

Abstract
The conserved oligomeric Golgi (COG) complex is a heterooctameric complex that regulates intraGolgi trafficking and the integrity of the Golgi compartment in eukaryotic cells. Here, we describe a patient with a mild form of congenital disorder of glycosylation type II (CDG-II) that is caused by a deficiency in the Cog1 subunit of the complex. This patient has a defect in both N- and O-glycosylation. Mass spectrometric analysis of the structures of the N-linked glycans released from glycoproteins from the patient's serum revealed a reduction in sialic acid and galactose residues. Peanut agglutinin (PNA) lectin staining revealed a decrease in sialic acids on core 1 mucin type O-glycans, indicating a combined defect in N- and O-glycosylation. Sequence analysis of the COG1 cDNA and gene identified a homozygous insertion of a single nucleotide (2659–2660insC), which is predicted to lead to a premature translation stop and truncation of the C terminus of the Cog1 protein by 80 amino acids. This mutation destabilizes several other COG subunits and alters their subcellular localization and hence the overall integrity of the COG complex. This results in reduced levels and/or altered Golgi localization of α-mannosidase II and β-1,4 galactosyltransferase I, which links it to the glycosylation deficiency. Transfection of primary fibroblasts of this patient with the full length hemagglutinin-tagged Cog1 indeed restored β-1,4 galactosyltransferase Golgi localization. We propose naming this disorder CDG-II/Cog1, or CDG-II caused by Cog1 deficiency.
Keywords: glycosyltransferases, Golgi trafficking
The congenital disorders of glycosylation (CDG) are a group of rare genetic disorders characterized by defects in one of the many enzymes, transporters, or other as-yet-unknown proteins, required for the normal glycosylation of proteins (1, 2). Two major types of CDG can be distinguished based on defects within the N-glycosylation pathway. The larger group of disorders, CDG type I (CDG-I), results from defects in the assembly of the common oligosaccharide precursor in the cytosol and endoplasmic reticulum (ER) and/or its transfer onto nascent proteins. The decreased production of the Glc3Man9GlcNAc2-PP-Dolichol precursor leads to underglycosylation of proteins containing N-linked oligosaccharides. On the other hand, CDG type II (CDG-II) arises from defects in the processing and maturation in the ER and Golgi apparatus of the N-linked glycans attached to proteins. This leads to the appearance of glycans with abnormal structures on glycoproteins (3). To date, a dozen different genetic defects have been identified in the CDG-I group but only five in the CDG-II group (for a review see refs. 2 and 4). The first CDG-II defects discovered were either in glycosyltransferases or in nucleotide sugar transporters, but recent research has suggested that many cases of CDG-II do not involve primary defects in these proteins.
The predominant method for rapid screening for CDG disorders still is isoelectric focusing (IEF) of serum transferrin (5). The correct processing of N-glycan structures depends not only on the levels of activity of the glycosyltransferases and glycosidases but also on the exposure of the glycan to these activities in the correct order. In vivo, this sequence of glycosylation is determined by the substrate specificity of the different glycosyltransferases and their precise distribution in the cis-, medial-, and transGolgi cisternae (6). Therefore, the correct localization as well as activity of the processing glycosyltransferases and glycosidases in the Golgi are crucial determinants of the carbohydrate structures present on proteins.
Similarities in the cellular phenotypes of fibroblasts from some patients with CDG of unknown etiology with the phenotypes of cells with defects in the conserved oligomeric Golgi (COG) complex raised the possibility that mutations in the genes encoding the subunits of COG might be responsible for some cases of CDG. The COG complex is an eight-subunit (Cog1–8) peripheral Golgi protein involved in Golgi-associated membrane trafficking and glycoconjugate synthesis. The COG complex was first proposed to contain two subcomplexes, lobe A (Cog1–4) and lobe B (Cog5–8) (7, 8). Recent studies (9, 10) have further refined this model (see below). In Cog1- or Cog2-deficient Chinese hamster ovary (CHO) cell mutants, multiple Golgi cisternae are dilated (7), and there are pleiotropic defects in Golgi-associated reactions affecting virtually all N-, O-, and lipid-linked glycoconjugates (11). Recent studies have also shown that depletion of Cog5 in HeLa cells by RNA interference results in dilated Golgi cisternae and glycosylation defects (9), and that transient depletion of Cog3 can apparently result in the formation of Golgi-derived vesicles distributed throughout the cytoplasm (12). A subset of Golgi type II membrane proteins, called GEARs, is mislocalized and/or abnormally rapidly degraded in Cog1- and Cog2-deficient CHO mutants (13). Yeast mutants with defects in COG subunits have also been identified and characterized (8, 14–18). It has been proposed that COG may directly or indirectly play a role in resident Golgi proteins' transport to, retention at, or retrieval to appropriate sites, or that it might otherwise determine the Golgi's structure and/or luminal environment without necessarily disrupting overall secretion or endocytosis (11). The relevance of COG for CDG was first illustrated in 2004 by Wu et al. (19), who showed that the underlying cause in a case of CDG-II was a defect in Cog7.
