

Abstract
The comparative analysis of protein sequences depends crucially on measures of amino acid similarity or distance. Many such measures exist, yet it is not known how well these measures reflect the operational exchangeability of amino acids in proteins, since most are derived by methods that confound a variety of effects, including effects of mutation. In pursuit of a pure measure of exchangeability, we present (1) a compilation of data on the effects of 9671 amino acid exchanges engineered and assayed in a set of 12 proteins; (2) a statistical procedure to combine results from diverse assays of exchange effects; (3) a matrix of “experimental exchangeability” values EXij derived from applying this procedure to the compiled data; and (4) a set of three tests designed to evaluate the power of an exchangeability measure to (i) predict the effects of amino acid exchanges in the laboratory, (ii) account for the disease-causing potential of missense mutations in the human population, and (iii) model the probability of fixation of missense mutations in evolution. EX not only captures useful information on exchangeability while remaining free of other effects, but also outperforms all measures tested except for the best-performing alignment scoring matrix, which is comparable in performance.
MEASURES of the pairwise distance (or similarity) of amino acids provide the basis for scoring schemes in the alignment of sequences (Henikoff and Henikoff 1993) and in other types of comparative analysis (Wen et al. 1996; Yang et al. 1998; Atchley et al. 2001; Alexandre and Zhulin 2003). A great many such matrices exist: an incomplete listing available from the AAIndex database (Kawashima and Kanehisa 2000) includes 83 matrices of pairwise amino acid similarity or distance and 494 indices of amino acid properties. Formally related to these are various schemes to distinguish “conservative” from “radical” amino acid changes (Hughes et al. 1990; Hughes 1992; Rand et al. 2000; Zhang 2000).
A tacit assumption has been that the ultimate yardstick for measuring amino acid similarity is the propensity for evolutionary change from one amino acid to another. However, evolutionary transition probabilities, although they must reflect the operational exchangeability of amino acids in proteins, must also reflect rates of mutation from one kind of codon to another (in principle, they may also reflect subtle fitness effects unrelated to protein operation, due to different metabolic costs of different amino acids or to different translational efficiencies of different codons, and so on). In the simplest case in which new mutations are rare, and regardless of whether one is considering random or selective fixations, the rate of evolution is directly proportional to the rate at which mutation introduces new alleles (Kimura 1983).
Although mutational effects are rarely treated as important phenomena in their own right, they appear to be extremely important. For instance, each nonidentical amino acid pair can be assigned a “genetic code distance” Gij ∈ {1, 2, 3} equal to the minimum number of nucleotides that must be changed to switch from amino acid i to amino acid j; the different categories sometimes are referred to as “singlet,” “doublet,” and “triplet” exchanges. The practical importance of genetic code distance is amply demonstrated by the effectiveness of Fitch's matrix of “mutational” distance (Fitch 1966) as a source of match scores for protein sequence alignment (Feng et al. 1985). Furthermore, pairs of amino acids with the same genetic code distance may differ in the density of minimum-length mutational paths connecting them. Finally, specific mutational paths between codons may differ in rate due to nucleotide mutation biases. For instance, both Ala-Gly and Ala-Val are singlet exchanges with the same singlet path density (1 per codon), but Ala and Val are interconverted by a nucleotide transition mutation (GCN ↔ GTN) with a severalfold higher rate (Schaaper and Dunn 1991; Nachman and Crowell 2000) than the nucleotide transversion mutation that interconverts Ala and Gly (GCN ↔ GGN). Likewise, a bias favoring AT (or GC) base pairs would affect the relative rates of Glu → Gly (GAR → GGR) vs. Glu → Val (GAR → GTR), and such effects seem to have played an important role in the evolution of amino acid composition (Singer and Hickey 2000).
The ability to distinguish such mutational effects from selective effects relating to protein operation is of considerable interest. To address this issue requires a reliable measure of the exchangeability of amino acids in proteins that is free of mutational effects. Such a measure would not be easy to find. The distance measures of Grantham (1974) and Miyata et al. (1979) are used commonly as though they were pure measures of physicochemical distance (e.g., Li 1997; Krawczak et al. 1998; Yang et al. 1998; Graur and Li 2000), yet they are not. The approach taken by Grantham (1974) was to identify, from a large number of potentially relevant physicochemical properties, a set of three—volume, polarity, and composition—that, when their differences are assigned appropriate weights, provide an unusually good fit with observed evolutionary transition probabilities. Thus, Grantham's distances (and the derived measure of Miyata et al. 1979) represent a physicochemical parameterization of evolutionary propensities. Pure measures of amino acid exchangeability exist, but they are theoretical; e.g., Miyazawa and Jernigan (1993) computed expected effects of exchanges in proteins of known structure using a contact-energy model. However, because the validity of the theoretical model is uncertain, one does not know how well the results will apply to real exchanges in proteins.
An alternative strategy for deriving a mutationally unbiased measure based on actual amino acid exchanges would be to use data from the experimental manipulation of proteins. The published literature includes many studies in which a large pool of variant proteins, each differing from a reference protein (typically a wild type) by a single amino acid residue, is assayed for the effects of this change. Such exchanges clearly reflect the complexities of real proteins (albeit operating under laboratory conditions in vitro or in vivo). Furthermore, such exchanges can be produced and assayed systematically. For instance, Rennell et al. (1991) placed nonsense codons at every position (except the start codon) in a T4 lysozyme gene and then used a set of 13 suppressor strains to insert (separately) 13 different amino acids at each position. The resulting nonsense-site/suppressor combinations were assayed by growth of the bacteriophage in vivo, with the effect of the exchange being assigned to one of four categories (“−,” “+ −,” “+,” “+ +”) on the basis of the size of the bacteriophage plaque. When such studies do not introduce biases on the retention or loss of activity (e.g., by altering only active-site residues), they may serve as the basis for deriving a measure of exchangeability.
In this work, a set of such systematic exchange studies is identified, comprising 9671 amino acid exchanges in 12 different target proteins. An analysis of the activity distribution of variants suggests a common severity-of-effect distribution that can be used to combine results of diverse studies, so as to yield a measure of the mean effect of exchanging amino acid i to amino acid j. The resulting measure is here called “experimental exchangeability” (EX). EX and diverse other measures were evaluated for their power to (1) predict effects of experimental amino acid exchanges, (2) account for the disease-causing potential of different types of missense mutations in humans, and (3) model the acceptance of missense mutations in protein evolution. In these tests, the performance of EX exceeds or closely follows the best of the other measures tested, which include matrices based on sequence alignments, computational structural modeling of proteins, and ad hoc statistical measures incorporating physicochemical properties of amino acids. Of the available measures, EX seems to be the best-performing measure that is free of mutational biases, and it is the only measure that is likely to be substantially improved in the future by modest increases in the amount of available experimental data.
MATERIALS AND METHODS
Identification of studies for inclusion:
An initial set of three widely cited exchange studies (Axe et al. 1998; Kleina and Miller 1990; Rennell et al. 1991) was expanded by tracking citations forward and backward (utilizing the “Web of Science” service from the Institute for Scientific Information). To identify further studies, surveys were carried out by inspection of entries in the Protein Mutant Database (PMD) (Kawabata et al. 1999), as well as by keyword searches in PubMed. Candidate studies were then screened individually to identify studies with at least 20 single-exchange variants generated and assayed under conditions that do not appear to impose strong biases on (i) the set of sites subject to exchanges, (ii) the set of exchanges imposed on each site, and (iii) the set of exchange variants assayed for biological or biochemical effects. Ideally, each set should be a complete set, a randomly chosen subset, or, at the very least, an arbitrary subset based on some factor (typically, experimental convenience) extraneous to the issues raised by exchangeability.
Some judgment was exercised in the application of the above criteria. For instance, the choice to alter only conserved binding motifs by Slack et al. (2000) was considered a problematic bias, while the choice of Hortnagel et al. (1999) to alter only a continuous block of 20 sites in RecA (or the similar focus of Cunningham and Wells 1989 on a set of three blocks comprising 54 sites) was considered tolerable, partly because a 20-residue block is a sizeable block and partly because the experimenters were otherwise highly systematic in producing and assaying all 20 × 19 = 380 possible single-exchange variants within this block. The second criterion was not interpreted to exclude alanine-scanning studies, to the extent that alanine is an arbitrary subset of all amino acids. The third criterion does not necessarily exclude studies that rely on selective growth of a subset of variants, because in some cases (see supplementary materials at http://www.genetics.org/supplemental/) the membership of the subset of variants that failed to grow can be inferred, so that the composition of both the “inactive” and “active” classes is known.
Structural analysis:
Protein structure data are from the Protein Data Bank (Berman et al. 2000). For proteins with a known structure (see results), surface accessibility is calculated using the web interface to ASC (Eisenhaber and Argos 1993; Eisenhaber et al. 1995) provided by the Peer Bork group at EMBL (http://www.bork.embl-heidelberg.de/ASC/scr1-form.html). A site is classified as “buried” if the wild-type amino acid is <20% accessible relative to its accessibility in an Ala-X-Ala tripeptide.
The correction for sampling error in the frequencies of exposure for each amino acid (used in computing the context-averaged exchangeability, explained below) is based on accessibilities computed for a large and nonredundant set of structural data, namely the PDB_SELECT25 (Hobohm and Sander 1994) subset of 2216 structurally characterized chains with an upper limit of 25% pairwise sequence similarity. For this set of chains, computed surface accessibilities for 342,785 residues in 2159 chains are available in the Definition of Secondary Structures of Proteins (DSSP) database (Hooft et al. 1996). The resulting frequencies with which amino acids are exposed (>20% surface accessibility relative to an Ala-X-Ala tripeptide) are, in increasing order: Cys, 21.7%; Ile, 24.7%; Phe, 27.8%; Leu, 28.8%; Val, 29.0%; Trp, 34.3%; Met, 37.3%; Ala, 42.9%; Tyr, 42.9%; Gly, 56.9%; Thr, 59.9%; His, 60.0%; Ser, 62.0%; Pro, 66.6%; Asn, 72.8%; Gln, 76.8%; Arg, 77.9%; Asp, 78.1%; Glu, 83.8%; and Lys, 88.0%. These values correlate strongly (R2 = 0.93) with the values presented in a previous study with a 100-fold smaller set of data (Holbrook et al. 1990, Table II).
