

Abstract
The Drosophila trithorax group gene brahma (brm) encodes the ATPase subunit of a 2-MDa chromatin-remodeling complex. brm was identified in a screen for transcriptional activators of homeotic genes and subsequently shown to play a global role in transcription by RNA polymerase II. To gain insight into the targeting, function, and regulation of the BRM complex, we screened for mutations that genetically interact with a dominant-negative allele of brm (brmK804R). We first screened for dominant mutations that are lethal in combination with a brmK804R transgene under control of the brm promoter. In a distinct but related screen, we identified dominant mutations that modify eye defects resulting from expression of brmK804R in the eye-antennal imaginal disc. Mutations in three classes of genes were identified in our screens: genes encoding subunits of the BRM complex (brm, moira, and osa), other proteins directly involved in transcription (zerknullt and RpII140), and signaling molecules (Delta and vein). Expression of brmK804R in the adult sense organ precursor lineage causes phenotypes similar to those resulting from impaired Delta-Notch signaling. Our results suggest that signaling pathways may regulate the transcription of target genes by regulating the activity of the BRM complex.
NUCLEOSOMES and other components of chromatin can block the access of transcription factors and other regulatory proteins to DNA. Chromatin is not merely a passive barrier to transcription; eukaryotic cells exploit the repressive effects of chromatin to regulate gene expression. Chromatin repression is regulated via two general mechanisms: the covalent modification of nucleosomal histones and ATP-dependent chromatin remodeling (Narlikar et al. 2002). Histone-modifying enzymes alter the acetylation, methylation, phosphorylation, or ubiquitination of N-terminal histone tails and other regions on the surface of the nucleosome. These modifications modulate interactions between nucleosomes and a wide variety of structural and regulatory proteins (Berger 2002; Peterson and Laniel 2004). By altering the structure or positioning of nucleosomes, chromatin-remodeling complexes can directly regulate the access of transcription factors and other proteins to DNA in the context of chromatin (Becker and Horz 2002; Martens and Winston 2003; Flaus and Owen-Hughes 2004). The coordinated actions of histone-modifying and chromatin-remodeling enzymes are critical for transcription in a chromatin environment.
Histone-modifying enzymes and ATP-dependent chromatin-remodeling complexes have been implicated in a broad range of biological processes, including transcription, DNA repair, recombination, viral integration, and malignant transformation (Martens and Winston 2003). Alterations in chromatin structure underlie many developmental processes, including the maintenance of cell fates and other epigenetic phenomena. In Drosophila and other metazoans, the identities of body segments are specified by transcription factors encoded by homeotic (Hox) genes (Gellon and McGinnis 1998). The initial patterns of Hox transcription are established in response to positional information in the early embryo. Once established, these patterns are maintained throughout development by two groups of regulatory proteins: the Polycomb group (PcG) of repressors and the trithorax group (trxG) of activators (Simon 1995; Gellon and McGinnis 1998; Francis and Kingston 2001). Counterparts of Drosophila PcG and trxG proteins play highly conserved roles in transcription and development in other metazoans, including humans (Gould 1997; Schumacher and Magnuson 1997).
A growing body of evidence suggests that PcG and trxG proteins regulate transcription via the covalent modification or remodeling of chromatin (Simon and Tamkun 2002). Two major complexes of PcG proteins have been identified: Polycomb repressor complex 1 (PRC1) and the enhancer of Zeste/extra sex combs [E(Z)/ESC] complex (Cao and Zhang 2004; Levine et al. 2004). The E(Z)/ESC complex has histone methyltransferase activity that promotes the binding of PRC1 to its target genes and is required for PcG repression in vivo. The trxG proteins Drosophila Absent, small or homeotic 1 (ASH1) and Trithorax (TRX) also have histone methyltransferase activity that is required for their function in vivo (Beisel et al. 2002; Smith et al. 2004). Other trxG proteins appear to regulate transcription via ATP-dependent chromatin remodeling. For example, the trxG genes brahma (brm), moira (mor), osa, and kismet (kis) were identified in genetic screens for Polycomb antagonists and were subsequently found to encode subunits of ATP-dependent chromatin-remodeling complexes (Simon and Tamkun 2002).
Chromatin-remodeling complexes are large (up to 2 MDa), multisubunit protein machines with a catalytic subunit belonging to the SNF2 family of ATPases (Lusser and Kadonaga 2003). The trxG protein BRM is highly related to yeast SWI2/SNF2 and STH1, the ATPase subunits of the SWI/SNF and RSC chromatin-remodeling complexes, respectively. mor encodes a conserved SANT-domain protein related to yeast SWI3 and RSC8, while osa encodes a conserved ARID-domain protein related to SWI1 (Papoulas et al. 1998; Collins et al. 1999; Crosby et al. 1999; Vazquez et al. 1999).
How do SWI/SNF complexes regulate gene expression? In vitro, these complexes use the energy of ATP hydrolysis to influence many aspects of chromatin structure. Examples of in vitro activities associated with SWI/SNF complexes include the distortion of DNA on the nucleosomal surface, nucleosome sliding, H2A/H2B dimer exchange, nucleosome transfer or eviction, nucleosome assembly, and the disruption or creation of regularly spaced nucleosomal arrays (Lusser and Kadonaga 2003; Eberharter and Becker 2004). The ATPase subunits of chromatin-remodeling complexes facilitate these reactions by functioning as ATP-dependent DNA translocases (Saha et al. 2002; Whitehouse et al. 2003). The ability of SWI/SNF complexes to remodel chromatin in vitro is inhibited by PRC1, suggesting a potential mechanism for PcG repression in vivo (Francis et al. 2001).
In spite of the tremendous progress toward understanding the mechanism of action of SWI/SNF complexes in vitro, much remains to be learned about their mechanism of action and biological functions in vivo. In yeast, SWI/SNF has been shown to be involved in both transcriptional activation and repression (Martens and Winston 2003). However, the genes encoding most of the SWI/SNF subunits are not essential and the SWI/SNF complex is required only for the expression of a small percentage of genes (Holstege et al. 1998; Sudarsanam et al. 2000). By contrast, the RSC chromatin-remodeling complex, which contains the STH1 ATPase, is both abundant and essential. RSC is required for transcription of several groups of genes (Angus-Hill et al. 2001) as well as sister chromatin cohesion during mitosis (Huang et al. 2004). Like their yeast and Drosophila counterparts, the human BAF and PBAF complexes regulate transcription by catalyzing ATP-dependent alterations in chromatin structure (Narlikar et al. 2002).
Although early work on the Drosophila BRM complex focused on its roles in Hox regulation, subsequent studies revealed that it plays a surprisingly general role in transcription. The BRM complex is essential for cell viability and extremely abundant; one copy of the BRM complex is present for every 20 nucleosomes in many cell types (Elfring et al. 1998). Furthermore, the BRM complex is associated with virtually all transcriptionally active regions of chromatin in salivary gland nuclei and the loss of brm function leads to a dramatic reduction in RNA polymerase II transcription (Armstrong et al. 2002). These findings raise many questions about the function of SWI/SNF-like complexes in higher eukaryotes. How are these complexes targeted to sites of active transcription? Which step(s) in the transcription cycle are dependent on their activity? Finally, how are the activities of these abundant and extremely stable complexes regulated?
The targeting of chromatin-remodeling complexes to specific chromosomal locations may involve interactions with both gene-specific transcriptional activators and components of the basal transcription machinery. The yeast SWI/SNF complex physically interacts with a variety of transcriptional activators (Peterson and Logie 2000; Peterson and Workman 2000; Neely et al. 2002), as do the human SWI/SNF-like complexes (Kadam et al. 2000; Kadam and Emerson 2003). It is possible that the Drosophila BRM complex is targeted via analogous mechanisms. Indeed, the transcription factor Zeste recruits the BRM complex to chromatin in vitro (Kal et al. 2000) and in vivo (Dejardin and Cavalli 2004). SWI/SNF also interacts with the general transcriptional machinery (Sharma et al. 2003; Yoon et al. 2003), and mutations in genes encoding components of RNA polymerase II impair the recruitment of SWI/SNF to the GAL1 promoter (Lemieux and Gaudreau 2004).
Recent studies suggest that signal transduction pathways may also be important for the regulation and targeting of chromatin-remodeling complexes. The EBV latency C promoter binding factor (CBF-1) and the intracellular domain (ICD) of Notch both physically interact with human BRM and are perhaps responsible for the targeting of BRM to the promoters of Notch target genes (Kadam and Emerson 2003). Human SWI/SNF complexes are targeted to muscle-specific genes by the MAP kinase p38 (Simone et al. 2004). Interactions between chromatin-remodeling factors and signal transduction pathways are not limited to SWI/SNF complexes. Drosophila NURF, an ISWI-containing complex, is a negative regulator of the JAK/STAT signal transduction pathway (Badenhorst et al. 2002). Additional signal transduction pathways may also participate in the regulation of chromatin-remodeling factors since the activities of several chromatin-remodeling complexes, including Drosophila NURF and yeast SWI/SNF, are regulated by inositol polyphosphate second messengers (Shen et al. 2003; Steger et al. 2003). Several chromatin-remodeling complexes, including the BRM complex, play a general role in transcription; these complexes are therefore logical targets for regulation by signal transduction pathways. Targeting, inactivation, or activation of these complexes could have profound consequences for gene expression.
To gain insight into the function and regulation of the Drosophila BRM complex, we conducted genetic screens for modifiers of brmK804R. This conservative substitution in the ATP-binding site of the BRM protein renders it catalytically inactive without disrupting its ability to interact with other proteins (Elfring et al. 1998). As a result, expression of brmK804R counteracts brm function in vivo. We therefore reasoned that mutations in genes that are important for brm function would strongly enhance phenotypes resulting from brmK804R expression, while mutations in brm antagonists would suppress them. In addition to mutations in subunits of the BRM complex and other proteins involved in transcription, our screens led to the recovery of mutations in genes involved in both the Notch and EGF receptor signal transduction pathways. These findings suggest that signal transduction pathways may regulate the activity of the BRM chromatin-remodeling complex to affect transcription of target genes.
