

Abstract
The incidence of marginal biotin deficiency in normal human gestation is approximately one in three. In ICR mice, maternal biotin deficiency results in cleft palate, micrognathia, microglossia and limb hypoplasia. However, the relationships among the severity of maternal biotin deficiency, fetal biotin status and malformations have not been reported. This study utilized validated indices of biotin status to investigate the relationships among maternal biotin status, fetal biotin status and the rate of fetal malformations in ICR mice. Biotin status was controlled by feeding diets with varying egg white concentration. In dams and fetuses, biotin status was assessed by hepatic biotin content and hepatic activity of the biotin-dependent enzyme propionyl-CoA carboxylase; in dams, status was also assessed by urinary excretion of biotin and 3-hydroxyisovaleric acid. Malformations were assessed morphologically. Biotin was measured by HPLC/avidin-binding assay. Propionyl-CoA carboxylase (PCC) activity was determined by H14CO3 incorporation. 3-Hydroxyisovaleric acid concentration was determined by GC/MS. Although no overt signs of deficiency appeared, metabolic disturbances caused by biotin deficiency were detectable in dams and fetuses. These disturbances increased with increasing egg white. Fetal biotin status correlated significantly with maternal biotin status (fetal vs. dam hepatic biotin, r = 0.671; fetal vs. dam PCC activity, r = 0.70). The incidences of malformations were strikingly dependent on egg white concentration. We conclude that in ICR mice, marginal maternal biotin deficiency causes fetal biotin deficiency. We speculate that the fetal malformations are primarily the consequence of fetal biotin deficiency. Because murine malformations appeared at degrees of biotin deficiency that are similar to those in human gestation, we speculate that some human fetal malformations may be caused by biotin deficiency.
Keywords: biotin deficiency, mice, birth defects, nutrition
Recent studies of biotin status during human pregnancy provide evidence that a marginal degree of biotin deficiency develops in a substantial proportion of women during normal pregnancy (1,2). Although the degree of deficiency is not severe enough to produce the classic cutaneous and behavioral manifestations of biotin deficiency, the deficiency is severe enough to produce metabolic derangements in these women (1,2). Our reanalysis of data from a published multivitamin supplementation study indirectly indicates that the marginal degree of biotin deficiency that occurs spontaneously in normal human gestation might be teratogenic (3).
Previous work in mice has demonstrated that the addition of egg white or avidin to the diet can produce birth defects including cleft palate, microglossia and micromelia (4,5). Such additions also produce defects in Syrian (6) or Golden (7) hamsters, but not in rats (6). Strain differences in the susceptibility to these effects have been reported; the A/Jax strain of mice is less susceptible than either the ICR or C57BL/6N/Jcl strains to the teratogenic effects of maternal biotin deficiency (6). A dose response was also observed; increasing the concentration of avidin added to the diet produced increasing incidences of defects in both mice and hamsters (4,7). Avidin and egg white, which contains avidin, are presumed to produce biotin deficiency in the dams and/or fetuses, but the biotin status of the fetus has only rarely been documented (8). For the studies described here, we chose ICR mice to investigate whether biotin deficiency of severity similar to that observed early in human gestation causes increased rates of fetal malformations. This question has not been addressed previously because sensitive, specific techniques for measuring urinary biotin and 3-hydroxyisovaleric acid (3HIA),4 an organic acid that arises from the decreased activity of the biotin-dependent enzyme methylcrotonyl-CoA carboxylase, have only recently been developed and validated in humans (9-11). This is the first application of these indices of biotin status to this mouse model of teratogenesis caused by biotin deficiency.
MATERIALS AND METHODS
Diets
Defined diets containing egg white were purchased from Teklad (Harlan Teklad, Madison, WI); all diets were formulated to be nutritionally adequate for all components except biotin. The dietary protein casein was replaced isonitrogenously gram for gram by egg white. The egg white concentration was 1, 1.3, 2, 3, 5, 10 or 25 g egg white/100 g diet. These particular diets are referred to hereafter as a 1% egg white diet, 1.3% egg white diet, and so on. Three control diets were used: 1) a standard rodent diet (ProLab M-R-H Diet, Syracuse, NY); 2) a defined diet with 0 g egg white/100 g diet (Teklad); or 3) a diet with 25 g egg white/100 g diet with added biotin (25% + biotin) sufficient to occupy all of the biotin binding sites on avidin in the egg white and provide sufficient excess biotin to meet the estimated biotin requirement of mice (Teklad).
Animals and animal husbandry
Virgin female and proven breeder male ICR mice were obtained from Harlan (Indianapolis, IN). Animal rooms were maintained at a constant temperature and humidity under a 12-h light:dark cycle. Females were group housed in plastic shoe box cages, and males were housed individually. Mice were provided a standard diet for at least 5 d to stabilize their nutritional status.
For breeding, two virgin female mice were placed with a male for 12 h overnight. The next morning, those females with a copulation plug were caged individually in wire-bottomed stainless steel metabolic cages (LabProducts, Seaford, DE), weighed, rank ordered and assigned randomly to one of the control diets or to one of the egg white diets using a balanced, stratified design. This day was denoted gestation day zero (GD 0). Food consumption was monitored daily throughout the entire experiment and was adjusted to maintain equivalent mean group weights.
Developmental toxicity
Before breeding, timed urine collections (usually 24 h) were made while the mice were in a food-deprived state; this collection is referred to as the “GD 0” collection. A 12-h collection occurred during the light cycle on GD 7, again from food-deprived mice, and a final 24-h collection was made from GD 16 to GD 17; this collection is referred to as the “GD 17” collection.
On GD 17, the dams were killed by cervical dislocation; the liver and the uterus were removed from each dam for subsequent analysis. The liver was perfused with 154 mmol/L NaCl, weighed and frozen at −70°C until analysis.
