

Abstract
Duchenne muscular dystrophy is an X-linked muscle disease characterized by mutations in the dystrophin gene. Many of these can be corrected at the posttranscriptional level by skipping the mutated exon. We have obtained persistent exon skipping in mdx mice by tail vein injection with an adeno-associated viral (AAV) vector expressing antisense sequences as part of the stable cellular U1 small nuclear RNA. Systemic delivery of the AAV construct resulted in effective body-wide colonization, significant recovery of the functional properties in vivo, and lower creatine kinase serum levels, suggesting an overall decrease in muscle wasting. The transduced muscles rescued dystrophin expression and displayed a significant recovery of function toward the normal values at single muscle fiber level. This approach provides solid bases for a systemic use of AAV-mediated antisense-U1 small nuclear RNA expression for the therapeutic treatment of Duchenne muscular dystrophy.
Keywords: exon skipping, adeno-associated virus vectors, antisense, small nuclear RNA, dystrophin
Deletions and point mutations in the human 2.5-Mb-long dystrophin gene cause either the severe progressive myopathy Duchenne muscular dystrophy (DMD) or the milder Becker muscular dystrophy (BMD), depending on whether the translational reading frame is lost or maintained (1).
The mdx mouse, carrying a stop codon inside exon 23 of the dystrophin gene, provides a useful system to study the effectiveness of different therapeutic strategies for the cure of this disease (2). The two challenging issues in the gene therapy of DMD are related on one side to the type of therapeutic gene to use and on the other to the delivery system suitable for efficient muscular transduction. Due to the large size of the protein, a traditional gene replacement approach is difficult: only adenoviral vectors could allow the expression of a full-length dystrophin cDNA. However, treatments of mdx mice with “gutted” vectors carrying the full-length dystrophin cDNA induced a weak immune reaction and transduction was not very effective (3, 4).
A different approach took advantage of adeno-associated viral (AAV) vectors for the delivery of microdystrophin gene (5). AAV vectors with an AAV8 capsid (or the very similar but not identical AAV1, or AAV6) have been shown to transduce efficiently and widely murine muscles after administration in the tail vein of WT mice (6). Those with an AAV6 capsid have been shown to correct the phenotype of dystrophic mdx mice when carrying microdystrophin (7).
Recently, other alternative ways of correcting the DMD phenotype have been achieved that consist in the delivery of antisense sequences able to induce exon skipping and cure the genetic alteration at the posttranscriptional level. Many of the internal in-frame deletions fall in the region encoding the spectrin-like central rod domain that is largely dispensable and produce only mild myopathic symptoms; therefore, for the out-of-frame mutations, it should be possible, by preventing the inclusion of specific mutated exons in the mature dystrophin mRNA, to obtain an in-frame mRNA and rescue the synthesis of a shorter, but still functional, dystrophin protein (8, 9).
Exon-specific skipping has been accomplished through the use of synthetic antisense oligonucleotides (AONs) against specific splice junctions or exonic enhancers in in vitro cultured DMD myoblasts of both human and murine origin (10). Subsequently, the same approach was successfully used in the mdx mouse model by intramuscular or i.v. delivery of AONs (11–14). The antisense approach resulted in very efficient rescue of the dystrophin protein even though the effect of AONs is limited in time, thus requiring multiple administrations. Therefore, a growing interest has been devoted to the transduction of muscle fibers with vectors stably producing antisense sequences. The strategy consists in the expression of these sequences as part of stable cellular RNAs and, in particular, in RNA backbones, ensuring the specific localization of the antisense sequence in the nuclear compartment where splicing occurs. In this direction, we have pioneered the use of an antisense-U1 small nuclear RNA (snRNA), in human DMD myoblasts carrying the 48–50 deletion, to induce efficient exon-51 skipping and dystrophin rescue (15). The U7 snRNA was also used as a backbone for antisense expression and was shown to be able to induce correct exon skipping both when transduced into mammalian dystrophic cells (15, 16) and when injected in muscles of mdx mice as part of AAV constructs (17). Similar effectiveness, upon local muscular injection, was also shown for the U1 snRNA backbone (M.A.D., data not shown).
In this article, we show that systemic delivery of antisense-AAV constructs through injection into the tail vein of mdx dystrophic mice leads to body-wide rescue of dystrophin synthesis and recovery of muscular strength. Several parameters, diagnostic for systemic recovery, indicate the effectiveness of this approach in the whole animal.