Assignment of scores to a common scale:
Some experimental exchange studies provide information on the proportion of variants pt observed to have activity below some threshold level, t. Such {t, pt} pairs are points on a cumulative frequency distribution function C(t), a nondecreasing function of t delimited by C(0) = 0 and C(∞) = 1. If a common function can be found to fit all studies, then it may be used to assign scores on the common scale of t, even for experiments that provide only a ranking of results with no activity measurements. To define a suitable cumulative frequency distribution function C(t) in the absence of a clear a priori expectation of its mathematical form, we find an empirical fit to C(t) = B(t)/(a + B(t)), where the basis function B(t) is chosen from a small number of simple functions (linear, exponential, or power function). In practice (see results), the power law allows the best fit, and thus the frequency distribution function is defined as
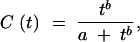 |
1 |
where a and b are estimated from the data (see Statistical procedures, below). Differentiating the cumulative distribution function, C(t), yields a frequency density function:
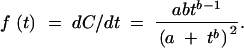 |
2 |
This distribution function may be used to convert ordinal values to continuous-activity values, as illustrated in Figure 1. First, given the frequencies of variants in each ordinal category, the lower and upper thresholds, tL and tU, may be estimated from the results of the regression of C on t (Equation 1). Second, given the values of t that define a category of effect (values that may be computed from Equation 2 or known empirically), observations in a category may be assigned a continuous value AL,U defined as the mean value for the category:
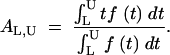 |
3 |
This has no analytical solution given the definition of f(t) in Equation 2; therefore, it is evaluated numerically (using Mathematica 4.0). As a numerical example, one may consider the hypothetical “minus” category depicted in Figure 1, which includes the 47% of variants with activities ranging from tL = 0 to the unknown upper threshold tU. If the regression (1) indicates that a = 0.28 and b = 0.67, then from Equation 1, tU = 0.12, or 12% wild-type activity, and the activity value assigned (by way of Equation 3) to variants in the minus category would be Aminus = 0.037, or 3.7% wild-type activity.
Figure 1.—

Assigning activity scores to categories based on a frequency distribution. (a) A hypothetical distribution of variants, with 47% in the “minus” class and the remainder in the “plus” class. (b) The fit of this classification to a known frequency distribution of effects on activity. For any given frequency distribution, there is a unique value T that divides the density into a minus class of 47% and a plus class with the remainder. Then, for this frequency distribution, variants in each class can be assigned a unique mean activity value (e.g., Aminus for variants in the minus category). This approach generalizes to any number of ranked categories.
In some cases, the effects of amino acid exchanges may be given, not by assignment to categories, but by a continuous value x from some measurement that does not have the units of relative wild-type activity (e.g., xi is a Kd or ΔΔG value). In such a case, the observed values can be ranked and treated as a set of categories (i.e., one category for each distinct value of xi) for the purpose of assigning activity scores. In general, this method makes the implicit assumptions (see discussion) that the scale of experimental values has a monotonic relationship with activity and that the polarity of this relationship is known (i.e., one knows which end of the scale represents low activity and which end represents high activity).
Statistical procedures:
Unless otherwise indicated, statistical analysis was performed with the Macintosh program JMP (SAS Institute 1999).
Frequency-activity regression:
The regression in Equation 1 is estimated (by nonlinear regression) from any {t, pt} pairs available from the set of 12 studies. Data points are weighted so that each study contributes equally. To assign weights by study size would be inappropriate, because most of the uncertainty is in the value of t, which is independent of study size, not pt, which is more reliable for larger studies. For example, in the T4 lysozyme study, the reported threshold value of t = 3% for the minimal activity of “+” variants might be off by a factor of two in either direction, whereas the range of the 95% confidence interval for pt = 328/1918 = 0.171, which is 0.154–0.189, is only a factor of 1.1 in either direction. In two cases (HIV-RT and insulin), quantitative activity values for all variants have been determined experimentally, and therefore the entire observed cumulative frequency distribution may be used in the regression.
Missing data, weights, and uncertainties:
For 6 of the 380 off-diagonal cells of the 20 ×20 exchangeability matrix, no observations are available, while the remaining cells represent varying numbers of observations (see results, Table 2). The missing EX values are excluded from all analyses, and the remaining values are analyzed using the number of observations as a weight. For instance, the symmetric form of exchangeability is computed with the formula Sij = (nijEXij + njiEXji)/(nij + nji); that is, the mean is weighted by the relevant numbers of observations n. When a standard error is given for an individual EXij value, this is the standard error derived by bootstrapping across studies (results are available from the authors).
TABLE 2.
Counts of exchanges by source (row) and destination (column)
| C | S | T | P | A | G | N | D | E | Q | H | R | K | M | I | L | V | F | Y | W | Total | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C | . | 7 | 2 | 7 | 7 | 9 | 1 | 1 | 7 | 6 | 6 | 8 | 6 | 3 | 1 | 7 | 2 | 8 | 9 | 2 | 99 |
| S | 45 | . | 13 | 45 | 61 | 46 | 8 | 5 | 40 | 39 | 37 | 44 | 40 | 4 | 7 | 42 | 5 | 42 | 44 | 3 | 570 |
| T | 33 | 56 | . | 54 | 60 | 44 | 16 | 7 | 34 | 35 | 35 | 43 | 39 | 5 | 21 | 34 | 9 | 32 | 33 | 5 | 595 |
| P | 21 | 33 | 21 | . | 45 | 30 | 5 | 6 | 22 | 26 | 29 | 39 | 26 | 5 | 6 | 41 | 5 | 22 | 20 | 6 | 408 |
| A | 52 | 68 | 16 | 71 | . | 80 | 5 | 12 | 59 | 53 | 54 | 56 | 55 | 4 | 2 | 61 | 25 | 53 | 54 | 3 | 783 |
| G | 57 | 59 | 15 | 46 | 77 | . | 11 | 24 | 53 | 45 | 43 | 63 | 48 | 10 | 14 | 45 | 43 | 42 | 43 | 13 | 751 |
| N | 27 | 39 | 17 | 31 | 40 | 34 | . | 16 | 27 | 29 | 40 | 31 | 36 | 7 | 17 | 35 | 9 | 28 | 35 | 7 | 505 |
| D | 30 | 33 | 7 | 34 | 58 | 57 | 24 | . | 53 | 33 | 46 | 33 | 33 | 5 | 5 | 35 | 25 | 32 | 49 | 4 | 596 |
| E | 25 | 34 | 6 | 31 | 58 | 49 | 5 | 15 | . | 32 | 27 | 29 | 33 | 4 | 3 | 32 | 13 | 25 | 28 | 3 | 452 |
| Q | 31 | 37 | 5 | 46 | 44 | 37 | 3 | 1 | 44 | . | 42 | 43 | 37 | 4 | 5 | 46 | 5 | 32 | 30 | 5 | 497 |
| H | 12 | 14 | 7 | 17 | 21 | 18 | 11 | 8 | 13 | 18 | . | 19 | 14 | 7 | 7 | 19 | 6 | 16 | 17 | 6 | 250 |
| R | 36 | 46 | 9 | 40 | 44 | 45 | 3 | 4 | 31 | 33 | 41 | . | 37 | 1 | 8 | 43 | 6 | 31 | 30 | 3 | 491 |
| K | 25 | 38 | 24 | 29 | 55 | 50 | 22 | 5 | 38 | 45 | 28 | 46 | . | 15 | 14 | 31 | 7 | 26 | 27 | 7 | 532 |
| M | 15 | 14 | 3 | 15 | 22 | 19 | 4 | 3 | 14 | 14 | 15 | 17 | 16 | . | 7 | 18 | 5 | 15 | 14 | 2 | 232 |
| I | 32 | 52 | 27 | 37 | 44 | 40 | 17 | 4 | 35 | 33 | 33 | 46 | 37 | 23 | . | 60 | 30 | 49 | 30 | 4 | 633 |
| L | 55 | 74 | 14 | 72 | 86 | 73 | 10 | 7 | 61 | 68 | 63 | 79 | 57 | 15 | 19 | . | 33 | 68 | 56 | 14 | 924 |
| V | 47 | 49 | 7 | 48 | 70 | 68 | 4 | 11 | 52 | 46 | 48 | 48 | 45 | 5 | 17 | 58 | . | 54 | 45 | 4 | 726 |
| F | 21 | 21 | 4 | 15 | 23 | 15 | 4 | 4 | 13 | 11 | 13 | 14 | 14 | 2 | 13 | 24 | 11 | . | 21 | 4 | 247 |
| Y | 29 | 27 | 0 | 16 | 26 | 22 | 11 | 12 | 14 | 15 | 26 | 18 | 14 | 1 | 0 | 16 | 0 | 31 | . | 2 | 280 |
| W | 11 | 8 | 1 | 6 | 6 | 12 | 0 | 0 | 5 | 6 | 5 | 9 | 6 | 1 | 0 | 11 | 1 | 6 | 6 | . | 100 |
| Total | 604 | 709 | 198 | 660 | 847 | 748 | 164 | 145 | 615 | 587 | 631 | 685 | 593 | 121 | 166 | 658 | 240 | 612 | 591 | 97 | 9671 |
Measures used for comparative evaluation:
The AAIndex database (Kawashima and Kanehisa 2000) lists nearly 100 measures of amino acid similarity or distance, not including some recently derived matrices (e.g., Venkatarajan and Braun 2001; Xia and Xie 2002). Rather than testing all available measures, we defined a set of six categories, drawing one or two representative measures from each category as explained in results. The BLOSUM series of matrices were taken from Henikoff and Henikoff's (1992) supplementary material, which provides five digits of precision (better than the reduced-precision matrices widely used by sequence alignment software). The VB (Venkatarajan and Braun 2001) matrix was obtained in electronic form from the authors, and the XX matrix was entered manually from Xia and Xie (2002). Other measures are from the AAIndex database (Kawashima and Kanehisa 2000).