Gravid uteri were removed and weighed. Uterine contents were examined for viability; the number, position and status (live, dead or resorbed) of each implant was noted. Each live fetus was weighed, examined grossly for defects and killed by decapitation. Heads were placed in Bouin's fixative for free-hand sectioning and examination as described by Wilson et al. (12). Fetuses were examined for visceral abnormalities and sexed by internal exam employing the Staples' fresh tissue dissection technique (13,14). Fetal livers were removed at this time, weighed and frozen at −70°C until analyzed. All fetal carcasses were stained with alizarin red S and alcian blue for subsequent examination of cartilage and bone in fetal skeletons (15). Outcome measures included gravid uterine weight, number of implantation sites per litter, number and percentage of dead implants per litter and number, percentage and types of malformations per litter.
Analytical methods
Hepatic biotin content
The mass of an aliquot of the frozen liver was determined gravimetrically. The aliquot was immediately homogenized in water using a sintered-glass homogenizer as described previously (16). Biotin was released from covalent binding to proteins by acid hydrolysis using 1.5 mol/L HCl as described previously (16). This method releases >95% of the bound biotin with destruction of <10% of the biotin (17). Biotin was then separated by HPLC from biotin analogs and other substances that interfere with the avidin-binding assay (18). The HPLC fractions containing biotin were assayed directly using an avidin-binding assay as described previously (18). Biotin was normalized by protein determined by the bicinchoninic acid assay (Pierce, Rockford, IL), by unit mass of liver and by total liver. Loss of sample due to technical problems reduced the number of pooled fetal livers available for analysis.
Urinary excretion of biotin and 3HIA
Biotin contents in urine were measured using an avidin-binding assay after HPLC separation as described above. Urinary 3HIA was quantitated by GC/MS using unlabeled 3HIA as the external standard and deuterated 3HIA as the internal standard (19). Urinary excretion rates were expressed as the ratio of 3HIA mass to creatinine mass. Creatinine was measured by the picric acid method (20,21) using a Beckman Creatinine Analyzer 2 (Beckman Instruments, Brea, CA). These urine collections were timed. When results were expressed per unit of time, conclusions and statistical significance were not different, but variability within groups increased somewhat, suggesting that some urine was lost occasionally during collections in metabolic cages.
Liver propionyl-CoA carboxylase (PCC) activity
A portion of liver was cut from the whole frozen liver and homogenized in 25 mmol/L sucrose, 50 mmol/L Tris buffer, pH 7.9, 5 mmol/L glutathione and 1 mmol/L EDTA at a ratio of 1 g liver to 5 mL buffer. While maintaining the crude homogenate on ice, a probe sonicator was used to release membrane-bound enzymes by sonicating three times for a total of 30 s. The 105,000 × g postsupernatant was retained for enzymatic and protein assay as described previously (11,22). Activity was normalized by protein, by mass of liver and by total liver.
Statistics
The litter was used as the experimental unit for all fetal analyses. For continuous variables (hepatic biotin, enzyme assays and fetal weight), ANOVA was used to test for significance of differences between diet groups. For urinary excretion of biotin and 3HIA, ANOVA with repeated measures was used. If the ANOVA was significant, Fisher's post-hoc test with corrections for multiple testing was used for pairwise comparisons. For binomial data (rates of malformations and rates of dead implants), the data were transformed using the arcsin transformation before analysis by ANOVA; if significant, between-group differences were tested by Dunnett's pairwise comparisons. Differences were considered significant if the P-value was ≤0.05.
RESULTS
Control groups
Indicators of tissue biotin status such as hepatic biotin and PCC activity in dams fed the standard rodent diet and those fed 0% egg white did not differ, providing evidence of biotin sufficiency. Therefore, values for all biotin status indicators and rates of malformations were pooled for these two groups and are referred to as “control” in subsequent statistical comparisons with two exceptions. Urinary excretion rates for biotin and 3HIA were significantly different, and values for the two groups were not pooled. The same grouping was used for analysis of fetal biotin status indicators.
Maternal biotin status
Maternal hepatic biotin content decreased with increasing egg white concentration (Table 1, P = 0.025 by ANOVA). Hepatic biotin content was lower in groups fed 5 or 25% egg white than in controls (P < 0.05 by Fisher's post-hoc test). Liver weights did not differ among the groups. The depletion of hepatic biotin was completely prevented by the 25% + biotin diet. These data are not included in the ANOVA assessing the effect of increasing egg white on biotin status.
TABLE 1.
Hepatic biotin content in mouse dams fed increasing concentrations of dietary egg white and concentrations in their fetuses1
| Dietary egg white, % | Dams | Fetuses | ||
|---|---|---|---|---|
| n | μmol/liver | n | pmol/g liver | |
| 25 + Biotin | 3 | 9.5 ± 2.1 | 2 | 960 ± 390 |
| Control2 | 8 | 3.6 ± 4.6 | 7 | 350 ± 50 |
| 1 | 4 | 2.4 ± 0.3 | 2 | 460 ± 1 |
| 1.3 | 3 | 2.0 ± 0.7 | ||
| 2 | 5 | 2.3 ± 0.2 | 3 | 410 ± 20 |
| 5 | 2 | 1.4 ± 0.1* | 2 | 13 ± 30 |
| 10 | 2 | 1.7 ± 0.2 | 2 | 90 ± 1* |
| 25 | 4 | 1.5 ± 0.1* | 3 | 65 ± 3* |
| ANOVA, P-value | <0.025 | <0.0025 |
Values are means ± SEM.
Different from the control, P ≤ 0.05.
Values from the standard diet group and the 0% egg white diet group were pooled to make the control group.
Urinary excretion of biotin decreased with duration of gestation (P < 0.0001 by repeated-measures ANOVA), but did not differ among the groups (Fig. 1). On GD 7, the decrease with time was significant (P < 0.05, Fisher's post-hoc test) for each diet containing at least 1% egg white. By GD 17, urinary biotin was lower (P < 0.003) even for the two control groups (the standard diet and 0% egg white) and for the 25% + biotin group. The substantial decreases in urinary biotin for these groups suggest that at least some degree of maternal biotin depletion developed late in pregnancy despite the provision of biotin in amounts that are adequate for nonpregnant mice. However, for the standard diet and the 25% + biotin diet groups, hepatic biotin was normal at GD 17, providing evidence that biotin status was only marginally decreased in these groups even at term.