Results
We previously showed that, to have efficient exon skipping, antisense sequences against both the 5′ and 3′ splice sites of the target exon should be coexpressed (15). In the chimeric construct U1#23, eight nucleotides at the 5′ end of U1 snRNA, required for recognition of the 5′ splice sites on pre-mRNAs, were substituted with a 54-nt-long region, complementary to both splice junctions of the dystrophin exon 23 (see Fig. 1A). The chimeric U1snRNA with such a long extension was previously shown to be efficiently expressed in vivo and to accumulate in a stable form as a small nuclear ribonucleoprotein (snRNP) (15, 18).
Fig. 1.

AAV2/1 efficiently delivers antisense constructs to muscles. (A) Schematic representation of the mdx dystrophin mutation and of the AAV-U1#23 antisense construct. Antisense sequences complementary to the 5′ and 3′ splice sites of exon 23 (black bars) were cloned consecutively in the 5′ portion of U1 snRNA (black box). The corresponding antisense sequence is shown underneath the dystrophin pre-mRNA. (B) Muscle transduction after AAV2/1 systemic delivery. Green fluorescence was evident in several muscle districts 12 weeks after tail vein administration of 3–4 × 1012 genome copies/mouse of AAV-U1#23. Intensively transduced fiber groups flank less or nonfluorescent muscle fibers possibly reflecting heterogeneous perfusion inside the muscle. Heart and diaphragm are also widely transduced although to a lower level than voluntary striated muscle.
To efficiently express the antisense constructs in murine muscles in vivo, a hybrid AAV vector containing the genome of AAV2 encoding for the transgene and packaged into an AAV1 capsid [vector AAV2/1 (19)] was used. This hybrid vector seemed to be very efficient for muscle gene transfer among those derived from the AAV serotypes isolated to date after intramuscular (20) or systemic injection (6)
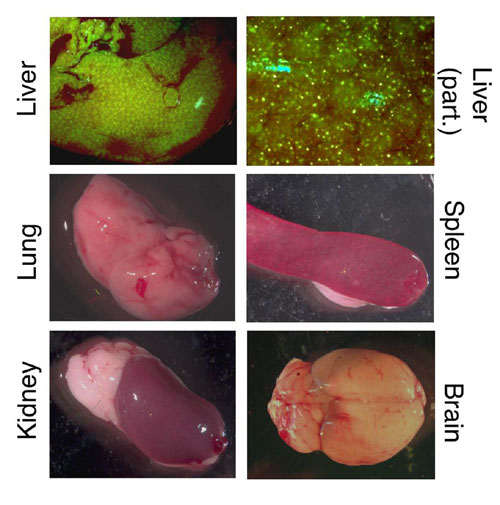
To asses whether the expression of the antisense RNAs could be widely spread to muscles, 3–4 × 1012 genome copies of the AAV derivative (AAV-U1#23), coexpressing the antisense U1 snRNA as well as the EGFP, were injected into the tail vein of 6-week-old mdx mice. This dose of vector was previously shown to efficiently transduce muscle fibers independently from the administration of the permeabilizing VEGF (7). In our experience, too, at these vector doses, no differences were found between injection of vector alone or in the presence of VEGF (data not shown). In the overall, 11 mdx mice were injected. They were killed 6 and 12 weeks after injection. For six of them the different tissues were also tested for EGFP expression. Fig. 1B shows an example of different muscular districts from a single transduced animal after 12 weeks of treatment. EGFP expression was widely spread to striated muscles over the whole animal. Fluorescence intensity was heterogeneous among the different muscles in which heavily green fluorescent fibers flanked EGFP-negative fibers. Notably, muscles from hind limb and forelimb showed an extensive transduction, whereas diaphragm and heart displayed a less uniform expression. EGFP-positive cells were also found in liver, whereas other tissues analyzed (lung, spleen, kidney, intestine, and brain) did not show any detectable transgene expression (see Fig. 5, which is published as supporting information on the PNAS web site). The observed transduction features were similar among the various animals systemically injected with the antisense-AAV construct.
For molecular analysis, individual muscles were resected and subdivided for RNA, protein, and immunofluorescence analyses. It is important to underline that the three samples are not perfectly comparable because of the heterogeneous AAV colonization in the different districts. Fig. 2A shows RT-PCR performed with primers corresponding to the 5′ and internal portions of the U1#23 construct. Because the 5′ oligo is specific for the antisense sequences, only U1 chimeric transcripts are detected. Specific amplification products of the correct size were identified upon AAV-mediated transduction, in various quantities, in the different muscles analyzed. Exon skipping was tested in the same districts by RT-PCR analysis, performed with oligos specific for exons 22 and 26, followed by nested PCR with oligos specific for exons 22 and 24. The results indicated that skipping of exon 23 had occurred (Fig. 2B). Sequencing of the skipped products showed the correct fusion between exon 22 and 24 (Fig. 2B).