MATERIALS AND METHODS
Drosophila stocks and crosses:
Flies were raised on a cornmeal-molasses-yeast-agar medium containing Tegosept and propionic acid at 25° unless otherwise indicated. The autosomal deficiency kit and many other Drosophila stocks were obtained from the Bloomington Drosophila Stock Center (http://flystocks.bio.indiana.edu). Information concerning many of the preexisting mutations and chromosome aberrations used in this study can be found at FlyBase (www.flybase.org).
Male-specific lethality screens:
To screen for dominant enhancers of brmK804R on the second chromosome, al b cn sp males were fed 20 mm ethylmethane sulfonate (EMS) in 1% sucrose for 12 hr and crossed to either w; al b cn ISWI1 sp/SM5, Cy sp or Df(2R)vg-C/SM5, Cy sp females (Figure 1A). Individual al b cn sp/SM5, Cy sp males were mated to Df(1)w67c2 y, P[w+, brmK804R]22D/Df(1)w67c2 y females and their progeny were scored for the absence of males bearing both the mutagenized chromosome and the P[w+,brmK804R]22D transgene (Df(1)w67c2 y, P[w+,brmK804R]22D/Y; al b cn sp/+). Potential enhancer mutations were recovered by mating Df(1)w67c2 y/Y; al b cn sp/+ males to virgin females heterozygous for the second chromosome balancer. To identify dominant enhancers of brmK804R on the third chromosome, ru h st ry e males were fed EMS as described above and mated to w; CxD/TM3, Sb females (Figure 1B). Individual male progeny of the genotype w/Y; ru h st ry e/CxD or w/Y; ru h st ry e/TM3, Sb were mated to Df(1)w67c2 y, P[w+, brmK804R]22D/Df(1)w67c2 y females. Progeny of these crosses were scored for the absence of males carrying both the mutagenized chromosome and the P[w+,brmK804R]22D transgene (Df(1)w67c2 y, P[w+,brmK804R]22D/Y; ru h st ry e/+). Potential enhancer mutations were recovered by mating Df(1)w67c2 y/Y; ru h st ry e/+ males to virgin females heterozygous for the third chromosome balancer.
Figure 1.—

Outline of F2 screens for mutations in the (A) second and (B) third chromosomes that result in male-specific lethality in combination with the X-linked brmK804R transgene. Asterisks indicate mutagenized chromosomes.
To identify potential dominant suppressors of brmK804R on the second and third chromosomes Df(3L)th102 ri Sb/TM6B, Hu e Tb ca males were fed EMS as described above and mated to Df(1)w67c2 y, P[w+,brmK804R]22D virgins. The progeny of this cross were scored for Df(1)w67c2 y, P[w+,brmK804R]22D/Y; +/+; Df(3L)th102 ri Sb/+ males.
Eye-based modifier screens:
To screen for dominant modifiers of brmK804R on the third chromosome, Df(1)w67c2 y males were fed EMS as described above and mated to w; CxD/TM3, Sb females (Figure 3). Individual female progeny of the genotype Df(1)w67c2 y/w; +/TM3, Sb were mated to w; P[w+,ey-GAL4], P[w+,UAS-brmK804R]/TM3, Sb males. The resulting F2 progeny were screened as described below for mutations that modify the rough eye phenotype resulting from expression of brmK804R. Candidate mutations were recovered by balancing the third chromosome of w/Y; +/TM3, Sb siblings. The P[w+,ey-GAL4], P[w+,UAS-brmK804R] chromosome was generated by recombination between w; P[w+, ey-GAL4] and w; P[w+, UASGALhsp70:brmK804R]2-2 as described (Papoulas et al. 2001).
Figure 3.—

Outline of eye-based screen for dominant modifiers of brmK804R. Asterisks indicate mutagenized chromosomes.
To quantify the severity of eye defects, we assigned individual eyes a score from 1 to 6 as follows: (1) eye is wild type; (2) 50% or less of the eye is rough (as determined by disordered ommatidia under the light microscope); (3) >50% of the eye is rough; (4) the eye is rough and reduced in size by ≤50%; (5) the eye is rough and reduced in size by >50%; and (6) the eye is absent. To assay enhancement or suppression of the brmK804R rough eye phenotype, eye scores for individuals of the genotype mutation/P[w+,ey-GAL4], P[w+,UAS-brmK804R] were compared to eye scores for siblings of the genotype balancer/P[w+,ey-GAL4], P[w+,UAS-brmK804R]. A mutation was designated as an Enhancer of brmK804R [E(brmK804R)] if the cumulative frequency distributions of the eye scores of the two progeny classes were statistically different (P < 0.05), using the Kolmogorov-Smirnov two-sample test. The lowest P-value given by this test is P < 0.001 and the highest is P > 0.1. This GAL4-based assay is inherently temperature sensitive. The screens and subsequent crosses were done at 24°.
As a specificity control, potential E(brmK804R) mutations were assayed for their ability to modify eye defects caused by expression of ISWIK159R in the developing eye. Eye scores for individuals of the genotype mutation/P[w+,ey-GAL4], P[w+,UAS-ISWIK159R] were compared to eye scores for siblings of the genotype balancer/P[w+,ey-GAL4], P[w+,UAS-ISWIK159R]. The P[w+,ey-GAL4], P[w+,UAS-ISWIK159R] chromosome was generated by recombination between w; P[w+, ey-GAL4] and w; P[w+, UASGALhsp70:ISWIK159R]11-4 as described (Papoulas et al. 2001).
Complementation analysis and genetic mapping:
Meiotic mapping was accomplished using either the W Sb or the ru h th st cu sr e ca chromosome, which do not themselves modify brmK804R phenotypes. Mapping by site-specific male recombination was carried out as previously described (Chen et al. 1998). All E(brmK804R) alleles were tested for the ability to complement each other as well as candidate genes and deficiencies. Alleles were placed into the same complementation group if trans-heterozygotes were not viable.
Antibody staining and electron microscopy:
For immunofluorescent staining, animals of the genotypes neuralized-GAL4/TM3 (control) or neuralized-GAL4/UAS-brmK804R (experimental) were allowed to develop at 18° until 22 or 30 hr after pupal formation (APF), hand dissected, fixed, and stained as described previously (Manning and Doe 1999). We used the following primary antibodies: mouse anti-Pros ascites at 1:1000, rat anti-SuH 24E at 1:3000 (F. Schweisguth), and goat anti-HRP-FITC at 1:200 (Jackson ImmunoResearch, West Grove, PA). Fluorescently conjugated secondary antibodies were used at 1:200 (Jackson ImmunoResearch). Imaging was done on a Bio-Rad (Hercules, CA) Radiance confocal microscope and processed in Adobe Photoshop. For scanning electron microscopy, flies were air dried for several days in a fume hood, mounted, and sputter coated with gold/palladium. Imaging was done on an ISI WB-6 scanning electron microscope at 10 kV.
RESULTS
Genetic screens for dominant modifiers of an X-linked dominant-negative brm transgene:
To gain insight into regulation and function of the BRM chromatin-remodeling complex, we screened for modifiers of an engineered dominant-negative allele, brmK804R. This lysine-to-arginine substitution in the ATP-binding site of the BRM protein renders it catalytically inactive without disrupting its incorporation into the BRM complex. brmK804R therefore acts as a potent dominant-negative brm allele (Elfring et al. 1998; Armstrong et al. 2002; Corona et al. 2004). The fly is extremely sensitive to changes in the relative ratio of the wild-type and BRMK804R proteins. Individuals expressing a 1:2 ratio of dominant-negative to wild-type BRM protein are phenotypically normal; individuals expressing a 1:1 ratio of the two proteins display a mild haltere-to-wing homeotic transformation due to decreased expression of Ubx; and a further doubling of the ratio of dominant-negative to wild-type BRM protein to 2:1 is lethal (Elfring et al. 1998).
As reported previously, an X-linked transgene expressing brmK804R under control of the brm promoter (P[w+, brmK804R]22D) forms the basis of an unusual genetic screen (Papoulas et al. 1998). Females that carry the X-linked brmK804R transgene and are heterozygous for a brm null allele express a BRMK804R to BRM+ ratio of 1:1 and are therefore viable. In males of the same genotype, dosage compensation of the X-linked transgene increases the ratio of BRMK804R to BRM+ from 1:1 to 2:1. As a result, these individuals do not survive to adulthood. Thus, brm mutations or deficiencies cause male-specific lethality in individuals heterozygous for the X-linked brmK804R transgene. Alleles of mor, a trxG gene that encodes the BAP155 subunit of the BRM complex (Crosby et al. 1999), also cause male-specific lethality in combination with the X-linked brmK804R transgene, suggesting that this genetic assay could be used to identify other genes that are critical for BRM function in vivo (Papoulas et al. 1998).
Previous studies showed that this genetic assay is highly selective; alleles of other trxG genes (including ash1, ash2, dev, kis, kto, Trl, urd, and vtd) and the majority of autosomal deficiencies present in deficiency kits provided by the Bloomington Stock Center failed to cause male-specific lethality in combination with the X-linked brmK804R transgene (Papoulas et al. 1998). Another advantage of this assay is that it is biased against recovery of mutations that merely reduce the expression of the BRM protein, since any mutation that decreases brm expression would similarly affect the expression of brmK804R. As a result, the relative levels of the two proteins would not change and no male-specific lethality would be observed.
To identify additional genes that functionally interact with brm in vivo, we screened for EMS-induced mutations that cause male-specific lethality in combination with the X-linked brmK804R transgene (Figure 1). We screened 6108 mutagenized second chromosomes and 3569 mutagenized third chromosomes and recovered five E(brmK804R) mutations that were placed into three lethal complementation groups. Complementation tests with existing alleles of candidate genes in addition to a combination of meiotic and site-specific male recombination mapping (Chen et al. 1998) allowed us to identify the modifiers of brmK804R as two moira alleles (mor1736 and mor2403), two osa alleles (osa2823 and osa3276), and one Delta allele (Dl2321) (Table 1). Thus, of the 9677 chromosomes screened, we recovered mutations in only three genes.
TABLE 1.
