

Abstract
Human replication protein A (hRPA), a heterotrimeric single-stranded DNA (ssDNA) binding protein, is required for many cellular pathways including DNA damage repair, recombination, and replication as well as the ATR-mediated DNA damage response. While extensive effort has been devoted to understanding the structural relationships between RPA and ssDNA, information is currently limited to the RPA domains, the trimerization core, and a partial cocrystal structure. In this work, we employed a mass spectrometric protein footprinting method of single amino acid resolution to investigate the interactions of the entire heterotrimeric hRPA with ssDNA. In particular, we monitored surface accessibility of RPA lysines with NHS-biotin modification in the contexts of the free protein and the nucleoprotein complex. Our results not only indicated excellent agreement with the available crystal structure data for RPA70 DBD-AB-ssDNA complex but also revealed new protein contacts in the nucleoprotein complex. In addition to two residues, K263 and K343 of p70, previously identified by cocrystallography as direct DNA contacts, we observed protection of five additional lysines (K183, K259, K489, K577, and K588 of p70) upon ssDNA binding to RPA. Three residues, K489, K577, and K588, are located in ssDNA binding domain C and are likely to establish the direct contacts with cognate DNA. In contrast, no ssDNA-contacting lysines were identified in DBD-D. In addition, two lysines, K183 and K259, are positioned outside the putative ssDNA binding cleft. We propose that the protection of these lysines could result from the RPA interdomain structural reorganization induced by ssDNA binding.
Replication protein A (RPA), the eukaryotic single-stranded DNA (ssDNA) binding protein (SSB), is an essential protein required for DNA replication, repair, and recombination (1, 2, 6) as well as in ATR-mediated DNA damage response (3, 4). For damage recognition in nucleotide excision repair, RPA binds to the ssDNA region of DNA induced by bulky DNA adducts (7, 8). Human RPA (hRPA) is a heterotrimeric protein comprised of three subunits, p70, p32, and p14 (1-5). The ssDNA binding ability of hRPA is derived from its four DNA binding domains (DBDs) located in p70 and p32. Mutational and biochemical analyses of recombinant RPA have localized the major ssDNA binding affinity to the DBD-A, DBD-B, and DBD-C in p70, with the DBD-D of p32 required for higher order binding modes. The p14 subunit is not implicated in ssDNA binding but is required for stable trimer formation (1, 9, 18, 19). Each DBD is constructed around a central oligonucleotide/oligosaccharide-binding fold (OB-fold) characterized as a five-strand β-sheet coiled to form a β-barrel capped by an α-helix between the third and fourth strands (20-23). RPA binding to ssDNA occurs via a sequential and hierarchal pathway with a polarity of 5′ to 3′ in three separate modes, resulting in a total of 30 nucleotides occluded. The interaction begins with an initial binding of 8-10 nucleotides (nt) by DBD-AB located in the central region of p70, followed by an intermediate binding mode of 12-23 nt by DBD-AB plus DBD-C (C-terminal, p70). Finally, a stable complex occluding 30 nt is formed when DBD-D, located in the central region of p32, engages the nucleic acids (1-2, 9, 14, 15, 18-21, 35, 36). RPA binds ssDNA with high affinity (Ka) 109-1011 M-1) and low cooperativity with a preference for pyrimidine-rich sequence and binds dsDNA and RNA with much less affinity (1, 9, 12, 16). RPA also interacts with a variety of nuclear factors, including but not limited to DNA polymerase R, p53, XPA, XPG, ERCC1/XPF, Rad51, Rad52, UNG2, BRCA1, and BRCA2 (1, 10, 25-30).
Although much work has been done to resolve the structures of RPA and the RPA/ssDNA complex, how the entire protein interacts with ssDNA remains elusive. This issue becomes even more interesting as dramatic global structural changes have been suggested for RPA to facilitate ssDNA binding (15, 18, 24). NMR and X-ray crystallography have yielded important advancements in our understanding of RPA/ssDNA interactions. However, these techniques have been limited to characterization of the domains and sub structures of RPA (21, 22, 28, 33, 34). This limitation has hampered our ability to understand how the intact RPA protein behaves during binding and how conformational shifts occur during binding mode transitions. Recently, a method has been developed by Kvaratskhelia et al. for accurate mapping of protein-nucleic acid contacts using mass spectrometry (31, 32). This method is based on the principle of protection of lysine residues in the binding sites from chemical modification when nucleic acid is present (31). Although trypsin digestion methods have previously been used to investigate RPA-nucleic acid interactions, these were limited by their low-resolution detection methods, such as SDS-PAGE (23, 24). In contrast, the mass spectrometric approach allows identification of specific protein-nucleic acid contacts at single amino acid resolution under physiologically relevant conditions where integrity of the entire nucleoprotein structure is fully preserved (31, 32).