In this study, we present a previously uncharacterized type of CDG-II with a defect in the Cog1 subunit of this complex. This mutation introduces a premature stop codon, resulting in a truncated Cog1 protein in the patient's fibroblasts. It affects not only the stability and localization of other COG subunits but also the levels, Golgi localization, and activities of glycosylation enzymes. It also points out a crucial role for the Cog1 C terminus for stability and complex formation. Our findings support the concept that there is a pivotal requirement for an intact COG complex for the proper targeting of Golgi enzymes. We suggest that deficiencies in other COG subunits will be responsible for other cases of CDG, and that COG deficiencies will compose an important cohort of all CDG-II cases. Therefore, we propose that COG-defective CDG-II be considered a distinct category of CDG, and that individual forms of COG-defective CDG-II be designated “CDG-II/COGx,” where “x” represents the defective COG subunit (e.g., CDG-II/Cog1, or CDG-II due to Cog1 deficiency).
Results
Identification of a CDG-II Patient with Defects in both N- and O-Glycosylation.
The patient described here is a girl, born by cesarean section after an uneventful pregnancy of 35 weeks. She is the second child of remotely (third level) consanguineous and healthy parents. She had a birth weight of 2,370 g, a length of 40 cm, and a head circumference of 37.5 cm. She had feeding problems since birth and failure to thrive by the end of the first month. She was admitted to the hospital at 2 months and 3 weeks. Clinical examination showed generalized hypotonia, mainly of the proximal limbs, but with normal muscular strength. She had small hands and feet, straightened bitemporal space, and antimongoloid eyelids. There was no hepatosplenomegaly. Cardiac ultrasound revealed slight left ventricular hypertrophy, with diastolic abnormalities of the left ventricle. Cerebral ultrasound was normal. At the age of 7 months, she developed growth retardation with a rhizomelic short stature. At 15 months, her growth was below the fifth percentile, and head circumference followed the fifth percentile. As of the last observation (21 months of age), she had only mild psychomotor retardation. During the last observation, a progressive microcephaly and mildly enlarged liver and spleen were registered. Brain imaging by MRI indicated only a slight cerebral and cerebellar atrophy.
The diagnosis of CDG in this patient was first indicated by IEF of serum transferrin. As can be seen in Fig. 1A, the serum sialotransferrin of this patient, when compared with the control, showed a marked reduction in the intensity of the penta- and hexasialotransferrin glycoforms and an increase in the intensity of the asialo-, mono-, di-, and trisialotransferrins. This pattern is clearly different from that characteristic of CDG-I patients (Fig. 1A), where it is known that the transferrin lacks either one or two complete N-glycans (20). The pattern observed for the patient is consistent with the presence of shortened N-glycans indicating a CDG type II (4).
Fig. 1.

IEF of serum transferrin and plasma ApoC-III, and PNA lectin staining of fibroblasts. (A) IEF of serum transferrin from a control, the patient under investigation, and a CDG-I patient are shown. The number of negative charges is indicated on the right. (B) IEF of plasma ApoC-III. Serum samples from a healthy control and the patient are shown. The ApoC-III forms with two, one, or zero neuraminic acids are indicated as ApoC-III2, -III1, and -III0, respectively. (C–F) PNA–Alexa Fluor 588 staining of control fibroblasts (C and E) and patient fibroblasts (D and F) before (C and D) and after (E and F) treatment with neuraminidase. All images were obtained at ×20.
Interestingly, the IEF of the patient plasma ApoC-III also showed an abnormal profile compared to the control (Fig. 1B). A hypoglycosylation profile was observed with the appearance of an ApoC-III0 band, which was absent in control plasma. This form corresponds to nonglycosylated ApoC-III, ApoC-III with only one GalNAc residue, or ApoC-III with a GalNAc-Gal oligosaccharide (21). Because ApoC-III is O- (mucin type glycan) but not N-glycosylated, O-glycan biosynthesis as well as N-glycosylation is disturbed in this patient.
The O-glycosylation deficiency was confirmed by staining control and patient fibroblasts with fluorescent PNA lectin, which binds specifically to the terminal galactose residues of O-glycans (22). PNA lectin fluorescence was barely detectable in control fibroblasts, because terminal sialic acid residues block access of the lectin to the penultimate galactose residues (Fig. 1C). Neuraminidase treatment, which removes terminal sialic acid residues of oligosaccharides, before PNA lectin binding considerably increased the staining (Fig. 1E). PNA lectin staining of the patient's fibroblasts was significantly greater than that of the control (Fig. 1D, top row), indicating reduced siaylation of surface O-linked glycans. Neuraminidase treatment of the patient's fibroblasts before PNA lectin binding also increased the staining (Fig. 1F), indicating that there is a partial O-glycan sialylation defect in the patient.
Structural Analysis of N-Glycans Reveals Undersialylation and Undergalactosylation of the Patient's Serum Proteins.