Interactions of selected mutations with the X-linkedbrmK804R transgene
| Mutant male |
Control male |
Mutant female |
Control female |
% survival |
|
|---|---|---|---|---|---|
| ru h st mor1736 ry e | 0 | 85 | 100 | 136 | 0 |
| ru h st mor2403 ry e | 1 | 20 | 54 | 46 | 5 |
| ru h st ry osa2823 e | 5 | 49 | 42 | 72 | 9 |
| ru h st ry osa3276 e | 1 | 38 | 43 | 74 | 3 |
| ru h st ry Dl2321 e | 2 | 30 | 60 | 53 | 7 |
| brm2 e ca | 1 | 25 | 98 | 53 | 4 |
| osa2 | 1 | 25 | 23 | 28 | 4 |
| arl1 | 32 | 34 | 95 | 63 | 48 |
| ru h st ry e | 56 | NA | 188 | NA | NA |
Virgin females homozygous for the X-linked P[w+, brmK804R] 22D transgene were mated to males bearing the mutant chromosome in trans to a balancer. The numbers of male and female progeny carrying either the mutant (mutant) or the balancer (control) chromosome are indicated. We determined percentage of survival by dividing the number of male mutant progeny by the total number of male progeny (male mutant + male control) and multiplying by 100. A percentage of survival of <10% was considered to be male-specific lethal in combination with the X-linked brmK804R transgene. arl1 is an allele of the essential arflike gene that resides next to brm (Tamkun et al. 1991) and is included as an example of a mutation that does not cause male-specific lethality in combination with the X-linked brmK804R transgene. brm2 is included as a positive control. ru h st ry e is the parent chromosome that was mutagenized to screen for mutants on the third chromosome that interact with brm. NA, not applicable.
This level of selectivity was not completely unexpected since a deficiency screen of the second and third chromosomes revealed only three interacting regions, one of which spanned the brm gene (Papoulas et al. 1998). Our EMS screens did not identify interacting genes in the remaining two regions. We did not identify Df(3R)Dl-BX12 (a deficiency that uncovers Dl) in our deficiency screen (Papoulas et al. 1998). This deficiency interacted weakly with brmK804R, but did not pass the stringent cutoff used in the screen (data not shown). It is possible that other genes uncovered by this deficiency obscured the genetic interaction between brm and Dl. Neither mor nor osa was uncovered by deficiencies tested in our deficiency screen (Papoulas et al. 1998).
The recovery of multiple alleles of osa, which encodes an ARID-domain protein found in a subset of BRM complexes (Collins et al. 1999; Mohrmann et al. 2004), and of mor, which encodes a subunit common to all BRM complexes, confirmed the utility of our screen for identifying factors that are critical for BRM function in vivo. The recovery of an allele of Dl, which encodes a ligand of the Notch receptor, may therefore reflect a close functional connection between Notch signaling and the BRM complex; this possibility is discussed at length below.
Genetic screens for dominant suppressors of the X-linked brmK804R transgene:
The recovery of histone mutations in screens for suppressors of snf2/swi2 mutations provided the first evidence that SWI/SNF counteracts chromatin repression in yeast (Winston and Carlson 1992). The success of these screens motivated us to employ a similar approach in flies. To identify potential antagonists of BRM function in vivo, we performed an F1 screen for mutations that allow P[w+,brmK804R]22D/Y; Df(3L)th102/+ individuals to survive to adulthood. The th102 deficiency spans cytological region 72A2–72D10, including the brm gene. We used this deficiency for our suppressor screen instead of a brm null allele because it results in a lower background level of surviving males, which are invariably sterile (Papoulas et al. 1998). Mutagenized Df(3L)th102 ri Sb/TM6B, Hu e Tb ca males were mated to Df(1)w67c2 y, P[w+,brmK804R]22D virgins and their progeny were scored for Df(1)w67c2 y, P[w+,brmK804R]22D/Y; Df(3L)th102 ri Sb/+ males. We recovered 73 males with potential suppressor mutations vs. 7001 sibling males of the genotype Df(1)w67c2 y, P[w+,brmK804R]22D /Y; +/TM6B, Hu Tb. We were unable to recover any potential suppressors, however, as each one of the 73 males was sterile. The recovery of 73 males (1%) is comparable to our background levels of male survival in a mock screen conducted without mutagen. The failure to recover mutations in genes encoding nucleosomal histones in this screen may be due to the presence of numerous copies of the histone gene cluster in flies. The failure to recover other dominant suppressors of brmK804R suggests that brm antagonists are either a relatively rare class of genes or not dosage sensitive.
Development of an eye-based screen for dominant modifiers of brmK804R:
Due to the relatively small number of mutations recovered in the above screens, we developed a more sensitive, eye-based modifier screen to identify additional genes that interact with brm. We chose this approach because similar screens have been successfully used to study a wide variety of biological processes (Thomas and Wassarman 1999). The expression of a UAS-brmK804R transgene in the eye-antennal disc using the eyeless-GAL4 (ey-GAL4) driver leads to the development of adults with eyes that are slightly rough and reduced in size. This phenotype is enhanced by mutations in genes encoding subunits of the BRM complex, including brm, mor, and snr1 (Table 2 and Figure 4) and BAP111 (Papoulas et al. 2001). We reasoned that additional factors that are critical for BRM function in vivo could be identified using this eye-based assay.
TABLE 2.
Dominant interactions in thebrmK804R eye-based assay
| Eye score
|
|||||||
|---|---|---|---|---|---|---|---|
| Progeny expressing brmK804R |
1 | 2 | 3 | 4 | 5 | 6 | P-value |
| Df(1)w67c2 | 0 | 82 | 10 | 14 | 3 | 1 | NA |
| brm2 e ca | 0 | 10 | 30 | 63 | 19 | 2 | <0.001 |
| TM6B, Sb Hu Tb | 0 | 93 | 12 | 9 | 0 | 0 | |
| snr1r3 | 5 | 47 | 34 | 19 | 9 | 4 | <0.001 |
| TM6B, Hu Tb | 21 | 26 | 6 | 2 | 1 | 0 | |
| brm3369 | 0 | 9 | 7 | 6 | 3 | 7 | <0.005 |
| TM3, Sb | 4 | 27 | 6 | 7 | 0 | 0 | |
| mor3090 | 0 | 0 | 1 | 6 | 14 | 9 | <0.001 |
| TM3, Sb | 0 | 16 | 16 | 2 | 0 | 0 | |
| zen436 | 0 | 11 | 17 | 16 | 6 | 3 | <0.001 |
| TM3, Sb | 2 | 30 | 10 | 9 | 1 | 0 | |
| zen714 | 2 | 10 | 7 | 12 | 3 | 2 | <0.025 |
| TM3, Sb | 4 | 19 | 7 | 4 | 0 | 0 | |
| zen2 pp | 0 | 18 | 6 | 1 | 4 | 15 | <0.001 |
| TM3, Sb | 9 | 40 | 3 | 11 | 4 | 2 | |
| vn643 | 0 | 4 | 4 | 14 | 12 | 8 | <0.001 |
| TM3, Sb | 0 | 10 | 13 | 3 | 0 | 0 | |
| P[ry+] vn10567 | 0 | 1 | 10 | 17 | 7 | 9 | <0.001 |
| TM3, Sb | 0 | 14 | 20 | 10 | 0 | 2 | |
| Dl2371 | 0 | 0 | 7 | 14 | 9 | 8 | <0.001 |
| TM3, Sb | 0 | 14 | 6 | 5 | 1 | 0 | |
| ru h th st cu sr Dl9P e ca | 0 | 1 | 11 | 19 | 8 | 5 | <0.001 |
| TM3, Sb | 2 | 24 | 15 | 1 | 0 | 0 | |
| RpII140brie | 0 | 0 | 6 | 22 | 7 | 1 | <0.001 |
| TM3, Sb | 1 | 11 | 10 | 1 | 1 | 0 | |
| RpII140A5 red e | 0 | 2 | 51 | 47 | 2 | 0 | <0.001 |
| TM3, Sb | 6 | 78 | 36 | 2 | 0 | 0 | |
With the exception of the snr1r3 stock, virgin females of the mutant stock of interest were mated to ey-GAL4,UAS-brmK804R/balancer males. Males of snr1r3/TM6B, Hu Tb were mated to ey-GAL4,UAS-brmK804R/balancer virgin females. Df(1)w67c2 was the parent stock mutagenized to screen for dominant enhancers of brmK804R on the third chromosome. Eyes of ey-GAL4,UAS-brmK804R progeny heterozygous for either a mutation of interest or the balancer chromosome were scored on a scale from 1 to 6, with 1 being wild type and 6 being complete absence of the eye as described in materials and methods. P-values were determined using the Kolmogorov-Smirnov two-sample test.
Figure 4.—
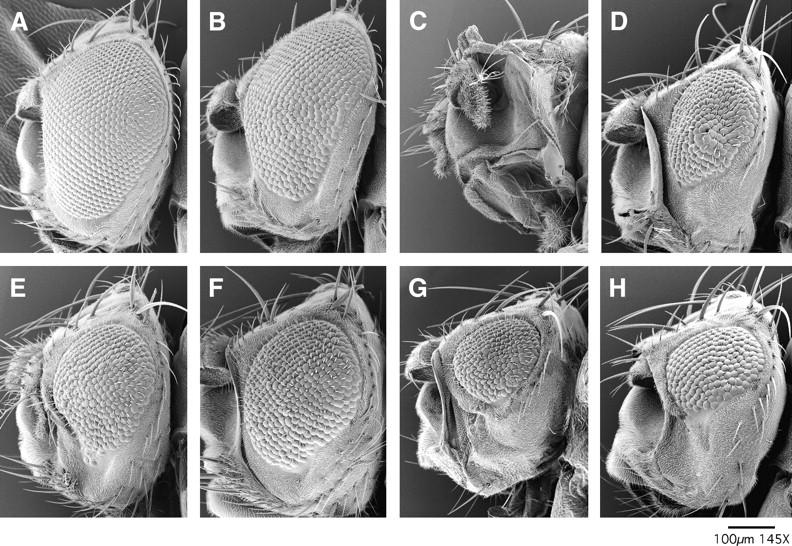
Examples of enhancement of eye defects resulting from brmK804R expression by mutations in mor, zen, Dl, and vn. Scanning electron micrographs of eyes of (A) Oregon R (wild-type) and (B) ey-GAL4, UAS-brmK804R/+ individuals are shown. Note the slight roughness resulting from the expression of brmK804R. This phenotype is enhanced in individuals heterozygous for mor3090 (C), zen436 (D), Dl2371 (E), Dl9P (F), vn643 (G), or vn10567 (H). Representative examples are shown. Additional information on the strength of these interactions is presented in Table 2.
