

Abstract
Silent synapses, or excitatory synapses that lack functional α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs), are thought to be critical for regulation of neuronal circuits and synaptic plasticity. Here, we report that SynGAP, an excitatory synapse-specific RasGAP, regulates AMPAR trafficking, silent synapse number, and excitatory synaptic transmission in hippocampal and cortical cultured neurons. Overexpression of SynGAP in neurons results in a remarkable depression of AMPAR-mediated miniature excitatory postsynaptic currents, a significant reduction in synaptic AMPAR surface expression, and a decrease in the insertion of AMPARs into the plasma membrane. This change is specific for AMPARs because no change is observed in synaptic NMDA receptor expression or total synapse density. In contrast to these results, synaptic transmission is increased in neurons from SynGAP knockout mice as well as in neuronal cultures treated with SynGAP small interfering RNA. In addition, activation of the extracellular signal-regulated kinase, ERK, is significantly decreased in SynGAP-overexpressing neurons, whereas P38 mitogen-activated protein kinase (MAPK) signaling is potentiated. Furthermore, ERK activation is up-regulated in neurons from SynGAP knockout mice, whereas P38 MAPK function is depressed. Taken together, these data suggest that SynGAP plays a critical role in the regulation of neuronal MAPK signaling, AMPAR membrane trafficking, and excitatory synaptic transmission.
Keywords: trafficking, Ras, glutamate, receptor, plasticity
Excitatory synaptic transmission in the mammalian forebrain is primarily mediated by activation of both α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and NMDA receptors (1–3). AMPA receptors (AMPARs) mediate the bulk of ion flux during excitatory postsynaptic currents (EPSCs). In contrast, the NMDA receptor is restricted in its ability to pass current because of a voltage-dependent block of its ion channel by magnesium ions, which precludes it from participating in fast synaptic transmission at normal resting membrane potentials. However, when NMDA receptors are activated under depolarized conditions, its high calcium permeability triggers a cascade of signaling events responsible for inducing long-lasting changes in synaptic transmission.
Recently, studies have demonstrated that stimuli that elicit long-term potentiation (LTP), a cellular correlate to learning and memory, also result in AMPAR delivery to synapses (4, 5). These studies have led to the idea that AMPARs are highly dynamic and the number of AMPARs at synaptic sites is tightly controlled. With this concept in mind, one can envision that AMPAR concentration at synapses is a major determinant of synaptic “weight” or “strength.” Thus, molecules and signaling pathways that regulate AMPAR trafficking are likely to directly influence LTP and may be key effectors in neuronal circuit plasticity and information storage.
SynGAP, a neuronal specific RasGAP that binds to the PDZ domains of PSD-95 and SAP102 (6), is exclusively localized to excitatory synapses and is a major constituent of the PSD (7). Structurally, SynGAP consists of a PH and C2 domain, which are common motifs present in proteins regulating membrane trafficking, a myosin VI-like coiled-coil domain, a type-I PDZ ligand, and a highly conserved RasGAP domain. SynGAP mutant mice have reduced LTP and perform poorly in spatial memory tasks (8, 9). In addition, SynGAP is phosphorylated during periods of heightened synaptic activity by kinases including calcium/calmodulin-dependent protein kinase (CaMKII) (7, 10, 11). As a result, SynGAP is ideally positioned to be a potential regulator of AMPAR function and thus synaptic strength.
Here, we report that SynGAP is a potent negative regulator of excitatory synaptic transmission and surface AMPAR expression. Overexpression of SynGAP results in a dramatic loss in synaptic efficacy and surface AMPAR expression, resulting in a greater number of silent synapses. Conversely, neurons from mice lacking SynGAP have enhanced synaptic transmission, as do neurons in which SynGAP protein is acutely disrupted by using RNA interference. We also report that the loss of synaptic AMPARs in SynGAP-overexpressing cells is a consequence of deficient AMPAR membrane insertion. Finally, we show that SynGAP is a potent regulator of the mitogen-activated protein kinase (MAPK) pathway, because extracellular signal-regulated kinase (ERK) activation is suppressed in SynGAP-overexpressing neurons and enhanced in neurons with little or no SynGAP protein. Conversely, P38 MAPK is up-regulated by SynGAP overexpression and is suppressed by SynGAP down-regulation. These results suggest that the MAPK signaling is a likely mechanism through which SynGAP affects synaptic AMPARs, LTP expression, and behavior.
