

Abstract
The mevalonate pathway produces cholesterol and nonsterol isoprenoids, such as geranylgeraniol. In the brain, a fraction of cholesterol is metabolized in neurons by the enzyme cholesterol 24-hydroxylase, and this depletion activates the mevalonate pathway. Brains from mice lacking 24-hydroxylase excrete cholesterol more slowly, and the tissue compensates by suppressing the mevalonate pathway. Here we report that this suppression causes a defect in learning. 24-Hydroxylase knockout mice exhibit severe deficiencies in spatial, associative, and motor learning, and in hippocampal long-term potentiation (LTP). Acute treatment of wild-type hippocampal slices with an inhibitor of the mevalonate pathway (a statin) also impairs LTP. The effects of statin treatment and genetic elimination of 24-hydroxylase on LTP are reversed by a 20-min treatment with geranylgeraniol but not by cholesterol. We conclude that cholesterol turnover in brain activates the mevalonate pathway and that a constant production of geranylgeraniol in a small subset of neurons is required for LTP and learning.
Keywords: cholesterol 24-hydroxylase, isoprenoids, neurons, synaptic plasticity, cytochrome P450
The mevalonate pathway is composed of 35 enzymes that synthesize cholesterol and more than a dozen other isoprenoids from mevalonate, the product of 3-hydroxy-3-methylglutaryl (HMG) CoA reductase (ref. 1; Fig. 1). Among the nonsterol isoprenoids generated by the pathway are farnesyl diphosphate and geranylgeranyl diphosphate, which are covalently linked to a large number of proteins, including GTP binding proteins that play diverse roles within the cell ranging from intracellular signaling to vesicular transport (2). The mevalonate pathway is regulated at multiple levels to prevent the buildup of cholesterol and to ensure a constant supply of nonsterol isoprenoids. The accumulation of cholesterol in membranes activates a negative feedback loop that reduces the levels of transcription factors required for the expression of multiple enzymes in the mevalonate pathway, including HMG CoA reductase (1). Cholesterol and isoprenoids also act postranscriptionally to accelerate the degradation of HMG CoA reductase, thereby potentially limiting the synthesis of all isoprenoids (3–5). A class of drugs called statins inhibits HMG CoA reductase directly and therefore has the ability to reduce synthesis of isoprenoids (ref. 6; Fig. 1).
Fig. 1.

Schematic of the mevalonate pathway. Arrows, flow of intermediates; brakes, pharmacologic and genetic inhibition of the indicated steps. PP, diphosphate.
Although the human brain accounts for only a small percentage of body mass (≈2%), it contains ≈25% of the body's cholesterol. A majority of brain cholesterol is located in the myelin membranes of oligodendrocytes that surround axons, whereas smaller amounts are found in the plasma membranes of neurons and other support cells. Cholesterol in the adult brain is largely metabolically inert; however, a small fraction of the pool, estimated to be ≈0.02% in humans and somewhat larger in the mouse (≈0.4%), turns over each day (7). The major mechanism by which cholesterol is metabolized in the brain is by conversion to 24(S)-hydroxycholesterol (8). This reaction is catalyzed by the enzyme cholesterol 24-hydroxylase (24-hydroxylase), a cytochrome P450 (CYP46A1) that is selectively expressed in the brain (ref. 9; Fig. 1). The 24(S)-hydroxycholesterol diffuses out of cells, crosses the blood–brain barrier, and is cleared by the liver (10, 11). Knockout (KO) mice lacking 24-hydroxylase have an ≈50% reduction in brain cholesterol excretion (12). This decrease is compensated for by a reduction in de novo synthesis such that steady state levels of cholesterol in the brains of KO mice are the same as those of wild-type (WT) mice. This reduced synthesis is likely to be mediated by a decrease in the activity of HMG CoA reductase.
24-Hydroxylase is expressed in only a small subset of neurons in the brain, including pyramidal cells of the cortex and hippocampus, granule cells of the dentate gyrus, and Purkinje cells of the cerebellum (9), but it is responsible for a majority of cholesterol turnover in the tissue. When calculated on a per cell basis, 24-hydroxylase-expressing neurons turn over cholesterol as fast as any other cell type in the body (13), suggesting that cholesterol catabolism in these neurons is of crucial importance. In the current study, we use learning tests in the whole animal and electrophysiological experiments with hippocampal slices in vitro to show that cholesterol turnover via the 24-hydroxylase enzyme ensures activation of the mevalonate pathway and the constant synthesis of geranylgeraniol, which, in turn, is essential for learning.
Results
Behavioral Assessment of Learning.