To further assess the feasibility of this approach, we screened the Bloomington Stock Center third chromosome deficiency kit for deficiencies that modify the brmK804R rough eye phenotype. We tested 62 deficiencies covering ∼70% of the chromosome. Eyes were scored on a six-point scale for roughness and reduction in size as described in materials and methods. Data for representative interacting and noninteracting deficiencies are presented in Table 3. Of the 62 deficiencies tested, 14 enhanced the brmK804R rough eye phenotype [Df(3L)emc-E12, Df(3L)ZN47, Df(3L)XD198, Df(3L)vin2, Df(L3)vin5, Df(3L)fz-GFb, Df(3L)brm11, Df(3R)st-f13, Df(3R)Scr, Df(3R)by10, Df(3R)T-32, Df(3R)Dl-BX12, Df(3R)crb-F89-4, and Df(3R)3450] (Figure 2). By comparison, a screen of the third chromosome deficiency kit for regions that cause male-specific lethality in combination with the X-linked brmK804R transgene identified only two deficiencies: Df(3L)ZN47 and Df(3L)th102 (Papoulas et al. 1998). Both the brm11 and th102 deficiencies span the brm gene. Thus, the eye-based assay uncovers regions previously identified as important for brm function, as well as additional regions.
TABLE 3.
Representative deficiencies assayed for dominant interactions withbrmK804R
| Eye score
|
|||||||
|---|---|---|---|---|---|---|---|
| Progeny expressing brmK804R |
1 | 2 | 3 | 4 | 5 | 6 | P-value |
| Df(3L)vin2 | 0 | 3 | 12 | 20 | 7 | 0 | <0.001 |
| TM3, Sb | 0 | 20 | 7 | 3 | 0 | 0 | |
| Df(3R)3450 | 0 | 5 | 5 | 14 | 12 | 0 | <0.05 |
| TM3, Ser | 0 | 7 | 3 | 4 | 0 | 0 | |
| Df(3L)lxd6 | 0 | 41 | 7 | 4 | 0 | 0 | >0.1 |
| Tm3, Sb Ser | 1 | 16 | 8 | 0 | 1 | 0 | |
| Df(3R)mbc-R1 | 16 | 50 | 1 | 3 | 2 | 0 | >0.1 |
| TM3, Sb | 8 | 52 | 6 | 2 | 0 | 0 | |
Virgin females heterozygous for a deficiency of interest were mated to balanced ey-GAL4,UAS-brmK804R males. Eyes of ey-GAL4, UAS-brmK804R progeny heterozygous for either the deficiency or balancer were scored as described in materials and methods. P-values were determined using the Kolmogorov-Smirnov two-sample test. The representative interacting deficiencies are Df(3L)vin2, ru1 h1 gl2 e4 ca1/TM3, Sb (67F2–3;68D6) and w1118; Df(3R)3450/TM6B, Tb (98E3;99A6–8). Representative noninteracting deficiencies are cn; Df(3R)mbc-R1, ry506/TM3, Sb ry (95A5–7;95D6–11) and y; Df(3L)lxd6/TM3, Sb Ser (67E1–2;68C1–2).
Figure 2.—

Deficiency screen of the third chromosome identified eight regions that dominantly enhance eye defects resulting from expression of brmK804R. Solid regions are deficiencies that specifically interact with brmK804R and not with ISWIK149R in this assay. Shaded regions indicate deficiencies that fail to enhance brmK804R. Hatched regions indicate deficiencies that enhance eye defects resulting from expression of either brmK804R or ISWIK149R. Genes identified in subsequent eye-based screens as Enhancers of brmK804R are indicated.
Rather than interacting with brm, it is possible that some of the deficiencies nonspecifically affect the GAL4 driver system. To control for this possibility, we performed a secondary screen in which the interacting deficiencies were tested for their ability to modify eye defects resulting from the expression of a dominant-negative form of ISWI (ISWIK159R), a chromatin-remodeling factor that is functionally distinct from BRM (Deuring et al. 2000). Four of the 14 deficiencies enhanced the ISWIK159R rough eye phenotype [Df(3R)st-f13, Df(3R)by10, Df(3R)Dl-BX12, and Df(3R)crb-F89-4] (data not shown). The remaining 10 deficiencies specifically interact with brm and define at least eight interacting regions (Figure 2). By comparing interacting deficiencies to overlapping, but noninteracting deficiencies, the regions containing potential enhancers of brmK804R were determined to be 61A–C, 64C–65E, 68C1–11, 70C–D, 71F–72D, 84A1–5, 87B1–13, and 98E3–99A. From the results of this third chromosome deficiency kit screen, we concluded that our eye-based assay represents a sensitive but selective approach for identifying factors that functionally interact with the BRM complex.
Most trithorax group genes do not enhance the brmK804R rough eye phenotype:
brm genetically interacts with several trxG genes, including trx and ash1. Flies doubly heterozygous for alleles of brm and trx display an increase in the incidence of homeotic transformations (including fifth abdominal segment to fourth and haltere to wing) (Tamkun et al. 1992), while flies doubly heterozygous for brm and ash1 display homeotic transformations including third to second leg (Tripoulas et al. 1994). Recent work suggests that methylation of histone tails by ASH1 may recruit the BRM complex to target promoters (Beisel et al. 2002). We were therefore interested in whether mutations in trxG genes interacted with brm in our eye-based assay. Alleles of breathless/devenir (btldev2), verthandi (vtd2), urdur (urd2), skuld (skd1 and skd2), trithorax (trxE2), and kismet (kis1) failed to modify the brmK804R rough eye phenotype (data not shown). Several trxG genes are uncovered by deficiencies that fail to interact with brm in the developing eye. These include kohtalo and ash1 [Df(3L)kto2], tonalli [Df(3L)lxd6], and osa [Df(3R)DG2]. Thus, with the exception of mor it appears that the majority of trxG genes may not directly function with brm in the developing eye.
Alleles of genes encoding subunits of RNA polymerase II interact with brm:
The BRM complex is required for global transcription by RNA polymerase II (pol II) on larval salivary gland polytene chromosomes (Armstrong et al. 2002). To address whether mutations in genes encoding general regulators of transcription functionally interact with brm, we assayed alleles of genes that encode the two largest subunits of RNA polymerase II. RpII140A5, RpII140Z45, and RpII2154 all enhanced the brmK804R rough eye phenotype (Table 2 and data not shown). RpII2154 enhanced eye defects resulting from expression of ISWIK159R (data not shown). By contrast, RpII140Z45 and RpII140A5 failed to enhance ISWIK159R eye defects (data not shown), suggesting that these interactions are specific to brm. Mutations in genes encoding components of the mediator complex (Trap80s9256, Trap100BG01670, Trap150KG00948, and paprK760) failed to modify eye defects resulting from expression of brmK804R, as did alleles of genes encoding TBP-associated factors (Taf41 and Taf10bKG01574) (data not shown). Although negative results in the eye assay should be interpreted cautiously, these data suggest that brm functionally interacts with RpII140, but not with subunits of TFIID or mediator.
Genetic screen for dominant modifiers of brmK804R:
As an unbiased approach to identify factors that functionally interact with the BRM complex, we screened for EMS-induced mutations on the third chromosome that enhance eye defects resulting from the expression of brmK804R (Figure 3). An F2 screen was necessary for two reasons. First, the severity of the eye defects observed in ey-GAL4, UAS-brmK804R individuals was variable. Most of the eyes had disordered ommatidia covering less than half of the eye, but 5% (14 of 304 eyes) were severely reduced in size or completely absent. In an F1 screen, this background would result in a high number of false positives. Second, expression of the ey-GAL4 driver is not limited to the developing eye. Mutations that strongly enhance brmK804R can lead to pupal lethality, as previously observed for the BAP111 subunit of the BRM complex (Papoulas et al. 2001); such mutations would be irretrievable in an F1 screen.
We screened 7469 EMS-mutagenized third chromosomes for dominant modifiers of the eye defects observed in ey-GAL4, UAS-brmK804R adults. We simultaneously screened for mutations that were lethal in combination with ey-GAL4, UAS-brmK804R (Figure 3). Although the severity of the ey-GAL4, UAS-brmK804R eye phenotype would allow the identification of suppressor mutations, none were identified in our screen. We recovered 47 E(brmK804R) mutations, 13 of which were homozygous viable and were not pursued further. The remaining 34 mutations were placed into 20 lethal complementation groups. A combination of meiotic mapping and complementation tests with interacting deficiencies and alleles of candidate genes allowed us to identify six alleles of brm (brm795, brm963, brm2419, brm3244, brm3369, and brm1630), two alleles of mor (mor880 and mor3090), two alleles of zerknullt (zen436 and zen714), eight alleles of Dl (Dl266, Dl470, Dl681, Dl2371, Dl1918, Dl1946, Dl4386, and Dl4585), one allele of vein (vn643), and one allele of RpII140 (RpII140brie) (Figure 4 and Table 2). The mutations in brm, mor, zen, RpII140, and vn failed to enhance the rough eye phenotype resulting from overexpression of ISWIK159R, suggesting that these genes specifically interact with brm. By contrast, the Dl alleles did enhance the ISWIK159R rough eye phenotype (data not shown). Dl, brm, zen, and vn all map to interacting deficiencies (Figure 2), suggesting that these alleles behave as loss-of-function mutations. As discussed below, other loss-of-function alleles of these genes—including brm2, mor4, zen2, vn10567, RpII140A5, and Dl9P—also dominantly enhance the brmK804R rough eye phenotype (Figure 4 and Table 2). The remaining 14 E(brm) alleles fall into single complementation groups and are currently under investigation.