In our present work, we have used the mass spectrometry footprinting technique (31) to identify specific lysine residues within the RPA binding clefts involved in ssDNA binding. Using the cocrystal structure of DBDs-AB-dC8mer (22) as an internal control, we have identified three new ssDNA-contact points within DBD-C of RPA70. These lysines are similar to those found in DBD-AB in location, orientation, and solvent accessibility in the unbound state. Also, we have located two possible interdomain contact points between the N-terminal domain and DBD-A of p70 upon ssDNA binding of RPA.
MATERIALS AND METHODS
Protein Purification and Substrate Labeling. Recombinant human RPA was expressed in Escherichia coli BL21(DE3)-RP (Stratagene) cells and purified as previously described (17). Protein concentration was determined by Bradford assay using Bio-Rad protein assay reagent. Oligonucleotide substrate was radiolabeled with [γ-32P]ATP (>5000 mCi/mmol; Amersham) using T4-PNK (New England Biolabs), and unincorporated radioactive nucleotides were removed by a P6 spin column (Bio-Rad).
Gel Mobility Shift Assays. Effects of the biotin modification on RPA-ssDNA binding were determined by gel mobility shift assays. Typically, the labeled oligonucleotide dT50mer (MWG Genetic) substrate (20 μM) was incubated with unmodified RPA (2 μM) at a molar ratio of DNA:RPA) 10:1 in binding buffer (40 mM HEPES-KOH, pH 7.9, 75 mM KCl, 8 mM MgCl2, 1 mM DTT, 4% glycerol) for 30 min at room temperature. The preformed complex was then exposed to modification with increasing concentrations of N-hydroxysuccinimidobiotin (NHS-biotin, Pierce) for another 30 min. The reactions were quenched by adding lysine in its free amino acid form (10 mM of final concentration). In parallel experiments, RPA was modified first with NHS-biotin, and then ssDNA was added to the reaction mix. Reactions were immediately loaded onto 3.5% native polyacrylamide gels and electrophoresed in TBE buffer at room temperature.
Biotin Modification and In-Gel Proteolysis. Purified hRPA was modified with NHS-biotin in the presence and absence of the single-stranded oligonucleotide dT30mer. Typically, RPA (2 μM) was incubated ±dT30mer (20 μM) in the binding buffer at room temperature for 30 min and then modified by adding NHS-biotin (400 μM of final concentration) for an additional 30 min incubation at room temperature. Modifications were quenched by addition of 10 mM lysine, followed by separation of the RPA subunits by SDS-PAGE. The subunits were visualized by Coomassie Blue staining, excised from the gel, and extensively destained in 50% methanol/10% acetic acid. SDS was removed by washing the gel pieces with ammonium bicarbonate, dehydrated with 100% acetonitrile, and vacuum desiccated. Samples were digested with 1 μg of trypsin (Roche) in 50 mM ammonium bicarbonate overnight at room temperature. The supernatant was then recovered for MS and MS/MS analysis.
Mass Spectrometric Analysis. MS spectra were obtained using matrix-assisted laser desorption time-of-flight (MALDI-TOF) and quadrupole time-of-flight (Q-TOF) techniques. MALDI-TOF experiments were performed using a Kratos Axima-CRF instrument (Kratos Analytical Instruments) with an α-cyano-4-hydroxycinnamic acid matrix. MS and MS/MS analyses were performed on a Micromass (Manchester, U.K.) Q-TOF-II instrument equipped with an electrospray source and Micromass cap-LC. Peptides were separated with a Waters Symmetry300 5 μm precolumn (Waters, Milford, MA) and a Micro-Tech Scientific (Vista, CA) ZC-10-C18SBWX-150 column using two sequential gradients of 5-40% acetonitrile for 35 min and 40-90% acetonitrile for 10 min. MS/MS sequence data and the MASCOT automated peptide search engine (http://www.matrixscience.com) were used to identify RPA peptide peaks from the NCBInr primary sequence database, and matched peaks were then located in the primary MS spectra. Protection events were qualitatively assigned as the appearance of a peak corresponding to a biotin-modified peptide in the modified protein spectrum and the absence of the modification peak in the modified nucleoprotein complex spectrum. A protection was considered to be significant only when the intensity of a modifiable peak was reduced by at least 85% in the nucleoprotein complex spectrum. To accurately identify protection events, at least two peaks that are not affected by procedures and present in all three spectra (unmodified protein, modified protein, and modified nucleoprotein complex) were used as control peaks. These control peaks served to standardize the peak intensities in each spectrum for accurate qualitative assignment of protection. Data were reproducibly compiled and analyzed from six independent experimental groups.