Fig. 2.

Analysis of antisense construct expression and exon 23 skipping. (A) Expression of chimeric antisense U1 construct in body-wide muscles 12 weeks after tail vein injection of the AAV-U1#23 construct. Nested RT-PCR was performed on 200 ng of total RNA with primers U1 + 130 and mAnti5′F (Fig. 1A). The products were run on a 2% agarose gel. Lanes were as follows: M, 100-bp DNA ladder (Invitrogen); Tr, triceps; Q, quadriceps; Gl, gluteus; G, gastrocnemius; Ti, tibialis; D, diaphragm; H, heart; −, RNA-minus control. (B) Skipping of exon 23. In the schematic representation of a portion of the dystrophin pre-mRNA, the oligos used for the nested RT-PCRs are indicated. Lanes are as in A. The chromatogram indicates that correct fusion between exon 22 and 24 has occurred. A schematic representation of the unskipped and skipped products is shown on the side of the gel.
Dystrophin expression was analyzed by Western blot on total protein extracts. Fig. 3A shows that AAV-U1#23 constructs rescued the translation of dystrophin. Note that the skipping of exon 23 alone would produce a protein 73 amino acids shorter; this decrease in size cannot be detected for such huge a protein (427 kDa) on this type of gel. Because RNA and protein samples derive from a mixture of transduced and untransduced fibers, it is impossible to assess the levels of exon skipping and dystrophin rescue in individual fibers. Therefore, the observed signals represent an average of the effect obtained in the entire muscle sample.
Fig. 3.

Analysis of dystrophin and DAPC rescue. (A) Two hundred micrograms of total proteins from different muscles were analyzed 12 weeks after injection by Western blotting with anti-dystrophin antibodies in parallel with 5 μg of proteins extracted from WT skeletal muscle cells (lane WT). P, pectoralis; Tr, triceps; Q, quadriceps; Gl, gluteus; G, gastrocnemius; Ti, tibialis; D, diaphragm; H, heart. (B) The same muscles were also sliced and analyzed by immunofluorescence with anti-dystrophin antibodies. Pictures were taken with a confocal microscope. (C) Sections of the quadriceps of AAV-U1#23-injected mice were also reacted with anti-α- and -β-sarcoglycan antibodies. Middle and Bottom panels indicate the reactivity of quadriceps sections of an untreated mdx mouse and of a WT mouse, respectively, with the same antibodies as above. Pictures were taken with a confocal microscope. (D) Rescue of dystrophin synthesis (dys) corresponded to the expression of EGFP protein (gfp). (DAPI) Bottom panel shows the merging of dystrophin signals with those of the DAPI staining. (Scale bars: 100 μm.)
Immunofluorescence analysis confirmed that AAV-U1#23 was able to rescue synthesis of dystrophin, which correctly localized at the periphery of the fibers as in WT. Fig. 3B shows an example of dystrophin immunostaining of a collection of muscle sections obtained from mdx mice treated with AAV-U1#23. Notably, rescue of dystrophin expression was associated with the correct expression and localization of α- and β-sarcoglycans, components of the dystrophin-associated protein complex (DAPC, Fig. 3C). Immunohistochemical analysis also indicated the correspondence between the expression of the EGFP marker protein and rescue of dystrophin synthesis (Fig. 3D). The enlargement shown in Fig. 3D clearly shows that the “rescued” dystrophin correctly localizes to the submembrane compartment. The microscopic analysis confirmed the heterogeneous colonization of each muscular district, as already shown in Fig. 1 by the EGFP analysis on total body, because extensive regions of positive dystrophin staining and EGFP expression were interspersed with negative bundles of fibers. No significant differences in terms of colonization and effectiveness of the antisense treatment were observed in mice treated for 6 weeks.
The diaphragm represents one of the most compromised muscles in both mdx mice and human patients due to its heavy functional demand. Despite the low expression level of EGFP in the diaphragm of transduced mice (Fig. 1B), we demonstrated, by Western blot (Fig. 3A) and immunostaining analyses (Fig. 3B) that dystrophin synthesis was successfully rescued. Similar results were obtained with the activation of dystrophin protein expression in the heart (Fig. 3 A and B). Because heart has been reported as an organ in which it is difficult to achieve widespread gene transfer, the restoration of dystrophin synthesis observed in our experimental conditions is particularly encouraging, suggesting that even low transduction is sufficient to provide enough gene expression to rescue dystrophin synthesis.