Results
SynGAP Gain-of-Function Reduces Synaptic Strength.
To investigate the role of SynGAP in synaptic transmission, we examined the effect of overexpressing GFP-SynGAP in neuronal cultures. SynGAP contains a PH and a C2 domain, a highly conserved RasGAP domain, and a type I PDZ ligand (Fig. 1A). At the cellular level, GFP-SynGAP exhibited an extremely punctuate distribution while occasionally exhibiting a moderate level of perinuclear signal (Fig. 1Bb1). When analyzed at higher resolution, GFP-SynGAP puncta colocalized with NMDA receptors, suggesting that the protein is targeted to excitatory synapses (Fig. 1Bb2). These data indicate that targeting of GFP-SynGAP is similar to the endogenous protein and that this construct can be used as a tool to probe SynGAP targeting and function in neurons. We also found that both myc-SynGAP and untagged SynGAP are also efficiently targeted to synapses when overexpressed in neurons (data not shown).
Fig. 1.

SynGAP overexpression results in decreased synaptic function. (A) Schematic of GFP-SynGAP fusion protein outlining various domains of interest. (B) Expression of GFP-SynGAP in cultured neurons. (Bb1) Low magnification of a cultured neuron expressing GFP-SynGAP for 16 h. (Bb2) GFP-SynGAP-expressing neurons were immunolabeled for GFP and NR1. (Bb3) Similar neurons were also labeled with antibodies detecting endogenous SynGAP (endo-SynGAP) and NR1. (Cc1) Whole-cell patch-clamp recording from an untransfected (control) cultured neuron. Recording solution allowed isolation of mEPSCs only from AMPARs. (Cc2) Whole-cell patch-clamp recording from a GFP-SynGAP-expressing neuron. This recording was obtained from a neuron adjacent to that in Cc1. (Calibration: 250 ms, 100 pA.) (D) Normalized AMPAR-mEPSCs from GFP-SynGAP- or eGFP-expressing neurons. To arrive at the normalized values, averages of frequency and amplitudes of mini events were taken from each recorded neuron. The average frequency (Hz) and amplitude (pA) from each neuron were then averaged to give a value for the entire population (untransfected = 2.59 ± 0.46 Hz, 18.8 ± 0.91 pA, n = 11; GFP-SynGAP = 0.37 ± 0.09 Hz, 9.77 ± 0.91 pA, n = 11). The average of the transfected population was normalized to the average of the untransfected (control) population to illustrate the overall effect of the expressed protein on each mEPSC parameter. Hence, values less than one represent a decrease in overall synaptic function, whereas values greater than one represent an increase. Statistical significance was determined by a Student's t test (two-tailed). n corresponds to the number of neurons in each population. This methodology is applied to all subsequent mEPSC plots. ∗, P < 0.001.
To directly assess the effect of SynGAP on AMPAR-mediated synaptic transmission, we expressed GFP-SynGAP in primary neurons and isolated AMPAR-mediated miniature EPSCs (mEPSCs). When we compared neurons expressing GFP-SynGAP with neighboring untransfected neurons, there was a striking depression in both the amplitude and frequency of mEPSCs (Fig. 1 C and D). We obtained identical results from neuronal cultures transfected with either myc-SynGAP or untagged SynGAP (data not shown). There were no significant changes in the kinetic properties of AMPAR-mediated mEPSCs including both rise and decay times, or in series resistance (Rs) or input resistance (Ri) between populations (data not shown). Importantly, there was no effect on AMPAR mEPSCs when eGFP was transfected into cultured neurons (Fig. 1D).
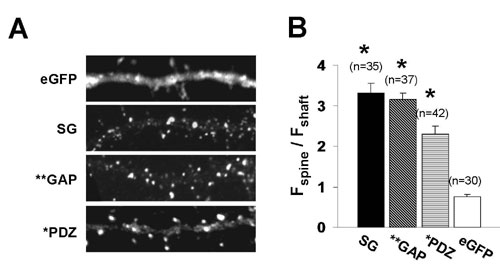
Considering that SynGAP overexpression results in a decrease in synaptic transmission, we wanted to determine the critical motifs within the protein that may be necessary for this effect. SynGAP specifically interacts with both PSD95 and SAP102 in neurons (6, 7). To investigate whether PDZ interactions are necessary for SynGAP function, we mutated the last residue of the type I PDZ ligand (GFP-SynGAP_QTRE). We confirmed that this mutation decreases the affinity for PDZ domains by coimmunoprecipitation in heterologous cells (Fig. 7, which is published as supporting information on the PNAS web site). To test the necessity of this interaction for SynGAP function, we expressed this mutation in cultured neurons and recorded AMPAR-mediated mEPSCs. Interestingly, mEPSCs were not statistically different between GFP-SynGAP_QTRE and untransfected neighbors (Fig. 2A, frequency) indicating that SynGAP-PDZ interactions are necessary for its effect on synaptic transmission in cultured neurons. The PDZ mutation in SynGAP did not significantly alter subcellular targeting of the expressed protein, suggesting that the interaction with PSD-95 is not critical for the synaptic targeting of SynGAP (Fig. 8, which is published as supporting information on the PNAS web site).