FIGURE 1.
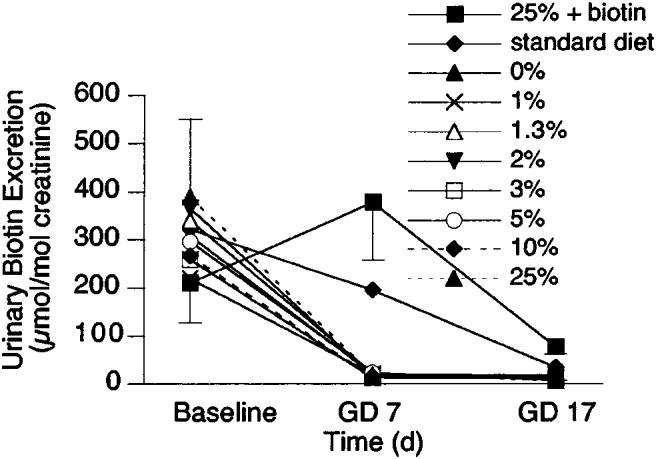
Effect of duration of gestation and increasing concentrations of egg white in the diet on urinary excretion of biotin in pregnant mice. Biotin excretion decreased significantly with duration of gestation (P < 0.001 by repeated-measures ANOVA), but diet had no effect. At baseline, biotin excretion did not differ among the diet groups. By gestational day (GD) 7, biotin excretion was lower (P < 0.05 by Fisher's post-hoc test) for all diet groups consuming egg white. By GD 17, biotin excretion was lower (P < 0.003) for all diet groups, including the control groups. Values are means ± sem, n ≥ 4.
Propionyl-CoA carboxylase (PCC) activity in dam livers decreased with increasing egg white in the diet (Table 2, P = 0.008 by ANOVA). In mice fed 25% egg white, PCC activity was 44% of the control value. Enzyme activities in those fed 10 and 25% egg white were lower than those of control (P < 0.002 by Fisher's post-hoc test).
TABLE 2.
Hepatic propionyl-CoA carboxylase (PCC) activity in mouse dams and fetuses with increasing concentrations of dietary egg white1
| Dietary egg white, % | Dams | Fetuses | ||
|---|---|---|---|---|
| n | nmol/(min · g liver) | n | nmol/(min · g liver) | |
| 25 + Biotin | 6 | 2350 ± 200 | 4 | 460 ± 90 |
| Control2 | 8 | 1110 ± 150 | 5 | 320 ± 90 |
| 1 | 4 | 1030 ± 70 | 4 | 430 ± 20 |
| 1.3 | 5 | 1060 ± 120 | 2 | 110 ± 60 |
| 2 | 5 | 840 ± 50 | ||
| 3 | 5 | 840 ± 80 | 4 | 85 ± 26* |
| 5 | 7 | 780 ± 130 | 7 | 46 ± 11* |
| 10 | 6 | 510 ± 110* | 6 | 39 ± 3* |
| 25 | 5 | 490 ± 190* | 5 | 65 ± 30* |
| ANOVA, P-value | 0.008 | <0.0001 |
Values are means ± SEM.
Different from the control diet, P ≤ 0.01 for dams; P ≤ 0.003 for fetuses.
Values from the standard diet group and the 0% egg white diet group were pooled to make the control group.
Maternal excretion of 3HIA increased strikingly with duration of gestation in some diet groups (Fig. 2). The increase was significant for time, diet and the interaction of time and diet (P < 0.0001 for all by repeated-measures, two-way ANOVA). At GD 7, 3HIA excretion rates for mice fed 5, 10 and 25% egg white differed from the two control groups (the standard diet and 0% egg white, P < 0.005). By GD 17, excretion of 3HIA in mice fed 3% egg white also differed from that of mice fed the control diet.
FIGURE 2.
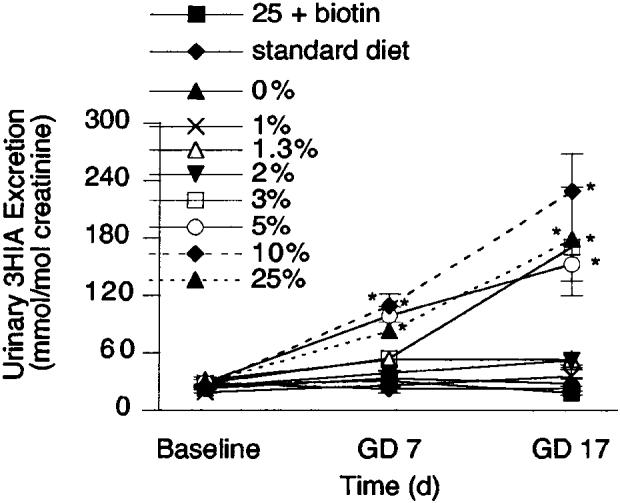
Effect of duration of gestation and increasing concentrations of egg white in the diet on urinary excretion of 3-hydroxyisovaleric acid (3HIA) in pregnant mice. Symbols denote values from at least four animals. At gestational day (GD) 0, 3HIA excretion did not differ among the diet groups. Values are means ± sem, n ≥ 4. *Different from the 0% egg white diet group, P < 0.005.
Taken together, these indices of biotin status provide strong evidence that the egg white feeding successfully induced biotin deficiency in the dams. The severity of the deficiency increased as egg white in the diet increased. In the control and the 25% + biotin groups, dietary biotin intake was sufficient to maintain normal hepatic biotin and hepatic carboxylase activity, although urinary biotin excretion decreased significantly late in gestation (Tables 1 and 2).