To assess whether dystrophin expression sustained a recovery of function, we analyzed the specific force of single skinned muscle fibers from gastrocnemius muscles of control C57BL/6, mdx, and AAV-U1#23 systemically treated mdx mice. Because in such muscles most fibers were found to be type 2X and 2B, we focused on such fiber types for comparisons. We pooled the data from type 2X and 2B because no difference in force (Po)/cross-sectional area (CSA) was observed between the two types of fibers in any of the conditions studied. First of all, we analyzed a large population (n = 120, mice n = 3 for each group) of muscle fibers of control C57BL/6, mdx, and mdx AAV-U1#23-treated mice. Specific force (Po/CSA) was significantly lower in mdx mice and recovered toward normal values in U1-treated mice (+30%; Fig. 4A). Although recovery was not complete, a statistically significant difference was observed in the specific force of single muscle fibers of AAV-U1#23-treated versus mdx mice. To confirm that the recovery of force was due to the expression of dystrophin per se, we analyzed Po/CSA of a second population of muscle fibers (n = 150, mice n = 3 for each group) obtained from the vastus of C57BL/6 and mdx, and from small EGFP-positive and EGFP-negative bundles (≈50 fibers each) of AAV-U1#23-treated muscles, dissected under a fluorescent stereomicroscope. This approach was also justified by the highly stereotyped EGFP-dystrophin expression in the vastus. Interestingly, EGFP-positive fibers from AAV-U1#23-treated mdx mice were stronger than both EGFP-negative fibers from treated mice and fibers from untreated mdx mice (Fig. 4B). The latter result strongly suggests that dystrophin expression is responsible for the functional recovery in treated mice.
Fig. 4.

Functional tests on mdx mice injected systemically with AAV-U1#23. Controls (black), untreated mdx (blue), and AAV-U1#23 (red)-treated mdx mice. (A) Specific force of single fibers from the gastrocnemius (n = 120, mice n = 3 for each group) was significantly lower in mdx mice in comparison with controls (P ≤ 0.01) and recovered toward normal values in AAV-U1#23 (+30%, P ≤ 0.01). (B) Po/CSA of fibers (n = 150, mice n = 3 for each group) from the vastus of C57BL/6 and mdx and from small GFP-positive and GFP-negative bundles of AAV-U1#23-treated muscles. GFP-positive fibers from treated mice showed Po/CSA values significantly higher (P ≤ 0.001) than those obtained from GFP-negative fibers of treated mice and fibers of untreated mdx mice (P ≤ 0.001). (C) CKnac (units/liter) serum levels were monitored 3 and 6 weeks after AAV-U1#23 injections in parallel to C57BL/6 and mdx mice. (D) Untreated mdx mice showed a significant reduction (P ≤ 0.001) of the tolerance to treadmill exhaustion test compared with C57BL/6, whereas a partial but significant recovery was observed after AAV-U1#23 treatment (P ≤ 0.01). ∗, significantly different from the other groups; bullet, significantly different from C57BL/6 and AAV-U1#23 GFP-positive. Data are mean values ± SEM. Statistical analysis was assessed by ANOVA followed by the Student–Newman–Keuls test. P ≤ 0.05 was considered significant.
To assess whether the expression of dystrophin improved sarcolemmal integrity, creatine kinase concentration in the serum (CKnac), whose blood levels increase after sarcolemma damage, was measured 3 and 6 weeks after injection. Fig. 4C shows that systemic delivery of AAV-U1#23 (red bars) was followed by a progressive decrease of Cknac, which, 6 weeks after injection, reached intermediate values between the control C57BL/6 (black bars) and mdx mice (blue bars).
Because systemic treatment reached body-wide muscles, we hypothesized that it was able to improve function of treated mice in vivo. Six weeks after injection, treadmill exhaustion tests were performed to assess the level of recovery in vivo. Untreated mdx mice showed a reduced tolerance to exercise compared with C57BL/6, whereas a partial recovery was observed after AAV-U1#23 treatment, indicating an overall amelioration of the physical performance (Fig. 4D).
In conclusion, despite the only partial AAV muscular transduction efficiency, the colonization was sufficient to provide a conspicuous functional amelioration.
Discussion
In consideration of the effectiveness of the antisense strategy, which can be applied to the cure of the majority of dystrophin mutations, parallel approaches have focused on the possibility of obtaining transduced muscles constitutively expressing antisense sequences as part of stable cellular RNAs. This strategy was successfully applied by local transduction of AAV vectors carrying antisense sequences as part of the U7 snRNA by muscular injection or intraarterial perfusion of the lower limb (17). However, a significant obstacle in designing effective vector-mediated replacement gene therapy for myopathies is the necessity to reach the entire musculature, a problem that cannot be easily overcome unless systemic delivery systems are improved.