Comparative evaluation using experimental exchanges:
The power of various measures of exchangeability was evaluated by predicting the results of experimental exchanges. Circularity in predicting experimental exchange effects using an EX measure is avoided by using a jackknife method in which the predictor applied to a set of results for a target protein T is the independent predictor EX−T based on results from all proteins except T. For target studies in which the effects of exchanges are given on a continuous scale (e.g., percentage of activity), a linear regression with the predictor is used. When the target results are ordinal (e.g., “−,” “+”), logistic regression is used. For testing the entire set of 9671 results from all 12 target studies, each result is paired with its independent predictor EX−T, and linear regression is used, with ordinal values converted to continuous-activity values as described (see above, Assignment of scores to a common scale). For purposes of interpreting the results of this test, it is helpful to define the power of a study as the number of variants assayed multiplied by the mean information content of an assay result. The mean information content of an ordinal assignment is −∑(fi log fi), where fi is the frequency of the ith category, ignoring uncertainty in the assignment itself, which reduces information content to an unknown degree. Continuous-valued results are ranked and treated as ordinal data for the purpose of computing information content (again, ignoring uncertainty in the ranking).
Comparative evaluation using data on human variation:
EX and other measures are used as predictors of disease-causing potential, defined (for each source-destination combination) as the ratio of the number of Human Gene Mutation Database (HGMD) entries (Stenson et al. 2003) to the number of HGV Base entries (Fredman et al. 2002; http://hgvbase.cgb.ki.se/), using only those HGVBase SNPs with “proven” status. This ratio of the number of entries for particular categories of variant is not the same as a ratio of population frequencies; nevertheless, it can be used as an estimator (e.g., as in Krawczak et al. 1998) under conditions in which genetic sites that contribute to the category are not saturated with detected variants.
Comparative evaluation using evolutionary probability of acceptance:
For use in a model of sequence evolution, similarity measures are converted to distances by subtracting each value of the measure from its maximum value. The minimum distance is thus zero, and in the Goldman-Yang model as implemented in the PAML package (Yang et al. 1998), amino acid pairs with this distance will be accepted with the highest probability possible for a nonsynonymous change. The nucleotide alignments used for evolutionary analysis are from two sources: the concatenated mitochondrial gene data from Yang et al. (1998) and the 10 eukaryotic sequence families analyzed by Qiu et al. (2004). Sites in the latter data sets with noncanonical codons (due to protist genes with noncanonical genetic codes) were removed from the analysis. The PAML software was executed using a control file specifying the codon model (seqtype = 1, model = 0, as in Yang et al. 1998) and the appropriate genetic code.
RESULTS
Studies chosen for inclusion:
From an initially broad literature search, the number of candidate studies was reduced by eliminating small studies (<20 variants) and then by eliminating studies with methodological biases, as described in materials and methods. These criteria reduced the number of candidate studies by roughly two orders of magnitude, to a total of 15. The most common grounds for eliminating a sufficiently large study were that the study aimed to identify only “critical” residues by focusing on a handful of predefined target residues (e.g., suspected active site residues) or that the study provided assay results only for a highly nonrandom subset of variants (e.g., variants that gained some crucial activity).
The studies selected for analysis, listed in Table 1 by target protein, comprise 12 different target proteins, 1266 altered sites, and 9671 individual exchanges. The exchanges are tabulated by source and destination amino acid in Table 2. No data are available for the doublet exchanges Y → T, Y → I, and Y → V nor for the triplet exchanges W → N, W → D, and W → I. For the remaining exchanges, the mean numbers of instances are 32.4 (range, 1–80) for singlets, 22.6 (range, 1–86) for doublets, and 12.4 (range, 1–32) for triplets.
TABLE 1.
Summary of experimental exchange studies
| Protein | Source species | Methoda | Sitesb | Variants | Exchange effects assayed | Citation |
|---|---|---|---|---|---|---|
| LacI | Escherichia coli | Sup | 328 (360) | 4038 | Operon repression, in vivo |
Markiewicz et al. (1994) |
| Lysozyme | Phage T4 | Sup | 155 (164) | 1918 | Plaque size, in vivo | Rennell et al. (1991) |
| Interleukin-3 | Homo sapiens | Sat | 103 (152) | 754 | Cell proliferation, in vivo |
Olins et al. (1995) |
| Barnase | E. coli | Sat | 109 (110) | 676 | RNAse activity, in vivo | Axe et al. (1998) |
| β-Lactamase | Pseudomonas | Sat | 27 (246) | 513 | Ampicillin resistance, in vivo |
Materon and Palzkill (2001) |
| RecA | E. coli | Sat | 20 (323) | 380 | Plaque size, in vivo | Hortnagel et al. (1999) |
| Reverse transcriptase | HIV | Sat | 109 (300) | 366 | RNA-dependent DNA polymerase activity, in vivo |
Wrobel et al. (1998) |
| Protease | HIV | Sat | 99 (99) | 336 | Protease activity, in vivo |
Loeb et al. (1989) |
| Protein V | Phage f1 | Sat | 86 (87) | 313 | Host inhibition, in vivo |
Zabin et al. (1991) |
| Nuclease | Staphylococcus | Scan | 143 (149) | 290 | ΔΔG, in vitro | Green et al. (1992); Meeker et al. (1996); Shortle et al. (1990) |
| Growth hormone | H. sapiens | Scan | 50 (191) | 50 | Dissociation constant, in vitro |
Cunningham and Wells (1989) |
| Insulin | H. sapiens | Scan | 37 (51) | 37 | Receptor affinity, in vitro |
Kristensen et al. (1997) |
| Total | 1266 | 9671 |
Sup, nonsense suppression; sat, saturation mutagenesis; scan, alanine scanning (Ala and Gly scanning in the case of Nuclease).
Amino acid positions altered (in parentheses, total number of positions in the protein).
The exchanges were engineered using three different methods: nonsense suppression (Miller 1991), that is, the introduction of a nonsense mutation at a site, followed by expression using a nonsense-suppressor tRNA that inserts some amino acid; cassette-based saturation mutagenesis (Reidhaar-Olson et al. 1991), resulting in a set of randomly generated codons at some position; and site-directed mutagenesis, resulting in a specific alternative at some position, as in alanine-scanning studies. For each of these 12 studies, the supplementary material at http://www.genetics.org/supplemental/ describes the experimental design (the method for generating and assaying variants) and the form of the results, which may be continuous values (e.g., percentage of wild-type activity) or ordinal values (i.e., ranked categories, such as −, + −, and +).
A common severity-of-effect distribution as the basis for combining results:
The 12 studies differ in the target protein, the type of assay performed, and the form of the results (Table 1; supplementary material at http://www.genetics.org/supplemental/). Results from different studies are not directly comparable: a value of + might mean “phage growth” in one study, and “drug resistance” in another study; a value of 0.4 might mean ΔΔG of 0.4 in one study and an activity of 40% of wild type in another study.
A method for combining such data would seem to require: (i) a precise physical model relating protein thermodynamics and chemistry to assays of enzymological or biological activity, (ii) an arbitrary rescaling of diverse results (e.g., each result converted to 0 or 1) on the assumption that arbitrary biases will cancel out given sufficient data, or (iii) a heuristic model that relates the results of different studies through some common parameter(s). The first approach is not possible, and the assumptions of the second approach are not justified, given the relatively small number of studies.
The possibility of a heuristic approach based on a common severity-of-effect distribution can be illustrated by reference to the barnase study of Axe et al. (1998), the T4 lysozyme study of Rennell et al. (1991), and the β-lactamase study of Materon and Palzkill (2001) (see Table 1). In each case, a large number of variants were produced and assayed to yield a + or − outcome relative to an arbitrary threshold level of activity. However, the studies differ dramatically in their outcomes: in the barnase case, only 4.9% of variants were inactive; in the lysozyme case, 20%; and in the β-lactamase case, 93%. This might be due to inherent differences between barnase, lysozyme, and β-lactamase. However, in the barnase case, the experimenters define inactive as having <∼0.2% of the wild-type protein activity (Mossakowska et al. 1989; Axe et al. 1998); in the lysozyme case, the threshold is 3% activity (Rennell et al. 1991); and in the β-lactamase case, the threshold is ∼50% activity (T. Palzkill, personal communication). Thus, the higher the threshold is, the fewer the variants are that surpass it, suggesting an underlying distribution of effects on activity that applies across studies. To the extent that such a common distribution applies, it can serve as the basis for a method to assign scores on a common scale, as illustrated in Figure 1 (see materials and methods).
Available data on the distribution of activity effects are plotted in Figure 2. A total of eight threshold values are available from five studies. For two additional studies (insulin and HIV-RT), activity values for all variants are reported, providing a finely discretized distribution. As described in materials and methods, these data may be used to estimate the shape of a common frequency distribution, on the assumption that such a distribution exists. This assumption is borne out by the close empirical fit to a power law (specifically, y = 0.91x0.374), the R2 value for which is 0.78 (residual sum-of-squares, 0.19). On the basis of this result, the data were then fit to Equation 1, a function that is based on the power law, but that has the properties of a cumulative frequency distribution (see materials and methods). The curved line in Figure 2 shows the best fit to Equation 1 (a = 0.278, b = 0.666), with a residual sum-of-squares of 0.13. Possible cumulative frequency distribution equations based on other candidate functions (e.g., linear, exponential) showed a worse empirical fit. This common severity-of-effect distribution may be used as the basis for transforming results of any experimental study to a common scale, nominally a scale of activity.