Fetal biotin status
Fetal hepatic biotin concentration decreased significantly with increasing dietary egg white feeding (Table 1). Loss of sample due to technical problems reduced the number of pooled fetal livers available for analysis. Despite the reduced sample size, the decrease in fetal hepatic biotin concentration was significant (P < 0.0025). The means for the 10 and 25% groups were significantly different from the control group by post-hoc test. Fetal PCC activity also was lower (P < 0.0001, Table 2). PCC activity in all diet groups fed ≥3% egg white differed from the control (P < 0.003).
Relationship of dam to fetus
The relationship between dam biotin status and fetal biotin status was examined for hepatic biotin and PCC activity. Hepatic biotin in pooled liver homogenates from the same litter correlated with hepatic biotin of the corresponding dam (Fig. 3A); r = 0.67, P < 0.0001). Fetal PCC activity correlated with PCC activity of the corresponding dam (Fig. 3B); r = 0.70, P < 0.0001.
FIGURE 3.
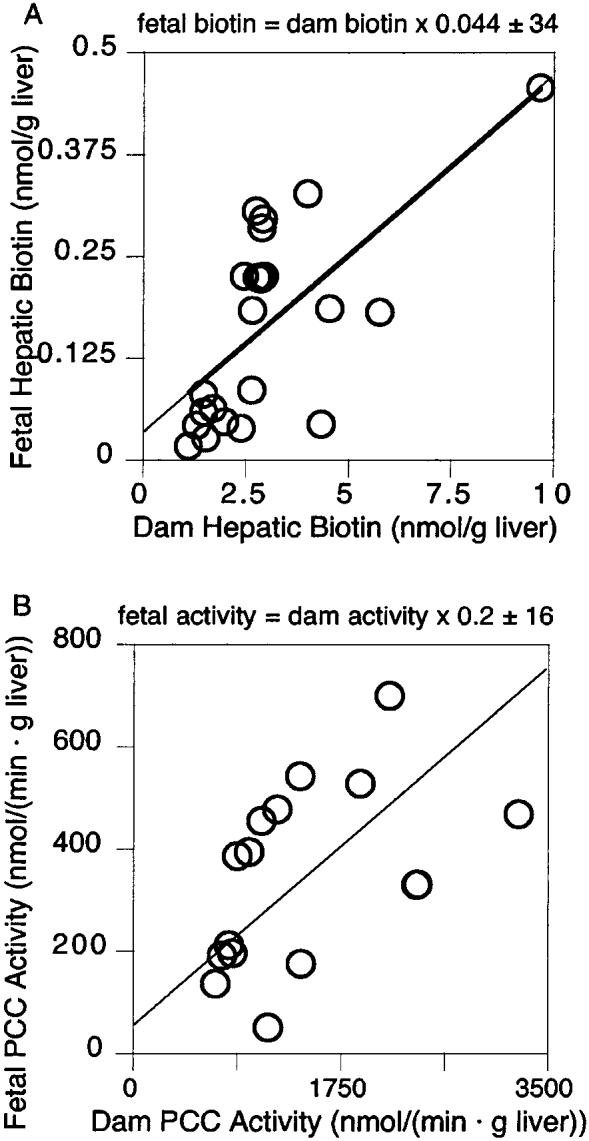
Correlation between fetal biotin status and dam biotin status in mice dams and pups when the dams were fed increasing concentrations of egg white in the diet. (A) The hepatic concentration of individual litter pools increased as the hepatic biotin of the corresponding dam increased (r = 0.67, P < 0.0001). (B) Fetal propionyl-CoA carboxylase (PCC) activity increased as the hepatic PCC of the corresponding dam increased (r = 0.70, P < 0.0001).
Pregnancy outcome
Maternal reproduction success and fetal viability were not affected by the egg white diet (Table 3). Neither the number of implantation sites nor the number of dead fetuses was altered by egg white concentration. Mean fetal weight was lower only with the 25% egg white diet. To assess whether the teratogenic events observed in this study could have been due to a specific teratogen present in the egg white diet rather than due to biotin deficiency per se, a 25% egg white diet with excess biotin was investigated. This diet produced biotin status that was significantly increased relative to the control group. Malformations were not observed in this group. The numbers of implants, dead fetuses and fetal weight did not differ between the 25% + biotin group and the combined control groups (Table 3).
TABLE 3.
Viability of fetuses and reproductive efficiency of mouse dams fed increasing concentrations of dietary egg white1
| Dietary egg white, % | Control2 | 1 | 1.3 | 2 | 3 | 5 | 10 | 25 | 25 + Biotin |
|---|---|---|---|---|---|---|---|---|---|
| Litters, n | 8 | 4 | 5 | 5 | 6 | 8 | 6 | 5 | 5 |
| n/litter | |||||||||
| Implantations | 12 ± 1 | 12 ± 1 | 13 ± 1 | 13 ± 1 | 11 ± 1 | 11 ± 1 | 11 ± 1 | 13 ± 0.4 | 12 ± 0.2 |
| Nonliving | 0.80 ± 0.40 | 0.50 ± 0.30 | 0.60 ± 0.40 | 0.20 ± 0.20 | 0.20 ± 0.20 | 1.1 ± 0.6 | 0.70 ± 0.30 | 0.60 ± 0.20 | 0.60 ± 0.40 |
| Living | 11 ± 1 | 12.0 ± 0.1 | 13 ± 1 | 12 ± 1 | 11 ± 1 | 10 ± 1 | 11 ± 1 | 12 ± 0.4 | 12 ± 0.3 |
| g/litter | |||||||||
| Fetal weight | 0.79 ± 0.03 | 0.83 ± 0.01 | 0.80 ± 0.03 | 0.82 ± 0.03 | 0.77 ± 0.04 | 0.73 ± 0.04 | 0.70 ± 0.02 | 0.68 ± 0.03* | 0.81 ± 0.3 |
Values are means ± SEM.
Different from control, P < 0.05.
