

Abstract
The 3′-UTR (untranslated region) of bcl-2 mRNA contains an ARE (AU-rich element) that potentially regulates the stability of bcl-2 mRNA in a cell specific fashion. Previous studies have demonstrated that multiple proteins interact with bcl-2 mRNA in HL-60 (human leukaemia-60) cells, potentially contributing to the overexpression of Bcl-2 protein. Treatment of HL-60 cells with taxol or okadaic acid has been shown to induce destabilization of bcl-2 mRNA, which was associated with decreased binding of trans-acting factors to bcl-2 mRNA. Nucleolin has been identified as one of the bcl-2 mRNA-binding proteins [Sengupta, Bandyopadhyay, Fernandes and Spicer (2004) J. Biol. Chem. 279, 10855–10863]. In an effort to identify additional bcl-2 mRNA-binding proteins, two polypeptides of approx. 45 kDa and 60 kDa were isolated from HL-60 cells by AREbcl-2 (transcripts that contain bcl-2 AREs) RNA affinity chromatography. These proteins were identified as the human proliferation associated protein, Ebp1, and human DRBP76 (double stranded RNA-binding protein 76) respectively, by MALDI (matrix-assisted laser-desorption ionization)-MS. RNA electrophoretic mobility shift assays indicated that recombinant Ebp1 binds to AREbcl-2 RNA but not to the group 1 ARE present in GM-CSF (granulocyte macrophage-colony stimulating factor) mRNA in vitro. Antibody supershift assays demonstrated that Ebp1 is present in protein–AREbcl-2 RNA complexes formed with cytosolic HL-60 extracts. The interaction of Ebp1 with bcl-2 mRNA in HL-60 cells was also demonstrated by RNA co-immunoprecipitation assays. This interaction was not detected in extracts of taxol-treated HL-60 cells. Immunoprecipitation assays further revealed that Ebp1 co-precipitates with nucleolin from HL-60 cytoplasmic extracts. The observation that co-precipitation was decreased when extracts were treated with RNase suggests that Ebp1 and nucleolin are present in the same bcl-2 mRNP (messenger ribonucleoprotein particle) complexes. RNA-decay assays further demonstrated that Ebp1 decreased the rate of decay of β-globin–AREbcl-2 transcripts in HL-60 cell extracts. Collectively, these results indicate a novel function for Ebp1 in contributing to the regulation of bcl-2 expression in HL-60 cells.
Keywords: bcl-2 expression, double stranded RNA-binding protein 76 (DRBP76), Ebp1, human leukaemia-60 cells (HL-60), mRNA binding, nucleolin
Abbreviations: AEBSF, 4-(2-aminoethyl) benzenesulphonyl fluoride hydrochloride; ARE, AU-rich element; DRBP76, double stranded RNA-binding protein 76; DTT, dithiothreitol; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; GST, glutathione S-transferase; HL-60, human leukaemia-60; IPTG, isopropylthio-galactoside; mRNP, messenger ribonucleoprotein particle; MALDI, matrix-assisted laser desorption ionization; NP-40, Nonidet P40; RT, reverse transcriptase; rRNA, ribosomal RNA; UTR, untranslated region
INTRODUCTION
Although the molecular mechanisms that regulate apoptosis are not yet completely understood, multiple lines of evidence point to a role for the Bcl-2 protein in the regulation of cell death [1]. The mammalian bcl-2 gene [2,3] encodes a 29 kDa protein that resides in mitochondrial membranes and functions as an inhibitor of programmed cell death or apoptosis [1]. bcl-2 was one of the first proto-oncogenes that was found to promote carcinogenesis by prolonging cell survival rather than increasing cell replication [4]. Overall, it is estimated that approx. 50% of human cancers have increased levels of the Bcl-2 protein [5]. Moreover, increased Bcl-2 expression in some malignant cells is an obstacle to chemotherapeutic treatment [6,7]. Accordingly, the identification of factors that regulate the expression of the bcl-2 gene has the potential to aid in the development of novel therapeutic approaches for treating cancers in which increased Bcl-2 protein levels contribute to cell survival.
There are a number of reports that describe the effects of apoptotic and chemotherapeutic agents on bcl-2 mRNA and protein levels. For example, Liu and Priest [8] found that treatment of OV2008 ovarian tumour cells with taxol leads to decreased stability of bcl-2 mRNA and decreased Bcl-2 protein levels. Subsequently, Riordan et al. [9] reported that okadaic acid treatment of HL-60 (human leukemia-60) cells, which contain a high level of the Bcl-2 protein, leads to destabilization of bcl-2 mRNA, as well as downregulation of Bcl-2 protein level. Previous studies in our laboratory have demonstrated that treatment of HL-60 cells with taxol, as well as okadaic acid, leads to the downregulation of bcl-2 mRNA followed by the induction of apoptosis [10]. Using both agents, downregulation of bcl-2 expression was associated with decreased bcl-2 mRNA stability. Recently, all-trans retinoic acid was found to induce differentiation, and subsequent apoptosis, of HL-60 cells through a mechanism that involved destabilization of bcl-2 mRNA [11]
The biochemical mechanisms underlying the downregulation of bcl-2 mRNA stability in response to chemotherapeutic agents have not been fully elucidated. In general, mRNA stability is governed by sequence and/or structural elements in mRNAs and by trans-acting factors that interact with these elements. Among the prominent mRNA elements that play a role in the modulation of mRNA stability are the AREs (AU-rich elements) found in the 3′-UTRs (untranslated regions) in a number of cytokine and oncogene mRNAs [12,13]. Schiavone et al. [14] have reported that there is a conserved ARE motif in the 3′-UTR of bcl-2 mRNA. The ARE (nucleotides 921–1057 of bcl-2 cDNA) has been shown to exhibit destabilizing activity when fused with the β-globin gene in transfected NIH3T3 cells [10,14]. Additionally, Schiavone et al. [14] found that the destabilizing activity of the AREbcl-2 (transcripts that contain bcl-2 the ARE) was enhanced by the apoptotic agent C2-ceramide. This suggested that the ARE motif could be involved in the downregulation of bcl-2 mRNA that is induced by apoptotic agents. Interestingly, Lapucci et al. [15] have found that the mRNA-destabilizing protein AUF1 (AU rich RNA binding factor 1) binds to bcl-2 mRNA in vitro. It was also observed that UVC irradiation-induced apoptosis in Jurkat cells was associated with an increase in the level of cytoplasmic AUF1 [15], suggesting a role for AUF1 in modulating the level of bcl-2 mRNA in these cells.
Previous studies in our laboratory have demonstrated that taxol and okadaic acid treatment led to decreased binding of HL-60 cytoplasmic proteins to AREbcl-2 RNA [10]. UV cross-linking assays revealed that approx. 6 proteins in extracts of untreated HL-60 cells bind to AREbcl-2 RNA. Proteins of approx. 40–60, 70 and 100 kDa were found to cross-link specifically with AREbcl-2 RNA. Interestingly, RNA cross-linking with these proteins was decreased in extracts of HL-60 cells treated with taxol or okadaic acid. Accordingly, these proteins were proposed to play a role in the modulation of bcl-2 mRNA stability that is observed following treatment with taxol or okadaic acid. Subsequent efforts to characterize the AREbcl-2-binding proteins led to the identification of the 100 and 70 kDa proteins as nucleolin and a proteolytic fragment of nucleolin respectively [16]. However, the identity of the 40–60 kDa protein–RNA complexes has so far remained unknown. The focus of the present study was to identify additional bcl-2 mRNA-binding proteins in order to better understand the mechanisms involved in post-transcriptional regulation of bcl-2 expression. As a result, we have identified a novel function for the ErbB3 binding protein, Ebp1, in binding to bcl-2 mRNA and possibly contributing to the regulation of bcl-2 expression in HL-60 cells.