Spatial learning in 24-hydroxylase KO mice was assessed in Morris water maze tests (14). As indicated in Fig. 2A, the time required for WT mice to find a submerged platform (the latency period) progressively decreased as the animal learned to navigate by visual cues supplied around the tank. In contrast, 24-hydroxylase KO mice never learned the location of the platform and instead swam aimlessly until they encountered the platform by chance. When the platform was removed, WT mice spent much more time swimming in the area where the platform had been than did KO mice (Fig. 2B). Control experiments showed that the KO mice were not blind and swam normally (data not shown).
Fig. 2.

Assessment of behavioral learning. (A) Spatial learning in Morris water maze tests. Mean time to find a submerged platform (latency) is shown as a function of trial day. Data were collected in four different experiments involving 40 WT (+/+) and 30 KO (−/−) mice. Error bars in this experiment and all other experiments represent SEM values. (B) After removal of the platform on the indicated day, the number of times animals of the two genotypes swam across the former location of the platform was determined. (C) Associative learning in cued conditioning fear tests. Percent freezing was measured in response to an auditory cue 1 h after the training period (Upper left) and 24 h after training period (Lower left), or in response to the context in which the training was performed after 1 h (Upper right) or 24 h (Lower right). Data were collected in two different experiments involving 30 +/+ mice and 30 −/− mice. (D) Motor learning in rotating rod tests. The mean times mice of the indicated genotypes stayed on the rotating rod are plotted as a function of trial day. Data were collected from 15 +/+ and 15 −/− mice.
Associative learning requires an animal to relate an environmental cue to a mild aversive stimulus such as an electric shock. As shown in Fig. 2C Left, WT mice learned to associate a tone with an electric shock and manifest a typical behavior (freezing, or cessation of involuntary movement) upon hearing the sound in the absence of a shock. In contrast, 24-hydroxylase KO mice were defective in this behavior and stopped moving in response to a tone only half as frequently as did control mice. This defect was manifest at both short (1 h) and long (24 h) intervals after the initial training period. The ability of mice to associate a context (box color and odor) with an electric shock was determined next. When WT mice were placed in the scented box in which they had been trained to associate the sound and shock, they froze a high percentage of the time when tested at short- and long-term intervals after training (Fig. 2C Right). The KO mice were again defective in this form of contextual learning. Control experiments revealed that the mutant mice were not deaf and that their pain thresholds were similar to those of WT mice (data not shown).
The cerebellum is associated with motor coordination and learning. The highly innervated Purkinje cells of this tissue express 24-hydroxylase (9). The histology and size of the cerebellum is normal in the KO mice, and they do not exhibit hypotonia, ataxia, or tremor based on observational and open field tests (12); however, as indicated by the data of Fig. 2D, the mutant mice displayed defects in motor learning. Whereas WT mice were able to double the amount of time they stayed on a rotating rod after a 4-day training period, KO mice increased their stay times to a lesser extent, and, by day 3, the difference between the two genotypes was significant.
Absence of Anatomical Defects in KO Mice.
Cued, contextual, and associative learning require an intact amygdala and hippocampus (15), suggesting that the substandard performance of the 24-hydroxylase KO mice in the behavioral tests might be because of an anatomical defect in these regions of the brain. To assess this possibility, we performed histological and protein expression analyses. The cellular architecture of the hippocampus and dentate gyrus was indistinguishable between WT and KO mice (Fig. 3A Left), as well as in other regions of the brain. Immunohistochemical staining for synaptogyrin, a widely distributed synaptic vesicle protein (16), and for microtubule-associated protein 2 (MAP2), a protein expressed in neuronal cell bodies and dendrites (17), were similar in WT and KO mice (Fig. 3A Center). The morphology of the synapse, as examined by electron microscopy, also did not differ between mice of different 24-hydroxylase genotypes (Fig. 3A Right).
Fig. 3.

Histology and protein expression in the hippocampus. (A Left) Hematoxylin and eosin staining of hippocampal sections from WT (+/+) and KO (−/−) mice. CA, cornu ammonis; DG, dentate gyrus. (Scale bar, 50 μm.) (A Center) Histochemical staining for cell nuclei (DAPI, blue), and double immunofluorescent staining for microtubule-associated protein 2 (MAP2) (antibody AP20, green) and synaptogyrin (antibody P925, red) in the CA1 region of the hippocampus. (Scale bar, 20 μm.) (A Right) Electron microscopic analyses of synaptic contacts on pyramidal cell dendrites in CA1 stratum radiatum. Arrows indicate synaptic clefts. (Scale bar, 400 nm.) (B) Protein expression assessed in hippocampal extracts by immunoblotting. The levels of three glutamate receptor subunits (NR1, GluR1, and GluR2/3), a small GTP binding protein (Rab3A), two synaptic vesicle proteins (synaptotagmin and synaptogyrin), glutamic acid decarboxylase (GAD67), and calnexin were determined.