The molecular mechanism of the glycosylation defect was investigated by determining the structures of the N-linked glycans on serum glycoproteins from controls and from the patient by using mass spectrometry (23). The proposed structures of the glycans released from total serum glycoproteins in three normal controls and the CDG-II patient are shown in Table 1, which is published as supporting information on the PNAS web site. Spectra for a control and the patient showing peak masses and predicted structures with letters corresponding to rows in Table 1 are also shown in Fig. 6, which is published as supporting information on the PNAS web site. Comparison of the glycan profiles for the CDG-II patient and the controls revealed differences in the relative amounts of individual oligosaccharides and the presence of abnormal oligosaccharide structures in the patient. There are relative increases in the patient of the undersialylated forms (Fig. 6 B, C, E, and G) and the appearance of an undergalactosylated and undersialylated glycan (Fig. 6A), which was not detected in the controls. Additional experiments, based on the incorporation of [3H]UDP-galactose and [3H]CMP-neuraminic acid in homogenates from the patient's cells, showed a reduced incorporation of these sugars (Fig. 7, which is published as supporting information on the PNAS web site). These data indicate a partial lack of sialic acid and galactose residues in the N-glycans linked to serum glycoprotein from the patient, suggesting a defect in both sialylation and galactosylation in this patient.
The Defect in Glycosylation Is Caused by a Single-Nucleotide Insertion in the hCOG1 Gene.
The biochemical data suggest a marked defect in the galactosylation and sialylation of serum proteins in the patient. A similar defect in glycosylation was recently described in another case of CDG-II, in which a mutation in the COG7 gene was identified (19). In view of the similarity in the glycosylation defect in the two cases, the levels of the eight COG subunits in lysates of fibroblasts from two controls and our patient were analyzed by quantitative Western blotting (Fig. 2A). The levels of several COG subunits (1, 2, 3, 4, and 8) were significantly lower in the patient than in the control fibroblasts, suggesting that a mutation in one of the COG subunits might be responsible for CDG in the patient. Strikingly, only one subunit, namely Cog1, displayed an immunoreactive band with a clearly altered (higher) mobility, indicating a smaller size. The Cog1 in control fibroblasts had an apparent molecular mass of 110 kDa, whereas the form in the patient had a molecular mass of ≈100 kDa (Fig. 2A, top). This suggested that not only was the concentration of this subunit decreased (weaker band intensity), but also its structure was altered, perhaps due to a premature translational stop mutation. To test this, the hCOG1 gene was sequenced at the genomic and cDNA levels. A homozygous insertion of a single nucleotide (c.2659–2660insC) was found in the cDNA from the patient (Fig. 2B). This insertion was confirmed by sequencing at the genomic DNA level (data not shown). Both parents were shown to be heterozygous for this mutation at the cDNA (Fig. 2B) as well as at the genomic level. The insertion is predicted to shift the ORF from amino acid position 888 onwards, thereby introducing a stop codon at position 900. This would result in the production of a truncated Cog1 protein containing 12 missense residues at the C terminus and lacking the normal C-terminal 80 amino acids.
Fig. 2.

Identification of a 2559–2660insC mutation in the genomic DNA of the patient. (A) The Western blots of whole-cell lysates from control and patient fibroblasts were quantified by using an Odyssey integrator, and the relative intensities of the bands in the patient's sample are represented as mean ± SD (n = 3). Actin levels were used as a loading control. (B) Sequence alignment of the cDNA fragment from a control, a patient homozygous for the 2559–2660insC mutation, and the heterozygous parents. The arrow indicates the location of the mutation. (C) Indirect immunofluorescence staining of control (Upper) and patient (Lower) fibroblasts using antibodies for Cog1 (Left) and GM130 (Center), and the merged images (Right). The images of control and patient fibroblast were collected with the same laser power and settings. (Scale bar, 30 μm.) (D) Western blot of membrane and cytosol fractions from control (C) and patient (P) fibroblasts.
We next compared the subcellular localizations of wild-type and truncated Cog1 in fibroblasts by confocal microscopy (Fig. 2C). As demonstrated earlier in cultured mammalian cells (7, 13, 24), the COG complex, including its wild-type Cog1 subunit, colocalizes with marker proteins of the Golgi compartment such as GM130 (Fig. 2C). As expected from Western blot analysis, a much weaker signal was detected for the mutant Cog1 in the patient's fibroblasts. However, and despite this weak signal, a colocalization of Cog1 and GM130 was observed (Fig. 2C), suggesting that the truncated Cog1 subunit can associate with Golgi membranes. This was confirmed by Western blotting of microsomal membrane and cytosolic fractions from a lysate of the patient's fibroblasts, which showed that a significant amount of the truncated Cog1 subunit was associated with the membrane fraction (Fig. 2D).
Stability of Other COG Subunits.
To date, only one other defect in a COG subunit has been associated with CDG-II: a mutation in the COG7 gene (19). The mutant CHO cell lines, denoted ldlB and ldlC, exhibit deficiencies in Cog1 and Cog2 subunits, respectively (7, 9, 12, 13, 24, 25). Recently, the COG complex has been examined in HeLa cells that have been rendered Cog3- or Cog5-deficient by RNA interference methods (9, 12). In these reported cases, the defect in one COG subunit resulted in a destabilization of other COG subunits. Quantitative Western blotting of the COG subunits in our patient's fibroblasts (Fig. 2A) showed that the steady-state levels of the Cog2, -3, -4, and -8 subunits were significantly reduced, whereas those of Cog5, -6, and -7 remained unchanged. Therefore, indirect immunofluorescence was used to study the localization of the other COG subunits in the patient's fibroblasts (Fig. 8, which is published as supporting information on the PNAS web site). Altogether, these data indicate that the Cog1 mutation affects the stability/expression levels of other COG subunits, resulting in a concomitantly lower association of these subunits with the Golgi compartment. Moreover, overexpression of full length hemagglutinin (HA)-tagged Cog1 in the patient fibroblast is sufficient to restore the Golgi association of affected COG subunits, as was demonstrated for Cog3 (Fig. 9, which is published as supporting information on the PNAS web site), indicating a causative effect of the Cog1 mutation on Cog3 localization (and most likely by extension on other subunits).