RESULTS
Effects of NHS-Biotin Modification on ssDNA Binding. The protein footprinting method we employed is based on the modification of solvent-accessible lysine residues with the primary amine specific reagent NHS-biotin and the protection of the contact lysines from modification by ssDNA binding. To properly identify protein-nucleic acid contacts, the modification procedure must be optimized to ensure the efficiency of NHS-biotin modification and that the integrity of the RPA-ssDNA complex is preserved. Therefore, we examined the effects of increasing concentrations of NHS-biotin on the ssDNA binding activity of RPA by gel mobility shift assays. Figure 1 shows the results of NHS-biotin modification of RPA before (Figure 1A) or after (Figure 1B) the addition of the dT50-mer. As NHS-biotin was added in increasing concentrations prior to nucleoprotein complex formation, nucleic acid binding affinity was reduced until it was completely lost at a concentration of 400 μM (Figure 1A, lane 6). This is an indication that lysine residues being modified are necessary for the formation of a stable RPA-ssDNA complex. However, when RPA was occupied by ssDNA prior to addition of NHS-biotin (Figure 1B), subsequent modification of the complex with 400 μM NHS-biotin (Figure 1B, lane 6) did not disrupt ssDNA binding. This indicates that the lysine residues necessary for stable ssDNA-binding are now shielded from modification in the nucleoprotein complex. From these experiments, we have determined a concentration of 400 μM NHS-biotin for efficient modification of the RPA-ssDNA complex while preserving the structural integrity of the complex.
Figure 1:
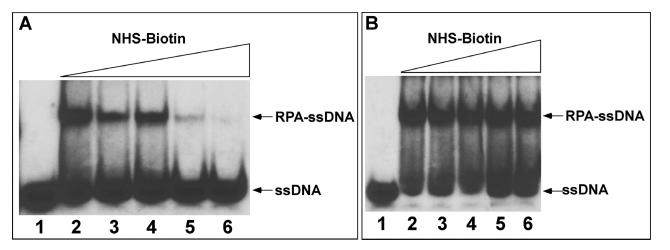
Effects of biotinylation on RPA-ssDNA binding. Lanes: 1, free ssDNA; 2-6, ssDNA bound by RPA pre- or posttreated with 0, 50, 100, 200, and 400 μM NHS-biotin. Panel A: Addition of increasing amounts of NHS-biotin prior to addition of ssDNA blocked critical lysine residues for ssDNA interaction and thus abolished binding affinity. Panel B: Addition of ssDNA prior to NHS-biotin, however, shielded critical lysine residues from modification and showed little effect of biotin on the formed RPA-ssDNA complex.
MS and MS/MS Analysis of the hRPA-ssDNA Complex. RPA is a heterotrimer with OB-folds located in all three subunits. To analyze each subunit individually, unmodified protein, modified protein, and the modified protein-ssDNA complex were subjected to SDS-PAGE prior to trypsin proteolysis. SDS-PAGE electrophoresis served two purposes: it allowed for the complete separation of the three subunits and rendered the protein in a linear, denatured form that equally exposed all possible trypsin proteolytic sites, ensuring a complete and reproducible hydrolysis of the protein. The hRPA lysines modified with NHS-biotin were readily detected by MS and MS/MS analysis of trypsin-generated peptide fragments. Figure 2A shows a representative MALDI-TOF spectrum for NHS-biotin-modified p70 with mass/charge peaks assigned to proteolysis-generated peptide fragments. Panels B and C of Figure 2 exhibit Q-TOF analysis of modified fragment 325-335 (K331 + biotin) of p70. Internal fragmentation of this parent peak primarily yielded y ions (Figure 2C), providing sequence information read from the C-terminus to the N-terminus. The mass increment between the y4 and y5 ions corresponded to the mass of a biotinylated lysine residue while all remaining y ions corresponded with the p70 sequence (Figure 2C).
Figure 2:

Mass spectrometric analysis of biotinylation of RPA protein. (A) A typical MALDI-TOF spectrum of tryptic digestion of biotinylated RPA70. Monoisotopic resolution for all of the peptide peaks was obtained, allowing unequivocal assignment of singly charged unmodified and biotinylated peptide fragments. (B) A typical Q-TOF spectrum of the doubly charged biotinylated p70 peptide fragment. The molecular mass of this ion in the form of m/z (mass per charge) corresponds to peptide fragment 325-335 + biotin. (C) MS/MS analysis of the parent ion (m/z) 754.91) shown in (B) confirms that lysine 331 is biotinylated. The y ion series were derived from internal fragmentation of the peptide bonds, providing sequence information read from the C-terminus (left) to the N-terminus (right). The mass increment between y4 and y5 ions corresponds to a biotinylated lysine while the masses of the remaining y ions correspond perfectly to the amino acid sequence of the fragment.