MATERIALS AND METHODS
Cell culture
HL-60 cells (ATCC) were grown in RPMI-1640 medium (GIBCO-BRL) supplemented with 10% (v/v) heat-inactivated foetal bovine serum, 100 units/ml penicillin and 100 μg/ml streptomycin. Cells were maintained at 37 °C in 95% air/5% CO2, in a fully humidified incubator. Cells were treated with taxol (200 nM) or okadaic acid (20 nM) as previously described [10].
Preparation of cell extracts
HL-60 cells were harvested by centrifugation at 100 g for 5 min at 4 °C. For whole-cell extracts, cells were washed twice with PBS and suspended in 50 ml of buffer A [16]. Cells were lysed with three 10 s bursts using a Virsonic sonicator (Virtis). Lysates were subjected to low-speed centrifugation (10000 g) for 10 min, followed by centrifugation at 100000 g for 1 h at 4 °C to produce an S100 total cell extract.
For cytoplasmic extracts, cells were washed twice in PBS and then suspended in 200 μl of buffer D [10 mM Hepes (pH 8.0), 3 mM MgCl2, 40 mM KCl, 0.1% protease inhibitor cocktail (Sigma), 0.2% NP-40 (Nonidet P40), 10% glycerol and 1 mM DTT (dithiothreitol)] and incubated on ice for 10 min. The lysate was centrifuged at 10000 g for 2 min in order to pellet nuclei, followed by centrifugation at 100000 g for 1 h at 4 °C to produce a cytoplasmic S100 extract. Aliquots of the samples were stored at −80 °C after flash-freezing on solid CO2.
Heparin–sepharose column chromatography
A 5 ml Hi-Trap™ heparin–sepharose column (Amersham-Pharmacia) was equilibrated with 50 ml of start buffer {50 mM Tris/HCl (pH 7.5), 100 mM NaCl, 10% glycerol, 0.2 mM EDTA, 0.5 mM DTT and 1 mM AEBSF [4-(2-aminoethyl) benzenesulphonyl fluoride hydrochloride]}. An aliquot (30 ml) of the HL-60 S100 total cell extract, dialysed against start buffer, was loaded on to the pre-equilibrated column with a peristaltic pump at a rate of approx. 0.5 ml/min. The column was washed with 15 ml of start buffer and bound proteins were eluted with a 50 ml linear gradient of 0–1.0 M NaCl in start buffer (0.5 ml/min flow rate).
Preparation of RNA transcripts
AREbcl-2 and β-globin–AREbcl-2 transcripts were synthesized using polymerase from plasmids pCR4-ARE and pCR4-β-globin-ARE respectively, as previously described [16]. AREbcl-2 transcripts contain the 137 nt AREbcl-2 motif (nt 921–1057 of bcl-2 cDNA) [10] flanked by 60 nt of the vector pCR4 sequence. β-globin–AREbcl-2 transcripts contain 284 nt of the β-globin coding region followed by the 137 nt AREbcl-2 sequence [16]. Transcripts containing the group 1 ARE motif, (AUUU)5A, [17] were synthesized using SP6 RNA polymerase from plasmid pSP70-(AUUU)5A [18] (a gift from Dr B. Tholanikunnel, Department of Nephrology, Medical University of South Carolina). Radio-labelled RNAs were synthesized in transcription reactions containing 40 μCi of 32P-UTP (Amersham Biosciences). The purity of 32P-RNA transcripts was monitored by analysis on 6% polyacrylamide/7 M urea gels, and the amount of full-length products was generally approx. 90%. The amount of unlabelled RNA was determined by absorbance at 260 nm.
RNA electrophoretic mobility shift assays
RNA mobility shift assays were performed as described previously [10]. Briefly, the column fraction or purified protein (15–30 nM) was mixed with 32P-AREbcl-2 RNA transcripts (approx. 20000 c.p.m.) in 20 μl of RNA binding buffer [10] and incubated on ice for 10 min. Samples were analysed on a 0.8% agarose/TAE (Tris/acetate/EDTA) gel, which was dried on nitrocellulose paper, and analysed by phosphorimaging using STORM™ phosphorimager and ImageQuant™ 1.1 software (Molecular Dynamics). For competition assays, 10×(150 nM) or 20×(300 nM) concentrations of homologous RNA (unlabelled AREbcl-2) or non-homologous RNA were added to samples containing 15 nM 32P-AREbcl-2 RNA before the addition of purified recombinant Ebp1. Non-homologous RNAs included poly(A) (Ambion) and a transcript containing (AUUU)5A. RNA concentrations were determined at A260. For antibody supershift assays, 32P-AREbcl-2 RNA was incubated with recombinant Ebp1 protein or S100 cytoplasmic extracts on ice for 10 min. Rabbit polyclonal anti-(mouse Ebp1) antibody (0.3 μg) (Upstate Biologicals) was added, followed by incubation for 20 min at room temperature and the products were then separated by agarose gel electrophoresis.
UV-induced RNA cross-linking
UV cross-linking assays were performed as described previously [10]. Briefly, 5 μl of 32P-AREbcl-2 RNA transcripts (2×105 c.p.m.) was incubated with 10 μl of the concentrated column fraction in RNA-binding buffer (without BSA) at 4 °C for 10 min followed by UV irradiation (254 nm) for 30 min. After UV cross-linking, reactions were treated with RNase A (Ambion, Inc.) (0.01 units/μl) and RNase T1 (0.4 units/μl) at 37 °C for 30 min. Samples were separated on a 12% polyacrylamide SDS gel, which was stained with Coomassie Blue. RNA-cross-linked protein bands were visualized by phosphorimaging of dried gels. Stained molecular mass markers were visualized with radioactive ink.
AREbcl-2 RNA affinity column chromatography
In vitro-transcribed AREbcl-2 RNA was polyadenylated using a poly(A) kit (Ambion) according to the manufacturer's protocol. Oligo(dT)-sepharose beads (Sigma) were equilibrated with buffer B [10 mM Tris/HCl (pH 8.0), 100 mM NaCl, 1 mM DTT and 0.1% protease inhibitor cocktail]. Polyadenylated AREbcl-2 RNA was incubated with the oligo(dT)-sepharose beads (100 μg in 200 μl) for 2 h at 4 °C with gentle shaking to produce an ARE-poly(A)-oligo(dT) sepharose matrix. After incubation the sample was centrifuged at 10000 g for 1 min in a microcentrifuge, and the matrix pellet was washed four times with 2 bed-volumes of buffer B to remove unbound RNA. Pooled heparin–sepharose column fractions were dialysed against buffer B and concentrated approx. 5-fold. The protein sample (300 μl) was then incubated with the ARE-RNA-oligo(dT) matrix for 2 h at 4 °C with gentle shaking followed by centrifugation at 10000 g for 30 s. The matrix pellet was washed 5 times with 5 bed-volumes of buffer B. Proteins were eluted from the matrix with a 0.2 M step gradient of NaCl (0.2 to 1.0 M) in buffer B.
MS
Samples eluted from the RNA affinity column were separated on a 12% polyacrylamide/0.1% SDS gel. The gel was stained with Coomassie Blue, and individual bands corresponding to RNA-cross-linked proteins were excised from the gel. Gel slices were prepared for MS as described previously [16]. Briefly, gel slices were digested with trypsin and tryptic peptides were extracted with 50 μl of 50% acetonitrile/5% formic acid [16]. For MALDI (matrix-assisted laser desorption ionization) analysis, desalted peptides were mixed with an α-cyano-4-hydroxycinnamic acid matrix in acetonitrile and spotted on to a MALDI plate. MALDI mass spectra were acquired with a Voyager-DE instrument (Applied Biosystems). Peptide sequencing was accomplished by tandem MS on an LCQ Classic ion-trap mass spectrometer (Finnigan). Desalted peptides were ionized via a custom nano-spray source. Peptides were selected for analysis according to their m/z ratios and then fragmented to produce patterns that were sequence informative. Short sequences deduced from tandem mass spectra were used in a search against the NCBI non-redundant PDB through the internet algorithm PepSea (www.unb.br/cbsp/paginiciais/pepseaseqtag.htm).