Effects of Truncated Cog1 on Golgi Matrix Proteins and Enzymes.
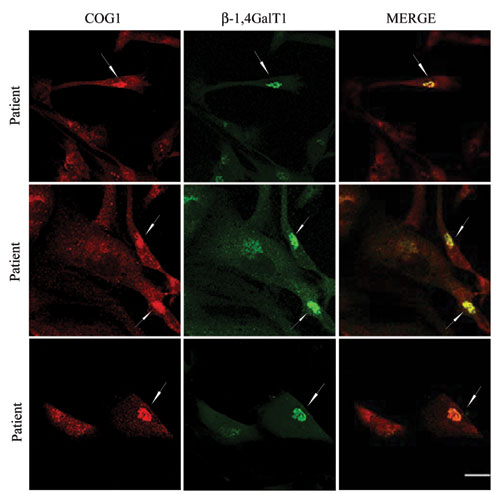
The abnormal glycosylation observed in the mutant Cog1 patient raised the possibility that the Cog1 mutation affects the normal sequence of Golgi glycosylation. This could be due to a defect in the localization or the level of individual glycosylation enzymes and was previously observed in the CHO ldlB (Cog1-deficient) and ldlC (Cog2-deficient) mutant cell lines in which Golgi proteins, such as Golgi α-mannosidase II and GPP130, exhibited a significant reduction in their steady-state levels (11, 13, 24). To test this hypothesis, the intracellular distribution and relative abundances of β-1,4 galactosyltransferase I and Golgi α-mannosidase II were analyzed by confocal microscopy. As can be seen in Fig. 3, both Golgi α-mannosidase II and β-1,4 galactosyltransferase I exhibit a significant reduction in perinuclear Golgi region staining intensity in the patient's cells compared with the control wild-type fibroblasts (55% decrease for the Golgi Mannosidase II and 67% decrease for the β-1,4 galactosyltransferase I; Fig. 4A Lower). However, and in contrast to what has been reported in CHO ldlB cells, no difference in the steady-state level of the heavily glycosylated Golgi protein GPP130 was observed (Fig. 3A). There was no effect on the levels or localization of the Golgi matrix protein GM130 (Fig. 3A), as previously observed for the Cog1- and Cog2-deficient CHO cells (13). These results were confirmed by Western blot analysis (Fig. 3B) and demonstrate at least one difference in effect on the Golgi apparatus between the truncated Cog1 protein (this study) and the Cog1 deficiency in ldlB cells (13). Finally, to demonstrate a direct relationship between an impaired Cog1 and the glycosylation disorder, the patient fibroblasts were transfected with an expression vector for the wild-type HA-Cog1 subunit followed by analysis by confocal microscopy. Fig. 4 shows that an intense Golgi staining for β-1,4 galactosyltransferase was restored in the transfected cells (arrows), whereas low levels remained in adjacent untransfected cells, as described above. This result has also been confirmed by transfecting the patient's cells with a wild-type untagged Cog1 (Fig. 10, which is published as supporting information on the PNAS web site). This indicates that overexpression of wild-type Cog1 in the patient's fibroblasts could restore the localization of the β-1,4 galactosyltransferase, thereby directly coupling Cog1 function to the subcellular targeting of glycosylation enzymes.
Fig. 3.

Expression of Golgi α-mannosidase II (ManII), β-1,4 galactosyltransferase I (B1,4 GalT1), GPP130, and GM130 in control and patient fibroblast: intracellular distribution (A) and steady-state levels of expression (B). (A Upper) Control and patient fibroblasts were processed for double indirect immunofluorescence microscopy by using antibodies against the indicated proteins. Images were collected under identical confocal settings. (Lower) Quantification of immunofluorescence intensities in the patient fibroblasts as a percentage of control. Results are mean ± SD, n = 10 cells. (Scale bar, 30 μm.) (B) Western blots of whole-cell lysates from control (C) and patient (P) fibroblasts. Actin levels were used as a loading control.
Fig. 4.

Rescue of β-1,4 galactosyltransferase I levels in patient fibroblasts by transient transfection with HA-COG1 cDNA. Double-immunofluorescent staining for HA-Cog1 and β-1,4 galactosyltransferase I in patient fibroblasts transiently transfected with an expression vector encoding Cog1 with an HA tag at the C terminus for two independent transfections. (Upper) Intermediate expression of the HA-Cog1; (Lower) high overexpression. All cells overexpressing HA-Cog1 (arrows) displayed an increase in immunostaining for β-1,4 galactosyltransferase.