Genetic interactions between brm and mutations in genes involved in cell signaling:
One of the most surprising outcomes of our screens was the recovery of mutations in genes involved in signal transduction pathways. With eight alleles, Delta (Dl) was the largest complementation group recovered in the eye-based screen. Furthermore, Dl was the only gene recovered in the male-specific lethality screen that did not encode a subunit of the BRM complex. Dl encodes a ligand for the Notch receptor and is critical for development (Lai 2004). We also recovered one allele of vein (vn), which encodes a secreted ligand for the epidermal growth factor receptor (EGFR) (Schnepp et al. 1996). To determine whether genetic interactions between brm and these signaling pathways are limited to the developing eye, we examined trans-heterozygotes for adult phenotypes. This approach has proven useful for uncovering genes important for brm function. For example, individuals heterozygous for alleles of either brm or mor appear normal, while trans-heterozygous adults display loss of humeral bristles, duplicated or extra macrochaetae, ectopic wing veins, rough eyes, and held-out wings (Table 4) (Brizuela and Kennison 1997). Likewise, individuals heterozygous for alleles of brm and osa display held-out wings (Vazquez et al. 1999). These genetic interactions provided early evidence that the BRM protein functionally interacts with MOR and OSA. We asked whether similar genetic interactions could be observed between brm and Dl or vn.
TABLE 4.
Genetic interactions betweenbrm andDl
| Genotype | % with loss of humeral bristles |
% with ectopic or duplicated macrochaetae |
% with rough eyes |
No. of flies |
|---|---|---|---|---|
| brm2/+ | 0 | 3.5 | 0 | 57 |
| brm2/mor4 | 22 | 35 | 10 | 49 |
| +/mor4 | 0 | 12 | 0 | 56 |
| brm2/Dl266 | 14 | 42 | 58 | 48 |
| +/Dl266 | 0 | 10 | 1.7 | 58 |
| brm2/Dl2371 | 19 | 52 | 69 | 42 |
| +/Dl2371 | 0 | 27 | 2.4 | 41 |
| brm2/Dl9P | 0 | 28 | 76 | 49 |
| +/Dl9P | 0 | 20 | 0 | 50 |
Individuals trans-heterozygous for brm2 and Dl266, Dl2371, or Dl9P display a variety of phenotypes including loss of humeral bristles, duplicated or extra macrochaetae, and rough eyes (Table 4, Figure 5). Individuals heterozygous for only one of the alleles display some of these phenotypes at low penetrance, but the penetrance of phenotypes was greatly enhanced in the trans-heterozygotes (Table 4). Individuals trans-heterozygous for brm and vn also display a variety of adult phenotypes including held-out wings, loss of humeral bristles, duplicated or extra macrochaetae, and mildly rough eyes (Table 5, Figure 5). In the single heterozygotes, these phenotypes either are not observed or are present at low penetrance (Table 5). Thus, genetic interactions between brm and both Dl and vn are not limited to the developing eye and are not dependent on either the GAL4 driver system or the dominant-negative brmK804R allele.
Figure 5.—

Adult phenotypes illustrate genetic interactions between brm and Dl and brm and vn. (A) Arrows indicate wild-type humeral bristles in Oregon R flies. Bristles are lost in flies trans-heterozygous for (B) brm2 and Dl2371 or (C) brm2 and vn643. (D) Wild-type Oregon R flies have four scutellar bristles. Extra scutellar bristles (indicated by arrows) are shown in flies that are trans-heterozygous for (E) brm2 and Dl2371 or (F) brm2 and vn643. (G) Wild-type Oregon R flies do not hold out their wings while (H) brm2/vn643 flies display held-out wings.
TABLE 5.
Genetics interactions betweenbrm andvn
| Genotype | % with held-out wings |
% with loss of humeral bristles |
% with ectopic or duplicated macrochaetae |
No. of flies |
|---|---|---|---|---|
| brm2/+ | 0 | 0 | 3.5 | 57 |
| brm2/vn643 | 96 | 92 | 21 | 53 |
| +/vn643 | 0 | 0 | 0 | 49 |
| brm2/vnc221 | 13 | 49 | 5.7 | 53 |
| +/vnc221 | 0 | 2.1 | 0 | 47 |
| brm2/vn10567 | 0 | 93 | 8.9 | 56 |
| +/vn10567 | 0 | 0 | 0 | 47 |
| brm2/Egfrf2 | 0 | 16 | 2.0 | 49 |
| +/Egfrf2 | 0 | 0 | 0 | 50 |
| brm2/grk3 | 0 | 0 | 0 | 36 |
| brm2/spi1 | 0 | 0 | 2.0 | 51 |
| brm2/Df(3L)ZN47 | 0 | 92 | 62 | 50 |
| +/Df(3L)ZN47 | 0 | 2.4 | 7.3 | 41 |
The signaling pathways involving Dl and Vn are complex since their respective receptors (Notch and EGF receptor) respond to more than one ligand. Vn signals via the EGF receptor, the Drosophila homolog of the epidermal growth factor receptor (Schnepp et al. 1996). We observed genetic interactions between brm2 and Egfrf2, a loss-of-function allele of Egfr. Of the trans-heterozygotes, 16% displayed loss of one or more humeral bristles (Table 5). This phenotype was not observed in either brm2 or Egfrf2 heterozygotes. The EGF receptor responds to four different receptor ligands: Vn, Gurken (Grk), Spitz (Spi), and Keren (Krn) (Shilo 2003). We failed to detect similar types of genetic interactions between brm2 and alleles of grk or spi (Table 5). Furthermore, grk3, spi1, spiS3547, and Df(3L)81k19 (a deficiency covering krn) all failed to modify eye defects resulting from expression of brmK804R (data not shown). These data suggest that the BRM complex is important for signaling by the Vn ligand. The Notch receptor receives signals from one of two ligands, Dl or Serrate (Ser) (Lai 2004). Ser is not expressed in the developing eye (Verheyen et al. 1996), and so we would not expect Ser alleles to modify eye defects resulting from loss of brm function. Mutations in Notch (N) would not have been identified in any of our screens since it is located on the X chromosome. However, a loss-of-function N allele, N264-39, dominantly enhances the rough eye phenotype resulting from expression of brmK804R (data not shown). These data suggest that BRM interacts with the Vn-Egfr and Dl-Notch signaling pathways.
The identification of genetic interactions between brm and Dl was intriguing since previous studies had revealed a role for brm in the development of the peripheral nervous system (Elfring et al. 1998). The external sense organs of the Drosophila peripheral nervous system are formed by the adult sense organ precursor (SOP) cells, which undergo asymmetric cell divisions to produce five different cell types: socket, bristle, sheath, neuron, and glia (Figure 6A). Cell fate in the SOP lineage is controlled by the level of Dl-Notch signaling: high Dl-Notch signaling promotes pIIA, pIIIB, socket, and sheath cell fates, whereas low Dl-Notch signaling results in pIIB, bristle, neuron, and glial cell fates (Hartenstein and Posakony 1990; Parks and Muskavitch 1993; Guo et al. 1996). If the BRM complex is required for Dl-Notch signaling, the loss of brm function should cause lineage defects similar to those resulting from reduced Dl-Notch signaling, i.e., an increase in bristles, glia, or neurons at the expense of other cell types.
Figure 6.—

Reduced BRM levels generate phenotypes similar to reduced Delta signaling in the adult SOP lineage. (A) Summary of the adult external sense organ lineage, with the cell fates requiring high Dl-Notch signaling shown in blue and the cell fates requiring low Dl-Notch signaling shown in red. S, socket; B, bristle; SH, sheath; N, neuron; and G, glial. (B and C) Pupal notum at 22 hr APF stained for socket cells (SuH, blue, large arrowhead), glia (strong Pros, red, thin arrows), and the pIIIB cell (weak Pros, red, small arrowheads). (B) Wild-type SOP lineage showing one socket, one pIIIB, and one glial cell visible. (C) The neuralized-GAL4/UAS-brmK804R SOP lineages contain fewer socket cells and extra glia (strong Pros, red, thin arrows). (D and E) Pupal notum at 30 hr APF stained for socket cells (SuH, blue, arrowhead), neurons (HRP, green, thick arrows) and sheath cells (Pros, red, asterisk). At this stage, the only Pros+ cell present is the sheath cell, because the Pros+ glial cell visible at 22 hr APF has migrated away (Gho et al. 1999; Reddy and Rodrigues 1999). (D) Wild-type SOP lineages at this stage showing one socket, one neuron, and one sheath cell. (E) The neuralized-GAL4/UAS-brmK804R SOP lineages contain fewer socket cells, fewer sheath cells, and extra neurons.
To test the prediction, we expressed brmK804R in the SOP lineage and used cell-specific markers to observe the resulting cell types. We used the following markers to score sense organ cell fates: Suppressor of Hairless (SuH) for socket cells, Prospero (Pros) for glia in early lineages and sheath cell in late lineages, and anti-horseradish peroxidase (HRP) for neuronal membranes (Jan and Jan 1982; Manning and Doe 1999). Expression of brmK804R in the SOP lineage using the neuralized-GAL4 driver at 20° resulted in high embryonic or larval lethality (78%; n = 803), early pupal lethality (17%; n = 803), and midpupal lethality (5%; n = 803); only the last class was used to score SOP phenotypes. Control genotypes containing the UAS-brmK804R transgene without a neuralized-GAL4 showed excellent viability (97%; n = 800). We found that neuralized-GAL4 UAS-brmK804R pupae contained variable-sized patches of tissue containing wild-type or defective SOP lineages, and we confined our analysis to the defective tissue. Early SOP lineages, 22 hr APF, showed a loss of SuH+ socket cells and an increase in Pros+ glial cells (Figure 6, B and C; n = 371 lineages examined). Late SOP lineages, 30 hr APF, showed a loss of SuH+ socket cells and Pros+ sheath cells but an increase in HRP+ neurons (Figure 6, D and E; n = 240). These two phenotypes were never observed in control 22-hr APF or 30-hr APF pupae containing the UAS-brmK804R transgene without the neuralized-GAL4 transgene (22 hr APF, n = 272; 30 hr APF, n = 137). Both the early and late-lineage phenotypes are similar to those seen following loss of Dl-Notch signaling, as summarized in Figure 6A (Hartenstein and Posakony 1990; Parks and Muskavitch 1993; Guo et al. 1996). We conclude that BRM and Dl act together to specify cell fate within the adult SOP lineage.