Figure 2.—

Empirical severity-of-effect distribution. The observed frequency of amino acid exchange variants pT that fall below some threshold of activity T is shown as a function of the threshold, on a double-log scale. Data on this relationship are available from seven studies (for details, see supplementary materials at http://www.genetics.org/supplemental/): the lysozyme (pink dot), barnase (black dot), and β-lactamase (yellow dot) studies each contribute a single point; two points are available from the interleukin-3 study (brown dots); three points from the LacI study (gray dots); and the observed discretized frequency distribution is available for 37 insulin variants (blue dots) and 366 HIV-RT variants (orange dots). The sizes of dots represent weights assigned to each point for purposes of regression. The dashed line is the best fit (residual sum-of-squares, 0.13) to a cumulative frequency distribution based on the power law (Equation 1).
EX:
The “experimental exchangeability from i to j,” or EXij, is the mean activity of variants with an exchange from amino acid i to amino acid j. For instance, Table 2 indicates that data on 34 T → E exchanges are available, and thus EXT,E will be the average of 34 values, each representing the fraction of wild-type activity in the variant protein, including both experimentally determined values (e.g., for variants of HIV-RTase) and estimates assigned using Equation 3 (materials and methods). Individual EX values have considerable uncertainty. The mean standard deviation of individual EX values, computed by bootstrap resampling (results not shown), is 0.071 for resampling among studies and 0.056 for resampling among individual exchanges.
By taking advantage of available structure data (see materials and methods and supplementary material at http://www.genetics.org/supplemental/), it is possible to classify most of the 1266 experimentally altered protein sites into surface sites or buried sites, so that exchangeability can be computed separately for each class of sites. The resulting pair of matrices, not presented here, provides a means to correct for statistical error in sampling surface and buried sites for each amino acid. The corrected “context-averaged” exchangeability value is the weighted average of the surface and buried exchangeability, where the weights for a specific exchange are based on the background frequency with which the source amino acid is found in each context (see materials and methods). The corrected values are very similar to the uncorrected values, performing only slightly better in the tests described below. Henceforth, the context-averaged exchangeability is treated as the definitive version of experimental exchangeability and is referred to simply as EX.
The values of EX (context averaged, as just noted) are given to three decimal places in Table 3, with source amino acids by row and destination amino acids by column. The mean exchangeability-as-source is given for each amino acid in the last column of Table 3, with the mean exchangeability-as-destination in the last row. The grand mean of exchangeability is 0.29. Alanine is the best replacement for other residues, with an exchangeability-as-destination of 0.41. The amino acid that is the most readily replaced is Lysine, with EXsrc = 0.41, though its exchangeability-as-destination is notably poor, 0.23.
TABLE 3.
Exchangeability (×1000) by source (row) and destination (column)
| C | S | T | P | A | G | N | D | E | Q | H | R | K | M | I | L | V | F | Y | W | EXsrc | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C | . | 258 | 121 | 201 | 334 | 288 | 109 | 109 | 270 | 383 | 258 | 306 | 252 | 169 | 109 | 347 | 89 | 349 | 349 | 139 | 280 |
| S | 373 | . | 481 | 249 | 490 | 418 | 390 | 314 | 343 | 352 | 353 | 363 | 275 | 321 | 270 | 295 | 358 | 334 | 294 | 160 | 351 |
| T | 325 | 408 | . | 164 | 402 | 332 | 240 | 190 | 212 | 308 | 246 | 299 | 256 | 152 | 198 | 271 | 362 | 273 | 260 | 66 | 287 |
| P | 345 | 392 | 286 | . | 454 | 404 | 352 | 254 | 346 | 384 | 369 | 254 | 231 | 257 | 204 | 258 | 421 | 339 | 298 | 305 | 335 |
| A | 393 | 384 | 312 | 243 | . | 387 | 430 | 193 | 275 | 320 | 301 | 295 | 225 | 549 | 245 | 313 | 319 | 305 | 286 | 165 | 312 |
| G | 267 | 304 | 187 | 140 | 369 | . | 210 | 188 | 206 | 272 | 235 | 178 | 219 | 197 | 110 | 193 | 208 | 168 | 188 | 173 | 228 |
| N | 234 | 355 | 329 | 275 | 400 | 391 | . | 208 | 257 | 298 | 248 | 252 | 183 | 236 | 184 | 233 | 233 | 210 | 251 | 120 | 272 |
| D | 285 | 275 | 245 | 220 | 293 | 264 | 201 | . | 344 | 263 | 298 | 252 | 208 | 245 | 299 | 236 | 175 | 233 | 227 | 103 | 258 |
| E | 332 | 355 | 292 | 216 | 520 | 407 | 258 | 533 | . | 341 | 380 | 279 | 323 | 219 | 450 | 321 | 351 | 342 | 348 | 145 | 363 |
| Q | 383 | 443 | 361 | 212 | 499 | 406 | 338 | 68 | 439 | . | 396 | 366 | 354 | 504 | 467 | 391 | 603 | 383 | 361 | 159 | 386 |
| H | 331 | 365 | 205 | 220 | 462 | 370 | 225 | 141 | 319 | 301 | . | 275 | 332 | 315 | 205 | 364 | 255 | 328 | 260 | 72 | 303 |
| R | 225 | 270 | 199 | 145 | 459 | 251 | 67 | 124 | 250 | 288 | 263 | . | 306 | 68 | 139 | 242 | 189 | 213 | 272 | 63 | 259 |
| K | 331 | 376 | 476 | 252 | 600 | 492 | 457 | 465 | 272 | 441 | 362 | 440 | . | 414 | 491 | 301 | 487 | 360 | 343 | 218 | 409 |
| M | 347 | 353 | 261 | 85 | 357 | 218 | 544 | 392 | 287 | 394 | 278 | 112 | 135 | . | 612 | 513 | 354 | 330 | 308 | 633 | 307 |
| I | 362 | 196 | 193 | 145 | 326 | 160 | 172 | 27 | 197 | 191 | 221 | 124 | 121 | 279 | . | 417 | 494 | 331 | 323 | 73 | 252 |
| L | 366 | 212 | 165 | 146 | 343 | 201 | 162 | 112 | 199 | 250 | 288 | 185 | 171 | 367 | 301 | . | 275 | 336 | 295 | 152 | 248 |
| V | 382 | 326 | 398 | 201 | 389 | 269 | 108 | 228 | 192 | 280 | 253 | 190 | 197 | 562 | 537 | 333 | . | 207 | 209 | 286 | 277 |
| F | 176 | 152 | 257 | 112 | 236 | 94 | 136 | 90 | 62 | 216 | 237 | 122 | 85 | 255 | 181 | 296 | 291 | . | 332 | 232 | 193 |
| Y | 142 | 173 | . | 194 | 402 | 357 | 129 | 87 | 176 | 369 | 197 | 340 | 171 | 392 | . | 362 | . | 360 | . | 303 | 258 |
| W | 137 | 92 | 17 | 66 | 63 | 162 | . | . | 65 | 61 | 239 | 103 | 54 | 110 | . | 177 | 110 | 364 | 281 | . | 142 |
| EXdest | 315 | 311 | 293 | 192 | 411 | 321 | 258 | 225 | 262 | 305 | 290 | 255 | 225 | 314 | 293 | 307 | 305 | 294 | 279 | 172 | 291 |
Italic and underlined values are one standard deviation above and below (respectively) the mean, where means and deviations are computed separately for EX, EXsrc (exchangeability as source), and EXdest (exchangeability as destination).
The exchangeability matrix has no diagonal values and no values for Y → T, Y → I, Y → V, W → N, W → D, and W → I exchanges, due to the lack of data noted earlier (Table 2). If needed, the missing values may be interpolated by averaging the exchangeability-as-source of the source amino acid, and the exchangeability-as-destination of the destination amino acid. A symmetric exchangeability matrix can be computed with EXSij = EXSji defined as the average of EXij and EXji, with each value weighted by the number of underlying observations in Table 2. The missing values are irrelevant to the tests of power below because: (i) the cross-validation obviously includes no observations of these types, since none are present in the original data set; and (ii) the other tests use only the values for singlet exchanges, whereas the six missing paths are doublet and triplet exchanges. The symmetric matrix EXSij has no missing values because exchange data are available for every possible unordered pair of amino acids.
Comparative evaluation of amino acid similarity measures:
Applying the heuristic procedure described above to available data on experimental exchanges yields an EX matrix that must reflect, to some unknown degree, the average effects of exchanging one amino acid for another. However, it remains to be demonstrated how well this procedure captures useful information about the “exchangeability” of amino acids or even if such a concept is valid. To address such questions there is neither a convenient existing benchmark nor a precise and validated theoretical model. For instance, on a priori grounds, one expects that greater exchangeability should correlate with greater similarity in crucial physicochemical properties such as volume and hydrophobicity, and indeed this correlation is observed for EX and various other measures (not shown). Yet, this relationship cannot be used to distinguish among such measures, because the prior expectation establishes only the polarity of the correlation, not its exact form.