Discussion
CDG-II patients suffer from multisystem abnormalities that arise from defects in the maturation of glycan chains on glycoproteins that normally occur during transport in the Golgi apparatus. In this study, we describe a patient diagnosed as CDG-II who has clear defects in N- and O-glycosylation. These defects most likely arise from the single homozygous nucleotide insertion in the patient's COG1 gene, which encodes the critical Cog1 subunit of the COG complex.
The heterooctameric COG complex plays a role in establishing or maintaining the normal structure and function of the Golgi apparatus (9, 11, 13, 24, 25). Mutations in COG complex subunits have been identified in mammalian (7, 9, 12, 13, 19), Drosophila melanogaster (26), and yeast (14–18) cells and are associated with defects in glycoconjugate synthesis, in the stability and localization of resident Golgi proteins, and in the ultrastructure of the Golgi apparatus (7, 9, 12, 13, 18, 19). The precise mechanism(s) by which COG influences Golgi structure and function remains to be established; however, it seems likely that COG influences either the retention or retrieval of cisternal specific Golgi proteins (12, 13), possibly directly by participating in intracellular membrane trafficking mediated by the COPI (coatomer) complex (12, 27). Although the ldlB and ldlC cells, both deficient in Cog1 and -2, respectively, are deficient in the low-density lipoprotein receptor (LDLR) trafficking pathway, the COG subunit deficiencies in the mammalian cells mutants examined to date (12, 13, 19) have not been clearly associated with defects in anterograde trafficking or endocytosis. The LDLR have been shown to be unstable in ldlB and ldlC cells (11); however, neither transport of secretory and membrane proteins (e.g., VSV-G) from the endoplasmic reticulum to the plasma membrane nor normal endocytic cycling is substantially disrupted in the mammalian Cog1 and -2 mutants (ldlB and ldlC) (7, 28, 29). Hence, a deficiency or functional defect in one of the eight COG subunits can alter the level and/or intraGolgi distribution of Golgi resident proteins that play a key role in the synthesis of glycoconjugates.
Indeed, the serum transferrin and apolipoprotein C-III of the mutant Cog1 patient examined here displayed abnormal N- and O-glycosylation. Moreover, detailed N-glycan structures from serum total N-glycoproteins showed a partial lack of galactose residues and sialic residues linked to galactose, which was accompanied in an in vitro assay by the low incorporation of UDP-Gal and CMP-NeuAc. Furthermore, there was a dramatic loss of Golgi localization of β-1,4 galactosyltransferase I immunoreactivity observed in the patient's cells that could be reversed by reintroducing wild-type COG1 cDNA. Golgi mannosidase II levels were substantially decreased in the patient's cells, although no hybrid N-glycan structures were detected, perhaps because of the compensatory activity of other Golgi mannosidases, such as mannosidase IIx (30). Thus, multiple abnormalities in the proteins associated with the glycosylation machinery of the Golgi apparatus are probably responsible for the global galactosylation and sialylation defects in the patient's cells and therefore the CDG-II phenotype.
Similar defects in galactosylation and sialylation were recently observed in another CDG-II patient in which a defect in the Cog7 subunit was detected (19). Whereas from the point of view of the glycosylation defect, no major difference was observed between this and our patient, the clinical description is radically opposite. Indeed, the Cog1 defect leads to a mild phenotype, in contrast with the one found in the Cog7 patient. Whereas the Cog7 patient died at 5 weeks after birth, the Cog1 patient is still alive and has only mild mental and motor retardation. Nevertheless, also in the case of the Cog7 defect, an alteration in the trafficking of glycosyltransferases, as demonstrated for hST3-Gal-I (19), appears to be the culprit. Whether in our patient the impaired sialylation is also due to a defect in the sialyltransferases remains to be investigated. However, we noted that, as cells are passaged in culture, a loss of the defect concerning the intracellular Golgi level of both the Golgi mannosidase II and the β-1,4 galactosyltransferase I is observed. This complicates further biochemical and cell biological studies in these cells. This phenomenon, which could be due to redundancy with other protein complexes mediating endoplasmic reticulum–Golgi trafficking, may also be observed by Wu et al. (19), who reported the cellular phenotype only in cells from one of the two affected siblings.
The direct link between COG function and glycosylation has also been documented in the identification of two CHO cell lines, ldlB- and ldlC-deficient in Cog1 and -2, respectively. These cell lines were originally identified in a genetic screen for mutants exhibiting defects in receptor mediated endocytosis of low-density lipoprotein (LDL). The LDL receptor-deficient phenotype observed in these mutant cell lines was due to aberrant Golgi processing of the receptor (11). Indeed, the ldlB and ldlC cell lines were consequently shown to exhibit pleiotropic defects in glycoconjugate synthesis with a notable deficiency in Golgi mannosidase II (11, 13).