Immunoblotting of proteins from the hippocampus of WT and KO mice revealed no differences in the levels of several glutamate receptor subunits involved in learning (NR1, GluR1, and GluR2/3), a GTP binding protein involved in synaptic vesicle trafficking (Rab3A), two synaptic vesicle proteins (synaptotagmin and synaptogyrin), an enzyme (glutamic acid decarboxylase, GAD67) involved in the biosynthesis of the neurotransmitter γ-aminobutyric acid (GABA), or calnexin, a chaperone of the endoplasmic reticulum (Fig. 3B).
Synaptic Transmission in KO Mice.
Changes in synaptic efficacy, or plasticity, are thought to occur during learning and to be required for memory storage. The leading examples of synaptic plasticity are the electrophysiological phenomena termed long-term potentiation (LTP) and long-term depression (LTD), which represent synaptic strengthening and weakening, respectively, in response to electrical stimulation (18–20). The neural transmission associated with these processes has both presynaptic and postsynaptic components, and defects in learning can be associated with either of these compartments.
We first examined the subcellular distribution of 24-hydroxylase at the synapse to determine whether the enzyme acted at the presynaptic or postsynaptic membrane. As indicated in Fig. 4A, 24-hydroxylase was distributed evenly between the presynaptic and postsynaptic membranes from WT mice. As expected, the glutamate receptor subunit GluR1 was detected only in postsynaptic membranes, whereas the synaptic vesicle protein synaptophysin was present only in presynaptic membranes. 24-Hydroxylase was not detected in membranes from KO mice, and more importantly, the subcellular distributions of GluR1 and synaptophysin were not altered in these membranes (Fig. 4A).
Fig. 4.

Synaptic transmission in WT and 24-hydroxylase KO mice. (A) The indicated hippocampal membrane fractions from WT (+/+) and KO (−/−) mice were probed for 24-hydroxylase, the GluR1 glutamate receptor subunit, and synaptophysin. (B) Spontaneous miniature synaptic currents in single CA1 pyramidal neurons in the presence of 1 μM tetrodotoxin. Representative traces from recordings made from 11 +/+ (○) and 14 −/− (•) neurons. (C) Input-output curves. Field potentials were determined in WT (○, n = 14) and KO slices (•, n = 13) over a stimulus intensity range of 5 to 40 μA. (D) Short term plasticity assessed by paired-pulse facilitation. Data from two different experiments with 43 +/+ (○) and 28 −/− (•) slices.
General synaptic transmission was assessed by recording miniature synaptic currents resulting from spontaneous fusion of synaptic vesicles in single pyramidal neurons of the hippocampal CA1 region (Fig. 4B). Measurement of spontaneous currents in WT neurons (n = 11) from nine hippocampal slices revealed an amplitude of 17.7 ± 1.1 pA (mean ± SEM) and a frequency of 0.8 ± 0.2 Hz. An amplitude of 19.7 ± 1.5 pA and a frequency of 1.0 ± 0.1 Hz were recorded in KO neurons (n = 14) from eight hippocampal slices. These values were not significantly different, which suggested that the strength of individual synapses, the number of synapses, and the probability of neurotransmitter release were similar between WT and KO mice.
Consistent with the intracellular measurements of synaptic transmission, excitatory postsynaptic field potentials (fEPSP) recorded at different stimulus intensities in the stratum radiatum of hippocampal slices were similar between animals of different 24-hydroxylase genotypes. No differences were observed between WT and KO slices when the initial slopes of the fEPSP measured in the input-output experiments were plotted against the fiber volley amplitudes (Fig. 4C).
The presynaptic component of plasticity was examined by measuring paired-pulse facilitation in hippocampal slices dissected from 35- to 40-day-old mice. As shown in Fig. 4D, no differences in paired-pulse facilitation responses were detected between WT and KO slices when the interpulse intervals varied between 25 and 200 ms. These data suggested that the presynaptic release probability and short term plasticity were unchanged in the mutant mice.
Defective LTP and Normal LTD in KO Mice.