Values from the standard diet group and the 0% egg white diet group were pooled to make the control group.
Fetal malformations and skeletal anomalies in the live fetuses increased with increasing egg white in the diet (Table 4). The incidence of cleft palate increased in a dose-dependent fashion for mice fed diets containing ≥2% egg white. Micrognathia and microglossia increased significantly in mice fed diets containing ≥3 and ≥5%, respectively. Hydrocephaly tended to increase when the diet contained egg white ≥3%, but the increase was significant (P < 0.05) only at 5% egg white concentration. The incidence of open eye tended to increase in mice fed diets containing ≥5% egg white and was significant (P < 0.05) at 25%. Incidences of hypoplasia of forelimbs, hind limbs, and the pelvic girdle were increased at all egg white concentrations ≥3%. Hypoplasia was determined relative to fetal size and was agreed on by two observers without knowledge of treatment.
TABLE 4.
Malformations per litter from mouse dams fed increasing concentrations of dietary egg white1
| Dietary egg white, % | Control2 | 1 | 1.3 | 2 | 3 | 5 | 10 | 25% |
|---|---|---|---|---|---|---|---|---|
| n/litter | ||||||||
| Cleft palate | 0.10 ± 0.13 | 0.25 ± 0.25 | 2 ± 2 | 4 ± 2* | 6 ± 2* | 10 ± 1* | 11 ± 1* | 12 ± 0.4* |
| Micrognathia | 0.0 | 0.0 | 0.0 | 0.2 ± 0.2 | 3 ± 2* | 9 ± 1* | 11 ± 1* | 12 ± 1* |
| Microglossia | 0.0 | 0.0 | 0.0 | 0.0 | 0.5 ± 0.5 | 5 ± 2* | 6 ± 2* | 9 ± 3* |
| Hydrocephaly | 0.0 | 0.0 | 0.0 | 0.0 | 0.3 ± 0.2 | 2 ± 1* | 3 ± 1 | 3 ± 2 |
| Open eye | 0.0 | 0.0 | 0.40 ± 0.40 | 0.0 | 0.0 | 0.80 ± 0.50 | 4 ± 1 | 5 ± 2* |
| Forelimb hypoplasia | 0.0 | 0.0 | 0.0 | 0.0 | 7 ± 2* | 9 ± 2* | 11 ± 1* | 12 ± 0.4* |
| Hindlimb hypoplasia | 0.0 | 0.0 | 0.0 | 0.0 | 5 ± 2* | 9 ± 2* | 10 ± 0.8* | 12 ± 0.4* |
| Pelvic girdle hypoplasia | 0.0 | 0.0 | 0.0 | 0.0 | 4 ± 2* | 9 ± 2* | 10 ± 1* | 12 ± 0.5* |
Values are means ± SEM.
Different from control, P < 0.05.
Values from the standard diet group and the 0% egg white diet group were pooled to make the control group.
The occurrence of cleft palate, micrognathia, and microglossia were clearly dose dependent (Fig. 4), i.e., the occurrence of cleft palate and micrognathia were ∼100% in mice fed diets containing ≥5% egg white. The effective dose for 50% occurrence (ED50) of clefting occurred when the diet contained 3% egg white. Similarly, at least half the fetuses demonstrated micrognathia and microglossia at ∼4% egg white in the diet.
FIGURE 4.
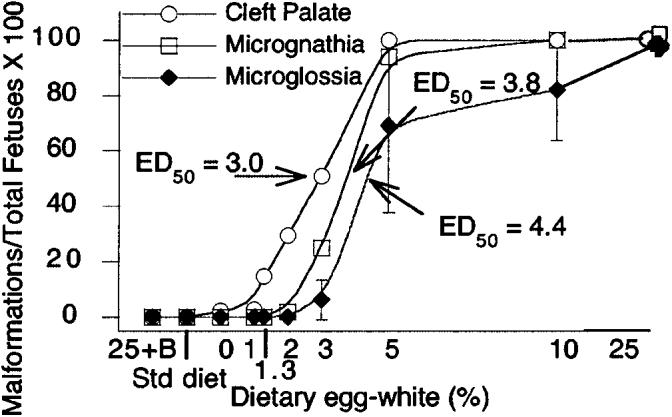
The incidence of cranial malformations increased with increasing concentrations of egg white in the diet. Fetuses whose dams were fed the 25% egg white diet had 100% incidence of clefting. The 50% effective dose (ED50) of clefting was 3% egg white. The ED50 for micrognathia and microglossia were at ∼4% egg white. For fetuses, the mean n for groups was 6; range 4–8.
The global reduction of skeleton size of the biotin-deficient fetuses was striking (Fig. 5). The incidence of limb hypoplasia in mice fed the 25% egg white diet was 100% and showed a dose-dependent increase at egg white concentrations ≥3% (Fig. 6).
FIGURE 5.
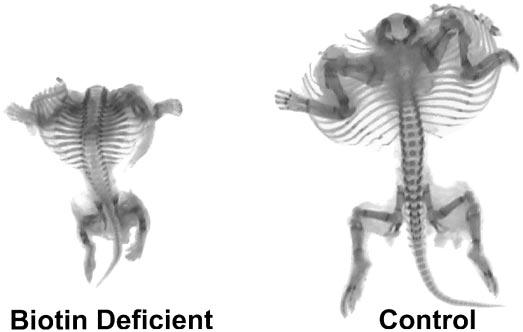
Skeletal malformations in fetuses whose dams were fed increasing concentrations of egg white in the diet. Fetuses whose dams were fed the 25% egg white diet exhibited striking shortening of the long bones of the limbs; control fetuses exhibited normal skeletal anatomy.
FIGURE 6.
