI] [PubMed] [Google Scholar]
- Desai, C. J., J. G. Gindhart, Jr., L. S. Goldstein and K. Zinn, 1996. Receptor tyrosine phosphatases are required for motor axon guidance in the Drosophila embryo. Cell 84: 599–609. [DOI] [PubMed] [Google Scholar]
- Entchev, E. V., A. Schwabedissen and M. Gonzalez-Gaitan, 2000. Gradient formation of the TGF-beta homolog Dpp. Cell 103: 981–991. [DOI] [PubMed] [Google Scholar]
- FlyBase, 2003. The FlyBase database of the Drosophila genome projects and community literature. Nucleic Acids Res. 31: 172–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbe, J. C., E. Yang and J. W. Fristrom, 1993. IMP-L2: an essential secreted immunoglobulin family member implicated in neural and ectodermal development in Drosophila. Development 119: 1237–1250. [DOI] [PubMed] [Google Scholar]
- Gomez-Skarmeta, J. L., and J. Modolell, 1996. araucan and caupolican provide a link between compartment subdivisions and patterning of sensory organs and veins in the Drosophila wing. Genes Dev. 10: 2935–2945. [DOI] [PubMed] [Google Scholar]
- Han, C., T. Y. Belenkaya, M. Khodoun, M. Tauchi and X. Lin, 2004. Distinct and collaborative roles of Drosophila EXT family proteins in morphogen signalling and gradient formation. Development 131: 1563–1575. [DOI] [PubMed] [Google Scholar]
- Harrison, D. A., and N. Perrimon, 1993. Simple and efficient generation of marked clones in Drosophila. Curr. Biol. 3: 424–433. [DOI] [PubMed] [Google Scholar]
- Hooper, J. E., 1994. Distinct pathways for autocrine and paracrine Wingless signalling in Drosophila embryos. Nature 372: 461–464. [DOI] [PubMed] [Google Scholar]
- Ingham, P. W., and A. P. McMahon, 2001. Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 15: 3059–3087. [DOI] [PubMed] [Google Scholar]
- Jia, J., K. Amanai, G. Wang, J. Tang, B. Wang et al., 2002. Shaggy/GSK3 antagonizes Hedgehog signalling by regulating Cubitus interruptus. Nature 416: 548–552. [DOI] [PubMed] [Google Scholar]
- Jia, J., C. Tong and J. Jiang, 2003. Smoothened transduces Hedgehog signal by physically interacting with Costal2/Fused complex through its C-terminal tail. Genes Dev. 17: 2709–2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, J., and G. Struhl, 1998. Regulation of the Hedgehog and Wingless signalling pathways by the F-box/WD40-repeat protein Slimb. Nature 391: 493–496. [DOI] [PubMed] [Google Scholar]
- Kehl, B. T., K. O. Cho and K. W. Choi, 1998. mirror, a Drosophila homeobox gene in the Iroquois complex, is required for sensory organ and alula formation. Development 125: 1217–1227. [DOI] [PubMed] [Google Scholar]
- Kisselev, L. L., and R. H. Buckingham, 2000. Translational termination comes of age. Trends Biochem. Sci. 25: 561–566. [DOI] [PubMed] [Google Scholar]
- Lambertsson, A., 1998. The minute genes in Drosophila and their molecular functions. Adv. Genet. 38: 69–134. [DOI] [PubMed] [Google Scholar]
- Lasko, P., 2000. The Drosophila melanogaster genome: translation factors and RNA binding proteins. J. Cell Biol. 150: F51–F56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecuit, T., W. J. Brook, M. Ng, M. Calleja, H. Sun et al., 1996. Two distinct mechanisms for long-range patterning by Decapentaplegic in the Drosophila wing. Nature 381: 387–393. [DOI] [PubMed] [Google Scholar]
- Lee, J. D., and J. E. Treisman, 2001. Sightless has homology to transmembrane acyltransferases and is required to generate active Hedgehog protein. Curr. Biol. 11: 1147–1152. [DOI] [PubMed] [Google Scholar]
- Lee, J. J., S. C. Ekker, D. P. von Kessler, J. A. Porter, B. I. Sun et al., 1994. Autoproteolysis in hedgehog protein biogenesis. Science 266: 1528–1537. [DOI] [PubMed] [Google Scholar]
- Lum, L., C. Zhang, S. Oh, R. K. Mann, D. P. von Kessler et al., 2003. Hedgehog signal transduction via Smoothened association with a cytoplasmic complex scaffolded by the atypical kinesin, Costal-2. Mol. Cell 12: 1261–1274. [DOI] [PubMed] [Google Scholar]
- Marti, E., and P. Bovolenta, 2002. Sonic hedgehog in CNS development: one signal, multiple outputs. Trends Neurosci. 25: 89–96. [DOI] [PubMed] [Google Scholar]
- McNeill, H., C. H. Yang, M. Brodsky, J. Ungos and M. A. Simon, 1997. mirror encodes a novel PBX-class homeoprotein that functions in the definition of the dorsal-ventral border in the Drosophila eye. Genes Dev. 11: 1073–1082. [DOI] [PubMed] [Google Scholar]