We next evaluated LTP and LTD in WT and KO mice. A θ burst stimulation protocol was used to induce LTP in the CA1 region of the hippocampus. Baseline responses recorded in WT slices in response to monophasic stimulation were stable, and the application of high frequency stimulation resulted in a robust and long lasting potentiation of synaptic activity (Fig. 5A). In contrast, although baseline responses were similar in hippocampal slices from KO mice, LTP was significantly impaired. An induction protocol consisting of a 1-Hz stimuli delivered for 15 min (900 pulses total) was used to produce LTD in hippocampal slices. As indicated in Fig. 5B, baseline responses were similar and synaptic currents were depressed to equal extents in WT and KO slices in response to this stimulation.
Fig. 5.

Impaired hippocampal LTP and normal LTD in 24-hydroxylase KO mice. (A) LTP in hippocampal slices from WT (○, n = 19) and KO (•, n = 13) mice. The arrow marks the point of high frequency stimulation (θ burst). Insets in this experiment and all subsequent experiments show representative recorded potentials immediately before (dashed lines) and 55 min after (solid lines) tetanization. (B) LTD in hippocampal slices from WT (○, n = 13) and KO (•, n = 10) mice.
Inhibitory Responses in KO Mice.
Synaptic activity reflects the balance of excitatory and inhibitory inputs mediated by different neurotransmitter receptors at the postsynaptic membrane. Inhibition in the hippocampus is chiefly mediated by GABA receptors of the A subtype (GABAA receptors), which are composed of multiple subunits, including those that bind pregnane steroids, which act as agonists of the GABAA receptor (21). Thus, it was conceivable that cholesterol or 24(S)-hydroxycholesterol binding to a GABAA receptor subunit might be altered in the KO mice, leading to increased inhibitory input and decreased LTP. To examine the contribution of inhibition, we performed LTP experiments in the presence of the GABAA receptor inhibitor, picrotoxin (22). This plant toxin increased the magnitude of the LTP response in WT slices as would be expected if inhibition were relieved but did not restore LTP in slices from KO mice (Fig. 7, which is published as supporting information on the PNAS web site), suggesting that excess inhibition was not the cause of the diminished LTP in the mutant mice.
Metabolic Basis of Impaired LTP in KO Mice.
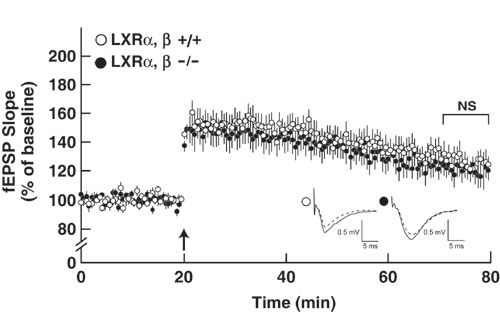
The 24-hydroxylase KO mice have two metabolic deficiencies. First, the animals do not synthesize or accumulate 24(S)-hydroxycholesterol in the central nervous system, and second, they exhibit an ≈50% decrease in de novo cholesterol synthesis owing to suppression of the mevalonate pathway (ref. 12; see Fig. 1). To explore the role of 24(S)-hydroxycholesterol in synaptic plasticity, we measured LTP in hippocampal slices from mice deficient in the liver X receptor (LXR) α and β genes (23). 24(S)-Hydroxycholesterol is a potent agonist of these nuclear receptors that together regulate numerous aspects of lipid metabolism (24), and it is possible that the absence of the oxysterol caused a decrease in LXR target gene activation and a concomitant reduction in LTP. As shown in Fig. 8, which is published as supporting information on the PNAS web site, LTP was induced to the same extent in hippocampal slices from WT and LXR α/β double KO mice. In agreement with this finding, microarray experiments and real-time PCR measurements of individual mRNAs failed to reveal a difference in LXR target gene expression in the KO mice (data not shown).
To determine the role of the mevalonate pathway in synaptic plasticity, we measured LTP in WT hippocampal slices incubated with compactin, a statin inhibitor of HMG CoA reductase, the rate-limiting enzyme in the pathway. Addition of 12.5 μM compactin inhibited de novo cholesterol synthesis ≈80%, as assessed by acetate incorporation, and decreased LTP in WT slices to an extent similar to that observed in untreated 24-hydroxylase KO tissue (Fig. 6A).
Fig. 6.