Our following efforts were focused to reveal lysines readily biotinylated in free RPA protein but protected from modification in the nucleoprotein complex. We assigned protection of lysine residues from modification based on the appearance of m/z peaks in the mass spectrum of the modified protein that are absent in both the modified nucleoprotein complex and the unmodified protein spectra. These m/z peaks correspond to the mass of a tryptically digested fragment plus the mass of the number of biotin molecules attached to the peptide. The number of biotin molecules attached to anytryptic peptide is equal to the number of missed lysine cleavage sites within the fragment. Figure 3 illustrates a typical assignment of lysine protection by Q-TOF analysis. When free RPA is biotinylated, a peak corresponding to p70 peptide fragment 260-267 (K263 + biotin) is observed. However, when RPA is modified following ssDNA binding, this peak is absent, indicating shielding of the lysine in the nucleoprotein complex. Biotin modification of lysine K139 on p32, however, is not protected when ssDNA is bound, and the peak corresponding to peptide fragment 139-145 (K139 + biotin) persists in both the free and bound RPA spectra. Peaks C1 and C2 appear as internal control peaks to provide reference for peak intensities in each spectrum.
Figure 3:
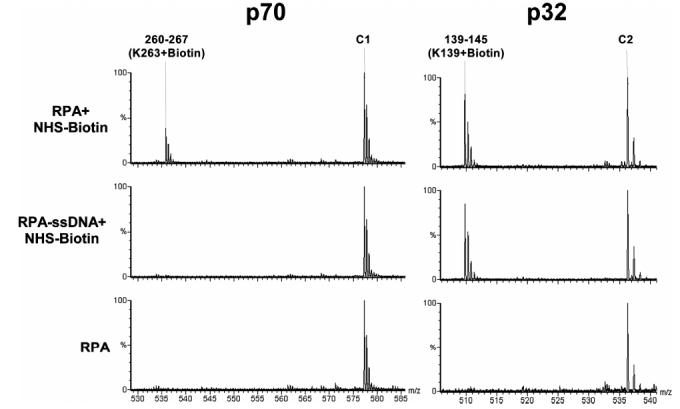
Identification of lysine residues protected from modification by nucleic acid. The typical Q-TOF data illustrate the protection of a lysine residue in p70 versus an unprotected lysine in p32. The peak corresponding to peptide fragment 260-267 + biotin in p70 is an example of protection from modification by binding of ssDNA. When ssDNA is not present, K263 is readily modified by NHS-biotin. In the presence of ssDNA, however, the modification peak disappears. In contrast, K139 in p32 is a lysine residue not protected by ssDNA binding. A modification peak appears upon treatment with NHS-biotin and persists following addition of ssDNA. Each multiply charged peptide resulted in clearly resolved peak clusters, indicating monoisotopic resolution; unmodified peaks C1 and C2 serve as controls.
In our analysis, seven lysine residues in p70 and none from p32 or p14 were found to be protected from NHS-biotin modification by ssDNA. Of those, two were previously observed as ssDNA contacts by X-ray crystallography (21), three were newly identified to be direct ssDNA contact points within DBD-C, and two were located outside of the binding clefts and believed to not be associated with ssDNA. Figure 4 illustrates the protection of K588 in DBD-C from biotin modification in the presence of the dT30mer. In the absence of ssDNA, K588 is readily biotinylated, and a peak corresponding to peptide fragment 587-600 + biotin is observed in the MALDI-TOF spectrum (Figure 4A). However, when the dT30mer is present, K588 is shielded from modification by direct contact with the ssDNA, and the peak corresponding to the biotinylated peptide fragment is significantly diminished (Figure 4B). Also shown in Figure 4 are similar results for residue K343 which is located in DBD-B and is a known ssDNA contact lysine (21). Data for this residue serve as an internal control to confirm the validity of our analysis. Figure 5 and ,Table 1 summarize the biotin modification data for all three subunits of RPA.
Figure 4:
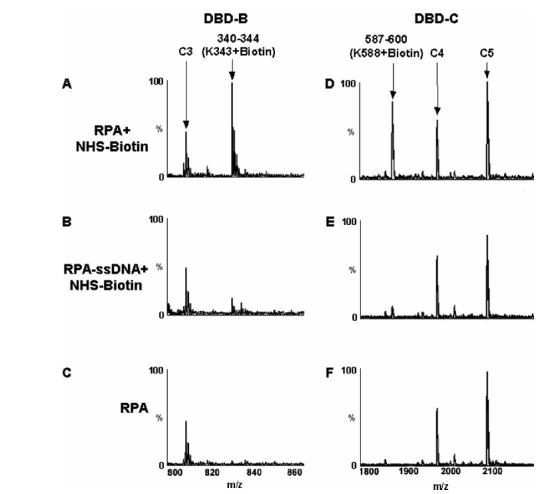
MALDI-TOF analysis of protection of lysine residues. The peptide peak containing modified K343 of DBD-B (A) is absent in the presence of the dT30mer (B). Similarly, K588 is located in DBD-C and is biotinylated in the absence of ssDNA (D) but is protected from modification by direct contact with the dT30mer (E). Panels C and F show the spectra of RPA without treatment of biotin. The unmodified peaks C3, C4, and C5 all serve as controls.
Figure 5:

Summary of the footprinting results in the context of the RPA sequence. Biotinylation sites in the three subunits of RPA are indicated in the primary amino acid sequence either as protected (boxed B) or as unprotected residues (unboxed B). The locations of domain structures are indicated by the shaded sequence with the name and amino acid numbering of the structure.