Expression and purification of recombinant Ebp1
A recombinant pGEX-4T plasmid, pGEX-Ebp1 [19], carrying the full-length Ebp1 gene and a GST (glutathione S-transferase) tag was provided by Dr A. Hamburger (Department of Pathology, University of Maryland, U.S.A.). This vector was transformed into Escherichia coli BL21 and expression of the GST–Ebp1 fusion protein was induced with IPTG (isopropylthio-galactoside). Although a high level of expression was obtained, the majority of GST–Ebp1 protein was present in inclusion bodies. To increase protein solubility, the Ebp1 coding region (cDNA nt 358–1380) was amplified from the pGEX-Ebp1 vector by PCR and cloned into the pBAD/Thio-TOPO® vector (Invitrogen). The resulting pBAD-Ebp1 was transformed into TOP 10 E. coli cells (Invitrogen). E. coli/pBAD-Ebp1 cells were grown until A600 reached 0.7 and were then induced overnight with 0.2% arabinose at 16 °C. Cell pellets were resuspended in buffer C [50 mM potassium phosphate (7.8), 500 mM KCl, 20 mM imidazole and 1 mM phenylmethane sulphonyl fluoride] and lysed by sonication. The lysate was centrifuged at 12000 g for 30 min. The supernatant was loaded on to a 1.0 ml cobalt Talon™ resin (BD Sciences) equilibrated with buffer C. After washing with buffer C, recombinant Thio–Ebp1–V5–His6 protein was eluted with a linear gradient of 0–200 mM imidazole in buffer C. Eluted proteins were concentrated and imidazole was removed by washing with buffer C using a Centricon spin column (Millipore). Protein purity was assessed by SDS/PAGE, and protein concentration was determined by Bradford assay (Bio-Rad), using BSA as a standard.
Immunoprecipitation and Western blot analysis
Approx. 400 μg of HL-60 cytosolic cell extract was mixed with 100 μl of Protein A-sepharose beads (pre-equilibrated with buffer A) for 1 h at 4 °C with gentle shaking. The resulting pre-cleared cell lysate was incubated with 2–5 μg of antibody overnight at 4 °C followed by incubation with 50 μl of Protein A sepharose beads for 1 h at 4 °C. Precipitations were performed using a monoclonal anti-(human nucleolin) antibody (C23) (Santa Cruz, Inc.) or normal mouse IgG. To test the dependence of protein co-precipitation on RNA interactions, RNase A (0.03 μg/reaction) was added to the pre-cleared lysate and the sample was incubated for 15 min at room temperature before the addition of the anti-nucleolin antibody. Following incubation with the antibody, the beads were collected by centrifugation at 6000 g for 1 min and washed 3 times with 1 ml of buffer A containing 0.1% NP-40, protease inhibitor cocktail and RNasin (Ambion, Inc.). Finally, beads containing immunoprecipitated proteins were resuspended in 50 μl of buffer A.
For Western blot analysis of precipitated protein, denaturation buffer was added to the sepharose beads, which were boiled for 5 min. Proteins were separated on a 12% polyacrylamide/SDS gel and then transferred to PVDF membrane. The membrane was blocked by incubation overnight in 5% non-fat dried milk in TBS-T buffer [10 mM Tris/HCl (pH 8.0), 150 mM NaCl and 0.05% Tween-20] at 4 °C. The membrane was then incubated at room temperature for 1 h with a polyclonal anti-Ebp1 antibody (0.2 μg/ml) (Upstate Biologicals). The unbound antibodies were removed by 4×10-min washes in TBS-T buffer. The membrane was then incubated with a peroxidase-conjugated secondary antibody (Santa-Cruz) for 1 h at room temperature followed by 4×10-min washes with TBS-T. The blot was developed with luminol reagent (Santa-Cruz) and visualized by autoradiography. Cruz markers (Santa-Cruz) were used as internal molecular mass standards.
For analysis of the effects of taxol on the cellular level of Ebp1, cells were treated for 0–45 h with 200 nM taxol. Equal amounts of total cellular protein (40 μg) were separated on an SDS/12% polyacrylamide gel. Proteins were transferred to a PVDF membrane, which was treated as described above. Membranes were incubated with a polyclonal anti-Ebp1 antibody, a monoclonal anti-GAPDH (glyceraldehyde-3-phosphate dehydrogenase) antibody (Chemicon) or a polyclonal anti-β-actin antibody (Santa Cruz).
In vitro mRNA decay assays
An SpeI-linearized pCR4-β-globin-ARE plasmid was used as a template for the synthesis of β-globin–AREbcl-2 transcripts. 5′-Capped, 32P-labelled transcripts were prepared with an mMessage mMachine kit (Ambion) using T7 RNA polymerase, as described previously [16]. A poly(A) tail was added to the 3′-end of the transcript using poly(A) polymerase (Ambion). Decay reactions were performed as described by Ford and Wilusz [20], with modifications as previously described [16]. S100 extracts were prepared from HL-60 cells, and treated with okadaic acid (20 nM) for 24 h. Decay reaction products contained approx. 150 nM 32P-labelled β-globin–AREbcl-2 RNA and 8 μg of HL-60 S100 cytosolic protein. To assay for the effects of Ebp1 on decay, the recombinant Thio–Ebp1 protein (approx. 310 nM) was added to the decay reaction before the addition of the cell extract. RNA recovery was assessed by electrophoresis on 7 M urea/6% polyacrylamide gels, which were analysed by phosphorimaging as described above.
RNA co-immunoprecipitation assay
Immunoprecipitation of protein–RNA complexes was performed as described by Niranjanakumari et al. [21]. Briefly, HL-60 cells (8×107) were harvested by centrifugation at 100 g for 5 min at 4 °C and then suspended in 10 ml of PBS. Formaldehyde (Sigma) was added to the cell suspension to a final concentration of 0.5% (v/v) and the reaction was incubated at room temperature for 10 min with slow mixing. Cross-linking was quenched by the addition of glycine (pH 7.0, 0.25 M final concentration), followed by incubation at room temperature for 5 min. The cells were harvested by centrifugation, followed by 2 washes with ice-cold PBS. Fixed cells were resuspended in 1 ml of RIPA buffer [50 mM Tris/HCl (pH 7.5), 1% NP-40, 0.05% SDS, 1 mM EDTA and 150 mM NaCl] containing protease inhibitors. The cells were lysed by 3 rounds of sonication for 20 s each. An aliquot of cell lysate (250 μl) was mixed with 20 μl of Protein A-sepharose beads for 1 h at 4 °C followed by centrifugation at 400 g for 5 min. The pre-cleared supernatant was diluted with RIPA buffer (250 μl) containing RNasin and protease inhibitors, mixed with polyclonal anti-Ebp1 (0.2 μg/μl), monoclonal anti-nucleolin (0.2 μg/ml) or IgG antibodies (0.2 μg/ml) and incubated overnight with shaking at 4 °C. Protein A-sepharose beads (100 μl) were added and the samples were incubated with shaking for 3 h at 4 °C. The sepharose beads were washed five times with RIPA buffer and then resuspended beads were incubated at 70 °C for 45 min to reverse cross-linking. RNA was extracted from the immunoprecipitates using TRIzol® (Invitrogen) according to the manufacturer's protocol and then treated with DNase I. Recovered nucleic acids were ethanol precipitated and resuspended in RNase-free water. The RNA was used as a template with which to synthesize cDNA using random hexamer primers and MMLV (Moloney-murine-leukaemia virus) RT (reverse transcriptase) (Invitrogen) according to the manufacturer's protocol. RT reactions were carried out for 1 h at 42 °C. PCR was performed using 20 μl of the cDNA sample for 30 cycles of amplification. An aliquot (20 μl) of each reaction mixture was analysed on a 2% agarose gel and visualized by ethidium bromide staining.