Restoration of LTP by mevalonate and isoprenoids. (A) Hippocampal slices from WT mice were incubated beginning at time 0 with no additions (○, n = 11) or 12.5 μM compactin (statin; •, n = 17), and LTP was induced after 20 min. (B) Reversal of compactin-mediated LTP inhibition by mevalonate (Mev) but not cholesterol (Chol). Hippocampal slices from WT mice were incubated with 12.5 μM compactin, 0.2 mM mevalonate, and 5 μg/ml cholesterol (○, n = 13), or 12.5 μM compactin and 5 μg/ml cholesterol (•, n = 10). (C) Mevalonate restores LTP in 24-hydroxylase KO mice. Hippocampal slices were incubated in the absence of additions (•, n = 10) or in the presence of 0.2 mM mevalonate (○, n = 12). (D) Geranylgeraniol (GG) restores LTP to WT levels. Hippocampal slices from WT mice (○, n = 18) were incubated with no additions. Slices from KO mice (•, n = 12) were incubated with 0.2 mM geranylgeraniol.
Mevalonate, the product of HMG CoA reductase, is converted into cholesterol and other biologically relevant end products, including the isoprenoids farnesyl diphosphate and geranylgeranyl diphosphate (Fig. 1). To determine whether decreased synthesis of cholesterol or isoprenoids caused loss of LTP, we assessed the effects of these end products on synaptic plasticity. As shown in Fig. 6B, the addition of cholesterol to compactin-treated hippocampal slices from WT mice had no effect on LTP; however, the addition of mevalonate plus cholesterol fully restored LTP. The addition of mevalonate to hippocampal slices from KO mice restored LTP to levels observed in WT tissue (Fig. 6C). In experiments not shown, the addition of cholesterol or compactin to KO slices had no effect on LTP, and the addition of pyruvate, which is converted into acetate for use in the mitochondrial tricarboxylic acid cycle, did not restore LTP. The 15-carbon isoprenoid farnesol is a direct intermediate in the synthesis of cholesterol, whereas the 20-carbon isoprenoid geranylgeraniol is synthesized from farnesol but cannot be converted into cholesterol (25). Fig. 6D shows that the addition of geranylgeraniol alone to hippocampal slices from KO mice restored LTP to levels observed in WT slices. A quantitative analysis of the LTP data presented in Fig. 6 appears in Fig. 9, which is published as supporting information on the PNAS web site.
Discussion
A striking aspect of the current data is that LTP can be suppressed by incubation of hippocampal slices with the HMG CoA reductase inhibitor compactin for only 20 min. It is highly unlikely that cholesterol becomes depleted in this short time period. Moreover, LTP is restored by the addition of geranylgeraniol, which cannot be converted to cholesterol. In mammalian cells, geranylgeraniol is activated to geranylgeranyl diphosphate by a salvage pathway (26). The major product derived from this activated intermediate is the geranylgeranyl group attached to small GTP binding proteins such as Rab, Rac, and Rho (27), which regulate vesicular transport and cytoskeletal/membrane interactions (28). We therefore hypothesize that one or more of these proteins are crucial for LTP. This protein must require a constant supply of freshly synthesized geranylgeranyl diphosphate, either because the protein turns over rapidly or because the geranylgeranyl group is constantly removed and degraded. Statin treatment interrupts the supply of geranylgeranyl diphosphate and leads to loss of function of the prenylated protein, which in turn disrupts LTP, and presumably behavioral learning in the whole animal. In the 24-hydroxylase KO mice, the block in cholesterol excretion leads to a slight buildup in neuronal cholesterol, which is expected to decrease transcription of the HMG CoA reductase gene and accelerate degradation of the protein (4, 5). The decrease in HMG CoA reductase limits the supply of geranylgeranyl diphosphate, reduces the amount of the crucial geranylgeranylated protein, and blocks LTP. This deficit can be reversed in vitro by addition of mevalonate or geranylgeraniol to hippocampal slices.
The LTP experiments reported here measure synaptic activity between pyramidal neurons of the CA3 and CA1 regions of the hippocampus. These cells normally express 24-hydroxylase (9), and, in the intact animal, they are implicated in spatial and associative learning (15). A major mechanism by which LTP is established involves the recruitment of new α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors to the synapse by a calcium-mediated signaling pathway (18–20). The movement of intracellular AMPA receptors to the cell surface involves membrane fusion, which requires several small G proteins that are posttranslationally modified by the addition of geranylgeranyl groups (29, 30). An isoprenoid requirement for AMPA receptor trafficking would presumably be selective for LTP as many aspects of membrane fusion, including those at the presynaptic terminal and LTD, appear normal in the KO mice. This hypothesis also cannot explain all of the phenotypes observed in these animals as spatial learning is normal in mice that lack an AMPA receptor subunit required for hippocampal LTP (31).