Thus, the task of evaluating a measure of amino acid exchangeability presents a challenge for which new methods are required. To confront this challenge, three distinct tests, each based on an independent source of data, have been developed and applied to EX and a set of measures chosen for purposes of comparison. The logic of each test is that, to the extent that predictable statistical regularities associated with effects of amino acid changes in proteins are captured by the procedure used here for deriving an EX matrix, the resulting EX matrix should be a statistically significant predictor of patterns involving amino acid changes in proteins. To reduce the number of statistical tests, rather than including all known matrices of similarity or distance, we define five categories of measures other than EX (which is in its own category as an empirical measure of pure exchangeability) and choose a prominent example from each:
Theoretical models of amino acid exchangeability in proteins. The MJ matrix of Miyazawa and Jernigan (1993) is used.
Empirical models of evolutionary transition probabilities. The WAG matrix (Whelan and Goldman 2001) provides maximum-likelihood estimates of transition probabilities for a Markov model of evolutionary amino acid replacement.
Physicochemical parameterizations of evolutionary transition probabilities. The so-called “biochemical distance” measures of Grantham (1974) and Miyata et al. (1979) are widely used.
Sequence alignment match-score matrices. Matrices that supply match scores for alignment algorithms are the most familiar type of amino acid similarity measure. Formally, the Sij values of such a matrix are odds ratios of true juxtaposition to false juxtaposition of residues i and j (Altschul 1991). The BLOSUM series of matrices based on conserved sequence blocks is widely used in homology searches and performs better than other measures in systematic tests (Henikoff and Henikoff 1992).
Miscellaneous heuristic measures. The VB matrix (Venkatarajan and Braun 2001) is derived from multidimensional scaling of a diverse set of 237 diverse amino acid properties. The XX matrix (Xia and Xie 2002) is based on observed neighbor frequencies of amino acids in protein sequences and represents a distinctive new approach.
Prediction of effects of experimental amino acid exchanges:
The data from experimental genetics collated for this study (9671 experimental exchanges) may be used as targets for prediction. Circularity in the use of EX is avoided by using, for each target protein T, the independent predictor EX−T (see materials and methods). Table 4 shows the results of this test, using logistic regressions for studies with ordinal results (e.g., −, +) and linear regressions for studies with continuous-valued results (e.g., percentage of activity). Given that the number of tests is on the order of 100 (12 target studies multiplied by eight types of predictors), a probability <5 × 10−4 is marginally significant when considering a result chosen ex posteriori, and a probability <10−6 is highly significant.
TABLE 4.
Comparative evaluation: prediction of experimental exchange effects
|
R2 and probability (in parentheses) for prediction with each target studya
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Study (typeb) | Power | EX−T | EXS−T | BLO100 | VB | XX | Miyata | WAG | MJ | Grantham |
| LacI (r) | 6241.5 |
0.030 (1.0E-63) |
0.020 (2.6E-40) |
0.016 (1.3E-31) |
0.018 (5.0E-35) |
0.013 (8.0E-27) |
0.012 (1.3E-24) |
0.011 (1.0E-22) |
0.008 (4.0E-16) |
0.0005 (3.2E-01) |
| T4 lysozyme (r) | 2736.9 |
0.049 (2.0E-42) |
0.048 (5.4E-37) |
0.020 (3.0E-18) |
0.016 (7.0E-15) |
0.017 (6.0E-16) |
0.011 (1.5E-10) |
0.016 (3.0E-15) |
0.013 (3.4E-12) |
0.008 (2.6E-08) |
| IL-3 (r) | 1013.7 |
0.040 (1.5E-11) |
0.040 (1.5E-12) |
0.026 (1.1E-09) |
0.027 (8.0E-10) |
0.029 (2.0E-10) |
0.023 (1.7E-08) |
0.013 (3.0E-04) |
0.023 (1.4E-08) |
0.021 (4.6E-08) |
| HIVProt (r) | 505.4 |
0.098 (1.0E-15) |
0.071 (2.0E-13) |
0.104 (1.7E-17) |
0.096 (2.0E-16) |
0.050 (3.0E-09) |
0.060 (8.0E-11) |
0.095 (3.6E-16) |
0.039 (1.4E-07) |
0.0522 (1.5E-09) |
| RecA (r) | 463 | 0.016 (9.0E-03) |
0.007 (8.7E-02) |
0.019 (5.0E-04) |
0.011 (7.0E-03) |
0.0147 (2.0E-03) |
0.024 (8.0E-05) |
0.009 (1.4E-02) |
0.000 (7.0E-01) |
0.0096 (9.1E-01) |
| β-Lac (r) | 191.8 | 0.009 (1.6E-01) |
0.008 (1.8E-01) |
0.023 (1.4E-02) |
0.011 (1.0E-01) |
0.012 (7.0E-02) |
0.013 (6.1E-02) |
0.012 (8.0E-02) |
0.050 (3.0E-04) |
0.019 (2.4E-02) |
| Barnase (r) | 190.2 | 0.047 (6.0E-04) |
0.060 (2.0E-05) |
0.053 (2.0E-04) |
0.050 (3.0E-04) |
0.045 (6.0E-04) |
0.031 (5.0E-03) |
0.038 (1.5E-03) |
0.012 (7.8E-02) |
0.022 (1.7E-02) |
| RTase (c) | 2170.1 |
0.163 (1.0E-15) |
0.165 (1.0E-12) |
0.164 (7.0E-16) |
0.099 (9.9E-02) |
0.072 (2.0E-07) |
0.110 (8.0E-11) |
0.150 (3.0E-15) |
0.024 (3.0E-04) |
0.091 (4.3E-09) |
| f1 pV (c) | 992.6 | 0.081 (3.0E-07) |
0.064 (6.5E-06) |
0.105 (4.1E-09) |
0.063 (6.3E-02) |
0.040 (4.0E-04) |
0.039 (5.0E-04) |
0.092 (4.1E-08) |
0.024 (6.0E-03) |
0.044 (2.0E-04) |
| Nuclease (c) | 1650 |
0.129 (3.0E-10) |
0.140 (5.0E-11) |
0.112 (5.5E-09) |
0.029 (3.4E-03) |
0.024 (8.3E-02) |
0.042 (4.0E-04) |
0.021 (1.4E-02) |
0.082 (6.8E-07) |
0.019 (1.8E-02) |
| hGH (c) | 254.7 | 0.017 (3.6E-01) |
0.001 (8.0E-01) |
0.015 (3.9E-01) |
0.010 (4.9E-01) |
0.034 (2.0E-01) |
0.018 (3.6E-01) |
0.013 (4.3E-01) |
0.062 (8.2E-02) |
0.016 (3.9E-01) |
| Insulin (c) | 184.8 | 0.026 (3.4E-01) |
0.045 (2.1E-01) |
0.087 (7.7E-02) |
0.002 (7.8E-01) |
0.038 (2.5E-01) |
0.036 (2.6E-01) |
0.079 (9.2E-02) |
0.061 (1.4E-01) |
0.006 (6.5E-01) |
| All |
0.0373 (8.0E-82) |
0.029 (2.0E-63) |
0.03631 (1.0E-79) |
0.02631 (5.0E-58) |
0.02458 (3.0E-54) |
0.02289 (1.0E-50) |
0.02204 (9.0E-49) |
0.01698 (7.0E-38) |
0.00918 (3.6E-21) |
|
Italic and underlined values indicate the best and next-best (respectively) predictors by R2 for each target study. EXS is not included in the rankings because it is redundant with EX.
Depending on whether the data are ordinal (r) or continuous (c), predictions use logistic or linear regression, respectively.
Overall predictive power reflected in R2 values is low, presumably due to crucial context effects not included in the model (i.e., every site has a specific context in a protein that is not addressed), such that the best predictor explains only 2.6–16.4% of variance in effects of individual substitutions. Systematic differences in predictability of target studies are explained predominantly by the power of the study (Table 4), defined as the number of assayed variants multiplied by the mean information content of an assay result. For instance, considering the best predictor, study power accounts for 82% of the between-target-study variance in predictability [i.e., R2 = 0.82 for the regression of −log(probability) on study power].
EX−T is the best predictor in the combined test, while BLOSUM100 (BLO100), the best of the five BLOSUM levels tested (30, 45, 62, 80, and 100), has a larger number of first-place and second-place results in the separate tests (Table 4). In general, the order of effectiveness of predictors is {EX−T, BLO100} > {VB, XX, Miyata, WAG} > Grantham. The symmetric matrix, EXS−T, performs better than the intermediate predictors but worse than EX−T and BLO100. In such comparative tests, the predictive power of EX−T is aided by its asymmetry and by the availability of statistical weights (the counts in Table 2). By contrast, all the other measures are symmetric and without weights. Of these two aspects, asymmetry is important, as indicated above by the reduced performance of EXS−T, while weighting has relatively little effect (results not shown).
Correlation with disease-causing potential of human missense mutants:
The analysis of deleterious human variants is a problem for which an asymmetric measure of exchangeability would be useful, since typically one can distinguish which allele is the ancestral wild-type allele and which is the mutant. The nearly 19,000 entries for missense mutants in HGMD (Stenson et al. 2003) can be categorized on the basis of the source (wild-type) amino acid and the destination (mutant) amino acid. In principle, as long as the probability of sampling remains low (both in regard to individuals bearing a particular haplotype and in regard to haplotypes within a particular amino acid exchange category), the number of entries in HGMD for some category (e.g., Arg → Thr) is an estimator of the total frequency of occurrence of disease-causing variants of that category, modulated by a likelihood of clinical characterization. However, the relative chance of clinical characterization of a point mutation in a protein-coding region is based solely on the disease-causing propensity and the population frequency and not on experimental detectability, given that all types of missense mutations are equally detectable by the standard experimental procedure of DNA sequencing. The population frequency, in turn, is a function of (i) the frequency of the source codon(s), (ii) the mutation rate to the destination codon(s), and (iii) the acceptability of the mutant alleles (i.e., due to their mean effects on survival and reproduction).