Table 1:
Proteolysis Fragments of hRPA Containing ModifiedLysinesa
| fragment | modified K | protection |
|---|---|---|
| RPA70 | ||
| 82-91 | 88 | - |
| 158-167 | 163 | - |
| 164-183 | 167 | - |
| 168-183 | 183 | + |
| 202-210 | 206 | - |
| 217-234 | 220 | - |
| 235-253 | 244 | - |
| 254-267 | 263 | + |
| 260-267 | 259 | + |
| 314-331 | 324 | - |
| 325-335 | 331 | - |
| 340-344 | 343 | + |
| 489-499 | 489 | + |
| 500-511 | 502 | - |
| 576-586 | 577 | + |
| 587-600 | 588 | + |
| 589-600 | 595 | - |
| RPA32 | ||
| 38-40 | 38 | - |
| 82-105 | 93 | - |
| 128-139 | 138 | - |
| 139-145 | 139 | - |
| RPA14 | ||
| 31-39 | 33 | - |
| 40-72 | 49 | - |
Tryptic digest fragments of hRPA subunits p70, p32, and p14 are shown with the modified lysine residue indicated.Lysines that are shown to be protected from modification in the presence of ssDNA are indicated by (+) while residues readily modified in the presence and absence of ssDNA are indicated by (-).Lysine residues that make direct contact with ssDNA are shown in bold.
DISCUSSION
Several crystal structures for RPA are currently available including the structure of the native form of the tandem DBD-AB (p70183-420) (22), the cocrystal structure of DBD-AB with a dC8 ssDNA (13), structures of the N-terminal domain of p70 (p708-105) (11), the C-terminal domain (p32171-270) of p32 (28), and the native form of the trimerization core containing DBD-C(p70439-616), DBD-D(p3243-171), and the entire p14 subunit (21). The cocrystal structure of DBD-AB with the dC8mer nucleotides provides an excellent internal control for the current analysis of lysine protection detected by mass spectrometry. Figure 6A shows the cocrystal structure of DBD-AB with the 8mer ssDNA (13) with modified lysine residues shown in blue and lysine residues protected from modification when dT30mer is present shown in red. Lysines residues K263 and K343 are located in DBD-A and DBD-B, respectively, and are both protected from biotin modification when the dT30mer is present. Both residues are oriented pointing into the binding clefts and are solvent accessible in the absence of ssDNA as shown by the crystal structure of free protein (22). These lysine residues directly contact ssDNA as it transverses the tandem binding domain. The confirmation of the mass spectrometry data with the cocrystal structure of DBD-AB with the 8mer ssDNA is an excellent control for supporting that the former method is capable of detecting nucleic acid contact points within RPA.
Figure 6:
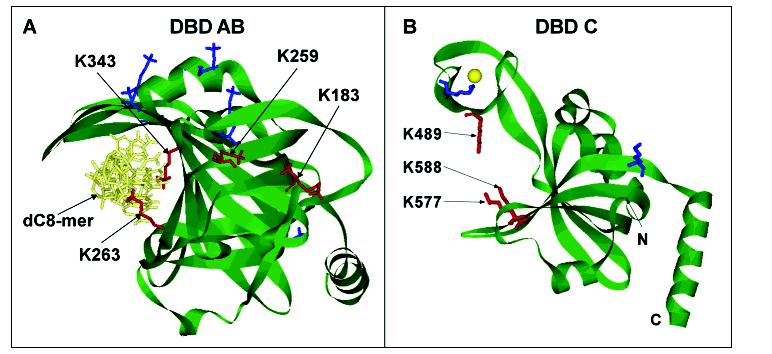
Structural exhibition of modified lysines in DBD-AB and DBD-C. (A) Structure of the DBD-AB-ssDNA complex (13). Lysine residues in the structures are presented in stick representation. K263 and K343 are found in the binding clefts of DBD-A and DBD-B, respectively, and are protected from modification by direct contact with the dC8mer. Lysine residues K183 and K259 are not in direct contact with the dC8mer but are protected from modification when ssDNA is present. (B) Structure of DBD-C (21). Lysine residues K489, K577, and K588 are located in the binding cleft of DBD-C and are protected from biotin modification when ssDNA is present, indicating direct contact with ssDNA as it transverses the binding cleft. Biotin-modified lysine residues without protection are shown in blue while the lysine residues protected from modification in the presence of ssDNA are shown in red in each structure.