The current data raise the question of why the brain relies on cholesterol excretion to ensure a constant supply of geranylgeraniol rather than regulating the expression of the prenyltransferase enzyme that synthesizes the isoprenoid? This reliance on de novo synthesis may reflect the need to coordinate the synthesis of isoprenoids with that of cholesterol to produce adequate amounts of both end products. Alternatively, the pathway may be a consequence of the blood–brain barrier, which prevents the removal of cholesterol from cells via their interactions with plasma lipoproteins. Learning in the intact mouse may also require both geranylgeraniol and the localized turnover of cholesterol at the synapse or elsewhere by 24-hydroxylase.
Statins are widely prescribed for the treatment of hypercholesterolemia and act by inhibiting the production of mevalonate (Fig. 1). Although it is abundantly clear that most of the therapeutic benefit derives from lowering plasma levels of cholesterol (32), it has been suggested that the associated reduction in nonsterol isoprenoid synthesis may also contribute to the beneficial outcome (33, 34). Anecdotal descriptions of amnesia and memory loss are reported in some patients on statins (35), which raises the question of whether this side effect may be caused by inhibition of cholesterol synthesis in the human brain producing an outcome similar to that observed in 24-hydroxylase KO mice. We think this scenario is unlikely for two reasons. First, the concentration of statin used to inhibit LTP in vitro is much higher than that obtained in therapy. Second, large clinical trials indicate no significant differences in cognitive abilities between placebo- and statin-treated subjects (36).
To conclude, cholesterol is typically thought of as an abundant component of membranes that plays a largely structural role in the lipid bilayer. The current studies expand this role and indicate that a constant breakdown of the molecule is required for learning. Hebb (37) postulated in 1949 that learning at the cellular level involved a “growth process or metabolic change” in neurons leading to synaptic strengthening. Geranylgeraniol synthesis appears to be one of the metabolic changes that must occur for learning.
Materials and Methods
Animals.
24-Hydroxylase KO mice harboring a null allele were of mixed strain background (C57BL/6J;129S6/SvEv) (12). LXR α−/−, β−/− double KO mice on a similar genetic background were from D. Mangelsdorf (University of Texas Southwestern Medical Center) (23).
Biochemistry.
Synaptic and microsomal membranes were isolated as described (11, 38). Immunoblotting used antibodies from Santa Cruz Biotechnology, Chemicon International (Temecula, CA), Bio-Rad, and Amersham Pharmacia.
Histology.
Mice were perfusion fixed via the heart with 0.9% saline followed by 4% paraformaldehyde in 0.1 M sodium phosphate, pH 7.4. Brains were postfixed overnight, embedded in paraffin, and coronally sectioned at 10 μm. Immunostaining used primary antibodies from BioDesign (Kennebunk, ME) and T. C. Südhof (University of Texas Southwestern Medical Center), and secondary antibodies from Molecular Probes. For EM, brains were perfusion fixed with 2% (vol/vol) glutaraldehyde and 0.1 M cacodylate, pH 7.4. The hippocampus was dehydrated, mounted in resin, and sectioned for transmission EM.
Behavioral Studies.
Contextual and cued fear conditioning tests were performed with separate groups of 3- to 4-month-old male mice (39, 40). Freezing (total immobilization) was scored at 10-s intervals in real time by an observer and by a video camera running freezeframe software (Actimetrics, Wilmette, IL). Statistical analyses used statview software (SAS Institute, Cary, NC) with Mann–Whitney U and Kolmogorov–Smirnov nonparametric tests. Threshold tests to determine relative sensitivity to electric shock used a Plexiglas chamber with a metal grid floor (Med Associates, Georgia, VT). Shock intensity was increased from 0.1 to 0.75 mA in four sessions performed at ≥1.5-h intervals over a 2-day period.
Morris water maze tests were done on 3- to 4-month-old male mice. Data were collected in four separate experiments with 40 WT and 30 KO mice by two experimentalists blinded to mouse genotypes. Latency to reach platform, distance traveled, swim speed, and time along walls were obtained. Probe trials in which the platform was removed were performed as the first trial on days 3, 5, 9, and 12 of each experiment with the same animals. Times spent swimming in target and opposite quadrants were measured together with the number of times mice crossed the former location of the platform. Statistical significance was assessed by Student's t test.
Motor learning and muscle strength were assessed by rotorod and hanging wire tests (40). Mean hang time in the latter test for WT (54.7 ± 1.5 s) and KO mice (42.1 ± 2.2 s) were not significantly different (Student's t test).
Electrophysiology.
Hippocampal slices (400 μm) were prepared from 5- to 6-week-old mice for LTP experiments, from 20- to 25-day-old mice for LTD experiments, and from 14- to 16-day-old mice for single cell recording experiments. LTD and LTP/picrotoxin experiments were done without the hippocampal CA3 region. Details regarding electrophysiological experiments appear in Supporting Materials and Methods, which is published as supporting information on the PNAS web site.