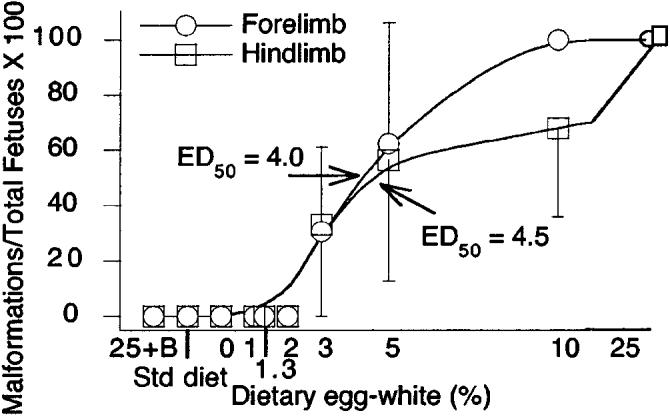
The incidence of limb hypoplasia in fetuses whose dams were fed increasing concentrations of egg white in the diet. The incidence was 100% in fetuses whose dams were fed the 25% egg white diet; incidence rates showed a dose-dependent increase at egg white concentrations >3%. The 50% effective dose (ED50) for limb hypoplasia was at ∼4% egg white. The n for each diet group was as in Figure 4.
DISCUSSION
The combination of decreased maternal hepatic biotin content, decreased hepatic PCC activity, increased 3HIA excretion and decreased urinary biotin excretion provides strong evidence that egg white diets consumed during pregnancy caused biotin deficiency in the dams. Decreased fetal hepatic biotin concentration and PCC activity indicate that the fetuses also became biotin deficient. The significant correlations between fetal and maternal hepatic biotin and between fetal and maternal hepatic PCC activity provide evidence that maternal biotin deficiency caused fetal biotin deficiency. The teratogenic effects observed in these studies are not attributable to a teratogen present in the egg white diet or to some other micronutrient deficiency. This conclusion is based on the observation that no malformations were seen in a control group that received a diet containing 25% egg white plus sufficient biotin to titrate all of the biotin-binding sites on avidin and excess biotin to meet the estimated biotin requirement of mice. The striking dose dependence of malformations on the amount of egg white in the diet and the complete reversibility with biotin supplementation of the highest egg white diet provide evidence that biotin deficiency is the cause of the observed fetal malformations. However, the results from the study described here do not exclude the possibility that maternal biotin deficiency could act through a different mechanism other than fetal biotin deficiency to produce teratogenic events in the fetus; such a mechanism could be sufficient by itself or could act in concert with fetal biotin deficiency.
Why were no malformations seen in the 0% egg white group or the 1% egg white group given that no biotin was added to either of these diets? Although no biotin was added to these diets, substantial amounts of biotin were present in the macronutrient components of the diets. In our laboratory, analysis of the diet with no added egg white (0%) detected 5 μg biotin/kg diet. The dietary requirement for biotin in mice is not known. For calculating the mouse requirement, we assume that the requirement per gram body weight is approximately that of humans [recommended dietary intake (RDI) 30 μg/d]. Because these mice consumed ∼0.15 g diet/(g body · d), biotin intake via the 0% egg white diet equaled 0.75 μg/kg body, which exceeds the human RDI by approximately twofold. This calculation is consistent with the observation that dietary avidin in egg white is required to render the biotin unavailable. Moreover, microbial biotin synthesis may contribute to absorbed biotin either as a primary process or as a consequence of coprophagia not prevented by individual housing and wire-bottomed cages. If so, dietary avidin may be required to interrupt this supply of biotin. Such an interpretation is consistent with the increase in 3HIA excretion and malformations noted in mice fed the diet containing ∼3% dietary egg white.
Given that biotin transport across the human placenta is apparently weak (23-25), how relevant is this mouse model to human teratogenesis? Several observations suggest that this mouse model is relevant. The cranial and skeletal birth defects occurred even though dams gained weight normally and showed no physical signs of biotin deficiency. The same is often true for these malformations when they occur as nonsyndromatic events in human gestation. Except for the highest egg white diet group, mouse fetuses gained weight normally, and few died in utero. Similarly, nonsyndromatic human cleft palate and limb shortening characteristically occur in the context of normal somatic growth and without a history of spontaneous abortions. The similarity of the proportional increases in 3HIA excretion also suggest that this egg white feeding produced a degree of biotin deficiency in our mouse model similar to that which occurs spontaneously in some women during normal human gestation. Specifically, compared with GD 0 mice that had not yet begun to consume an egg white diet, urinary 3HIA excretion increased two- to fourfold by GD 7 in pregnant mice fed the 25% egg white diet. This relative increase is similar to the two- to fourfold increase observed in women during the first trimester of pregnancy relative to the excretion rate in women who are not pregnant (26). In future studies, we hope to compare the relative decrease in activity of PCC in peripheral blood lymphocytes taken from pregnant women during the first trimester to values generated for hepatic PCC activity in rodents as an additional assessment of comparative biotin status.
However, comparison of biotin status across species may be problematic. For example, egg white feeding was not as effective in producing teratogenic events in some strains of mice as in ICR mice and was noticeably unsuccessful in producing teratogenic events in rats (6). The cause of the species difference is not clear. We speculate that the difference might be any one or a combination of several factors: 1) The dietary concentration and interval for egg white feeding may not have produced as severe or as early biotin deficiency in rats as in ICR mice. The study comparing species (6) did not assess the degree or time of onset of biotin deficiency, thus preventing comparison with the biotin status assessed in this study. 2) Biotin transport across the placenta may vary substantially among species. At a given degree of maternal deficiency, a higher affinity transporter or a higher Vmax for total transport at the brush border membrane of the epithelial cells of the cotyledon might have ameliorated biotin depletion in rat fetuses compared with mouse fetuses. 3) Fetal susceptibility to the teratogenic effects of biotin deficiency may vary from species to species. The mechanism by which biotin deficiency leads to teratogenic events remains unknown, but is a topic of active investigation in our laboratory.