In this article, we addressed this issue by showing a conspicuous colonization of many muscular districts through systemic delivery of antisense-AAV chimeric viruses; body-wide exon skipping, dystrophin rescue, restoration of the DAPC, and amelioration of functional parameters were obtained after the expression of U1 snRNA carrying antisense sequences against the mutated exon 23 of the mdx mouse.
Notably, a significant functional rescue was demonstrated by using two complementary approaches: analysis of the recovery of force of single muscle fibers in vitro and of mobility in vivo. The analysis of specific force of single muscle fibers has been shown to be a very sensitive method to assess the impairment of force in mice models of muscular dystrophy and the functional recovery after treatment (21, 22). The recovery of specific force of individual skeletal muscle fibers clearly indicates that systemic treatment was able to restore the integrity of the contractile machinery at the cellular level. Importantly, the recovery of function was directly related to the reconstitution of the DAPC because force recovery was observed in EGFP-positive and, therefore, dystrophin-positive fibers and not in EGFP-negative fibers within the same muscle. The reason why reconstitution of DAPC could avoid or minimize the force loss in skinned single muscle fibers remains obscure. It can be speculated that restoration of dystrophin expression at the periphery of the cells may exert a 2-fold advantage: from one side it can prevent myofibrillar hypercatabolism through the activation of calcium-activated intracellular proteases, thus minimizing reduction of sarcomeric density and force generation; from the other, it may exert a mechanical support to force transmission even in skinned conditions. Indeed, we found normally located dystrophin in skinned preparations (data not shown).
Finally, the extent of dystrophin expression and its body-wide distribution was such to determine a significant recovery of the mobility of the whole animal in an exercise test in vivo. Certainly, the recovery of function in vivo represents the goal of any muscular dystrophy therapy, and its extent (+20%) after a 12-week treatment is encouraging in light of the therapy of the disease. Altogether, these results show the clearest indication of functional recovery after DMD gene therapy to date.
Although AAV-mediated muscle transduction was widespread, it did not reach, at least as far as the EGFP staining was concerned, the entire population of muscle cells; nevertheless, the functional and molecular recovery was considerable, indicating that even partial muscle colonization could lead to important functional amelioration. These results are of clinical relevance and suggest a cautious optimism in the development of appropriate gene therapy approach for human patients, indicating that it is not necessary to correct the entire muscle to gain a functional benefit. Our data indicate that systemic delivery was efficient because a single virus administration was sufficient to produce amelioration of functional parameters and to sustain dystrophin expression for at least 12 weeks. The long persistence of expression of both transgenes also suggested that cell-mediated immune response was not occurring. This observation, together with the persistence of expression from AAV vectors in muscle of nonhuman primates for as long as 5 years (23), suggests that one-in-life treatment for muscular dystrophies might be feasible. Translating this approach to large animals or humans is not so obvious due to the high doses of vector required and to immunological responses in cases of multiple administrations. A careful evaluation of such risks and of prudent strategies might help understanding of the potential of such therapeutic approaches in human patients.
Methods
Plasmid and AAV Constructs.
Clone U1#23 was obtained by inverse PCR on the human U1 snRNA gene, with oligos mU1anti5′ (5′-CGAAATTTCAGGTAAGCCGAGGTTATGAGATCTTGGGCCTCTGC-3′) and mU1anti3′ (5′-GAACTTTGCAGAGCCTCAAAATTAAATAGGGCAGGGGAGATACCATGATC-3′). The antisense-containing insert was amplified from corresponding plasmid with oligos U1cas-up-NheI (5′-CTAGCTAGCGGTAAGGACCAGCTTCTTTG-3′) and U1cas-down-NheI (5′-CTAGCTAGCGGTTAGCGTACAGTCTAC-3′). The resulting fragment was NheI-digested and cloned in the forward orientation of the pAAV2.1-CMV-EGFP plasmid (19).
Vector Production and Characterization.
AAV-U1#23 vector was produced by triple transfection of 293 cells, purified by CsCl2 ultracentrifugation and titered by using both real-time PCR-based and dot-blot assays, as described (19, 24). Because the vector included the EGFP expression cassette, the number of green-forming units/ml was assessed by serial dilution on 293 cells, and the resulting infectious/physical particle was >1:100. AAV vector was produced by the AAV TIGEM Vector Core.
Vector Administration to mdx Mice.
All procedures on mice (including their euthanasia) were performed in accordance with institutional guidelines for animal research. Six-week-old mdx mice were administered with 3–4 × 1012 genome copies of AAV vector via tail vein. Six and 12 weeks after virus administration, animals were killed, and muscles from different districts were harvested. EGFP analysis and dissections were performed under a fluorescent stereomicroscope (Leica MZ16FA).