Supplementary Material
Acknowledgments
We thank R. Alpern, D. Head, R. Ram, and B. Thompson for technical assistance; S. McKnight for access to behavioral testing equipment; T. C. Südhof for antibodies; D. Mangelsdorf for LXR KO mice; and M. Brown, J. Dietschy, J. Goldstein, H. Hobbs, and J. Horton for critical reading of the manuscript. This work was supported by National Institutes of Health Grants HL20948 (to D.W.R.) and NS045711 (to K.M.H.) and the Perot Family Foundation (to D.W.R.).
Abbreviations
- 24-hydroxylase
cholesterol 24-hydroxylase
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- GABA
γ-aminobutyric acid
- HMG
3-hydroxy-3-methylglutaryl
- LTD
long-term depression
- LTP
long-term potentiation
- LXR
liver X receptor.
Footnotes
Conflict of interest statement: No conflicts declared.
References
- 1.Horton J. D., Goldstein J. L., Brown M. S. J. Clin. Invest. 2002;109:1125–1131. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pfeffer S., Aivazian D. Nat. Rev. Mol. Cell Biol. 2004;5:886–896. doi: 10.1038/nrm1500. [DOI] [PubMed] [Google Scholar]
- 3.Brown M. S., Goldstein J. L. J. Lipid Res. 1980;21:505–517. [PubMed] [Google Scholar]
- 4.Goldstein J. L., Brown M. S. Nature. 1990;343:425–430. doi: 10.1038/343425a0. [DOI] [PubMed] [Google Scholar]
- 5.Sever N., Song B.-L., Yabe D., Goldstein J. L., Brown M. S., DeBose-Boyd R. A. J. Biol. Chem. 2003;278:52479–52490. doi: 10.1074/jbc.M310053200. [DOI] [PubMed] [Google Scholar]
- 6.Endo A. J. Lipid Res. 1992;33:1569–1582. [PubMed] [Google Scholar]
- 7.Dietschy J. M., Turley S. D. J. Lipid. Res. 2004;45:1375–1397. doi: 10.1194/jlr.R400004-JLR200. [DOI] [PubMed] [Google Scholar]
- 8.Bjorkhem I., Meaney S. Arterioscler. Thromb. Vasc. Biol. 2004;24:806–815. doi: 10.1161/01.ATV.0000120374.59826.1b. [DOI] [PubMed] [Google Scholar]
- 9.Lund E. G., Guileyardo J. M., Russell D. W. Proc. Natl. Acad. Sci. USA. 1999;96:7238–7243. doi: 10.1073/pnas.96.13.7238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bjorkhem I., Andersson U., Ellis E., Alvelius G., Ellegard L., Diczfalusy U., Sjovall J., Einarsson C. J. Biol. Chem. 2001;276:37004–37010. doi: 10.1074/jbc.M103828200. [DOI] [PubMed] [Google Scholar]
- 11.Li-Hawkins J., Lund E. G., Bronson A. D., Russell D. W. J. Biol. Chem. 2000;275:16543–16549. doi: 10.1074/jbc.M001810200. [DOI] [PubMed] [Google Scholar]
- 12.Lund E. G., Xie C., Kotti T., Turley S. D., Dietschy J. M., Russell D. W. J. Biol. Chem. 2003;278:22980–22988. doi: 10.1074/jbc.M303415200. [DOI] [PubMed] [Google Scholar]
- 13.Xie C., Lund E. G., Turley S. D., Russell D. W., Dietschy J. M. J. Lipid Res. 2003;44:1780–1789. doi: 10.1194/jlr.M300164-JLR200. [DOI] [PubMed] [Google Scholar]
- 14.Morris R. J. Neurosci. Methods. 1984;11:47–60. doi: 10.1016/0165-0270(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 15.Sweatt J. D. Mechanisms of Memory. Amsterdam: Elsevier; 2003. [Google Scholar]
- 16.Stenius K., Janz R., Südhof T. C., Jahn R. J. Cell. Biol. 1995;131:1801–1809. doi: 10.1083/jcb.131.6.1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Caceres A., Payne M. R., Binder L. I., Steward O. Proc. Natl. Acad. Sci. USA. 1983;80:1738–1742. doi: 10.1073/pnas.80.6.1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Malinow R., Malenka R. C. Annu. Rev. Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- 19.Bredt D. S., Nicoll R. A. Neuron. 2003;40:361–379. doi: 10.1016/s0896-6273(03)00640-8. [DOI] [PubMed] [Google Scholar]