- Methot, N., and K. Basler, 1999. Hedgehog controls limb development by regulating the activities of distinct transcriptional activator and repressor forms of Cubitus interruptus. Cell 96: 819–831. [DOI] [PubMed] [Google Scholar]
- Methot, N., and K. Basler, 2000. Suppressor of fused opposes hedgehog signal transduction by impeding nuclear accumulation of the activator form of Cubitus interruptus. Development 127: 4001–4010. [DOI] [PubMed] [Google Scholar]
- Micchelli, C. A., I. The, E. Selva, V. Mogila and N. Perrimon, 2002. Rasp, a putative transmembrane acyltransferase, is required for Hedgehog signaling. Development 129: 843–851. [DOI] [PubMed] [Google Scholar]
- Minestrini, G., A. S. Harley and D. M. Glover, 2003. Localization of Pavarotti-KLP in living Drosophila embryos suggests roles in reorganizing the cortical cytoskeleton during the mitotic cycle. Mol. Biol. Cell 14: 4028–4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motzny, C. K., and R. Holmgren, 1995. The Drosophila cubitus interruptus protein and its role in the wingless and hedgehog signal transduction pathways. Mech. Dev. 52: 137–150. [DOI] [PubMed] [Google Scholar]
- Mullor, J. L., M. Calleja, J. Capdevila and I. Guerrero, 1997. Hedgehog activity, independent of decapentaplegic, participates in wing disc patterning. Development 124: 1227–1237. [DOI] [PubMed] [Google Scholar]
- Myrick, K. V., and C. R. Dearolf, 2000. Hyperactivation of the Drosophila Hop jak kinase causes the preferential overexpression of eIF1A transcripts in larval blood cells. Gene 244: 119–125. [DOI] [PubMed] [Google Scholar]
- Nellen, D., R. Burke, G. Struhl and K. Basler, 1996. Direct and long-range action of a DPP morphogen gradient. Cell 85: 357–368. [DOI] [PubMed] [Google Scholar]
- Neumann, C. J., and S. M. Cohen, 1997. Long-range action of Wingless organizes the dorsal-ventral axis of the Drosophila wing. Development 124: 871–880. [DOI] [PubMed] [Google Scholar]
- Ogden, S. K., M. Ascano, Jr., M. A. Stegman, L. M. Suber, J. E. Hooper et al., 2003. Identification of a functional interaction between the transmembrane protein Smoothened and the kinesin-related protein Costal2. Curr. Biol. 13: 1998–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou, C. Y., Y. F. Lin, Y. J. Chen and C. T. Chien, 2002. Distinct protein degradation mechanisms mediated by Cul1 and Cul3 controlling Ci stability in Drosophila eye development. Genes Dev. 16: 2403–2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pena-Rangel, M. T., I. Rodriguez and J. R. Riesgo-Escovar, 2002. A misexpression study examining dorsal thorax formation in Drosophila melanogaster. Genetics 160: 1035–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson, U., H. Izumi, S. Souchelnytskyi, S. Itoh, S. Grimsby et al., 1998. The L45 loop in type I receptors for TGF-beta family members is a critical determinant in specifying Smad isoform activation. FEBS Lett. 434: 83–87. [DOI] [PubMed] [Google Scholar]
- Pestova, T. V., V. G. Kolupaeva, I. B. Lomakin, E. V. Pilipenko, I. N. Shatsky et al., 2001. Molecular mechanisms of translation initiation in eukaryotes. Proc. Natl. Acad. Sci. USA 98: 7029–7036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter, J. A., D. P. von Kessler, S. C. Ekker, K. E. Young, J. J. Lee et al., 1995. The product of hedgehog autoproteolytic cleavage active in local and long-range signalling. Nature 374: 363–366. [DOI] [PubMed] [Google Scholar]
- Price, M. A., and D. Kalderon, 1999. Proteolysis of cubitus interruptus in Drosophila requires phosphorylation by protein kinase A. Development 126: 4331–4339. [DOI] [PubMed] [Google Scholar]
- Price, M. A., and D. Kalderon, 2002. Proteolysis of the Hedgehog signaling effector Cubitus interruptus requires phosphorylation by glycogen synthase kinase 3 and casein kinase 1. Cell 108: 823–835. [DOI] [PubMed] [Google Scholar]
- Quirk, J., M. van den Heuvel, D. Henrique, V. Marigo, T. A. Jones et al., 1997. The smoothened gene and hedgehog signal transduction in Drosophila and vertebrate development. Cold Spring Harbor Symp. Quant. Biol. 62: 217–226. [PubMed] [Google Scholar]