Fig. 2.

PDZ binding and GAP activity are required for SynGAP function. (Aa1) Representative recordings from either an untransfected (control) or GFP-SynGAP_QTRE-expressing neuron. Five sequential traces from each neuron are overlaid to illustrate relative mEPSC frequency and amplitudes between neurons. (Aa2) Normalized mEPSC amplitude and frequency from both GFP-SynGAP_QTRE and control populations (untransfected = 3.73 ± 0.57 Hz, 15.6 ± 1.1 pA, n = 10; GFP-SynGAP_QTRE = 4.09 ± 0.04 Hz, 15.3 ± 1.7 pA, n = 11). (Calibration: 600 ms, 20 pA.) (B) Representative recordings from either an untransfected (control) or GFP-SynGAP_AL-expressing neuron. Five sequential traces from each neuron are overlaid to illustrate relative mEPSC frequency and amplitudes. (Bb2) Normalized mEPSC amplitude and frequency from both GFP-SynGAP_AL and control populations (untransfected = 0.92 ± 0.19 Hz, 11.9 ± 1.3 pA, n = 8; GFP-SynGAP_AL = 1.05 ± 0.38 Hz, 12.9 ± 2.1 pA, n = 8). (Calibration: 600 ms, 20 pA.)
SynGAP contains a highly conserved RasGAP domain (Fig. 1A). We and others have previously shown that this portion of SynGAP exhibits GAP activity toward the small GTPase Ras in vitro (6, 7). To examine the role of this domain in neurons, we mutated a conserved region of the domain that has been shown previously to inhibit GAP function (12). This mutant (GFP-SynGAP_AL) had no significant effect on mEPSC frequency or amplitude (Fig. 2B). The lack of inhibition of AMPAR-mediated mEPSCs by this mutant indicates that the GAP activity of SynGAP is required for the regulation of synaptic transmission. Importantly, this mutation did not significantly alter the subcellular targeting of GFP-SynGAP (Fig. 8).
SynGAP Loss-of-Function Results in Increased Synaptic Strength.
To investigate the role of endogenous SynGAP in synaptic strength and AMPAR-dependent transmission, we analyzed AMPAR mEPSCs in neurons cultured from WT and SynGAP −/− (KO) mice. If SynGAP were a negative regulator of AMPAR function, then loss of protein function should lead to an enhancement of AMPAR responses. Neurons derived from KO mice exhibited a significant increase in AMPAR-mediated mEPSCs when compared with responses recorded from WT littermates (Fig. 3Aa1). This increase was manifested by an increase in frequency of events, although there was no difference in the amplitude of these responses (Fig. 3Aa2). A change in either frequency or amplitude is sufficient to modify overall synaptic strength. Therefore, these data show that loss of SynGAP function in neurons has the reverse effect of SynGAP overexpression. Next, we transfected GFP-SynGAP into neuronal cultures made from KO mice and analyzed miniature events. In KO neurons expressing GFP-SynGAP, synaptic transmission was significantly depressed relative to untransfected KO neurons (Fig. 3B). Interestingly, the mEPSC frequency was reduced to WT levels, whereas the amplitude was decreased to lower levels than that seen in WT cultures.
Fig. 3.