RESULTS
Identification of novel bcl-2 mRNA-binding proteins
Previous studies demonstrated that nucleolin binds specifically to bcl-2 mRNA, implicating nucleolin as a bcl-2 mRNA regulatory factor [16]. Additional proteins of approx. 40–60 kDa were found to bind bcl-2 RNA but they were not identified in these previous studies. To identify the 40–60 kDa proteins that interact with AREbcl-2 RNA, S100 total-cell extracts from HL-60 cells were fractionated by heparin–sepharose column chromatography. Column fractions were assayed for AREbcl-2 RNA binding by gel-shift and UV cross-linking assays, as employed in the previous study [16]. As shown in Figure 1(A), multiple fractions from the heparin–sepharose column contained proteins that bind a 32P-labelled 200 nt transcript containing AREbcl-2 in gel-shift assays. UV cross-linking assays (Figures 1B and 1C) indicated that fraction 3 contained proteins of approx. 40–75 kDa that cross-linked efficiently with AREbcl-2 RNA, as well as a strongly cross-linking band at approx. 90 kDa. Accordingly, proteins in fraction 3 were further purified by AREbcl-2 RNA affinity chromatography. In vitro-transcribed AREbcl-2 RNA was 3′-polyadenylated and then incubated with oligo(dT)-sepharose beads. The heparin-column fraction 3 was incubated with the ARE-RNA-poly(A):oligo(dT)-sepharose, and protein–ARE-RNA complexes were recovered by centrifugation. Proteins bound to ARE RNA were eluted with a 0.2 M stepwise gradient of 0.2–1.0 M NaCl. Eluted proteins were assayed for AREbcl-2 RNA binding by gel-shift assays (Figure 2A) and by UV cross-linking assays (Figures 2B and 2C). As shown in Figure 2(A), the 0.4–1.0 M NaCl fractions contained proteins that bound AREbcl-2 RNA in gel-shift assays. SDS/PAGE revealed that the 0.8 and 1.0 M NaCl fractions contained 2 predominant bands of 70 kDa and 100 kDa, which Western blot analysis revealed to be nucleolin and a nucleolin fragment (results not shown). Accordingly, these fractions were not examined further. As shown in Figure 2(B), the 0.4 and 0.6 M NaCl fractions contained numerous proteins, ranging in size from 45–110 kDa. However, UV cross-linking assays of these fractions (Figures 2B and 2C) revealed that proteins of approx. 45 kDa and 60 kDa were efficiently cross-linked to AREbcl-2 RNA, whereas other proteins cross-linked inefficiently or did not cross-link at all. Accordingly, the non-radiolabelled 45 kDa and 60 kDa bands (Figure 2B) were excised from the SDS gel and subjected to further analysis.
Figure 1. RNA binding and UV cross-linking assays of heparin–sepharose column fractions.

(A) RNA gel-shift assay. Aliquots of the indicated fractions were incubated with 32P-AREbcl-2 RNA in RNA binding buffer. Free and bound RNAs were separated by electrophoresis on a 0.8% agarose gel, which was dried and analysed by phosphorimaging. (B) and (C), aliquots of the indicated fractions were incubated with 32P-AREbcl-2 RNA in RNA binding buffer and exposed to UV light for 30 min. Complexes were digested with RNase A and T1, and proteins were separated by SDS/PAGE. (B) Image of the Coomassie Blue stained gel. (C) Phosphorimage of the same SDS gel. MW, molecular mass markers; S, sample applied to the column; FT, flow through fraction; W, wash fraction.
Figure 2. RNA affinity purification of heparin-sepharose column fraction.

Heparin–sepharose fraction 3 was incubated with ARE-RNA-poly(A):oligo(dT) beads. Proteins were eluted from the RNA affinity matrix with a step gradient of NaCl. Eluted fractions were examined by gel-shift and UV cross-linking assays. (A) RNA gel-shift assays of column fractions. Aliquots of the fractions were incubated with 32P-AREbcl-2 RNA in binding buffer and RNA–protein complexes were separated by electrophoresis on a 0.8% agarose gel. (B) Aliquots of the indicated fractions were separated by SDS/PAGE and detected by Coomassie Blue staining. (C) Aliquots of the fractions eluted with 0.4 and 0.6 M NaCl were incubated with 32P-AREbcl-2 RNA and exposed to UV light. Complexes were digested with RNase A and RNase T1 and proteins were separated by SDS/PAGE. Bands were detected by phosphorimaging of the SDS gel. MW, molecular mass markers; S, sample applied to the column; FT, flow through fraction; W, wash fraction. Arrows indicate the bands excised from the gel for MALDI-MS analysis.
To identify the 45 kDa and 60 kDa proteins, gel slices containing the corresponding proteins were digested with trypsin. Tryptic peptides were eluted from the gel slices and then submitted to MALDI-MS and subsequent peptide sequence analysis by capillary liquid chromatography tandem MS. From the 45 kDa band, four peptides were sequenced. A comparison of the sequence of one peptide with a mass of 2265.8 Da with the sequences of peptides in the NCBI PDB, using the program BLAST [22], indicated that it was identical to the sequence of residues 216–236 of human Ebp1 (44 kDa; accession numbers U59435 [23] and U87954 [24]). The sequences of three other peptides also matched the sequence of Ebp1 peptides, as shown in Table 1. Two peptides from the 60 kDa protein, that were 3333.1 Da and 985.4 Da respectively, were also sequenced by nanospray tandem MS. A comparison of sequences in the NCBI PDB indicated that these peptides match two peptides from DRBP76 (double stranded RNA-binding protein 76), (76 kDa; accession number AF147209) [25] (Table 1). Since the 45 and 60 kDa proteins were purified by ARE-RNA affinity chromatography and were shown to bind to ARE RNA by UV cross-linking assays, both Ebp1 and DRBP76 are potential bcl-2 mRNA binding factors. As a first step to determine whether Ebp1 plays a role in regulating bcl-2 mRNA expression, we have characterized the RNA binding properties of recombinant Ebp1 and probed for Ebp1–bcl-2 mRNA interactions in vivo. Future studies will explore the potential for DRBP76 in regulating bcl-2 expression.
Table 1. Summary of tryptic peptides from the 45 kDa and 60 kDa proteins sequenced by tandem MS.
| Protein | Theoretical mass (mr) | Theoretical mass (m/z) | Observed mass (m/z) | Charge state* | Sequence† | Residues | Missed cleavage‡ |
|---|---|---|---|---|---|---|---|
| 45 kDa | |||||||
| Peptide 1 | 2265.48 | 755.37+3 | 755.28 | 3+ | AEFEVHEVYAVDVLVSSGEG K | Ebp1: 216–236 | 0 |
| Peptide 2 | 1928.15 | 643.01+3 | 643.23 | 3+ | LVKPGNQNTQVTEAWNK | Ebp1: 156–172 | 0 |
| Peptide 3 | 1376.71 | 688.37+2 | 688.26 | 2+ | FTVLLMPNGPMR | Ebp1: 321–332 | 0 |
| Peptide 4 | 2628.94 | 876.09+3 | 876.36 | 3+ | ITSGPFEPDLYKSEMEVQDAELK | Ebp1: 333–355 | 1 |
| 60 kDa | |||||||
| Peptide 1 | 3330.82 | 832.95+4 | 833.28 | 4+ | VADNLAIQLAAVTEDKYEILQSVDDAAIVIK | Drbp76: 128–158 | 1 |
| Peptide 2 | 985.17 | 492.79+2 | 492.71 | 2+ | LAAFGQLHK | Drbp76: 324–332 | 0 |
* Charge state observed experimentally.