- 20.Malenka R. C., Bear M. F. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 21.Belelli D., Lambert J. J. Nat. Rev. Neurosci. 2005;6:565–575. doi: 10.1038/nrn1703. [DOI] [PubMed] [Google Scholar]
- 22.Kleschevnikov A. M., Belichenko P. V., Villar A. J., Epstein C. J., Malenka R. C., Mobley W. C. J. Neurosci. 2004;24:8153–8160. doi: 10.1523/JNEUROSCI.1766-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Repa J. J., Turley S. D., Lobaccaro J.-M. A., Medina J., Li L., Lustig K., Shan B., Heyman R. A., Dietschy J. M., Mangelsdorf D. J. Science. 2000;289:1524–1529. doi: 10.1126/science.289.5484.1524. [DOI] [PubMed] [Google Scholar]
- 24.Tontonoz P., Mangelsdorf D. J. Mol. Endocrinol. 2003;17:985–993. doi: 10.1210/me.2003-0061. [DOI] [PubMed] [Google Scholar]
- 25.Kellogg B. A., Poulter C. D. Curr. Opin. Chem. Biol. 1997;1:570–578. doi: 10.1016/s1367-5931(97)80054-3. [DOI] [PubMed] [Google Scholar]
- 26.Crick D. C., Andres D. A., Waechter C. J. Biochem. Biophys. Res. Commun. 1997;237:483–487. doi: 10.1006/bbrc.1997.7145. [DOI] [PubMed] [Google Scholar]
- 27.Colicelli J. Sci. STKE. 2004:re13. doi: 10.1126/stke.2502004re13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sinensky M. Biochim. Biophys. Acta. 2000;1529:203–209. doi: 10.1016/s1388-1981(00)00149-9. [DOI] [PubMed] [Google Scholar]
- 29.Zhu J. J., Qin Y., Zhao M., Aeist L. V., Malinow R. Cell. 2002;110:443–455. doi: 10.1016/s0092-8674(02)00897-8. [DOI] [PubMed] [Google Scholar]
- 30.Park M., Penick E. C., Edwards J. G., Kauer J. A., Ehlers M. D. Science. 2004;305:1972–1975. doi: 10.1126/science.1102026. [DOI] [PubMed] [Google Scholar]
- 31.Zamanillo D., Sprengel R., Hvalby O., Jensen V., Burnashev N., Rozov A., Kaiser K. M. M., Koster H. J., Borchardt T., Worley P., et al. Science. 1999;284:1805–1811. doi: 10.1126/science.284.5421.1805. [DOI] [PubMed] [Google Scholar]
- 32.Grundy S. M., Cleeman J. I., Merz C. N. B., Brewer J. H. B., Clark L. T., Hunninghake D. B., Pasternak R. C., Smith J. S. C., Stone N. J. Circulation. 2004;110:227–239. doi: 10.1161/01.CIR.0000133317.49796.0E. [DOI] [PubMed] [Google Scholar]
- 33.Liao J. K. J. Clin. Invest. 2002;110:285–288. doi: 10.1172/JCI16421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Topol E. J. N. Engl. J. Med. 2004;350:1562–1564. doi: 10.1056/NEJMe048061. [DOI] [PubMed] [Google Scholar]
- 35.Wagstaff L. R., Mitton M. W., Arvik B. M., Doraiswamy P. M. Pharmacotherapy. 2003;23:871–880. doi: 10.1592/phco.23.7.871.32720. [DOI] [PubMed] [Google Scholar]
- 36.Heart Protection Study Collaborative Group Lancet. 2002;360:7–22. doi: 10.1016/S0140-6736(11)61125-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hebb D. O. The Organization of Behavior: A Neuropsychological Theory. New York: Wiley; 1949. [Google Scholar]
- 38.Hell J. W., Jahn R. Methods Enzymol. 1998;296:116–124. doi: 10.1016/s0076-6879(98)96010-4. [DOI] [PubMed] [Google Scholar]
- 39.Garcia J. A., Zhang D., Estill S. J., Michnoff C., Rutter J., Reick M., Scott K., Diaz-Arrastia R., McKnight S. L. Science. 2000;288:2226–2230. doi: 10.1126/science.288.5474.2226. [DOI] [PubMed] [Google Scholar]
- 40.Erbel-Sieller C., Dudley C. A., Zhou Y., Wu X., Estil S. J., Han T., Diaz-Arrastia R., Brunskill E. W., Potter S. S., McKnight S. L. Proc. Natl. Acad. Sci. USA. 2004;101:13648–13653. doi: 10.1073/pnas.0405310101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}