Based on genetic and biochemical studies, the COG complex was initially proposed to be composed of two lobes, lobe A containing the Cog1, -2, -3, and -4 subunits and lobe B containing the Cog5, -6, -7, and -8 subunits (7–11, 16, 31). Recent studies in mammals have indicated that Cog2–4 and Cog5–7 and Cog1–4 plus Cog8 form stable subcomplexes, and that Cog1 and -8 play an important role in linking the Cog2–4 and Cog5–7 subcomplexes to each other and to the Golgi apparatus (9, 10). (For an alternative model of the subunit interaction map of COG, see refs. 31 and 33). The c.2659–2660insC COG1 allele, for which the patient is homozygous, encodes an apparently unstable polypeptide that is C-terminally truncated. The data reported here provide strong support for the recent revised model (Fig. 5, adapted from refs. 9 and 10) in that the defective Cog1 appears to be responsible for the reduction in cellular levels of Cog2–4 as well as Cog8, those subunits that form a subcomplex with Cog1, but has little influence on the cellular levels of the Cog5–7 subcomplex (7, 9, 11, 12). Cog1 mutants have been identified and characterized in mammalian (11, 24, 25) and yeast (8, 16) cells. In mammalian cells, the virtually complete Cog1 deficiency affects the stability of all lobe B subunits while not dramatically affecting Cog2, -3, and -4 levels (7, 9, 13). In yeast, the interaction of the Cog2–4 subunits with the Cog5–8 subunits is disrupted in either Cog1-deficient cells or cells expressing Cog1 with an 80-residue truncation at the N terminus (33). We found that the truncated C terminus of Cog1 in our patient's cells was associated with an instability in Cog2–4 and Cog8, but not the Cog5–7 subcomplex. Additional studies will be required to determine the molecular basis for the different effects on the stabilities of the subcomplexes of COG of complete deletion of COG1 versus truncation of its C terminus.
Fig. 5.

Model of the subunit architecture of the COG complex. This subunit connectivity map is adapted from studies of Oka et al. (9) and Ungar et al. (10). This model highlights the effects of the truncated Cog1 on the observed Cog8 expression.
Oka et al. (13) showed that the steady-state levels of a subset of Golgi resident proteins (GEARs) depended on the COG complex. We report additional evidence for a functional relationship between COG deficiency and the intracellular levels and/or localization of Golgi glycosyltransferases. We found that, in the Cog1-deficient patient, both Golgi α-mannosidase II and β-1,4 galactosyltransferase I Golgi localization were dramatically reduced. We have not determined the intracellular level of the Golgi sialyltransferases (19), but the residual activity observed in vitro suggests that the intracellular level/localization of these Golgi enzymes may also be altered in the patient's cells. Altogether, these data confirm the direct relationship between the COG complex and the intraGolgi levels of specific Golgi proteins. This relationship may be due to the possible role of COG in controlling intraGolgi trafficking of some resident Golgi proteins (e.g., COPI-dependent retrograde trafficking) (32). The analysis of human COG subunit mutations from CDG patients, together with mammalian somatic cell mutants, yeast mutants, and RNA interference knockdown studies (9, 12, 33), should help define the precise mechanism by which COG controls the structure and function of the Golgi apparatus.
According to the current CDG nomenclature, this new CDG patient should be classified as CDG-IIg. However, we consider that the current nomenclature system does not adequately address cases of CDG due to defects in Golgi trafficking. Therefore, we propose that individual forms of COG-defective CDG-II be designated “CDG-II/Cogx,” where “x” represents the defective COG subunit (e.g., CDG-II/Cog1 for the case reported here).
Materials and Methods
Cell Culture and Transfections.
Primary fibroblasts from patient and controls were cultured at 37°C under 5% CO2 in DMEM/F12 (Life Technologies, Paisley, U.K.) supplemented with 10% Fetal Clone III (HyClone). Transfections of 75–90% confluent cells were performed with FuGEN6 (Roche Diagnostics, Indianapolis), as recommended by the manufacturer's instructions.
Antibodies.
Affinity-purified or crude serum rabbit polyclonal antibodies (pab) against the Cog1–8 subunits (13) were used in dilutions ranging from 1:1,000 to 1:5,000 for Western blotting and in 1:50 to 1:200 dilutions for indirect immunofluorescence microscopy. Pab against Cog3 was a gift from V. Lupashin (University of Arkansas for Medical Sciences, Little Rock, AK). Pab against Golgi α-mannosidase II or β-1,4 galactosyltransferase I were gifts from K. Moremen (University of Georgia, Athens) and E. G. Berger (University of Zürich, Zürich), respectively. Pab anti-GPP130 antibody and mAb anti-GM130 were from Covance (Richmond, CA) and BD Biosciences (Franklin Lakes, NJ).
Western Blot Analysis.
Control and patient fibroblasts were rinsed twice with ice-cold PBS and then lysed for 60 min on ice in cell lysis buffer (Tris-buffered saline supplemented with 1% Triton X-100). Insoluble material was removed by centrifugation at 13,000 × g for 15 min at 4°C. Proteins were quantified by using the BCA Kit (Interchim, Montlucon, France). Unless specified, 20 μg of total protein was mixed with reducing 4× sample loading buffer (Invitrogen), incubated at 90°C for 5 min, and separated by electrophoresis on 4–12% precast gels SDS/PAGE (Invitrogen) during 1 h at 180 V. Proteins were transferred to nitrocellulose membranes and further processed for Western blotting by using the appropriate primary and horseradish peroxidase-conjugated secondary antibodies. Protein bands were detected by enhanced chemiluminiscence (Renaissance; PerkinElmer). For the quantification, anti-mouse and anti-rabbit secondary antibodies conjugated to Alexa Fluor 680 (Molecular Probes; 1:3,000) were used to probe primary antibodies. Proteins bands were detected and quantified by Western blotting with the Odyssey system (Li-Cor, Lincoln, NE).