† Underlining indicates sequence determined by MS.
‡ Note: trypsin does not cleave after K/R when proline is located on the C-terminal side of K/R residues.
Recombinant Ebp1 binds bcl-2 RNA
UV cross-linking assays (Figure 2) suggest that Ebp1 binds directly to bcl-2 RNA rather than binding to the AREbcl-2 affinity matrix through interactions with another protein. To confirm this conclusion, recombinant Ebp1 was purified and assayed for bcl-2 RNA binding activity. To express soluble Ebp1 in bacteria, cDNA encoding the full-length Ebp1 protein was subcloned from plasmid pGEX-GST-Ebp1 [19] into the pBAD/Thio-V5 vector. A fusion protein containing thioredoxin, the full-length Ebp1 protein, the V5 epitope and a His6 tag encoded by pBAD-Ebp1 was expressed in E. coli. The fusion protein was purified by Co+2 Talon-resin column chromatography to ≥90% purity. To test the RNA binding activity of Ebp1, recombinant Ebp1 was incubated with 32P-labelled AREbcl-2 RNA [10] and the formation of protein–RNA complexes was examined by RNA gel mobility-shift assays. As shown in Figure 3(A), the incubation of recombinant Ebp1 with AREbcl-2 RNA (in the presence of approx. 1000×molar ratio of tRNA) produced a shift in the RNA mobility (lanes 4 and 5). By contrast, thioredoxin did not form a complex with AREbcl-2 RNA (Figure 3A, lanes 2 and 3). When AREbcl-2 RNA and recombinant Ebp1 were incubated in the presence of a polyclonal anti-Ebp1 antibody, all of the complex was retarded on the gel (i.e. was supershifted) (Figure 3A, lane 6), indicating that the protein–RNA complex was formed with Ebp1 and was not due to binding of a minor contaminant in the Ebp1 sample.
Figure 3. RNA binding activity of recombinant Ebp1.

(A) 32P-AREbcl-2 RNA (15 nM) was incubated with purified recombinant thioredoxin (15 and 30 nM; lanes 2 and 3) or purified recombinant Ebp1 (Thio–Ebp1–His6) (15 and 30 nM; lanes 4 and 5). To confirm binding of Ebp1 to AREbcl-2 RNA, 32P-AREbcl-2 RNA was incubated with Ebp1 (30 nM) in the presence of a polyclonal anti-Ebp1 antibody (0.3 μg) (lane 6). (B) To examine the specificity of RNA binding, recombinant Ebp1 was incubated with 32P-AREbcl-2 RNA in the presence or absence (lane 2) of 10× or 20× molar excess of unlabelled competitor RNA as indicated (lanes 3–8). Competitor RNAs were: homologous AREbcl-2 RNA (lanes 3 and 4), poly(A) (lanes 5 and 6) and the (AUUU)5A transcript (lanes 7 and 8). After incubation in RNA binding buffer, samples were separated on a 0.8% agarose gel, which was dried and analysed by phosphorimaging. Rf, free RNA; Rc, Ebp1–RNA complex; Ss, supershifted complex.
To examine the specificity of Ebp1 binding to AREbcl-2 RNA, competition RNA gel-shift assays were performed. In the presence of a 10- or 20-fold molar excess of unlabelled AREbcl-2 RNA, a Ebp1–32P-AREbcl-2 RNA complex was not observed (Figure 3B, lanes 3 and 4). By contrast, the addition of the same concentrations of an RNA transcript containing the sequence (AUUU)5A, which is the group 1 ARE motif present in GM-CSF mRNA and a number of other mRNAs [17], did not inhibit the formation of Ebp1–AREbcl-2 RNA complexes (Figure 3B, lanes 7 and 8). Similarly, poly(A) (in addition to tRNA) did not decrease binding to AREbcl-2 RNA, as indicated by the absence of unbound 32P-AREbcl-2 RNA in the gel-shift assay (Figure 3B, lanes 5 and 6). Thus recombinant Ebp1 exhibits a ≥20-fold preference for AREbcl-2 RNA over a GM-CSF ARE-RNA transcript or poly(A) sequence, as well as a 1000-fold preference over tRNA.
Previous UV cross-linking assays [10] revealed that approx. 6 proteins in HL-60 cell extracts interact specifically with AREbcl-2 RNA in vitro. To determine if Ebp1 is one of the ARE-binding proteins detected in HL-60 cell extracts, RNA gel mobility supershift assays were performed using an anti-Ebp1 antibody. As shown in Figure 4, incubation of 32P-AREbcl-2 RNA with cytoplasmic S100 extracts from HL-60 cells produced a shift in the mobility of the RNA (lane 2), whereas the incubation of the cell extracts with AREbcl-2 RNA and a polyclonal anti-Ebp1 antibody produced a supershifted complex (lane 3). Thus Ebp1 is present in the protein–RNA complexes detected in extracts of untreated HL-60 cells.
Figure 4. Detection of Ebp1 in cytoplasmic protein–ARE-RNA complexes.
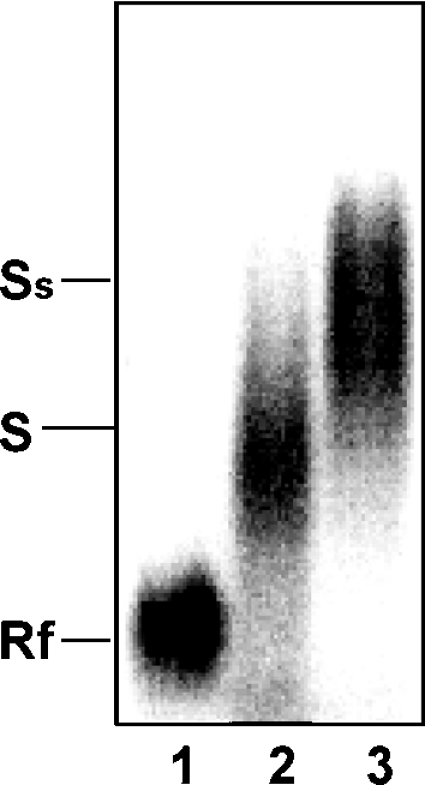
32P-AREbcl-2 RNA was incubated with 0 μg (lane 1) or 2 μg (lanes 2 and 3) of S100 cytoplasmic extract from HL-60 cells. The presence of Ebp1 in protein–RNA complexes was tested for by the addition of an anti-Ebp1 antibody to the reaction mixture (lane 3). Protein–RNA complexes were separated and analysed as described in Figure 3. Rf, free RNA; S, shifted RNA (protein–RNA complex); Ss, antibody supershifted complex.
Ebp1 stabilizes β-globin–AREbcl-2 mRNA in vitro
To investigate the potential functions of Ebp1 in regulating the expression of bcl-2, the effect of Ebp1 on bcl-2 RNA decay was examined, using cell-free decay assays [20]. We have previously observed that a transcript synthesized from a fusion gene containing the AREbcl-2 at the 3′-end of the β-globin gene (β-globin–AREbcl-2 transcripts) decays rapidly in extracts of taxol- or okadaic acid-treated HL60 cells [16]. The addition of recombinant nucleolin to the assays notably decreased the rate of decay of β-globin–AREbcl-2 transcripts. To monitor the stabilizing potential of Ebp1, 32P-labelled, capped and polyadenylated β-globin–AREbcl-2 transcripts were incubated in S100 extracts of okadaic acid-treated HL60 cells in the presence or absence of recombinant Ebp1. RNA was isolated after various times of incubation and the recovery of RNA was monitored by gel electrophoresis. As illustrated in Figure 5, when Ebp1 was added to the reactions β-globin–AREbcl-2 RNA decayed considerably more slowly than in the absence of exogenous Ebp1. Thus Ebp1 exhibits a stabilizing activity in cell-free decay assays of β-globin–AREbcl-2 transcripts.