On the basis of the available domain structures (11, 13, 21, 22, 28) as reference, our mass spectrometry data have shown modifications of about 60% of surface-accessible lysines in p70, ∼70% in p32, and ∼40% for p14, which include 80% of lysine residues found in the ssDNA binding domains. It should be noted that our experiments were performed on full-length hRPA, while X-ray structural data are available for the domain or partial hRPA. Therefore, it is possible that a significant number of lysines found to be exposed to solvent in crystallographic studies are in fact involved in the interdomain or intermotif interactions in the context of the full-length form of native hRPA. The lysines shielded by protein-protein interactions would not be susceptible to NHS-biotin modification. Our results clearly indicate that, in addition to K263 and K343, lysine residues K489, K577, and K588 located within DBD-C also are protected in the presence of the dT30mer. Figure 6B shows the crystal structure of DBD-C located in the C-terminal ofp70 (21). The biotin-modified lysine residues are shown in blue while protected lysine residues are indicated in red. Since lysines K489, K577, and K588 are located in the binding cleft of DBD-C, protection of these residues from biotin modification strongly suggests that these lysines are directly involved in ssDNA interactions. Although lysine residues K489 and K588 were previously implicated as ssDNA contact points by computer-assisted molecular modeling (21), lysine K577 was not. Similar to K263 and K343 in DBD-AB, the protected residues in DBD-C are oriented pointing into the binding cleft and are solvent accessible in the absence of ssDNA. The similarities between these protected lysine residues versus those found in DBD-AB such as orientation in the binding cleft and solvent accessibility strengthen the claim that these lysine residues are ssDNA contact points.
Two additional lysine residues, K183 and K259 in DBD-AB, were also protected from modification in the presence of the dT30mer. These two residues are not located in either binding cleft of DBD-AB nor do they directly contact the 8 nt ssDNA in the cocrystal structure (13). These lysine residues are shown to be oriented toward the N-terminal of the protein and parallel to the bound ssDNA, suggesting that these residues may be protected by conformational shift within the p70 subunit upon ssDNA binding, in which the N-terminal domain shields K183 and K259 from biotin modification. This interdomain protein-protein interaction may stabilize the protein-ssDNA complex. As no structure for the N-terminal domain together with DBD-A or DBD-AB is available, the exact nature of this protection is unclear, though internal protein-protein interactions stabilizing the nucleoprotein complex are a possibility.
Our observation that no protected residues were found in DBD-D (p32) or the OB-fold of p14 is consistent with previous biochemical data of the ssDNA binding affinities of these domains. Although K139 is located in DBD-D of p32, its orientation positions it out of the binding cleft. DBD-D has been implicated in the 30 nt binding mode, though its apparent ssDNA affinity is significantly lower than that of DBD-AB or DBD-C on p70 (1, 9, 18, 19). Kim et al. (9) reported the Ka for RPA binding to various lengths of oligonucleotides dT and found that when the length of ssDNA is increased from 12 to 20 nt (the length at which DBD-C engages the substrate), the Ka increases by >3-fold from 0.16 × 109 M-1 (resulting from DBD-AB binding) to 3.7 × 109 M-1 However, the difference between the dT20mer and dT30mer (the length at which DBD-D engages the substrate) is only ∼0.2-fold. In the present study we monitored only lysine contacts. It is possible that other p32 amino acids may contribute to ssDNA binding that could not be detected by our approach. It is currently believed based on biochemical evidence that p14 is not involved in ssDNA binding but is required for formation of the stable heterotrimer (1, 9, 18-21). Our results reflect this model in that we observed no protection of the two lysine residues located in the OB-fold of p14.
Footnotes
This study was supported by NCI Grant CA86927 (to Y.Z.).
Contributor Information
Sonja Hess, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Department of Health and Human Services, Bethesda, Maryland 20892.
Mamuka Kvaratskhelia, The Ohio State University Health Sciences Center, College of Pharmacy, Center for Retrovirus Research and Comprehensive Cancer Center, Columbus, Ohio 43210.