RNA Preparation and Analysis.
Total RNA was prepared from frozen sections homogenized in TRIzol solution (Invitrogen) with the Ultra-Turrax T8 homogenizer (IKA WERKE GmbH, Staufen, Germany). To monitor the antisense-U1 expression, RT-PCR was carried out with the Access RT-PCR system (Promega) on 200 ng of total RNA with primers U1 + 130 (5′-AGCACATCCGGAGTGCAATG-3′) and mAnti5′F (5′-CGGCTTACCTGAAATTTTCG-3′). For the analysis of dystrophin mRNA, 200 ng of total RNA were first reverse transcribed and amplified with oligos: Ex26Ro (5′-TTCTTCAGCTTGTGTCATCC-3′) and Ex22 (5′-TGCGCTATCAGGAGACAATG-3′) for 40 cycles with the Access RT-PCR system (Promega). Four microliters of the RT-PCR products were then used as template for the nested reaction performed in 50 μl with oligos Ex22 and Ex24 (5′-TTCAGGATTTCAGCATCCCC-3′).
Protein Preparation and Analysis.
Proteins were extracted from powdered tissue with 100 mM Tris·HCl (pH 7.4), 1 mM EDTA, 2% SDS, and a protease inhibitor mixture (Roche Diagnostics). Two hundred micrograms of proteins were loaded onto 6% polyacrylamide gels, electrophoresed, and transferred to nitrocellulose transfer membranes (Protran; Schleicher & Schuell). After blocking with 5% nonfat dry milk, membranes were incubated overnight with NCL-DYS1 (NovoCastra, Newcastle, U.K.) 1:125, washed in TBS-T, and incubated with goat anti-mouse IgG (H+L) horseradish peroxidase (HRP) secondary antibody (diluted 1:10,000 in TBS-T) for 1 h. Protein detection was carried out with Super Signal chemiluminescent substrate (Pierce).
Immunofluorescent Staining.
Seven-micrometer-thick muscle cryosections were treated with 15% goat serum before examination for dystrophin, α- and β-sarcoglycan expression. The following monoclonal antibodies were used: NCL-DYS2 (NovoCastra) 1:12.5; NCL-aSARC (NovoCastra) 1:20; and NCL-bSARC (NovoCastra) 1:20. As a secondary antibody, a goat anti-mouse Cy™3-conjugated IgG (H+L) antibody (Jackson ImmunoResearch) was used at dilution 1:600. Pictures were taken with an inverted confocal microscope (Leica TCS SP2).
CSA and Po of Single Fibers.
Absolute (Po) and specific (absolute force over the fiber cross-sectional area Po/CSA) force of single muscle fibers were measured by using skinned preparations. The procedures used have been described (25, 26) except for the solutions used. The skinning solution (Sk, 5 mM EGTA, pCa 9.0), prepared as described (26), was enriched with protease inhibitors (leupeptin 20 μM and E-64 10 μM). Preactivating (PA) and activating (A) solutions were of the following composition. For PA, solutions were as follows: 10 mM imidazole-propionate (Imid-p), 2.5 mM magnesium-propionate (Mg-p), 170 mM potassium-propionate (K-p), and 5 mM K2-EGTA. For A, solutions were as follows: 10 mM Imid-p, 2.2 mM Mg-p, 170 mM K-p, 0.11 mM K2-EGTA, and 4.8 mM Ca-EGTA. In both solutions, protease inhibitors leupeptin and E-64 were added.
Po was measured at pCa 4.5, 12°C and optimal sarcomeric length for force developing (2.5 μm). Po/CSA is expressed as kN/m2.
Fiber Typing and Myosin Isoform Identification.
At the end of the mechanical experiment, each fiber segment was immersed in a tube containing 20 μl of Laemmli solution and stored at −20°C until assayed for the myosin heavy chain (MHC) content by SDS/PAGE electrophoresis on 8% polyacrylamide slab gels as described (25, 26). In the MHC region, three bands corresponding to the adult MHC isoforms MHC 2A, MHC 2X, and MHC 2B were separated. The single fibers were classified pure fiber types (2A, 2X, and 2B) and hybrid fiber types (2AX and 2XB). Because the large majority of fibers contained MHC 2B (≈80%), only fibers of type 2B were used for comparison.
Exercise Tolerance Tests.