Conveniently, the latter three factors are subsumed in the corresponding category frequencies from HGVBase (Fredman et al. 2002), a database of human single-nucleotide polymorphisms (SNPs). To the extent that confounding cross-factor effects can be ignored, then, dividing the number (or frequency) of HGMD entries by the number of HGVBase entries for the same type of missense change would cancel out extraneous effects of codon usage and mutation, leaving only the disease-causing potential. Thus, the logic of this test is that, if the method for deriving EX captures useful information on effects of amino acid changes in proteins, EX should correlate negatively with the HGMD/HGVBase ratio. Note that this test scrutinizes only singlet exchanges, given that SNP data, by definition, are single-nucleotide polymorphisms.
In general, measures of amino acid similarity or distance show a better linear fit to the log of the disease-causing potential than to the untransformed value. Figure 3 shows the regression for the best predictor, EX, which explains half of the variance in the log(HGMD/HGVBase) ratio. The values of R2 for the various predictors are: EX, 0.494 (EXS, 0.499); BLOSUM100, 0.475; WAG, 0.368; Grantham, 0.352; Miyata, 0.330; XX, 0.325; VB, 0.299; and MJ, 0.076.
Figure 3.—

Relationship of disease-causing potential to EX. The vertical scale is the log of the disease-causing potential, defined as the ratio of the number of HGMD (Krawczak and Cooper 1997) entries for a given missense class, to the number of HGVBase (Fredman et al. 2002) entries for the same class. For reasons explained in the text, this ratio is expected to reflect disease-causing potential and to be free of confounding effects of mutation. The solid line shows the weighted least-squares regression, y = 4.08 − 6.38x, with weights based on Table 2 (weight of each point is reflected by its size). EX explains 49% of the variance in the log HGMD/HGVBase ratio, more than any other measure tested. Given the observed regression, one way to describe how HGMD is enriched (relative to HGVBase) in low-exchangeability variants is to note that the bottom one-third of the distribution of EX values is enriched 2.4-fold relative to the overall sample and ∼9-fold relative to the top one-third.
As for the previous test using experimental exchanges, the higher levels of the BLOSUM series of matrices performed better; e.g., R2 for BLOSUM62 was 0.444, and for BLOSUM30 it was 0.309 (the BLOSUM level is the upper limit of sequence identity among pairs of sequences used to compute the matrix). A potential explanation is that closely related proteins should more accurately reflect exchangeability due to the shared context in which the differences arise. That is, when the first exchange from i to j takes place in one of two initially identical proteins, the resulting difference occurs in an identical context, and thus the pairing of i and j in the aligned sequences is a more accurate indication of the exchangeability of i and j than the same pairing of residues in proteins that have diverged so that they are only 30% identical. Indeed, we find that, for singlet and doublet exchanges, the slope of the correlation of BLOSUM scores on EXS values is steepest for BLOSUM100 and becomes flatter with decreasing BLOSUM level (results not shown). This observation is not necessarily in conflict with the results of Benner et al. (1994), who argue [from a discrepancy that arises in extrapolating the percent accepted mutation (PAM) model] that the effect of minimal mutation distance on the pattern of divergence decreases as proteins diverge. The effect of minimum mutational distance also diminishes with the BLOSUM level (i.e., the mean BLOSUM scores for singlets, doublets, and triplets become less extreme).
Use as a model of acceptance of missense changes in evolution:
While the above results demonstrate that EX performs well in predicting the effects of a random sample of exchanges (the experimental data) or a sample enriched for damaging exchanges (the disease-associated variants), one might argue that a more subtle measure is needed for modeling evolutionary change, to the extent that (presumably) it is a sample enriched for benign or innocuous exchanges. The problem of compensating for mutational effects in evolution is addressed by the phylogenetic analysis by maximum likelihood (PAML) software of Yang (1997) for numerical analysis of maximum likelihood models. The basis for this approach is as follows. Molecular evolution is often characterized as an origin-fixation process with a steady-state rate equal to the product of the rate of introduction of new mutants, μN, and their probability of fixation, π: in the simplest case, for neutral variants, π = 1/N, while for significantly beneficial mutants, π ≈ 2s, where s is the coefficient of selection (Kimura 1983). Thus, Goldman and Yang (1994)(see also Muse and Gaut 1994) introduced a “mechanistic” model of codon change with separate factors for mutation and acceptance. The mutational factor is modeled using a nucleotide substitution mutation model applied to codons. The acceptance factor for different types of missense mutations is modeled using a linear or geometric transformation (with two fitted parameters) of a user-supplied, symmetric measure of amino acid distance. Yang et al. (1998) tested five symmetric measures of amino acid distance: differences in polarity, volume, and composition; Grantham's distances (Grantham 1974); and Miyata's distances (Miyata et al. 1979).
Here, to evaluate various measures, a likelihood analysis was carried out using PAML with the original data set of Yang et al. (1998) and with 10 additional data sets from Qiu et al. (2004), as described in materials and methods. Measures of similarity, such as EXS or BLOSUM, are converted into the required form of a distance measure by subtracting each value from the maximum. The likelihood score that results from an analysis can be used to evaluate the performance of a distance measure in comparison to other measures (all tests have the same number of parameters). As with the previous test involving disease-causing variants, this test evaluates power only with respect to singlet exchanges, because these are the only kind used in the Goldman-Yang model.
The results of this comparative analysis are shown in Table 5. For each of the 11 data sets, log(likelihood) values are shown for the geometric transformation only, which (as in Yang et al. 1998) generally yields higher likelihoods than the linear transformation. The best measure for use as an acceptance function is BLOSUM100, which outperforms the second-best measure, EXS, in terms of both the sum of log(likelihood) values and the number of first- and second-place results. The order of performance is BLO100 > EXS > {VB, Miyata, WAG, Grantham} > MJ.
TABLE 5.
Comparative evaluation: PAML acceptance function
| Log-likelihood of observed family data given an acceptance function based on the predictor at lefta
|
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Actin | AdhII | AdhI | Aldh | CuZnSOD | EF1Alpha | GAPDH | HSP70 | MnFeSOD | TPI | mtCDNA | Total | |
| BLO100 | −49,185 | −25,526 | −25,721 | −45,735 | −8,293 | −24,745 | −16,231 | −57,489 | −11,187 | −8,482 | −29,835 | −302,429 |
| EXS | −49,116 | −25,653 | −25,879 | −45,952 | −8,328 | −24,675 | −16,261 | −57,641 | −11,069 | −8,480 | −29,881 | −302,937 |
| VB | −49,350 | −25,618 | −25,818 | −45,900 | −8,353 | −24,857 | −16,297 | −57,711 | −11,222 | −8465 | −29,869 | −303,458 |
| Miyata | −49,358 | −25,643 | −25,864 | −46,086 | −8,401 | −24,770 | −16,321 | −57,790 | −11,199 | −8,508 | −29,890 | −303,829 |
| WAG | −49,400 | −25,654 | −25,872 | −46,020 | −8,358 | −24,868 | −16,285 | −57,708 | −11,248 | −8,528 | −29,887 | −303,830 |
| XX | −49,259 | −25,656 | −25,926 | −46,087 | −8,387 | −24,792 | −16,345 | −57,866 | −11,188 | −8,500 | −29,867 | −303,872 |
| Grantham | −49,578 | −25,720 | −25,931 | −46,285 | −8,410 | −24,915 | −16,398 | −57,971 | −11,226 | −8,538 | −29,912 | −304,883 |
| MJ | −49,561 | −25,785 | −26,062 | −46,423 | −8,481 | −24,896 | −16,332 | −57,998 | −11,242 | −8,517 | −29,907 | −305,204 |
Italic values, best predictor for this gene family (column); underlined values, next-best predictor.
DISCUSSION
A measure of the exchangeability of amino acids has been computed from results of 9671 exchanges in 12 proteins, based on a set of experimental studies chosen to avoid systematic biases in the assessment of exchange effects (Tables 1 and 2). In this set of studies, the relationship observed between frequency and severity of effect suggests a common distribution that provides a basis for combining results from different studies (Figures 1 and 2) to yield a measure of mean effect, called EX (Table 3). This measure has been evaluated, in comparison to a sample of other types of measures, by a statistical cross-validation using the data on experimental exchanges (Table 4), by measuring its correlation with the disease-causing potential of human missense mutants (Figure 3), and by testing its ability to serve as the basis for evolutionary probabilities of acceptance (Table 5). On the basis of the results of these tests, EX is recommended as the only measure of the mean effects of amino acid exchanges that performs well and that is known to avoid potentially confounding effects of mutation.
The concept of a pure measure of exchangeability is largely novel, as is the method of deriving EX, and this novelty raises a number of questions. How does the severity-of-effect model provide a basis for combining results from different studies? What are the most likely sources of bias and error in this approach? How could EX be improved with new data or methods? What, exactly, does EX represent, and how does it relate to concepts such as evolutionary acceptability or “functional” effect or to measures such as PAM? How can exchangeability be applied to scientific questions or technical challenges?
The concept of amino acid exchangeability:
Most of the questions listed in the previous paragraph relate to the general issue of what it means to seek out average tendencies among heterogeneous sets of data. The capacity of EX to represent average tendencies useful for any given purpose depends on three types of factors. First, EX is derived from individual assay results that reflect experimental biases and uncertainties, and thus the accuracy of EX depends on the strength of such effects. Second, EX is a measure of mean effect averaged over diverse contexts (diverse sites in different proteins operating under differing conditions in different assays), and thus the accuracy of EX depends on the strength of these context-dependent effects (relative to intrinsic effects) and how well the distribution of contexts has been sampled. The procedure used here compensates only for the differing severity of assays (via the frequency-activity regression, Figure 2) and for sampling error in regard to the distribution of amino acids among surface and buried sites. Third, EX is a measure of effects focused mainly on protein activity and stability as measured with biochemical or growth assays in the laboratory, and thus its utility in analyzing other phenomena of amino acid exchange (e.g., in natural variation and evolution) depends on the extent to which these effects, as opposed to others (e.g., metabolic cost of amino acids), are important.