Figure 5. Effect of Ebp1 on decay of β-globin–AREbcl-2 transcripts in HL-60 cell extracts.

5′-Capped polyadenylated 32P-β-globin–AREbcl-2 transcripts were incubated with S100 extracts of okadaic acid-treated HL-60 cells. +Ebp1, Indicates assays in which recombinant Ebp1 was added to reactions before the addition of cell extracts. RNA recovery was assessed by analysis on polyacrylamide gels and phosphorimaging (lower panel). The percentage RNA remaining indicates the amount of full-length RNA recovered as a function of incubation time (upper panel). Data points are the average of 2 independent assays. OA, okadaic acid.
Ebp1 binds bcl-2 mRNA in vivo
The fact that Ebp1 was purified from an AREbcl-2 RNA affinity column, and the observation that purified recombinant Ebp1 binds specifically to AREbcl-2 RNA, suggests that Ebp1 plays a role in regulating bcl-2 mRNA expression in vivo. To confirm that Ebp1 binds bcl-2 mRNA in vivo, RNA co-immunoprecipitation assays were performed. Freshly harvested HL-60 cells were treated with formaldehyde to induce covalent cross-linking of protein–nucleic acid complexes [21]. Total cell extracts prepared from cross-linked cells were incubated with polyclonal anti-Ebp1, monoclonal anti-nucleolin or polyclonal anti-(rabbit IgG) antibodies. Protein–antibody complexes were incubated with Protein A-sepharose beads and recovered by centrifugation. Immunoprecipitated complexes were then heat-treated to reverse the protein–nucleic acid cross-links. The precipitated nucleic acid was extracted and examined for the presence of bcl-2 mRNA by RT-PCR, using primers specific for the 3′-UTR of bcl-2 mRNA. As shown in Figure 6(A), a bcl-2 PCR product was observed in the sample precipitated with the anti-Ebp1 antibody (lane 1). As expected, bcl-2 mRNA was also recovered in reactions containing an anti-nucleolin antibody (Figure 6A, lane 3). However, bcl-2 RNA was not recovered when an anti-rabbit IgG antibody was used in the pulldown assay (Figure 6A, lane 5). Reactions were also performed, in which exogenous ARE-RNA was added to supplement endogenous bcl-2 RNA in cell extracts, as positive controls (lanes 2 and 4). PCR products were not observed in reactions performed with GAPDH cDNA-specific primers (results not shown), indicating that Ebp1 specifically associates with bcl-2 mRNA. Interestingly, bcl-2 RNA was not detected in immunoprecipitation reactions performed using extracts of HL-60 cells treated for 20 h with taxol, when either anti-Ebp1 or anti-nucleolin antibodies were used (Figure 6B, lanes 3 and 4). This is consistent with previous observations regarding the downregulation of HL-60 protein–bcl-2 mRNA interactions after 20 to 32 h of taxol treatment [10].
Figure 6. Detection of Ebp1–bcl-2 mRNA interactions in vivo.

Whole-cell lysates from formaldehyde cross-linked HL-60 cells were incubated with antibodies and RNA–protein–antibody complexes were recovered by incubation with Protein A sepharose beads. The presence of bcl-2 mRNA in immunoprecipitated RNP complexes was detected by RT-PCR. PCR products were analysed on a 2% agarose gel, which was stained with ethidium bromide. (A) Cell extracts from untreated HL-60 cells were incubated with an anti-Ebp1 antibody (lane 1), an anti-Ebp1 antibody and exogenous AREbcl-2 RNA (positive control, lane 2), a monoclonal anti-nucleolin antibody (lane 3), an anti-nucleolin antibody and exogenous ARE-RNA (positive control) (lane 4) and an anti-(rabbit IgG) antibody (lane 5). (B) HL-60 cells were grown in the absence (lanes 1, 2 and 5) or presence of taxol (lanes 3 and 4) for 20 h and then treated with formaldehyde. Cell extracts were precipitated with an anti-Ebp1 antibody (lanes 1 and 3), an anti-nucleolin antibody (lanes 2 and 4) or an anti-(mouse IgG) antibody (lane 5). MW, molecular mass markers.
We have previously observed that taxol or okadaic acid treatment of HL-60 cells leads to the proteolysis of nucleolin, after 20 to 34 h of treatment [16]. To determine if taxol also downregulates the level of Ebp1, Western blot analysis was performed. Cells were treated with taxol for 0 to 45 h and cytosolic extracts were analysed for the levels of Ebp1, and GAPDH (as a loading control) proteins. As shown in Figure 7, taxol treatment leads to a modest (approx. 20–25%) decrease in the cytoplasmic levels of Ebp1 compared with GAPDH after 20 to 32 h of treatment. A more pronounced decrease in the level of Ebp1 occurred after 32 to 45 h of treatment. Similar results were observed when the level of Ebp1 was compared with that of β-actin (which, like GAPDH, was unchanged during taxol treatment) (results not shown). Since Ebp1–bcl-2 mRNA interactions appear to be disrupted or decreased after 20 h of taxol treatment (Figure 6), this suggests that taxol treatment leads to the inactivation of Ebp1, possibly through phosphorylation, prior to downregulation. Alternatively, the previously observed downregulation of nucleolin after 20 h of taxol treatment [16] may be sufficient to allow for nuclease cleavage of bcl-2 mRNA, which ultimately leads to the disruption of Ebp1–bcl-2 mRNA complexes.
Figure 7. Western blot analysis of Ebp1 in extracts of taxol-treated HL-60 cells.

Cytoplasmic extracts were prepared from cells treated with taxol for 0 to 45 h, as indicated. Proteins were separated on a 12% polyacrylamide/SDS gel and then transferred to Immobilon P paper. Blots were probed with a polyclonal anti-Ebp1 antibody or a monoclonal anti-GAPDH antibody, as indicated. The normalized ratio of Ebp1 to GAPDH was determined by densitometry and is plotted versus incubation time in taxol.
Concurrent binding of Ebp1 and nucleolin to bcl-2 mRNA
The observation that both Ebp1 and nucleolin form complexes with bcl-2 mRNA in vivo suggests that the two proteins may be present in the same bcl-2 mRNP (messenger ribonucleoprotein particle) complexes. Alternatively, these two proteins may bind to overlapping sites on bcl-2 mRNA, in which case the individual proteins may be bound with separate bcl-2 mRNA transcripts in vivo. To distinguish between these possibilities, protein coimmunoprecipitation assays were performed using an anti-nucleolin antibody. S100 cytoplasmic extracts of HL-60 cells were incubated with a monoclonal anti-nucleolin antibody and protein–antibody complexes were recovered by incubation with Protein A-sepharose beads. The precipitated proteins were solubilized and examined for the presence of Ebp1 by Western blot analysis. As shown in Figure 8 (lane 2), Ebp1 was recovered from precipitates produced using the anti-nucleolin antibody. However, only trace amounts of Ebp1 were detected in control reactions precipitated with an anti-IgG antibody (lane 4). To determine whether the interaction between Ebp1 and nucleolin is mediated by binding to RNA, cell extracts were briefly treated with RNase before immunoprecipitation. Interestingly, the precipitation of Ebp1 was considerably decreased after RNase treatment (Figure 8, lane 3), suggesting that Ebp1–nucleolin interactions may be mediated through RNA binding, rather than occurring through direct protein–protein interactions.
Figure 8. Co-immunoprecipitation of Ebp1 and nucleolin from HL-60 cell extracts.