REFERENCES
- 1.Wold MS. Replication Protein A: A Heterotrimeric, Single-Stranded DNA-Binding Protein Required for Eukaryotic DNA Metabolism. Annu. ReV. Biochem. 1997;66:61–92. doi: 10.1146/annurev.biochem.66.1.61. [DOI] [PubMed] [Google Scholar]
- 2.Iftode C, Daniely Y, Borowiec JA. Replication Protein A (RPA): The Eukaryotic SSB. Crit. ReV. Biochem. Mol. Biol. 1999;34:141–180. doi: 10.1080/10409239991209255. [DOI] [PubMed] [Google Scholar]
- 3.Stigger E, Dean FB, Hurwitz J, Lee SH. Reconstitution of Functional Human Single-Stranded DNA-Binding Protein from Individual Subunits Expressed by Recombinant Baculoviruses. Proc. Natl. Acad. Sci. U.S.A. 1994;91:579–583. doi: 10.1073/pnas.91.2.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Henricksen LA, Umbricht CB, Wold MS. Recombinant Replication Protein A: Expression, Complex Formation, and Functional Characterization. J. Biol. Chem. 1994;269:11121–11132. [PubMed] [Google Scholar]
- 5.Burns JL, Daniely Y, Borowiec JA. Crit. ReV. Biochem. Mol. Biol. 1999;34:141–180. doi: 10.1080/10409239991209255. [DOI] [PubMed] [Google Scholar]
- 6.Sancar A. Annu. ReV. Biochem. 1996;65:43–81. doi: 10.1146/annurev.bi.65.070196.000355. [DOI] [PubMed] [Google Scholar]
- 7.Patrick SM, Turchi JJ. Replication Protein A (RPA) Binding to Duplex Cisplatin-Damaged DNA is Mediated Through the Generation of Single-Stranded DNA. J. Biol. Chem. 1999;274:14972–14978. doi: 10.1074/jbc.274.21.14972. [DOI] [PubMed] [Google Scholar]
- 8.Hermanson-Miller IL, Turchi JJ. Strand-Specific Binding of RPA and XPA to Damaged Duplex DNA. Biochemistry. 2002;41:2402–2408. doi: 10.1021/bi0112863. [DOI] [PubMed] [Google Scholar]
- 9.Kim C, Paulus BF, Wold MS. Interactions of Human Replication Protein A with Oligonucleotides. Biochemistry. 1994;33:14197–14206. doi: 10.1021/bi00251a031. [DOI] [PubMed] [Google Scholar]
- 10.Binz SK, Sheehan AM, Wold MS. Replication Protein A Phosphorylation and the Cellular Response to DNA Damage. DNA Repair. 2004;3:1015–1024. doi: 10.1016/j.dnarep.2004.03.028. [DOI] [PubMed] [Google Scholar]
- 11.Jacobs DM, Lipton AS, Isren NG, Daughdrill GW, Lowry DF, Gomes X, Wold MS. Human Replication Protein A: Global Fold of the N-Terminal RPA-70 Domain Reveals a Basic Cleft and Flexible C-Terminal Linker. J. Biomol. NMR. 1999;14:321–331. doi: 10.1023/a:1008373009786. [DOI] [PubMed] [Google Scholar]
- 12.Kim C, Snyder RO, Wold MS. Binding Properties of Replication Protein A from Human and Yeast Cells. Mol. Cell. Biol. 1992;12:3050–3059. doi: 10.1128/mcb.12.7.3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bochkarev A, Pfuetzner RA, Edwards AM, Frappier L. Structure of the Single-Stranded-DNA-Binding domain of Replication Protein A Bound to DNA. Nature. 1997;385:176–181. doi: 10.1038/385176a0. [DOI] [PubMed] [Google Scholar]
- 14.DeLaat WL, Appeldoorn E, Sugasawa K, Weterings E, Jaspers NGJ, Hoeijmakers JHJ. DNA-Binding Polarity of Human Replication Protein A Positions Nucleases in Nucleotide Excision Repair. Genes DeV. 1998;12:2598–2609. doi: 10.1101/gad.12.16.2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kolpashchikov DM, Weisshart K, Nasheuer HP, Khodyreva SN, Fanning E, Farve A, Lavrik OI. Interaction of the p70 Subunit of RPA with a DNA Template Directs p32 to the 3′-end of Nascent DNA. FEBS Lett. 1999;450:131–134. doi: 10.1016/s0014-5793(99)00484-6. [DOI] [PubMed] [Google Scholar]
- 16.Kim C, Wold MS. Recombinant Human Replication Protein A Binds to Polynucleotides with Low Cooperativity. Biochemistry. 1995;34:2058–2064. doi: 10.1021/bi00006a028. [DOI] [PubMed] [Google Scholar]
- 17.Yang ZG, Liu Y, Mao LY, Zhang JT, Zou Y. Dimerization of Human XPA and Formation of XPA2-RPA Protein Complex. Biochemistry. 2002;41:13012–13020. doi: 10.1021/bi026064z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bastin-Shanower SA, Brill SJ. Functional Analysis of the Four DNA Binding Domains of Replication Protein A. J. Biol. Chem. 2001;276:36446–36453. doi: 10.1074/jbc.M104386200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wyka IM, Khar K, Binz SK, Wold MS. Replication Protein A Interactions with DNA: Differential Binding of the Core Domains and Analysis of the DNA Interaction Surface. Biochemistry. 2003;42:12909–12918. doi: 10.1021/bi034930h. [DOI] [PubMed] [Google Scholar]