In principle, such sources of variance could prove so overwhelming that it would be pointless to pursue a general measure of exchangeability. In practice, this is not the case. The results presented here demonstrate conclusively that there is a general phenomenon of exchangeability in the sense of predictable statistical regularities seen across various types of data involving amino acid exchanges (or differences) from a diverse array of proteins and organisms. The three data sets used to evaluate EX and other measures are from three independent sources and correspond to random exchanges (experimental data), relatively damaging exchanges (HGMD data), and relatively benign exchanges (evolution). Yet, most predictors are significant in most tests, and the ranking of predictors shows considerable regularity between tests (e.g., EX and BLO are the best, and Grantham and MJ are among the worst). Among other things, this indicates that various sources of data could be used in deriving a measure of exchangeability, not just data from experimental genetics. The unique value of the latter data is that, if one wishes to disentangle mutational and selective effects in evolution, experimental exchanges provide independent data on amino acid exchange effects with no obvious risk of confounding mutational effects.
The common severity-of-effect distribution:
The success of the method of assigning activity scores from the regression in Figure 2 would seem to depend on the extent to which two principles apply: (i) regardless of the nature of the protein, the frequency distribution of activity effects in a random or arbitrary set of amino acid exchanges is the same, and (ii) regardless of how exchanges are assayed, the rank order of the mean severity-of-effect for all 380 source-destination pairs will be the same (e.g., whether the assay is for biological activity, biochemical activity, ΔΔG, or Kd).
Presumably neither principle is perfectly applicable, but applies only roughly. The first principle is supported by the strength of the regression shown in Figure 2. With respect to the second principle, one may consider the case of integrating the staphylococcal nuclease studies (Shortle et al. 1990; Green et al. 1992; Meeker et al. 1996) in which ΔΔG values (rather than activity effects) are measured for protein variants. To assign scores to exchanges in this study, the ΔΔG values are ranked from highest to lowest, and this is treated as a rank order of severity of effect, going from what are presumed to be the most disruptive effects (highest ΔΔG, greatest loss of stability) to the least disruptive or most benign. The implicit expectation is that if staphylococcal nuclease variants were assayed for activity rather than for thermostability, the rankings would tend to correspond, with the most destabilized variants being the least active. That is, activity is assumed to be an increasing function of stability. However, at the low end of the scale of ΔΔG, there are a few exchanges that actually increase stability (decrease ΔG), and one would not necessarily assume that such exchanges increase activity—the increased stability might make for a too-rigid protein unable to bind a substrate or release a product. An analogous interpretation could be applied to the problem of assigning scores to human growth hormone variants on the basis of a ranking of Kd values. To clarify this issue would require systematic data (not currently available, to our knowledge) in which a large set of variants is subjected both to assays for effects on biological activity and to effects on kinetic parameters or thermostability.
Future prospects for a measure of exchangeability:
Many have expressed surprise that EX performs better than other measures tested, given that its derivation reflects the results of relatively crude laboratory experiments with a small set of proteins that may not be representative of proteins in general. Clearly, individual EX values are highly imprecise relative to other measures, with relative standard deviations of 20–25%, on average. A major reason for this imprecision is that an individual EX value is based on an average of only 25 exchanges. The exchanges typically are assigned a highly discretized (thus imprecise) score, and the assignment itself may have considerable individual uncertainty.
However, though imprecise, EX values are focused on protein exchangeability per se, a concept that has not received much attention. Of the other measures tested, only MJ is derivationally a pure measure of protein-level effects. While other measures sometimes are used as if they were pure measures of operational exchangeability (e.g., Terwilliger 1995; Wen et al. 1996; Li 1997; Krawczak et al. 1998; Yang et al. 1998; Graur and Li 2000; Alexandre and Zhulin 2003; Pupko et al. 2003), they are not.
Thus, what EX lacks in precision and reliability, it makes up for in accuracy, because it is focused specifically on the operational exchangeability of amino acids in proteins, as opposed to being focused on something else. Precisely because of this combination of high accuracy with low precision and reliability, there is every reason to believe that EX can be improved simply by gathering more and better data. If future experimental studies can be designed so that measured exchange effects have high information content, and exchanges are distributed equally among the most practically relevant class—the singlet exchanges—a mere twofold addition to the amount of data (another 19,000 variants) would ensure >100 variants of each singlet type, which would yield a considerably more powerful measure. With modern high-throughput methods, producing such data could be much faster and cheaper than it was in the past.
SUPPLEMENTARY DATA
The supplementary data include a detailed description of experimental exchange studies (EX-studies.doc), which describes (and provides sources of data on) exchanges, assays, and structural models. A spreadsheet with EX values as in Table 3, and weights as in Table 2, is also available (EX-matrix.xls). Data for the computation of exchangeability from experimental results and for the comparative evaluation of exchangeability are available by contacting the authors.
Acknowledgments
The authors thank John Moult for comments on the manuscript; Douglas Axe and Tim Palzkill for providing unpublished data and interpretive comments; Douglas Axe, Clyde Hutchison, Jeffrey Miller, Tony Poteete, and the administrators of the Protein Mutant Database (T. Kawabata, m. Ota and K. Nishikawa) for providing electronic files with published experimental results, as well as for providing supporting information and interpretation; and Richard Goldstein for suggesting information content as a predictor of study power. This work was supported by the Center for Advanced Research in Biotechnology (a research institute jointly supported by the National Institute of Standards and Technology and the University of Maryland Biotechnology Institute). The identification of specific commercial software products in this article is for the purpose of specifying a protocol and does not imply a recommendation or endorsement by the National Institute of Standards and Technology.
References
- Alexandre, G., and I. B. Zhulin, 2003. Different evolutionary constraints on chemotaxis proteins CheW and CheY revealed by heterologous expression studies and protein sequence analysis. J. Bacteriol. 185: 544–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul, S. F., 1991. Amino acid substitution matrices from an information theoretic perspective. J. Mol. Biol. 219: 555–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atchley, W. R., T. Lokot, K. Wollenberg, A. Dress and H. Ragg, 2001. Phylogenetic analyses of amino acid variation in the serpin proteins. Mol. Biol. Evol. 18: 1502–1511. [DOI] [PubMed] [Google Scholar]
- Axe, D. D., N. W. Foster and A. R. Fersht, 1998. A search for single substitutions that eliminate enzymatic function in a bacterial ribonuclease. Biochemistry 37: 7157–7166. [DOI] [PubMed] [Google Scholar]
- Benner, S. A., M. A. Cohen and G. H. Gonnet, 1994. Amino acid substitution during functionally constrained divergent evolution of protein sequences. Protein Eng. 7: 1323–1332. [DOI] [PubMed] [Google Scholar]
- Berman, H. M., J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat et al., 2000. The Protein Data Bank. Nucleic Acids Res. 28: 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham, B. C., and J. A. Wells, 1989. High-resolution epitope mapping of hGH-receptor interactions by alanine-scanning mutagenesis. Science 244: 1081–1085. [DOI] [PubMed] [Google Scholar]
- Eisenhaber, F., and P. Argos, 1993. Improved strategy in analytic surface calculation for molecular-systems—handling of singularities and computational-efficiency. J. Comput. Chem. 14: 1272–1280. [Google Scholar]
- Eisenhaber, F., P. Lijnzaad, P. Argos, C. Sander and M. Scharf, 1995. The double cubic lattice method—efficient approaches to numerical-integration of surface-area and volume and to dot surface contouring of molecular assemblies. J. Comput. Chem. 16: 273–284. [Google Scholar]
- Feng, D. F., M. S. Johnson and R. F. Doolittle, 1985. Aligning amino acid sequences. Comparison of commonly used methods. J. Mol. Evol. 21: 112–125. [DOI] [PubMed] [Google Scholar]
- Fitch, W. M., 1966. An improved method of testing for evolutionary homology. J. Mol. Biol. 16: 9–16. [DOI] [PubMed] [Google Scholar]
- Fredman, D., M. Siegfried, Y. P. Yuan, P. Bork, H. Lehvaslaiho et al., 2002. HGVbase: a human sequence variation database emphasizing data quality and a broad spectrum of data sources. Nucleic Acids Res. 30: 387–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman, N., and Z. Yang, 1994. A codon-based model of nucleotide substitution for protein-coding DNA sequences. Mol. Biol. Evol. 11: 725–736. [DOI] [PubMed] [Google Scholar]
- Grantham, R., 1974. Amino acid difference formula to help explain protein evolution. Science 185: 862–864. [DOI] [PubMed] [Google Scholar]
- Graur, D., and W.-H. Li, 2000 Fundamentals of Molecular Evolution. Sinauer, Sunderland, MA.