DISCUSSION
In this study we report the results of two different screens designed to identify factors that are critical for the function of the Drosophila BRM chromatin-remodeling complex. We screened a total of 17,146 mutant chromosomes and recovered 39 mutations that genetically interact with a dominant-negative allele of brm (brmK804R). Of the 25 mutations that we positively identified, nearly half (48%) are alleles of genes encoding subunits of the BRM complex (brm, mor, or osa), suggesting that the other genes identified in our screens are also critical for brm function. Similar screens could be used to study any Drosophila chromatin-remodeling factor that functions as the ATPase subunit of a protein complex (Corona et al. 2004).
Interactions between brm and other factors involved in transcription:
Our screens identified a single allele of RpII140, which encodes the second largest subunit of RNA pol II. Other alleles of RpII140 also dominantly enhanced eye defects resulting from expression of brmK804R. This finding complements our observation that the BRM complex is required for global transcription by RNA pol II (Armstrong et al. 2002) and suggests that the BRM complex may interact more closely than previously thought with the general transcriptional machinery. These findings are consistent with the observation that yeast TFIID and RNA pol II are required for the recruitment of SWI/SNF to the RNR3 promoter (Sharma et al. 2003). We have been unable to detect a physical interaction between RNA pol II and the BRM complex by co-immunoprecipitation (Armstrong et al. 2002), however, and SWI/SNF recruitment does not depend upon RNA pol II at all yeast promoters (Hirschhorn et al. 1992; Gavin and Simpson 1997). Why the basal transcription machinery targets chromatin-remodeling complexes to some, but not all, promoters remains to be determined.
Two distinct BRM complexes (called BAP and PBAP) were recently identified in Drosophila (Mohrmann et al. 2004). Both complexes contain the BRM ATPase (related to the yeast SWI2/SNF2 and RSC ATPases), the SANT-domain protein Moira (MOR), the HMG-domain protein BAP111, the actin-related protein BAP55, actin, BAP60, and SNR1 (Papoulas et al. 1998; Collins et al. 1999; Mohrmann et al. 2004). The BAP complex contains OSA, while the PBAP complex lacks OSA and instead contains Polybromo and the ARID-domain, zinc-finger protein BAP170 (Collins et al. 1999; Mohrmann et al. 2004). BAP may represent the Drosophila counterpart of the yeast SWI/SNF and human BAF complexes, while PBAP appears more highly related to the yeast RSC and human PBAF complexes (Mohrmann and Verrijzer 2005). Both BAP and PBAP are abundant and are widely associated with transcriptionally active chromatin in larval salivary glands (Mohrmann et al. 2004). Both complexes use the BRM ATPase; the expression of BRMK804R should therefore interfere with the functions of both the BAP and PBAP complexes.
The presence or absence of the OSA subunit distinguishes the BAP complex from PBAP (Mohrmann et al. 2004). We isolated two osa alleles from the male-specific lethality screens, suggesting that this screen has the potential to identify factors important for BAP function. Our osa alleles fail to modify the eye defects caused by expression of dominant-negative brm (as does a deficiency spanning osa), suggesting that our eye-based screen may select for genes important for PBAP function. In agreement with these observations, Collins et al. (1999) found that while osa interacted with brm in the wing, it acted in opposition to brm in the eye. The elucidation of the relative roles of BAP and PBAP in vivo will require the isolation of mutations in genes encoding unique subunits of this complex, including polybromo and BAP170 (Mohrmann et al. 2004).
Interactions between BRM and other proteins that regulate chromatin structure and function:
Numerous recent studies have revealed close functional relationships between chromatin-remodeling complexes and histone-modifying enzymes (Hassan et al. 2002). For example, the MOF histone acetyltransferase functionally antagonizes the Drosophila ISWI chromatin-remodeling factor (Corona et al. 2002); bromodomains within the yeast RSC chromatin-remodeling complex recognize acetylated histone H3 (Kasten et al. 2004); and methylation of lysines 4 and 9 of H3 and lysine 20 of H4 by Ash1 may recruit the BRM complex (Beisel et al. 2002). Histone modification, including methylation of lysine 4 of H3, is also required for expression of Notch target genes (Bray et al. 2005).
However, to date we have not yet identified E(brm) mutations in genes encoding histone-modifying enzymes. We also failed to recover genes encoding structural components of chromatin or subunits of other chromatin-remodeling complexes. Why weren't mutations in these classes of genes recovered in our screens? We did not expect to recover mutations in histone genes in our screens since they are present in many copies in flies. Our eye-based screen was limited to the third chromosomes, and genes on the X chromosome would have escaped detection in both of our screens. Furthermore, we do not believe that either one of our genetic screens was taken to saturation. It is also possible that chromatin-remodeling and modifying enzymes that interact with brm are redundant or are not expressed in limiting quantities.
The BRM complex and Dl-Notch signaling:
Dl represented the largest E(brm) complementation group; over a third of the mutations (36%) were alleles of Dl. These findings suggest that the functions of the BRM complex and the Notch signaling pathway are intimately related. Notch signaling is one of the most extensively studied signaling pathways (Kadesch 2004). It is essential for the development of most tissues and is likely present in all metazoans, although here we focus on the pathway in Drosophila. A transmembrane ligand (either Delta or Serrate) on the signaling cell binds the Notch receptor on the signal-receiving cell, resulting in two proteolytic cleavages of the Notch transmembrane protein. This proteolysis causes the release of the Notch ICD, which translocates to the nucleus to regulate gene expression. Once in the nucleus, the ICD forms a complex with the Suppressor of Hairless [Su(H)] transcription factor (a CSL protein) to activate Notch target genes. In the absence of signaling (and therefore the absence of ICD), Su(H) complexes with corepressors that deacetylate histones to repress transcription of target genes (Lai 2004; Schweisguth 2004). The role of Notch signaling is particularly well understood in regard to cell fate determinations within the adult SOP lineage. Loss of Dl-Notch signaling can result in an increase of neurons or glia at the expense of other cell types (Hartenstein and Posakony 1990; Parks and Muskavitch 1993).
Previous work suggested that the BRM complex was critical for the development of the peripheral nervous system; somatic clones of brm mutant tissue throughout the fly showed duplicated, stunted, or fused mechanosensory bristles (Elfring et al. 1998). Expression of the dominant-negative allele of brm results in similar bristle defects, as well as alterations in the number and identities of campaniform sensilla, sensory organs used for flight (Elfring et al. 1998). The identification of numerous alleles of Dl in our screens as well as the observation of increased penetrance of a variety of phenotypes in individuals heterozygous for alleles of both brm and Dl is consistent with these observations and points to a close functional connection between the Notch signaling pathway and the BRM complex.
To explore further the connection between the BRM complex and Dl-Notch signaling, we investigated the role of the BRM complex in cell fate specification within the adult SOP lineage, where every stage of development is regulated by Dl-Notch signaling. Reduced Dl-Notch signaling within the imaginal disc proneural cluster that gives rise to the SOP leads to formation of ectopic SOPs that form perfectly normal sense organs, leading to bristle/socket duplications (Hartenstein and Posakony 1990; Parks and Muskavitch 1993), a phenotype similar to the bristle defects seen in brm mutant clones (Elfring et al. 1998). In contrast, reduced Dl-Notch specifically within the SOP lineage results in loss of external cell types and production of ectopic internal cell types such as glia or neurons (Hartenstein and Posakony 1990; Parks and Muskavitch 1993). This is precisely the phenotype we observe following expression of brmK804R within the SOP lineage.
What is the role of the BRM complex in the Notch signaling pathway? Since the BRM complex plays a global role in transcription by RNA pol II (Armstrong et al. 2002), it is possible that the genetic interactions and phenotypes that we have observed are the result of decreased Dl expression. We believe this is unlikely due to the selectivity of our screens. Indeed, we failed to observe genetic interactions between Dl and RpII140 mutations (data not shown). It is also possible that the BRM complex and the Dl-Notch pathway are independently regulating the same target genes. If both pathways are limiting, a reduction in Dl-Notch signaling may enhance a brm phenotype. A more intriguing possibility is that Dl-Notch signaling may regulate the activity or targeting of the BRM complex. As a ubiquitous complex that is critical for the transcription of most genes by RNA pol II genes, the BRM complex is a logical target for the signaling pathways. Once the ICD of Notch is in the nucleus, it may form complexes not only with Su(H), but also with the BRM complex, thus regulating its activity or its association with Notch target genes. Strong support for this model is provided by recent biochemical studies of the human BRM (hBRM) protein. hBRM physically interacts with the ICD of Notch and both hBRM and ICD are found to be associated with the promoters of Notch target genes (Kadam and Emerson 2003). On the basis of these findings, further analyses of the interactions between Dl-Notch signaling and the BRM chromatin-remodeling complex are clearly warranted.
Our data suggest that the BRM complex may play an important role in another signal transduction pathway. An allele of vn, which encodes a secreted protein related to the mammalian neuregulin family of ligands for the EGF receptor, was recovered as an enhancer of eye defects resulting from the expression of brmK804R. Many signal pathways intersect and complex interactions between EGF receptor signaling and the Notch pathway have been reported in Drosophila. EGF receptor signaling can work in concert with (Flores et al. 2000; Kumar and Moses 2001; Tsuda et al. 2002) or antagonistically to Notch signaling (Culi et al. 2001; Carmena et al. 2002; Rohrbaugh et al. 2002). Our findings suggest that the BRM complex interacts with one or both of these pathways during eye development, but the precise nature of these interactions remains to be determined.
In conclusion, our unbiased genetic screens led us to an unexpected connection between the BRM chromatin-remodeling complex and Dl-Notch signaling. Both the BRM complex and the Dl-Notch signaling pathway are conserved in mammals; our results therefore suggest that similar interactions may be critical for mammalian development. In mice, loss of Notch activity leads to tumor formation (Nicolas et al. 2003); similarly the genes encoding subunits of the mammalian BRM complexes also act as tumor suppressors (Dunaief et al. 1994; Versteege et al. 1998). Further work is required to determine the precise nature and extent of interactions between the BRM chromatin-remodeling complex and signaling pathways.