The study described here confirms the conclusions of Watanabe et al. concerning the relationship between maternal biotin deficiency and fetal malformations in mice (4-8,27,28). In certain strains of mice (6-8,27,28), biotin deficiency was associated with substantial increases in fetal malformations and mortality. Although questioned at one point (29), studies in mice (4,8,30) and mouse fetal culture (28) provided evidence that the teratogenic effect of the egg white diet is caused by biotin deficiency per se. This study extends the findings of Watanabe and co-workers by providing careful assessment of the dose dependence on egg white concentration, by providing new assessments of maternal and fetal biotin status and by revealing a significant relationship between maternal and fetal biotin status.
Conclusions concerning the teratogenic effect of biotin deficiency likely apply to other species. Hens with a marginal biotin deficiency produced eggs with higher embryonic mortality, reduced hatchability, chronodystrophy (“parrot beak” deformity), perosis (an abnormality of bone/tendon formation that results in a deformity similar to “club foot”), micromelia and syndactyly (31-34). Similar effects on hatchability and viability have been observed in turkey poults (35).
In conclusion, we have demonstrated that a constellation of malformations including cleft palate and short limb were caused by biotin deficiency and that the incidence increases with increasing biotin deficiency. These malformations appeared at degrees of biotin deficiency that are similar to those in human gestation. We speculate that some human fetal malformations may be caused by biotin deficiency.
Footnotes
Presented in part at North American Pediatric Gastroenterology and Nutrition Meeting, October 1999, Denver, CO [Mock, D. M., Mock, N. I., Stewart, C. W., LaBorde, J. B. & Hansen, D. K. (1999) Maternal biotin deficiency causes fetal malformations by inducing fetal biotin deficiency. J. Pediatr. Gastroenterol. Nutr. 29: 504 (abs.)]; at Experimental Biology 99, April 1999, Washington DC [Stewart, C. W., Mock, N. I., Mock, D. M. & Hansen, D. (1999) Maternal biotin deficiency (BD) causes fetal malformations (FM) by inducing fetal BD. FASEB J. 13 A227 (abs.)]; and at the Teratology Society Annual Meeting, June 1998, San Diego, CA [LaBorde, J. B., Wall, K. S., Mock, N. I., Mock, D. M. & Hansen, D. K. (1998) Embryotoxicity of dietary biotin deficiency. Teratology 57: 233–234a (abs.)].
This work was supported by National Institutes of Health National Institute of Diabetes, Digestive, and Kidney Diseases grant DK36823.
Abbreviations used: 3HIA, 3-hydroxyisovaleric acid; ED50, 50% effective dose; GD, gestation day; PCC, propionyl-CoA carboxylase; RDI, recommended dietary intake.
LITERATURE CITED
- 1.Mock DM, Stadler DD. Conflicting indicators of biotin status from a cross-sectional study of normal pregnancy. J. Am. Coll. Nutr. 1997;16:252–257. doi: 10.1080/07315724.1997.10718682. [DOI] [PubMed] [Google Scholar]
- 2.Mock DM, Stadler D, Stratton S, Mock NI. Biotin status assessed longitudinally in pregnant women. J. Nutr. 1997;127:710–716. doi: 10.1093/jn/127.5.710. [DOI] [PubMed] [Google Scholar]
- 3.Zempleni J, Mock DM. Marginal biotin deficiency is teratogenic. Proc. Soc. Exp. Biol. Med. 2000;223:14–21. doi: 10.1046/j.1525-1373.2000.22303.x. [DOI] [PubMed] [Google Scholar]
- 4.Watanabe T, Endo A. Teratogenic effects of avidin-induced biotin deficiency in mice. Teratology. 1984;30:91–94. doi: 10.1002/tera.1420300112. [DOI] [PubMed] [Google Scholar]
- 5.Watanabe T. Teratogenic effects of biotin deficiency in mice. J. Nutr. 1983;113:574–581. doi: 10.1093/jn/113.3.574. [DOI] [PubMed] [Google Scholar]
- 6.Watanabe T, Endo A. Species and strain differences in teratogenic effects of biotin deficiency in rodents. J. Nutr. 1989;119:255–261. doi: 10.1093/jn/119.2.255. [DOI] [PubMed] [Google Scholar]
- 7.Watanabe T. Dietary biotin deficiency affects reproductive function and prenatal development in hamsters. J. Nutr. 1993;123:2101–2108. doi: 10.1093/jn/123.12.2101. [DOI] [PubMed] [Google Scholar]
- 8.Watanabe T, Endo A. Teratogenic effects of maternal biotin deficiency in mouse embryos examined at midgestation. Teratology. 1990;42:295–300. doi: 10.1002/tera.1420420313. [DOI] [PubMed] [Google Scholar]
- 9.Mock N, Malik M, Stumbo P, Bishop W, Mock D. Increased urinary excretion of 3-hydroxyisovaleric acid and decreased urinary excretion of biotin are sensitive early indicators of decreased status in experimental biotin deficiency. Am. J. Clin. Nutr. 1997;65:951–958. doi: 10.1093/ajcn/65.4.951. [DOI] [PubMed] [Google Scholar]