C57BL/6, mdx, and AAV-U1#23-treated (n = 5) mice were subjected to an exhaustion treadmill test. Mice were placed on the belt of a six-lane motorized treadmill (Exer 3/6 Treadmill; Columbus Instruments, Columbus, OH) supplied with shocker plates. The treadmill was run at an inclination of 0° at 5 m/min for 5 min, after which the speed was increased 1 m/min every minute. The test was stopped when the mouse remained on the shocker plate for 20 s without attempting to reengage the treadmill, and the time to exhaustion was determined.
Creatine Kinase Assay.
Determination of creatine kinase in the serum (CKnac) was performed by using a CK reagent kit [creatine kinase (NAC) Assay; Diagnostic Chemicals Limited, Prince Edward Island, Canada) by evaluating the rate of increase in absorbance at 340 nm due to the formation of NADPH.
The change in absorbance at 1-min intervals for 10 min was recorded. CKnac activity is expressed as units/liter.
Statistical Analysis.
Data were expressed as means ± SEM, unless otherwise stated. Statistical significance of the differences between means was assessed by ANOVA followed by the Student–Newman–Keuls test or by t test (nonparametric). A probability of <5% was considered significant.
Supplementary Material
Acknowledgments
This work is dedicated to the memory of Professor Giorgio Tecce. We are grateful to M. Arceci for technical assistance and to A. Catizone, C. Ramissa, S. De Grossi, and E. Marchetti for confocal microscopy. This work was partially supported by Telethon Grants GGP030279 (to I.B.), GSP030543C (to A.M.), and GSP030543C (to I.B. and R.B.); a Duchenne Parent Project Italia grant (to I.B.); Ministero dell’Università e della Ricerca Scientifica e Technologica (MURST) Grant FIRB-p.n. RBNE015MPB and RBNE01KXC9 (to I.B.); a “Centro di Eccellenza Biologia e Medicina Molecolare” grant (to I.B.); a Fondazione Cariplo grant (to R.B.); and an Italian Ministry of Agriculture and “Progetto Malattie Rare” grant from the Istituto Superiore di Sanità (to A.A.). M.A. is recipient of a fellowship from the European School of Molecular Medicine (SEMM).
Abbreviations
- AAV
adeno-associated virus
- CSA
cross-sectional area
- DAPC
dystrophin-associated protein complex
- DMD
Duchenne muscular dystrophy
- Po
force
- MHC
myosin heavy chain
- snRNA
small nuclear RNA
- CKnac
creatine kinase concentration in the serum
Footnotes
Conflict of interest statement: No conflicts declared.
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Koenig M., Beggs A. H., Moyer M., Scherpf S., Heindrich K., Bettecken T., Meng G., Muller C. R., Lindlof M., Kaariainen H., et al. Am. J. Hum. Genet. 1989;45:498–506. [PMC free article] [PubMed] [Google Scholar]
- 2.Sicinski P., Geng Y., Ryder-Cook A. S., Barnard E. A., Darlison M. G., Barnard P. J. Science. 1989;244:1578–1580. doi: 10.1126/science.2662404. [DOI] [PubMed] [Google Scholar]
- 3.DelloRusso C., Scott J. M., Hartigan-O’Connor D., Salvatori G., Barjot C., Robinson A. S., Crawford R. W., Brooks S. V., Chamberlain J. S. Proc. Natl. Acad. Sci. USA. 2002;99:12979–12984. doi: 10.1073/pnas.202300099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jiang Z., Schiedner G., van Rooijen N., Liu C. C., Kochanek S., Clemens P. R. Mol. Ther. 2004;10:688–696. doi: 10.1016/j.ymthe.2004.07.020. [DOI] [PubMed] [Google Scholar]
- 5.Wang B., Li J., Xiao X. Proc. Natl. Acad. Sci. USA. 2000;97:13714–13719. doi: 10.1073/pnas.240335297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Z., Zhu T., Qiao C., Zhou L., Wang B., Zhang J., Chen C., Li J., Xiao X. Nat. Biotechnol. 2005;23:321–328. doi: 10.1038/nbt1073. [DOI] [PubMed] [Google Scholar]
- 7.Gregorevic P., Blankinship M. J., Allen J. M., Crawford R. W., Meuse L., Miller D. G., Russell D. W., Chamberlain J. S. Nat. Med. 2004;10:828–834. doi: 10.1038/nm1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Deutekom J. C., van Ommen G. J. Nat. Rev. Genet. 2003;4:774–783. doi: 10.1038/nrg1180. [DOI] [PubMed] [Google Scholar]