- Green, S. M., A. K. Meeker and D. Shortle, 1992. Contributions of the polar, uncharged amino acids to the stability of staphylococcal nuclease: evidence for mutational effects on the free energy of the denatured state. Biochemistry 31: 5717–5728. [DOI] [PubMed] [Google Scholar]
- Henikoff, S., and J. G. Henikoff, 1992. Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. USA 89: 10915–10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff, S., and J. G. Henikoff, 1993. Performance evaluation of amino acid substitution matrices. Proteins 17: 49–61. [DOI] [PubMed] [Google Scholar]
- Hobohm, U., and C. Sander, 1994. Enlarged representative set of protein structures. Protein Sci. 3: 522–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holbrook, S. R., S. M. Muskal and S. H. Kim, 1990. Predicting surface exposure of amino acids from protein sequence. Protein Eng. 3: 659–665. [DOI] [PubMed] [Google Scholar]
- Hooft, R. W., C. Sander, M. Scharf and G. Vriend, 1996. The PDBFINDER database: a summary of PDB, DSSP and HSSP information with added value. Comput. Appl. Biosci. 12: 525–529. [DOI] [PubMed] [Google Scholar]
- Hortnagel, K., O. N. Voloshin, H. H. Kinal, N. Ma, C. Schaffer-Judge et al., 1999. Saturation mutagenesis of the E. coli RecA loop L2 homologous DNA pairing region reveals residues essential for recombination and recombinational repair. J. Mol. Biol. 286: 1097–1106. [DOI] [PubMed] [Google Scholar]
- Hughes, A. L., 1992. Coevolution of the vertebrate integrin alpha- and beta-chain genes. Mol. Biol. Evol. 9: 216–234. [DOI] [PubMed] [Google Scholar]
- Hughes, A. L., T. Ota and M. Nei, 1990. Positive Darwinian selection promotes charge profile diversity in the antigen-binding cleft of class I major-histocompatibility-complex molecules. Mol. Biol. Evol. 7: 515–524. [DOI] [PubMed] [Google Scholar]
- Kawabata, T., M. Ota and K. Nishikawa, 1999. The protein mutant database. Nucleic Acids Res. 27: 355–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawashima, S., and M. Kanehisa, 2000. AAindex: amino acid index database. Nucleic Acids Res. 28: 374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura, M., 1983 The Neutral Theory of Molecular Evolution. Cambridge University Press, Cambridge, UK.
- Kleina, L. G., and J. H. Miller, 1990. Genetic studies of the lac repressor. XIII. Extensive amino acid replacements generated by the use of natural and synthetic nonsense suppressors. J. Mol. Biol. 212: 295–318. [DOI] [PubMed] [Google Scholar]
- Krawczak, M., and D. N. Cooper, 1997. The human gene mutation database. Trends Genet. 13: 121–122. [DOI] [PubMed] [Google Scholar]
- Krawczak, M., E. V. Ball and D. N. Cooper, 1998. Neighboring-nucleotide effects on the rates of germ-line single-base-pair substitution in human genes. Am. J. Hum. Genet. 63: 474–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristensen, C., T. Kjeldsen, F. C. Wiberg, L. Schaffer, M. Hach et al., 1997. Alanine scanning mutagenesis of insulin. J. Biol. Chem. 272: 12978–12983. [DOI] [PubMed] [Google Scholar]
- Li, W.-H., 1997 Molecular Evolution. Sinauer, Sunderland, MA.
- Loeb, D. D., R. Swanstrom, L. Everitt, M. Manchester, S. E. Stamper et al., 1989. Complete mutagenesis of the HIV-1 protease. Nature 340: 397–400. [DOI] [PubMed] [Google Scholar]
- Markiewicz, P., L. G. Kleina, C. Cruz, S. Ehret and J. H. Miller, 1994. Genetic studies of the lac repressor. XIV. Analysis of 4000 altered Escherichia coli lac repressors reveals essential and non-essential residues, as well as “spacers” which do not require a specific sequence. J. Mol. Biol. 240: 421–433. [DOI] [PubMed] [Google Scholar]
- Materon, I. C., and T. Palzkill, 2001. Identification of residues critical for metallo-beta-lactamase function by codon randomization and selection. Protein Sci. 10: 2556–2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeker, A. K., B. Garcia-Moreno and D. Shortle, 1996. Contributions of the ionizable amino acids to the stability of staphylococcal nuclease. Biochemistry 35: 6443–6449. [DOI] [PubMed] [Google Scholar]
- Miller, J. H., 1991. Use of nonsense suppression to generate altered proteins. Methods Enzymol. 208: 543–563. [DOI] [PubMed] [Google Scholar]
- Miyata, T., S. Miyazawa and T. Yasunaga, 1979. Two types of amino acid substitutions in protein evolution. J. Mol. Evol. 12: 219–236. [DOI] [PubMed] [Google Scholar]
- Miyazawa, S., and R. L. Jernigan, 1993. A new substitution matrix for protein sequence searches based on contact frequencies in protein structures. Protein Eng. 6: 267–278. [DOI] [PubMed] [Google Scholar]
- Mossakowska, D. E., K. Nyberg and A. R. Fersht, 1989. Kinetic characterization of the recombinant ribonuclease from Bacillus amyloliquefaciens (barnase) and investigation of key residues in catalysis by site-directed mutagenesis. Biochemistry 28: 3843–3850. [DOI] [PubMed] [Google Scholar]
- Muse, S. V., and B. S. Gaut, 1994. A likelihood approach for comparing synonymous and nonsynonymous nucleotide substitution rates, with application to the chloroplast genome. Mol. Biol. Evol. 11: 715–724. [DOI] [PubMed] [Google Scholar]
- Nachman, M. W., and S. L. Crowell, 2000. Estimate of the mutation rate per nucleotide in humans. Genetics 156: 297–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olins, P. O., S. C. Bauer, S. Braford-Goldberg, K. Sterbenz, J. O. Polazzi et al., 1995. Saturation mutagenesis of human interleukin-3. J. Biol. Chem. 270: 23754–23760. [DOI] [PubMed] [Google Scholar]
- Pupko, T., R. Sharan, M. Hasegawa, R. Shamir and D. Graur, 2003. Detecting excess radical replacements in phylogenetic trees. Gene 319: 127–135. [DOI] [PubMed] [Google Scholar]
- Qiu, W. G., N. Schisler and A. Stoltzfus, 2004. The evolutionary gain of spliceosomal introns: sequence and phase preferences. Mol. Biol. Evol. 21: 1252–1263. [DOI] [PubMed] [Google Scholar]
- Rand, D. M., D. M. Weinreich and B. O. Cezairliyan, 2000. Neutrality tests of conservative-radical amino acid changes in nuclear- and mitochondrially-encoded proteins. Gene 261: 115–125. [DOI] [PubMed] [Google Scholar]
- Reidhaar-Olson, J. F., J. U. Bowie, R. M. Breyer, J. C. Hu, K. L. Knight et al., 1991. Random mutagenesis of protein sequences using oligonucleotide cassettes. Methods Enzymol. 208: 564–586. [DOI] [PubMed] [Google Scholar]
- Rennell, D., S. E. Bouvier, L. W. Hardy and A. R. Poteete, 1991. Systematic mutation of bacteriophage T4 lysozyme. J. Mol. Biol. 222: 67–87. [DOI] [PubMed] [Google Scholar]
- SAS Institute, 1999 JMP version 3. Cary, NC.
- Schaaper, R. M., and R. L. Dunn, 1991. Spontaneous mutation in the Escherichia coli lacI gene. Genetics 129: 317–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shortle, D., W. E. Stites and A. K. Meeker, 1990. Contributions of the large hydrophobic amino acids to the stability of staphylococcal nuclease. Biochemistry 29: 8033–8041. [DOI] [PubMed] [Google Scholar]
- Singer, G. A., and D. A. Hickey, 2000. Nucleotide bias causes a genomewide bias in the amino acid composition of proteins. Mol. Biol. Evol. 17: 1581–1588. [DOI] [PubMed] [Google Scholar]
- Slack, J. L., K. Schooley, T. P. Bonnert, J. L. Mitcham, E. E. Qwarnstrom et al., 2000. Identification of two major sites in the type I interleukin-1 receptor cytoplasmic region responsible for coupling to pro-inflammatory signaling pathways. J. Biol. Chem. 275: 4670–4678. [DOI] [PubMed] [Google Scholar]
- Stenson, P. D., E. V. Ball, M. Mort, A. D. Phillips, J. A. Shiel et al., 2003. Human Gene Mutation Database (HGMD): 2003 update. Hum. Mutat. 21: 577–581. [DOI] [PubMed] [Google Scholar]
- Terwilliger, T. C., 1995. Engineering the stability and function of gene V-protein. Adv. Protein Chem. 46: 177–215. [DOI] [PubMed] [Google Scholar]
- Venkatarajan, M. S., and W. Braun, 2001. New quantitative descriptors or amino acids based on multidimensional scaling of a large number of physicochemical properties. J. Mol. Model 7: 445–453. [Google Scholar]
- Wen, J. A., X. Chen and J. U. Bowie, 1996. Exploring the allowed sequence space of a membrane protein. Nat. Struct. Biol. 3: 141–148. [DOI] [PubMed] [Google Scholar]
- Whelan, S., and N. Goldman, 2001. A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol. Biol. Evol. 18: 691–699. [DOI] [PubMed] [Google Scholar]
- Wrobel, J. A., S. F. Chao, M. J. Conrad, J. D. Merker, R. Swanstrom et al., 1998. A genetic approach for identifying critical residues in the fingers and palm subdomains of HIV-1 reverse transcriptase. Proc. Natl. Acad. Sci. USA 95: 638–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia, X., and Z. Xie, 2002. Protein structure, neighbor effect, and a new index of amino acid dissimilarities. Mol. Biol. Evol. 19: 58–67. [DOI] [PubMed] [Google Scholar]
- Yang, Z., 1997. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. 13: 555–556. [DOI] [PubMed] [Google Scholar]
- Yang, Z., R. Nielsen and M. Hasegawa, 1998. Models of amino acid substitution and applications to mitochondrial protein evolution. Mol. Biol. Evol. 15: 1600–1611. [DOI] [PubMed] [Google Scholar]
- Zabin, H. B., M. P. Horvath and T. C. Terwilliger, 1991. Approaches to predicting effects of single amino acid substitutions on the function of a protein. Biochemistry 30: 6230–6240. [DOI] [PubMed] [Google Scholar]
- Zhang, J., 2000. Rates of conservative and radical nonsynonymous nucleotide substitutions in mammalian nuclear genes. J. Mol. Evol. 50: 56–68. [DOI] [PubMed] [Google Scholar]