Immunofluorescence Microscopy.
Details on the fixation and processing of fibroblasts for double and triple immunofluorescence labeling and confocal microscopy are provided in Supporting Text, which is published as supporting information on the PNAS web site.
Mutation Analysis.
Details of mutation analysis are provided in Supporting Text. Primers were designed to amplify the 15 exons of COG1, including at least 50 bp of the flanking intronic regions, based on the genomic sequence of COG1 (NM_018714). Primer sequences are available on request. The exons were amplified by using standard PCR conditions, subsequently sequenced with Big Dye Terminator Ready reaction cycle sequencing kit ver. 3.1 (Applera), and analyzed on an ABI3100 Avant (Applera, Foster City, CA).
Transient Transfection of HA-Cog1 or Wild-Type Cog1 into the Patient's Fibroblasts.
Control COG1 cDNA was generated by using Pfu-Turbo-Polymerase (Stratagene). The blunt-ended PCR fragments were ligated into a pcDNA3.1 vector or pMH6 vector encoding Cog1 with an HA tag at the C terminus. The plasmid sequence was then sequenced. Twelve hours before transfection, the patient's cells were plated onto glass coverslips and cultured. Transient transfection of Cog1 or HA-Cog1 patient cells was performed with FuGEN6 by using 2 μg of vector DNA. Six hours after transfection, the cells were washed and the medium changed. Cells were then incubated for 48 h before harvesting. The relatively low efficiency of the transfection resulted in the presence of untransfected cells adjacent to the transfected cells expressing substantial levels of Cog1.
Supplementary Material
Acknowledgments
We acknowledge the support of Drs. D. Ungar and F. Hughson (Princeton University, Princeton); Dr. Toshihiko Oka (Kyushu University, Fukuoka, Japan); Ms. L. Keldermans; and Drs. R. Zeevaert (Katholieke Universiteit Leuven), R. Cacan (University of Lille, Lille, France), L. Diogo (Hospital Pediatrico de Coimba, Coimba, Portugal), and L. Vilarinho (Medical Genetics Institute, Porto). We are grateful to Drs. K. Moremen, E. G. Berger, and V. Lupashin for providing antibodies. This work was supported by the Fondation pour la Recherche Médicale and the community under a Marie Curie Intra-European Fellowship (to F.F.), the Körber Prize (to G.M.), Fonds voor Wetenschappelijk Onderzoek Flanders Grants G.0504.06 and G.0173.04 (to W.A. and G.M., respectively), National Institutes of Health Grant GM59115 (to M.K.), and the Sixth Framework program of the European Union (Euroglycanet: LSHM-2005-512131). P.M. acknowledges the support of the Ethan Batchelor Trust to P. Clayton.
Abbreviations
- CDG
congenital disorder of glycosylation
- COG
conserved oligomeric Golgi
- IEF
isoelectric focusing
- PNA
peanut agglutinin
- HA
hemagglutinin
Footnotes
Conflict of interest statement: No conflicts declared.
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Grunewald S., Matthijs G., Jaeken J. Pediatr. Res. 2002;52:618–624. doi: 10.1203/00006450-200211000-00003. [DOI] [PubMed] [Google Scholar]
- 2.Jaeken J. J. Inherit. Metab. Dis. 2003;26:99–118. doi: 10.1023/a:1024431131208. [DOI] [PubMed] [Google Scholar]
- 3.Aebi M., Helenius A., Schenk B., Barone R., Fiumara A., Berger E. G., Hennet T., Imbach T., Stutz A., Bjursell C., et al. Glycoconj. J. 1999;16:669–671. doi: 10.1023/a:1017249723165. [DOI] [PubMed] [Google Scholar]
- 4.Jaeken J., Matthijs G. Annu. Rev. Genomics Hum. Genet. 2001;2:129–151. doi: 10.1146/annurev.genom.2.1.129. [DOI] [PubMed] [Google Scholar]
- 5.de Jong G., van Eijk H.-G. Electrophoresis. 1988;9:589–598. doi: 10.1002/elps.1150090921. [DOI] [PubMed] [Google Scholar]
- 6.Schachter H. Glycobiology. 1991;1:453–461. doi: 10.1093/glycob/1.5.453. [DOI] [PubMed] [Google Scholar]
- 7.Ungar D., Oka T., Brittle E.-E., Vasile E., Lupashin V.-V., Chatterton J.-E., Heuser J.-E., Krieger M., Waters M.-G. J. Cell Biol. 2002;157:405–415. doi: 10.1083/jcb.200202016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Whyte J.-R., Munro S. Dev. Cell. 2001;1:527–537. doi: 10.1016/s1534-5807(01)00063-6. [DOI] [PubMed] [Google Scholar]