Cytosolic extracts were incubated with monoclonal anti-nucleolin or control anti-(mouse IgG) antibodies and protein complexes were recovered as described in Figure 6. The presence of Ebp1 in immunoprecipitates (IP) was determined by Western blotting using a polyclonal anti-Ebp1 antibody. Pulldown reactions were carried out using an anti-nucleolin antibody (lane 2), an anti-nucleolin antibody after pre-treatment with RNase A for 15 min (lane 3), or an anti-(mouse IgG) antibody (negative control) (lane 4). Total extracts (input control) were analysed in lanes 1 and 6. Supernatants (Sup) from the precipitation reactions were analysed in lanes 7–9 as indicated.
DISCUSSION
Previous studies revealed that nucleolin is one of a number of proteins present in HL-60 cell extracts that interact with the ARE motif of bcl-2 mRNA [16]. These proteins are thought to be components of a multi-protein complex that regulates the stability of bcl-2 mRNA in a cell-specific fashion. In this study, two polypeptides of 45 and 60 kDa were purified by AREbcl-2 RNA affinity chromatography. MALDI-MS indicated that the polypeptide with an apparent molecular mass of 60 kDa is the human double stranded RNA binding protein, DRBP76 [25]. The 45 kDa protein was identified as the proliferation associated protein Ebp1 [23,24]. RNA gel mobility-shift assays revealed that recombinant Ebp1 binds specifically to AREbcl-2 RNA, in the absence of other proteins. In addition, the presence of Ebp1 in protein–bcl-2 mRNA complexes, formed with cytoplasmic extracts of HL-60 cells, was demonstrated by RNA gel mobility supershift assays. The in vivo interaction of Ebp1 and nucleolin with bcl-2 mRNA in HL-60 cells was demonstrated by RNA co-immunoprecipitation assays. Interestingly, Ebp1 was found to co-precipitate with nucleolin in antibody pulldown assays, suggesting that they are both present in bcl-2 mRNP complexes. In addition, recombinant Ebp1 decreased the rate of decay of AREbcl-2 transcripts in cell-free decay assays.
Ebp1 is a member of the PA2G4 (proliferation-associated 2G4) family [23] and is the human homologue of the mouse cell cycle regulated protein p38-2G4 [26]. Ebp1 is conserved in yeast [27], as well as in mouse, and is expressed in a wide variety of cell types including both malignant and non-malignant cells [28]. Ectopic expression of Ebp1 inhibits the growth of breast and prostate cancer cell lines and induces the differentiation of breast carcinoma cell lines [29]. Ebp1 overexpression inhibits the growth of human fibroblasts, as well as the proliferation of tumour cells [30], demonstrating that both normal and transformed cells are sensitive to Ebp1 overexpression. Ebp1 was initially identified as an ErbB3 binding protein in a yeast two-hybrid screen [24]. It was subsequently observed that Ebp1 interacts with the tumour suppressor Rb (retino-blastoma) protein in cultured AU565 breast carcinoma cells and represses the activity of the E2F1-regulated cyclin E promoter in transiently transfected cells [19]. It has also been reported that Ebp1 repression of specific E2F-regulated promoters involves interactions with histone deacetylases [31].
Recently, Ebp1 was shown to be a growth-regulating protein involved in ribosome biogenesis [30]. In that study, Ebp1 was found to be located in both the cytoplasm and the nucleus of HeLa, NIH3T3 and EpH4 cells. Within the nucleus, Ebp1 was found to be localized to the nucleolus in association with rRNA (ribosomal RNA) [30]. Using recombinant GST–Ebp1, a direct interaction was demonstrated between Ebp1 and 5S rRNA. A study of proteins that interact with Ebp1, as examined by immunoprecipitation and MS, revealed that nucleolin is one of approx. 16 proteins that interact with Ebp1 in HeLa cells transfected with a FLAG–Ebp1-expressing construct. Interestingly, the treatment of extracts of transfected HeLa cells with RNase before immunoprecipitation eliminated the co-precipitation of Ebp1 with most of the proteins, including nucleolin. The requirement of RNA for protein association suggested that Ebp1 and nucleolin are present in RNP complexes.
Our own observations from antibody pulldown assays indicate that Ebp1–nucleolin associations are decreased when cytosolic extracts are treated with RNase. This suggests that Ebp1 and nucleolin bind to bcl-2 mRNA concurrently; thus, Ebp1 and nucleolin are present in the same bcl-2 mRNP complexes in the cytoplasm of HL-60 cells. Collectively, these studies reveal a novel role for Ebp1 in binding mRNA and potentially regulating the expression of bcl-2 in HL-60 cells. A possible role for Ebp1 in the post-transcriptional regulation of bcl-2 expression includes the modulation of mRNA stability; however, Ebp1 may also influence protein translation efficiency or the transport of bcl-2 mRNA from the nucleus to the cytoplasm. Future studies that address the functions of Ebp1 are expected to yield insights into the mechanisms by which bcl-2 expression is differentially regulated in normal and cancer cells.
Acknowledgments
The authors gratefully acknowledge the MALDI-MS analysis expertly performed by Dr Kevin Schey and Ms Jennifer Bethard (Medical University of South Carolina, MALDI Facility). The authors thank Dr Anne Hamburger (University of Maryland) for the gift of plasmid pGEX-Ebp1 and Dr Baby Tholanikunnel for the gift of plasmid pSP70-(AUUU)5A. The authors thank Dr Daniel Fernandes for many helpful discussions regarding this study. This work was supported by a Public Health Service Grant CA 87553 (to E. K. S) from the National Cancer Institute.