Acknowledgments
We thank the Bloomington Stock Center for numerous Drosophila strains. We thank Mark Mortin for RpII140 alleles, Jon Krupp for scanning electron microscopy, and Keith Maggert for assistance with statistical analyses. We thank members of our laboratories for helpful discussions and advice. A.S.S. was supported by a fellowship from the Beckman Scholars' Program. J.A.A was supported by the Damon Runyon Center Research Foundation Fellowship, DRG-1556. C.Q.D. is supported by the Howard Hughes Medical Institute. This work was supported by National Institutes of Health grant GM49883 to J.W.T.
References
- Angus-Hill, M. L., A. Schlichter, D. Roberts, H. Erdjument-Bromage, P. Tempst et al., 2001. A Rsc3/Rsc30 zinc cluster dimer reveals novel roles for the chromatin remodeler RSC in gene expression and cell cycle control. Mol. Cell 7: 741–751. [DOI] [PubMed] [Google Scholar]
- Armstrong, J. A., O. Papoulas, G. Daubresse, A. S. Sperling, J. T. Lis et al., 2002. The Drosophila BRM complex facilitates global transcription by RNA polymerase II. EMBO J. 21: 5245–5254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badenhorst, P., M. Voas, I. Rebay and C. Wu, 2002. Biological functions of the ISWI chromatin remodeling complex NURF. Genes Dev. 16: 3186–3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker, P. B., and W. Horz, 2002. ATP-dependent nucleosome remodeling. Annu. Rev. Biochem. 71: 247–273. [DOI] [PubMed] [Google Scholar]
- Beisel, C., A. Imhof, J. Greene, E. Kremmer and F. Sauer, 2002. Histone methylation by the Drosophila epigenetic transcriptional regulator Ash1. Nature 419: 857–862. [DOI] [PubMed] [Google Scholar]
- Berger, S. L., 2002. Histone modifications in transcriptional regulation. Curr. Opin. Genet. Dev. 12: 142–148. [DOI] [PubMed] [Google Scholar]
- Bray, S., H. Musisi and M. Bienz, 2005. Bre1 is required for Notch signaling and histone modification. Dev. Cell 8: 279–286. [DOI] [PubMed] [Google Scholar]
- Brizuela, B. J., and J. A. Kennison, 1997. The Drosophila homeotic gene moira regulates expression of engrailed and HOM genes in imaginal tissues. Mech. Dev. 65: 209–220. [DOI] [PubMed] [Google Scholar]
- Cao, R., and Y. Zhang, 2004. The functions of E(Z)/EZH2-mediated methylation of lysine 27 in histone H3. Curr. Opin. Genet. Dev. 14: 155–164. [DOI] [PubMed] [Google Scholar]
- Carmena, A., E. Buff, M. S. Halfon, S. Gisselbrecht, F. Jimenez et al., 2002. Reciprocal regulatory interactions between the Notch and Ras signaling pathways in the Drosophila embryonic mesoderm. Dev. Biol. 244: 226–242. [DOI] [PubMed] [Google Scholar]
- Chen, B., T. Chu, E. Harms, J. P. Gergen and S. Strickland, 1998. Mapping of Drosophila mutations using site-specific male recombination. Genetics 149: 157–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins, R. T., T. Furukawa, N. Tanese and J. E. Treisman, 1999. Osa associates with the Brahma chromatin remodeling complex and promotes the activation of some target genes. EMBO J. 18: 7029–7040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corona, D. F., C. R. Clapier, P. B. Becker and J. W. Tamkun, 2002. Modulation of ISWI function by site-specific histone acetylation. EMBO Rep. 3: 242–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corona, D. F., J. A. Armstrong and J. W. Tamkun, 2004. Genetic and cytological analysis of Drosophila chromatin-remodeling factors. Methods Enzymol. 377: 70–85. [DOI] [PubMed] [Google Scholar]
- Crosby, M. A., C. Miller, T. Alon, K. L. Watson, C. P. Verrijzer et al., 1999. The trithorax group gene moira encodes a brahma-associated putative chromatin-remodeling factor in Drosophila melanogaster. Mol. Cell. Biol. 19: 1159–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culi, J., E. Martin-Blanco and J. Modolell, 2001. The EGF receptor and N signalling pathways act antagonistically in Drosophila mesothorax bristle patterning. Development 128: 299–308. [DOI] [PubMed] [Google Scholar]
- Dejardin, J., and G. Cavalli, 2004. Chromatin inheritance upon Zeste-mediated Brahma recruitment at a minimal cellular memory module. EMBO J. 23: 857–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deuring, R., L. Fanti, J. A. Armstrong, M. Sarte, O. Papoulas et al., 2000. The ISWI chromatin-remodeling protein is required for gene expression and the maintenance of higher order chromatin structure in vivo. Mol. Cell 5: 355–365. [DOI] [PubMed] [Google Scholar]
- Dunaief, J. L., B. E. Strober, S. Guha, P. A. Khavari, K. Alin et al., 1994. The retinoblastoma protein and BRG1 form a complex and cooperate to induce cell cycle arrest. Cell 79: 119–130. [DOI] [PubMed] [Google Scholar]
- Eberharter, A., and P. B. Becker, 2004. ATP-dependent nucleosome remodelling: factors and functions. J. Cell Sci. 117: 3707–3711. [DOI] [PubMed] [Google Scholar]
- Elfring, L. K., C. Daniel, O. Papoulas, R. Deuring, M. Sarte et al., 1998. Genetic analysis of brahma: the Drosophila homolog of the yeast chromatin remodeling factor SWI2/SNF2. Genetics 148: 251–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaus, A., and T. Owen-Hughes, 2004. Mechanisms for ATP-dependent chromatin remodelling: Farewell to the tuna-can octamer? Curr. Opin. Genet. Dev. 14: 165–173. [DOI] [PubMed] [Google Scholar]
- Flores, G. V., H. Duan, H. Yan, R. Nagaraj, W. Fu et al., 2000. Combinatorial signaling in the specification of unique cell fates. Cell 103: 75–85. [DOI] [PubMed] [Google Scholar]
- Francis, N. J., and R. E. Kingston, 2001. Mechanisms of transcriptional memory. Nat. Rev. Mol. Cell. Biol. 2: 409–421. [DOI] [PubMed] [Google Scholar]
- Francis, N. J., A. J. Saurin, Z. Shao and R. E. Kingston, 2001. Reconstitution of a functional core polycomb repressive complex. Mol. Cell 8: 545–556. [DOI] [PubMed] [Google Scholar]
- Gavin, I. M., and R. T. Simpson, 1997. Interplay of yeast global transcriptional regulators Ssn6p-Tup1p and Swi-Snf and their effect on chromatin structure. EMBO J. 16: 6263–6271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gellon, G., and W. McGinnis, 1998. Shaping animal body plans in development and evolution by modulation of Hox expression patterns. BioEssays 20: 116–125. [DOI] [PubMed] [Google Scholar]
- Gho, M., Y. Bellaiche and F. Schweisguth, 1999. Revisiting the Drosophila microchaete lineage: a novel intrinsically asymmetric cell division generates a glial cell. Development 126: 3573–3584. [DOI] [PubMed] [Google Scholar]
- Gould, A., 1997. Functions of mammalian Polycomb group and trithorax group related genes. Curr. Opin. Genet. Dev. 7: 488–494. [DOI] [PubMed] [Google Scholar]
- Guo, M., L. Y. Jan and Y. N. Jan, 1996. Control of daughter cell fates during asymmetric division: interaction of Numb and Notch. Neuron 17: 27–41. [DOI] [PubMed] [Google Scholar]
- Hartenstein, V., and J. W. Posakony, 1990. A dual function of the Notch gene in Drosophila sensillum development. Dev. Biol. 142: 13–30. [DOI] [PubMed] [Google Scholar]
- Hassan, A. H., P. Prochasson, K. E. Neely, S. C. Galasinski, M. Chandy et al., 2002. Function and selectivity of bromodomains in anchoring chromatin-modifying complexes to promoter nucleosomes. Cell 111: 369–379. [DOI] [PubMed] [Google Scholar]
- Hirschhorn, J. N., S. A. Brown, C. D. Clark and F. Winston, 1992. Evidence that SNF2/SWI2 and SNF5 activate transcription in yeast by altering chromatin structure. Genes Dev. 6: 2288–2298. [DOI] [PubMed] [Google Scholar]
- Holstege, F. C., E. G. Jennings, J. J. Wyrick, T. I. Lee, C. J. Hengartner et al., 1998. Dissecting the regulatory circuitry of a eukaryotic genome. Cell 95: 717–728. [DOI] [PubMed] [Google Scholar]
- Huang, J., J. M. Hsu and B. C. Laurent, 2004. The RSC nucleosome-remodeling complex is required for Cohesin's association with chromosome arms. Mol. Cell 13: 739–750. [DOI] [PubMed] [Google Scholar]
- Jan, L. Y., and Y. N. Jan, 1982. Antibodies to horseradish peroxidase as specific neuronal markers in Drosophila and in grasshopper embryos. Proc. Natl. Acad. Sci. USA 79: 2700–2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadam, S., and B. M. Emerson, 2003. Transcriptional specificity of human SWI/SNF BRG1 and BRM chromatin remodeling complexes. Mol. Cell 11: 377–389. [DOI] [PubMed] [Google Scholar]
- Kadam, S., G. S. McAlpine, M. L. Phelan, R. E. Kingston, K. A. Jones et al., 2000. Functional selectivity of recombinant mammalian SWI/SNF subunits. Genes Dev. 14: 2441–2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadesch, T., 2004. Notch signaling: the demise of elegant simplicity. Curr. Opin. Genet. Dev. 14: 506–512. [DOI] [PubMed] [Google Scholar]
- Kal, A. J., T. Mahmoudi, N. B. Zak and C. P. Verrijzer, 2000. The Drosophila brahma complex is an essential coactivator for the trithorax group protein zeste. Genes Dev. 14: 1058–1071. [PMC free article] [PubMed] [Google Scholar]
- Kasten, M., H. Szerlong, H. Erdjument-Bromage, P. Tempst, M. Werner et al., 2004. Tandem bromodomains in the chromatin remodeler RSC recognize acetylated histone H3 Lys14. EMBO J. 23: 1348–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, J. P., and K. Moses, 2001. The EGF receptor and notch signaling pathways control the initiation of the morphogenetic furrow during Drosophila eye development. Development 128: 2689–2697. [DOI] [PubMed] [Google Scholar]