- 10.Mock DM, Henrich CL, Carnell N, Mock NI. Indicators of marginal biotin deficiency and repletion in humans: validation of 3-hydroxyisovaleric acid excretion and a leucine challenge. Am. J. Clin. Nutr. 2002;76:1061–1068. doi: 10.1093/ajcn/76.5.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mock DM, Mock NI. Lymphocyte propionyl-CoA carboxylase activity is an early and sensitive indicator of biotin deficiency in rats, but urinary excretion of 3-hydroxyisopropionic acid is not. J. Nutr. 2002;132:1945–1950. doi: 10.1093/jn/132.7.1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilson JG. Embryological considerations in teratology. In: Wilson JG, Warkany J, editors. Teratology: Principles and Techniques. University of Chicago Press; Chicago, IL: 1965. [Google Scholar]
- 13.Staples RE. Detection of visceral alterations in mammalian fetuses. Teratology. 1974;9:37A. (abs.) [Google Scholar]
- 14.Stuckhardt JH, Poppe SM. Fresh visceral examination of rat and rabbit fetuses used in teratogenicity testing. Teratog. Carcinog. Mutagen. 1984;4:181–188. doi: 10.1002/tcm.1770040203. [DOI] [PubMed] [Google Scholar]
- 15.LaBorde JD, Pipkin JL, Hinson WG, Anson JF, Sheehan DM, Young JF, Hansen DK. Retinoic acid-induced stress protein synthesis in the mouse. Life Sci. 1995;56:1767–1778. doi: 10.1016/0024-3205(95)00148-y. [DOI] [PubMed] [Google Scholar]
- 16.Mock DM. Evidence for a pathogenic role of ω6 polyunsaturated fatty acid in the cutaneous manifestations of biotin deficiency. J. Pediatr. Gastroenterol. Nutr. 1990;10:222–229. doi: 10.1097/00005176-199002000-00013. [DOI] [PubMed] [Google Scholar]
- 17.Mock DM, Malik MI. Distribution of biotin in human plasma: Most of the biotin is not bound to protein. Am. J. Clin. Nutr. 1992;56:427–432. doi: 10.1093/ajcn/56.2.427. [DOI] [PubMed] [Google Scholar]
- 18.Mock DM. Determinations of biotin in biological fluids. In: McCormick DB, Suttie JW, Wagner C, editors. Methods in Enzymology. Academic Press; New York, NY: 1997. [DOI] [PubMed] [Google Scholar]
- 19.Mock DM, Jackson H, Lankford GL, Mock NI, Weintraub ST. Quantitation of urinary 3-hydroxyisovaleric acid using deuterated 3-hydroxyisovaleric acid as internal standard. Biomed. Environ. Mass Spectrom. 1989;18:652–656. doi: 10.1002/bms.1200180903. [DOI] [PubMed] [Google Scholar]
- 20.Heingard D, Tiderstrom G. Determination of serum creatinine by a direct calorimetric method. Clin. Chim. Acta. 1973;43:305–310. doi: 10.1016/0009-8981(73)90466-x. [DOI] [PubMed] [Google Scholar]
- 21.Rose DB. Clinical physiology of acid-base and electrolyte disorders. In: Jeffers J, Navrozov M, editors. Renal Physiology. McGraw-Hill; New York, NY: 1994. [Google Scholar]
- 22.Zempleni J, Trusty TA, Mock DM. Lipoic acid reduces the activities of biotin-dependent carboxylases in rat liver. J. Nutr. 1997;127:1776–1781. doi: 10.1093/jn/127.9.1776. [DOI] [PubMed] [Google Scholar]
- 23.Schenker S, Hu Z, Johnson RF, Yang Y, Frosto T, Elliott BD, Henderson GI, Mock DM. Human placental biotin transport: normal characteristics and effect of ethanol. Alcohol. Clin. Exp. Res. 1993;17:566–575. doi: 10.1111/j.1530-0277.1993.tb00801.x. [DOI] [PubMed] [Google Scholar]
- 24.Hu Z-Q, Henderson GI, Mock DM, Schenker S. Biotin uptake by basolateral membrane of human placenta: normal characteristics and role of ethanol. Proc. Soc. Biol. Exp. Med. 1994;206:404–408. doi: 10.3181/00379727-206-43778. [DOI] [PubMed] [Google Scholar]
- 25.Karl P, Fisher SE. Biotin transport in microvillous membrane vesicles, cultured trophoblasts and the isolated perfused cotyledon of the human placenta. Am. J. Physiol. 1992;262:C302–C308. doi: 10.1152/ajpcell.1992.262.2.C302. [DOI] [PubMed] [Google Scholar]
- 26.Mock DM, Quirk JG, Mock NI. Marginal biotin deficiency during normal pregnancy. Am. J. Clin. Nutr. 2002;75:295–299. doi: 10.1093/ajcn/75.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Watanabe T, Endo A. Biotin deficiency per se is teratogenic in mice. J. Nutr. 1991;121:101–104. doi: 10.1093/jn/121.1.101. [DOI] [PubMed] [Google Scholar]
- 28.Watanabe T, Dakshinamurti K, Persaud TVN. Biotin influences palatal development of mouse embryos in organ culture. J. Nutr. 1995;125:2114–2121. doi: 10.1093/jn/125.8.2114. [DOI] [PubMed] [Google Scholar]
- 29.Heard GS, Blevins TL. Is biotin deficiency teratogenic in mice? J. Nutr. 1989;119:1348–1349. doi: 10.1093/jn/119.9.1348. [DOI] [PubMed] [Google Scholar]
- 30.Watanabe T, Endo A. Reply to the letter of G. S. Heard and T. L. Blevins. J. Nutr. 1989;119:1350. [Google Scholar]
- 31.Couch JR, Craven WW, Elvehjem CA, Halpin JG. Relation of biotin to congenital deformities in the chick. Anat. Rec. 1948;100:29–48. doi: 10.1002/ar.1091000104. [DOI] [PubMed] [Google Scholar]
- 32.Cravens WW, McGibbon WH, Sebesta EE. Effect of biotin deficiency on embryonic development in the domestic fowl. Anat. Rec. 1944;90:55–64. [Google Scholar]
- 33.Balnave D. Clinical symptoms of biotin deficiency in animals. Am. J. Clin. Nutr. 1977;30:1408–1413. doi: 10.1093/ajcn/30.9.1408. [DOI] [PubMed] [Google Scholar]
- 34.Whitehead CC. Effect of nutrient deficiencies in animals: biotin. In: Recheigl M, editor. CRC Handbook Series in Nutrition and Food. CRC Press; West Palm Beach, FL: 1978. [Google Scholar]
- 35.Ferguson TM, Whiteside TH, Creger CR, Jones ML, Atkinson AL, Couch JR. B-vitamin deficiency in the mature turkey hen. Poult. Sci. 1961;40:1151–1159. [Google Scholar]