- 9.Aartsma-Rus A., Janson A. A., Kaman W. E., Bremmer-Bout M., van Ommen G. J., den Dunnen J. T., van Deutekom J. C. Am. J. Hum. Genet. 2004;74:83–92. doi: 10.1086/381039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dunckley M. G., Manoharan M., Villiet P., Eperon I. C., Dickson G. Hum. Mol. Genet. 1998;7:1083–1090. doi: 10.1093/hmg/7.7.1083. [DOI] [PubMed] [Google Scholar]
- 11.Mann C. J., Honeyman K., Cheng A. J., Ly T., Lloyd F., Fletcher S., Morgan J. E., Partridge T. A., Wilton S. D. Proc. Natl. Acad. Sci. USA. 2001;98:42–47. doi: 10.1073/pnas.011408598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu Q. L., Mann C. J., Lou F., Bou-Gharios G., Morris G. E., Xue S. A., Fletcher S., Partridge T. A., Wilton S. D. Nat. Med. 2003;9:1009–1014. doi: 10.1038/nm897. [DOI] [PubMed] [Google Scholar]
- 13.Bremmer-Bout M., Aartsma-Rus A., de Meijer E. J., Kaman W. E., Janson A. A., Vossen R. H., van Ommen G. J., den Dunnen J. T., van Deutekom J. C. Mol. Ther. 2004;10:232–240. doi: 10.1016/j.ymthe.2004.05.031. [DOI] [PubMed] [Google Scholar]
- 14.Lu Q. L., Rabinowitz A., Chen Y. C., Yokota T., Yin H., Alter J., Jadoon A., Bou-Gharios G., Partridge T. Proc. Natl. Acad. Sci. USA. 2005;102:198–203. doi: 10.1073/pnas.0406700102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Angelis F. G., Sthandier O., Berarducci B., Toso S., Galluzzi G., Ricci E., Cossu G., Bozzoni I. Proc. Natl. Acad. Sci. USA. 2002;99:9456–9461. doi: 10.1073/pnas.142302299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brun C., Suter D., Pauli C., Dunant P., Lochmuller H., Burgunder J. M., Schumperli D., Weis J. Cell. Mol. Life Sci. 2003;60:557–566. doi: 10.1007/s000180300047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goyenvalle A., Vulin A., Fougerousse F., Leturcq F., Kaplan J. C., Garcia L., Danos O. Science. 2004;306:1796–1799. doi: 10.1126/science.1104297. [DOI] [PubMed] [Google Scholar]
- 18.Michienzi A., Conti L., Varano B., Prislei S., Gessani S., Bozzoni I. Hum. Gene Ther. 1998;9:621–628. doi: 10.1089/hum.1998.9.5-621. [DOI] [PubMed] [Google Scholar]
- 19.Auricchio A., Kobinger G., Anand V., Hildinger M., O’Connor E., Maguire A. M., Wilson J. M., Bennett J. Hum. Mol. Genet. 2001;10:3075–3081. doi: 10.1093/hmg/10.26.3075. [DOI] [PubMed] [Google Scholar]
- 20.Chao H., Liu Y., Rabinowitz J., Li C., Samulski R. J., Walsh C. E. Mol. Ther. 2000;2:619–623. doi: 10.1006/mthe.2000.0219. [DOI] [PubMed] [Google Scholar]
- 21.Sampaolesi M., Torrente Y., Innocenzi A., Tonlorenzi R., D’Antona G., Pellegrino M. A., Barresi R., Bresolin N., De Angelis M. G., Campbell K. P., et al. Science. 2003;301:487–492. doi: 10.1126/science.1082254. [DOI] [PubMed] [Google Scholar]
- 22.Torrente Y., Belicchi M., Sampaolesi M., Pisati F., Meregalli M., D’Antona G., Tonlorenzi R., Porretti L., Gavina M., Mamchaoui K., et al. J. Clin. Invest. 2004;114:182–195. doi: 10.1172/JCI20325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rivera V. M., Gao G. P., Grant R. L., Schnell M. A., Zoltick P. W., Rozamus L. W., Clackson T., Wilson J. M. Blood. 2005;105:1424–1430. doi: 10.1182/blood-2004-06-2501. [DOI] [PubMed] [Google Scholar]
- 24.Auricchio A., Hildinger M., O’Connor E., Gao G. P., Wilson J. M. Hum. Gene Ther. 2001;12:71–76. doi: 10.1089/104303401450988. [DOI] [PubMed] [Google Scholar]
- 25.Bottinelli R., Canepari M., Pellegrino M. A., Reggiani C. J. Physiol. (London) 1996;495:573–586. doi: 10.1113/jphysiol.1996.sp021617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.D’Antona G., Pellegrino M. A., Adami R., Rossi R., Carlizzi C. N., Canepari M., Saltin B., Bottinelli R. J. Physiol. (London) 2003;552:499–511. doi: 10.1113/jphysiol.2003.046276. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}