- 9.Oka T., Vasile E., Penman M., Novina C.-D., Dykxhoorn D.-M., Ungar D., Hughson F.-M., Krieger M. J. Biol. Chem. 2005;280:32729–32735. doi: 10.1074/jbc.M505558200. [DOI] [PubMed] [Google Scholar]
- 10.Ungar D., Oka T., Vasile E., Krieger M., Hughson F.-M. J. Biol. Chem. 2005;280:32736–32745. doi: 10.1074/jbc.M505558200. [DOI] [PubMed] [Google Scholar]
- 11.Kingsley D.-M., Kozarsky K.-F., Segal M., Krieger M. J. Cell Biol. 1986;102:1576–1585. doi: 10.1083/jcb.102.5.1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zolov S.-N., Lupashin V.-V. J. Cell Biol. 2005;168:747–759. doi: 10.1083/jcb.200412003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oka T., Ungar D., Hughson F.-M., Krieger M. Mol. Biol. Cell. 2004;15:2423–2435. doi: 10.1091/mbc.E03-09-0699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim D.-W., Sacher M., Scarpa A., Quinn A.-M., Ferro-Novick S. Mol. Biol. Cell. 1999;10:3317–3329. doi: 10.1091/mbc.10.10.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim D.-W., Massey T., Sacher M., Pypaert M., Ferro-Novick S. Traffic. 2001;2:820–830. doi: 10.1034/j.1600-0854.2001.21111.x. [DOI] [PubMed] [Google Scholar]
- 16.Ram R.-J., Li B., Kaiser C.-A. Mol. Biol. Cell. 2002;13:1484–1500. doi: 10.1091/mbc.01-10-0495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.VanRheenen S.-M., Cao X., Lupashin V.-V., Barlowe C., Waters M.-G. J. Cell Biol. 1998;141:1107–1119. doi: 10.1083/jcb.141.5.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.VanRheenen S.-M., Cao X., Sapperstein S.-K., Chiang E.-C., Lupashin V.-V., Barlowe C., Waters M.-G. J. Cell Biol. 1999;15:729–742. doi: 10.1083/jcb.147.4.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu X., Steet R.-A., Bohorov O., Bakker J., Newell J., Krieger M., Spaapen L., Kornfeld S., Freeze H.-H. Nat. Med. 2004;10:518–523. doi: 10.1038/nm1041. [DOI] [PubMed] [Google Scholar]
- 20.Yamashita K., Ideo H., Ohkura T., Fukushima K., Yuasa I., Ohno K., Takeshita K. J. Biol. Chem. 1993;268:5783–5789. [PubMed] [Google Scholar]
- 21.Wopereis S., Grunewald S., Morava E., Penzien J. M., Briones P., Garcia-Silva M.-T., Demacker P.-N., Huijben K.-M., Wevers R.-A. Clin. Chem. 2003;49:1839–1845. doi: 10.1373/clinchem.2003.022541. [DOI] [PubMed] [Google Scholar]
- 22.Chacko B.-K., Appukuttan P.-S. Int. J. Biol. Macromol. 2001;28:365–371. doi: 10.1016/s0141-8130(01)00139-8. [DOI] [PubMed] [Google Scholar]
- 23.Mills P.-B., Mills K., Mian N., Winchester B.-G., Clayton P.-T. J. Inherit. Metab. Dis. 2003;26:119–134. doi: 10.1023/a:1024476915278. [DOI] [PubMed] [Google Scholar]
- 24.Podos S.-D., Reddy P., Ashkenas J., Krieger M. J. Cell Biol. 1994;127:679–691. doi: 10.1083/jcb.127.3.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chatterton J.-E., Hirsch D., Schwartz J.-J., Bickel P.-E., Rosenberg R.-D., Lodish H.-F., Krieger M. Proc. Natl. Acad. Sci. USA. 1999;96:915–920. doi: 10.1073/pnas.96.3.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Farkas R.-M., Giansanti M.-G., Gatti M., Fuller M.-T. Mol. Biol. Cell. 2003;14:190–200. doi: 10.1091/mbc.E02-06-0343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Suvorova E.-S., Duden R., Lupashin V.-V. J. Cell Biol. 2002;157:631–643. doi: 10.1083/jcb.200111081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reddy P., Krieger M. Mol. Cell Biol. 1989;9:4799–4806. doi: 10.1128/mcb.9.11.4799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oka T., Krieger M. J. Biochem. Tokyo. 2005;137:109–114. doi: 10.1093/jb/mvi024. [DOI] [PubMed] [Google Scholar]
- 30.Misago M., Liao Y.-F., Kudo S., Eto S., Mattei M.-G., Moremen K.-W., Fukuda M.-N. Proc. Natl. Acad. Sci. USA. 1995;25:11766–11770. doi: 10.1073/pnas.92.25.11766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Loh E., Hong W. J. Biol. Chem. 2004;279:24640–24648. doi: 10.1074/jbc.M400662200. [DOI] [PubMed] [Google Scholar]
- 32.Ungar D., Oka T., Krieger M., Hughson F.-M. Trends Cell Biol. 2006 doi: 10.1016/j.tcb.2005.12.004. in press. [DOI] [PubMed] [Google Scholar]
- 33.Fotso P., Koryakina Y., Pavliv O., Tsiomenko A., Lupashin V. J. Biol. Chem. 2005;280:27613–27623. doi: 10.1074/jbc.M504597200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}