- 20.Bochkarev A, Bochkarev E. From RPA to BRCA2: Lessons From Single-Stranded DNA Binding by the OB--Fold. Curr. Opin. Struct. Biol. 2004;14:36–42. doi: 10.1016/j.sbi.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 21.Bochkarev E, Korolev S, Lees-Miller SP, Bochkarev A. Structure of the RPA Trimerization Core and Its Role in the Multistep DNA-Binding Mechanism of RPA. EMBO J. 2002;21:1855–1863. doi: 10.1093/emboj/21.7.1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bochkarev E, Visar B, Korolev S, Bochkarev A. Structure of the Major Single-Stranded DNA-Binding Domain of Replication Protein A Suggests a Dynamic Mechanism for DNA Binding. EMBO J. 2001;20:612–618. doi: 10.1093/emboj/20.3.612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bochkareva E, Frappier L, Edwards AM, Bochkarev A. The RPA32 Subunit of Human Replication Protein A Contains a Single-Stranded DNA-Binding Domain. J. Biol. Chem. 1998;273:3932–3936. doi: 10.1074/jbc.273.7.3932. [DOI] [PubMed] [Google Scholar]
- 24.Gomes XV, Henricksen LA, Wold MS. Proteolytic Mapping of Human Replication Protein A: Evidence for Multiple Structural Domains and a Conformational Change Upon Interaction with Single-Stranded DNA. Biochemistry. 1996;35:5586–5595. doi: 10.1021/bi9526995. [DOI] [PubMed] [Google Scholar]
- 25.Stauffer ME, Chazin WJ. Physical Interaction Between Replication Protein A and RAd51 Promotes Exchange on Single-Stranded DNA. J. Biol. Chem. 2004;279:25638–25645. doi: 10.1074/jbc.M400029200. [DOI] [PubMed] [Google Scholar]
- 26.Stigger E, Drissi R, Lee SH. Functional Analysis of Human Replication Protein A in Nucleotide Excision Repair. J. Biol. Chem. 1998;273:9337–9343. doi: 10.1074/jbc.273.15.9337. [DOI] [PubMed] [Google Scholar]
- 27.Jackson D, Dhar K, Wahl JK, Wold MS, Borgstahl GEO. Analysis of the Human Replication Protein A: Rad52 Complex: Evidence for Crosstalk Between RPA32, RPA70, Rad52 and DNA. J. Mol. Biol. 2002;321:133–148. doi: 10.1016/s0022-2836(02)00541-7. [DOI] [PubMed] [Google Scholar]
- 28.Mer G, Bochkarev A, Gupta R, Bochkarev E, Frappier L, Ingles CJ, Edwards AM, Chazin WJ. Structural Basis for the Recognition of DNA Repair Proteins UNG2, XPA, and RAD52 by Replication Factor RPA. Cell. 2000;103:449–456. doi: 10.1016/s0092-8674(00)00136-7. [DOI] [PubMed] [Google Scholar]
- 29.Daughdrill GW, Buchko GW, Botuyan MV, Arrowsmith C, Wold MS, Kennedy MA, Lowry DF. Chemical Shift Changes Provide Evidence for Overlapping Single-Stranded DNA- and XPA-Binding Sites on the 70kDa Subunit of Human Replication Protein A. Nucleic Acids Res. 2003;31:4176–4183. doi: 10.1093/nar/gkg451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reardon JT, Sancar A. Molecular Anatomy of the Human Excision Nuclease Assembled at Sites of DNA Damage. Mol. Cell. Biol. 2002;22:5938–5945. doi: 10.1128/MCB.22.16.5938-5945.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kvaratskhelia M, Miller JT, Budihas SR, Pannell LK, Le Grice SFJ. Identification of Specific HIV-1 Reverse Transcriptase Contacts to the Viral RNA:tRNA Complex by Mass Spectrometry and a Primary Amine Selective Reagent. Proc. Natl. Acad. Sci. U.S.A. 2002;99:15988–15993. doi: 10.1073/pnas.252550199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shkriabai N, Patil SS, Hess S, Budihas SR, Craigie R, Burke TR, Jr., Le Grice SFJ, Kvaratskhelia M. Identification of an Inhibitor-Binding Site to HIV-1 Integrase With Affinity Acetylation and Mass Spectrometry. Proc. Natl. Acad. Sci. U.S.A. 2004;101:6894–6899. doi: 10.1073/pnas.0400873101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee JH, Park CJ, Arunkumar AI, Chazin WJ, Choi BS. NMR Study On the Interaction Between RPA and DNA Decamer Containing cis-syn Cyclobutane Pyrimidine Dimer in the Presence of XPA: Implication for Damage Verification and Strand-Specific Dual Incision in Nucleotide Excision Repair. Nucleic Acids Res. 2003;31:4747–4754. doi: 10.1093/nar/gkg683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ikegami T, Kuraoka I, Saijo M, Kodo N, Kyogoku Y, Morikawa K, Tanaka K, Shirakawa M. Solution Structure of the DNA- and RPA-Binding Domain of the Human Repair Factor XPA. Nat. Struct. Biol. 1998;5:701–706. doi: 10.1038/1400. [DOI] [PubMed] [Google Scholar]
- 35.Iftode C, Borowiec JA. 5′f3′ Molecular Polarity of Human Replication Protein A (hRPA) Binding to Pseudo-origin DNA Substrates. Biochemistry. 2000;39:11970–11981. doi: 10.1021/bi0005761. [DOI] [PubMed] [Google Scholar]
- 36.Zou L, Elledge SJ. Sensing DNA Damage Through ATRIP Recognition of RPA-ssDNA Complexes. Science. 2003;300:1542–1548. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]