References
- 1.Hockenbery D., Nunez G., Milliman C., Schreiber R. D., Korsmeyer S. J. Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death. Nature (London) 1990;348:334–336. doi: 10.1038/348334a0. [DOI] [PubMed] [Google Scholar]
- 2.Tsujimoto Y., Finger L. R., Yunis J., Nowell P. C., Croce C. M. Cloning of the chromosome breakpoint of neoplastic B cells with the t(14;18) chromosome translocation. Science (Washington, DC) 1984;226:1097–1099. doi: 10.1126/science.6093263. [DOI] [PubMed] [Google Scholar]
- 3.Cleary M. L., Smith S. D., Sklar J. Cloning and structural analysis of cDNAs for bcl-2 and a hybrid bcl-2/immunoglobulin transcript resulting from the t(14;18) translocation. Cell. 1986;47:19–28. doi: 10.1016/0092-8674(86)90362-4. [DOI] [PubMed] [Google Scholar]
- 4.Vaux D. L., Cory S., Adams J. M. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature (London) 1988;335:440–442. doi: 10.1038/335440a0. [DOI] [PubMed] [Google Scholar]
- 5.Reed J. C. Bcl-2 family proteins: strategies for overcoming chemoresistance in cancer. Adv. Pharmacol. 1997;41:501–532. doi: 10.1016/s1054-3589(08)61070-4. [DOI] [PubMed] [Google Scholar]
- 6.Reed J. C. Bcl-2 family proteins: regulators of apoptosis and chemoresistance in hematologic malignancies. Semin. Hematol. 1997;34:9–19. [PubMed] [Google Scholar]
- 7.Gao G., Dou Q. P. G(1) phase-dependent expression of bcl-2 mRNA and protein correlates with chemoresistance of human cancer cells. Mol. Pharmacol. 2000;58:1001–1010. doi: 10.1124/mol.58.5.1001. [DOI] [PubMed] [Google Scholar]
- 8.Liu Y., Priest D. G. Enhancement of Bcl-2 mRNA degradation by taxol and other anticancer drugs that induce apoptotic cell death. Cellular Pharmacol. 1996;3:405–408. [Google Scholar]
- 9.Riordan F. A., Foroni L., Hoffbrand A. V., Mehta A. B., Wickremasinghe R. G. Okadaic acid-induced apoptosis of HL60 leukemia cells is preceded by destabilization of bcl-2 mRNA and downregulation of bcl-2 protein. FEBS Lett. 1998;435:195–198. doi: 10.1016/s0014-5793(98)01070-9. [DOI] [PubMed] [Google Scholar]
- 10.Bandyopadhyay S., Sengupta T. K., Fernandes D. J., Spicer E. K. Taxol- and okadaic acid-induced destabilization of bcl-2 mRNA is associated with decreased binding of proteins to a bcl-2 instability element. Biochem. Pharmacol. 2003;66:1151–1162. doi: 10.1016/s0006-2952(03)00453-2. [DOI] [PubMed] [Google Scholar]
- 11.Otake Y., Sengupta T. K., Bandyopadhyay S., Spicer E. K., Fernandes D. J. Retinoid-induced apoptosis in HL-60 cells is associated with nucleolin down-regulation and destabilization of Bcl-2 mRNA. Mol. Pharmacol. 2005;67:319–326. doi: 10.1124/mol.104.006080. [DOI] [PubMed] [Google Scholar]
- 12.Shaw G., Kamen R. A conserved AU sequence from the 3′ untranslated region of GM-CSF mRNA mediates selective mRNA degradation. Cell. 1986;46:659–667. doi: 10.1016/0092-8674(86)90341-7. [DOI] [PubMed] [Google Scholar]
- 13.Chen C. Y., Shyu A. B. AU-rich elements: characterization and importance in mRNA degradation. Trends Biochem. Sci. 1995;20:465–470. doi: 10.1016/s0968-0004(00)89102-1. [DOI] [PubMed] [Google Scholar]
- 14.Schiavone N., Rosini P., Quattrone A., Donnini M., Lapucci A., Citti L., Bevilacqua A., Nicolin A., Capaccioli S. A conserved AU-rich element in the 3′ untranslated region of bcl-2 mRNA is endowed with a destabilizing function that is involved in bcl-2 down-regulation during apoptosis. FASEB J. 2000;14:174–184. doi: 10.1096/fasebj.14.1.174. [DOI] [PubMed] [Google Scholar]
- 15.Lapucci A., Donnini M., Papucci L., Witort E., Tempestini A., Bevilacqua A., Nicolin A., Brewer G., Schiavone N., Capaccioli S. AUF1 Is a bcl-2 A+U-rich element-binding protein involved in bcl-2 mRNA destabilization during apoptosis. J. Biol. Chem. 2002;277:16139–16146. doi: 10.1074/jbc.M201377200. [DOI] [PubMed] [Google Scholar]
- 16.Sengupta T. K., Bandyopadhyay S., Fernandes D. J., Spicer E. K. Identification of nucleolin as an AU-rich element binding protein involved in bcl-2 mRNA stabilization. J. Biol. Chem. 2004;279:10855–10863. doi: 10.1074/jbc.M309111200. [DOI] [PubMed] [Google Scholar]
- 17.Shim J., Karin M. The control of mRNA stability in response to extracellular stimuli. Mol. Cells. 2002;14:323–331. [PubMed] [Google Scholar]
- 18.Tholanikunnel B. G., Raymond J. R., Malbon C. C. Analysis of the AU-rich elements in the 3′-untranslated region of beta 2-adrenergic receptor mRNA by mutagenesis and identification of the homologous AU-rich region from different species. Biochemistry. 1999;38:15564–15572. doi: 10.1021/bi9913348. [DOI] [PubMed] [Google Scholar]
- 19.Xia X., Cheng A., Lessor T., Zhang Y., Hamburger A. W. Ebp1, an ErbB-3 binding protein, interacts with Rb and affects Rb transcriptional regulation. J. Cell Physiol. 2001;187:209–217. doi: 10.1002/jcp.1075. [DOI] [PubMed] [Google Scholar]
- 20.Ford L. P., Wilusz J. An in vitro system using HeLa cytoplasmic extracts that reproduces regulated mRNA stability. Methods. 1999;17:21–27. doi: 10.1006/meth.1998.0703. [DOI] [PubMed] [Google Scholar]
- 21.Niranjanakumari S., Lasda E., Brazas R., Garcia-Blanco M. A. Reversible cross-linking combined with immunoprecipitation to study RNA-protein interactions in vivo. Methods. 2002;26:182–190. doi: 10.1016/S1046-2023(02)00021-X. [DOI] [PubMed] [Google Scholar]
- 22.Altschul S. F., Madden T. L., Schaffer A. A., Zhang J., Zhang Z., Miller W., Lipman D. J. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lamartine J., Seri M., Cinti R., Heitzmann F., Creaven M., Radomski N., Jost E., Lenoir G. M., Romeo G., Sylla B. S. Molecular cloning and mapping of a human cDNA (PA2G4) that encodes a protein highly homologous to the mouse cell cycle protein p38–2G4. Cytogenet. Cell. Genet. 1997;78:31–35. doi: 10.1159/000134621. [DOI] [PubMed] [Google Scholar]
- 24.Yoo J. Y., Wang X. W., Rishi A. K., Lessor T., Xia X. M., Gustafson T. A., Hamburger A. W. Interaction of the PA2G4 (EBP1) protein with ErbB-3 and regulation of this binding by heregulin. Br. J. Cancer. 2000;82:683–690. doi: 10.1054/bjoc.1999.0981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Patel R. C., Vestal D. J., Xu Z., Bandyopadhyay S., Guo W., Erme S. M., Williams B. R., Sen G. C. DRBP76, a double-stranded RNA-binding nuclear protein, is phosphorylated by the interferon-induced protein kinase, PKR. J. Biol. Chem. 1999;274:20432–20437. doi: 10.1074/jbc.274.29.20432. [DOI] [PubMed] [Google Scholar]
- 26.Radomski N., Jost E. Molecular cloning of a murine cDNA encoding a novel protein, p38–2G4, which varies with the cell cycle. Exp. Cell Res. 1995;220:434–445. doi: 10.1006/excr.1995.1335. [DOI] [PubMed] [Google Scholar]
- 27.Yamada H., Mori H., Momoi H., Nakagawa Y., Ueguchi C., Mizuno T. A fission yeast gene encoding a protein that preferentially associates with curved DNA. Yeast. 1994;10:883–894. doi: 10.1002/yea.320100704. [DOI] [PubMed] [Google Scholar]
- 28.Xia X., Lessor T. J., Zhang Y., Woodford N., Hamburger A. W. Analysis of the expression pattern of Ebp1, an ErbB-3-binding protein. Biochem. Biophys. Res. Commun. 2001;289:240–244. doi: 10.1006/bbrc.2001.5942. [DOI] [PubMed] [Google Scholar]
- 29.Lessor T. J., Yoo J. Y., Xia X., Woodford N., Hamburger A. W. Ectopic expression of the ErbB-3 binding protein ebp1 inhibits growth and induces differentiation of human breast cancer cell lines. J. Cell Physiol. 2000;183:321–329. doi: 10.1002/(SICI)1097-4652(200006)183:3<321::AID-JCP4>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 30.Squatrito M., Mancino M., Donzelli M., Areces L. B., Draetta G. F. EBP1 is a nucleolar growth-regulating protein that is part of pre-ribosomal ribonucleoprotein complexes. Oncogene. 2004;23:4454–4465. doi: 10.1038/sj.onc.1207579. [DOI] [PubMed] [Google Scholar]
- 31.Zhang Y., Woodford N., Xia X., Hamburger A. W. Repression of E2F1-mediated transcription by the ErbB3 binding protein Ebp1 involves histone deacetylases. Nucleic Acids Res. 2003;31:2168–2177. doi: 10.1093/nar/gkg318. [DOI] [PMC free article] [PubMed] [Google Scholar]