- Robbins, D. J., K. E. Nybakken, R. Kobayashi, J. C. Sisson, J. M. Bishop et al., 1997. Hedgehog elicits signal transduction by means of a large complex containing the kinesin-related protein costal2. Cell 90: 225–234. [DOI] [PubMed] [Google Scholar]
- Ruel, L., R. Rodriguez, A. Gallet, L. Lavenant-Staccini and P. P. Therond, 2003. Stability and association of Smoothened, Costal2 and Fused with Cubitus interruptus are regulated by Hedgehog. Nat. Cell Biol. 5: 907–913. [DOI] [PubMed] [Google Scholar]
- Ruiz i Altaba, A., P. Sanchez and N. Dahmane, 2002. Gli and hedgehog in cancer: tumours, embryos and stem cells. Nat. Rev. Cancer 2: 361–372. [DOI] [PubMed] [Google Scholar]
- Ryder, E., F. Blows, M. Ashburner, R. Bautista-Llacer, D. Coulson et al., 2004. The DrosDel collection: a set of P-element insertions for generating custom chromosomal aberrations in Drosophila melanogaster. Genetics 167: 797–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sisson, J. C., K. S. Ho, K. Suyama and M. P. Scott, 1997. Costal2, a novel kinesin-related protein in the Hedgehog signaling pathway. Cell 90: 235–245. [DOI] [PubMed] [Google Scholar]
- Strigini, M., and S. M. Cohen, 1997. A Hedgehog activity gradient contributes to AP axial patterning of the Drosophila wing. Development 124: 4697–4705. [DOI] [PubMed] [Google Scholar]
- Strigini, M., and S. M. Cohen, 2000. Wingless gradient formation in the Drosophila wing. Curr. Biol. 10: 293–300. [DOI] [PubMed] [Google Scholar]
- Tabata, T., and Y. Takei, 2004. Morphogens, their identification and regulation. Development 131: 703–712. [DOI] [PubMed] [Google Scholar]
- Taipale, J., M. K. Cooper, T. Maiti and P. A. Beachy, 2002. Patched acts catalytically to suppress the activity of Smoothened. Nature 418: 892–897. [DOI] [PubMed] [Google Scholar]
- Takei, Y., Y. Ozawa, M. Sato, A. Watanabe and T. Tabata, 2004. Three Drosophila EXT genes shape morphogen gradients through synthesis of heparan sulfate proteoglycans. Development 131: 73–82. [DOI] [PubMed] [Google Scholar]
- Teleman, A. A., and S. M. Cohen, 2000. Dpp gradient formation in the Drosophila wing imaginal disc. Cell 103: 971–980. [DOI] [PubMed] [Google Scholar]
- Thayer, S. P., M. P. di Magliano, P. W. Heiser, C. M. Nielsen, D. J. Roberts et al., 2003. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature 425: 851–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The, I., Y. Bellaiche and N. Perrimon, 1999. Hedgehog movement is regulated through tout velu-dependent synthesis of a heparan sulfate proteoglycan. Mol. Cell 4: 633–639. [DOI] [PubMed] [Google Scholar]
- Theodosiou, N. A., S. Zhang, W. Y. Wang and T. Xu, 1998. slimb coordinates wg and dpp expression in the dorsal-ventral and anterior-posterior axes during limb development. Development 125: 3411–3416. [DOI] [PubMed] [Google Scholar]
- van den Heuvel, M., and P. W. Ingham, 1996. smoothened encodes a receptor-like serpentine protein required for hedgehog signalling. Nature 382: 547–551. [DOI] [PubMed] [Google Scholar]
- Wang, G., K. Amanai, B. Wang and J. Jiang, 2000. Interactions with Costal2 and suppressor of fused regulate nuclear translocation and activity of cubitus interruptus. Genes Dev. 14: 2893–2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins, D. N., D. M. Berman, S. G. Burkholder, B. Wang, P. A. Beachy et al., 2003. Hedgehog signalling within airway epithelial progenitors and in small-cell lung cancer. Nature 422: 313–317. [DOI] [PubMed] [Google Scholar]
- Wu, J., and S. M. Cohen, 1999. Proximodistal axis formation in the Drosophila leg: subdivision into proximal and distal domains by Homothorax and Distal-less. Development 126: 109–117. [DOI] [PubMed] [Google Scholar]
- Zecca, M., K. Basler and G. Struhl, 1996. Direct and long-range action of a wingless morphogen gradient. Cell 87: 833–844. [DOI] [PubMed] [Google Scholar]
- Zhai, R. G., P. R. Hiesinger, T. W. Koh, P. Verstreken, K. L. Schulze et al., 2003. Mapping Drosophila mutations with molecularly defined P element insertions. Proc. Natl. Acad. Sci. USA 100: 10860–10865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, A. J., L. Zheng, K. Suyama and M. P. Scott, 2003. Altered localization of Drosophila Smoothened protein activates Hedgehog signal transduction. Genes Dev. 17: 1240–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]