- Lai, E. C., 2004. Notch signaling: control of cell communication and cell fate. Development 131: 965–973. [DOI] [PubMed] [Google Scholar]
- Lemieux, K., and L. Gaudreau, 2004. Targeting of Swi/Snf to the yeast GAL1 UAS G requires the Mediator, TAF IIs, and RNA polymerase II. EMBO J. 23: 4040–4050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine, S. S., I. F. King and R. E. Kingston, 2004. Division of labor in Polycomb group repression. Trends Biochem. Sci. 29: 478–485. [DOI] [PubMed] [Google Scholar]
- Lusser, A., and J. T. Kadonaga, 2003. Chromatin remodeling by ATP-dependent molecular machines. BioEssays 25: 1192–1200. [DOI] [PubMed] [Google Scholar]
- Manning, L., and C. Q. Doe, 1999. Prospero distinguishes sibling cell fate without asymmetric localization in the Drosophila adult external sense organ lineage. Development 126: 2063–2071. [DOI] [PubMed] [Google Scholar]
- Martens, J. A., and F. Winston, 2003. Recent advances in understanding chromatin remodeling by Swi/Snf complexes. Curr. Opin. Genet. Dev. 13: 136–142. [DOI] [PubMed] [Google Scholar]
- Mohrmann, L., and C. P. Verrijzer, 2005. Composition and functional specificity of SWI2/SNF2 class chromatin remodeling complexes. Biochim. Biophys. Acta 1681: 59–73. [DOI] [PubMed] [Google Scholar]
- Mohrmann, L., K. Langenberg, J. Krijgsveld, A. J. Kal, A. J. Heck et al., 2004. Differential targeting of two distinct SWI/SNF-related Drosophila chromatin-remodeling complexes. Mol. Cell. Biol. 24: 3077–3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narlikar, G. J., H. Y. Fan and R. E. Kingston, 2002. Cooperation between complexes that regulate chromatin structure and transcription. Cell 108: 475–487. [DOI] [PubMed] [Google Scholar]
- Neely, K. E., A. H. Hassan, C. E. Brown, L. Howe and J. L. Workman, 2002. Transcription activator interactions with multiple SWI/SNF subunits. Mol. Cell. Biol. 22: 1615–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolas, M., A. Wolfer, K. Raj, J. A. Kummer, P. Mill et al., 2003. Notch1 functions as a tumor suppressor in mouse skin. Nat. Genet. 33: 416–421. [DOI] [PubMed] [Google Scholar]
- Papoulas, O., S. J. Beek, S. L. Moseley, C. M. McCallum, M. Sarte et al., 1998. The Drosophila trithorax group proteins BRM, ASH1 and ASH2 are subunits of distinct protein complexes. Development 125: 3955–3966. [DOI] [PubMed] [Google Scholar]
- Papoulas, O., G. Daubresse, J. A. Armstrong, J. Jin, M. P. Scott et al., 2001. The HMG-domain protein BAP111 is important for the function of the BRM chromatin-remodeling complex in vivo. Proc. Natl. Acad. Sci. USA 98: 5728–5733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks, A. L., and M. A. Muskavitch, 1993. Delta function is required for bristle organ determination and morphogenesis in Drosophila. Dev. Biol. 157: 484–496. [DOI] [PubMed] [Google Scholar]
- Peterson, C. L., and M. A. Laniel, 2004. Histones and histone modifications. Curr. Biol. 14: R546–R551. [DOI] [PubMed] [Google Scholar]
- Peterson, C. L., and C. Logie, 2000. Recruitment of chromatin remodeling machines. J. Cell. Biochem. 78: 179–185. [DOI] [PubMed] [Google Scholar]
- Peterson, C. L., and J. L. Workman, 2000. Promoter targeting and chromatin remodeling by the SWI/SNF complex. Curr. Opin. Genet. Dev. 10: 187–192. [DOI] [PubMed] [Google Scholar]
- Reddy, G. V., and V. Rodrigues, 1999. A glial cell arises from an additional division within the mechanosensory lineage during development of the microchaete on the Drosophila notum. Development 126: 4617–4622. [DOI] [PubMed] [Google Scholar]
- Rohrbaugh, M., E. Ramos, D. Nguyen, M. Price, Y. Wen et al., 2002. Notch activation of yan expression is antagonized by RTK/pointed signaling in the Drosophila eye. Curr. Biol. 12: 576–581. [DOI] [PubMed] [Google Scholar]
- Saha, A., J. Wittmeyer and B. R. Cairns, 2002. Chromatin remodeling by RSC involves ATP-dependent DNA translocation. Genes Dev. 16: 2120–2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnepp, B., G. Grumbling, T. Donaldson and A. Simcox, 1996. Vein is a novel component in the Drosophila epidermal growth factor receptor pathway with similarity to the neuregulins. Genes Dev. 10: 2302–2313. [DOI] [PubMed] [Google Scholar]
- Schumacher, A., and T. Magnuson, 1997. Murine Polycomb- and trithorax-group genes regulate homeotic pathways and beyond. Trends Genet. 13: 167–170. [PubMed] [Google Scholar]
- Schweisguth, F., 2004. Notch signaling activity. Curr. Biol. 14: R129–R138. [PubMed] [Google Scholar]
- Sharma, V. M., B. Li and J. C. Reese, 2003. SWI/SNF-dependent chromatin remodeling of RNR3 requires TAF(II)s and the general transcription machinery. Genes Dev. 17: 502–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, X., H. Xiao, R. Ranallo, W. H. Wu and C. Wu, 2003. Modulation of ATP-dependent chromatin-remodeling complexes by inositol polyphosphates. Science 299: 112–114. [DOI] [PubMed] [Google Scholar]
- Shilo, B. Z., 2003. Signaling by the Drosophila epidermal growth factor receptor pathway during development. Exp. Cell Res. 284: 140–149. [DOI] [PubMed] [Google Scholar]
- Simon, J., 1995. Locking in stable states of gene expression: transcriptional control during Drosophila development. Curr. Opin. Cell Biol. 7: 376–385. [DOI] [PubMed] [Google Scholar]
- Simon, J. A., and J. W. Tamkun, 2002. Programming off and on states in chromatin: mechanisms of Polycomb and trithorax group complexes. Curr. Opin. Genet. Dev. 12: 210–218. [DOI] [PubMed] [Google Scholar]
- Simone, C., S. V. Forcales, D. A. Hill, A. N. Imbalzano, L. Latella et al., 2004. p38 pathway targets SWI-SNF chromatin-remodeling complex to muscle-specific loci. Nat. Genet. 36: 738–743. [DOI] [PubMed] [Google Scholar]
- Smith, S. T., S. Petruk, Y. Sedkov, E. Cho, S. Tillib et al., 2004. Modulation of heat shock gene expression by the TAC1 chromatin-modifying complex. Nat. Cell Biol. 6: 162–167. [DOI] [PubMed] [Google Scholar]
- Steger, D. J., E. S. Haswell, A. L. Miller, S. R. Wente and E. K. O'Shea, 2003. Regulation of chromatin remodeling by inositol polyphosphates. Science 299: 114–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudarsanam, P., V. R. Iyer, P. O. Brown and F. Winston, 2000. Whole-genome expression analysis of snf/swi mutants of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 97: 3364–3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamkun, J. W., R. A. Kahn, M. Kissinger, B. J. Brizuela, C. Rulka et al., 1991. The arflike gene encodes an essential GTP-binding protein in Drosophila. Proc. Natl. Acad. Sci. USA 88: 3120–3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamkun, J. W., R. Deuring, M. P. Scott, M. Kissinger, A. M. Pattatucci et al., 1992. brahma: a regulator of Drosophila homeotic genes structurally related to the yeast transcriptional activator SNF2/SWI2. Cell 68: 561–572. [DOI] [PubMed] [Google Scholar]
- Thomas, B. J., and D. A. Wassarman, 1999. A fly's eye view of biology. Trends Genet. 15: 184–190. [DOI] [PubMed] [Google Scholar]
- Tripoulas, N. A., E. Hersperger, D. La Jeunesse and A. Shearn, 1994. Molecular genetic analysis of the Drosophila melanogaster gene absent, small or homeotic discs1 (ash1). Genetics 137: 1027–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuda, L., R. Nagaraj, S. L. Zipursky and U. Banerjee, 2002. An EGFR/Ebi/Sno pathway promotes delta expression by inactivating Su(H)/SMRTER repression during inductive notch signaling. Cell 110: 625–637. [DOI] [PubMed] [Google Scholar]
- Vazquez, M., L. Moore and J. A. Kennison, 1999. The trithorax group gene osa encodes an ARID-domain protein that genetically interacts with the brahma chromatin-remodeling factor to regulate transcription. Development 126: 733–742. [DOI] [PubMed] [Google Scholar]
- Verheyen, E. M., K. J. Purcell, M. E. Fortini and S. Artavanis-Tsakonas, 1996. Analysis of dominant enhancers and suppressors of activated Notch in Drosophila. Genetics 144: 1127–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Versteege, I., N. Sevenet, J. Lange, M. F. Rousseau-Merck, P. Ambros et al., 1998. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature 394: 203–206. [DOI] [PubMed] [Google Scholar]
- Whitehouse, I., C. Stockdale, A. Flaus, M. D. Szczelkun and T. Owen-Hughes, 2003. Evidence for DNA translocation by the ISWI chromatin-remodeling enzyme. Mol. Cell. Biol. 23: 1935–1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winston, F., and M. Carlson, 1992. Yeast SNF/SWI transcriptional activators and the SPT/SIN chromatin connection. Trends Genet. 8: 387–391. [DOI] [PubMed] [Google Scholar]
- Yoon, S., H. Qiu, M. J. Swanson and A. G. Hinnebusch, 2003. Recruitment of SWI/SNF by Gcn4p does not require Snf2p or Gcn5p but depends strongly on SWI/SNF integrity, SRB mediator, and SAGA. Mol. Cell. Biol. 23: 8829–8845. [DOI] [PMC free article] [PubMed] [Google Scholar]