Decreased SynGAP expression results in enhancement of synaptic transmission. (Aa1) Representative recordings from either WT (+/+) or SynGAP-null (−/−) neurons. Five sequential traces from each neuron are overlaid to illustrate relative mEPSC frequency and amplitude. (Aa2) Normalized mEPSC amplitude and frequency from both populations (+/+ = 1.45 ± 0.25 Hz, 18.5 ± 1.0 pA, n = 17; −/− = 2.67 ± 0.45 Hz, 16.9 ± 0.79 pA, n = 17). (Calibration: 1 s, 20 pA.) ∗, P < 0.05. (Bb1) Representative recordings from either an untransfected neuron or GFP-SynGAP-expressing neurons derived from SynGAP-null (−/−) mice. Five sequential traces from each neuron are overlaid to illustrate relative mEPSC frequency and amplitudes. (Bb2) Normalized mEPSC amplitude and frequency from all cells in each population [−/− = 4.85 ± 0.78 Hz, 14.3 ± 1.9 pA, n = 5; GFP-SynGAP (−/−) = 2.34 ± 1.2 Hz, 9.45 ± 0.37 pA, n = 5]. (Calibration: 1 s, 20 pA.) ∗, P < 0.05. (Cc1) siRNAs and eGFP were cotransfected together, and recordings were performed after 72 h. These recordings were compared with neurons expressing eGFP alone for at least 72 h. In each condition, recordings illustrated are five, 5-sec traces that have been superimposed. (Cc2) si-PAN- or si-ALPHA-expressing neurons were normalized to eGFP-only-expressing neurons for both mEPSC frequency and amplitude (si-PAN: GFP = 1.50 ± 0.20 Hz, 12.5 ± 0.53 pA, n = 16; GFP + siRNA = 2.53 ± 0.27 Hz, 12.5 ± 0.62 pA, n = 17; si-ALPHA: GFP = 4.42 ± 0.99 Hz, 14.8 ± 1.5 pA, n = 13; GFP + siRNA = 8.42 ± 1.3 Hz, 15.8 ± 1.2 pA, n = 13). Black bars, mEPSC frequency; gray bars, mEPSC amplitude. (Calibration: 1 s, 20 pA.) ∗, P < 0.05; ∗∗, P < 0.01. (Dd1) siRNAs and eGFP were cotransfected together and recordings were carried out after 72 h by using neurons derived from SynGAP-null mice (−/−). These recordings were compared with neurons expressing only eGFP after 72 h. In each condition the recordings illustrated are five, 5-sec traces that have been superimposed. (Dd2) siRNA-expressing neurons were normalized to eGFP-only-expressing neurons for both mEPSC frequency and amplitude in SynGAP-null mice [(−/−) GFP = 7.59 ± 2.03 Hz, 12.2 ± 2.27 pA, n = 6; (−/−) siRNA + GFP = 7.41 ± 1.67 Hz, 22.8 ± 1.27 pA, n = 6]. (Calibration: 500 ms, 20 pA.)
The observed enhancement of AMPAR transmission in SynGAP KO mice could be due to unknown indirect changes that occur during synapse formation. Therefore, we used small interfering RNA (siRNA) to disrupt SynGAP expression after completion of synaptogenesis. We generated siRNAs targeted toward a sequence in SynGAP that is present in all known splice variants and is conserved between rat and mouse (bases 3512–3531 from AF_058790). siRNAs corresponding to this sequence (si_PAN) potently inhibited expression of transfected and endogenous SynGAP protein (Fig. 9, which is published as supporting information on the PNAS web site). siRNAs and eGFP were cotransfected in primary neurons to enable visualization of neurons with a high probability of siRNA expression. When we compared neurons expressing eGFP plus siRNAs with neurons expressing only eGFP, we observed a significant increase in the frequency of AMPAR mEPSCs while detecting no change in amplitude of events (Fig. 3C). We also obtained siRNAs that specifically knock down only SynGAP-α [si_ALPHA (13)], the isoform of SynGAP that binds to synaptic PDZ proteins. These oligonucleotides were equally effective in knocking down SynGAP protein (data not shown). Transfection of these siRNAs also significantly enhanced the frequency of AMPAR-mediated mEPSCs in cultured neurons, indicating that this isoform is dominant in regulating synaptic AMPAR function. Although the effects of both siRNAs were strikingly similar to data obtained from SynGAP null mice, we wanted to test for potential off-target effects of these siRNAs. To achieve this, we transfected siRNAs targeted against SynGAP into SynGAP KO neurons. We observed no detectable changes in mEPSCs (Fig. 3D), suggesting that these siRNAs are highly specific. Together, these data suggest SynGAP loss of function causes an increase in synaptic strength through enhanced AMPAR function and further support the hypothesis that SynGAP acts as a negative regulator of AMPAR function.
SynGAP Regulates Synaptic Strength by Altering AMPAR Trafficking.
Our data indicate that SynGAP acts as a potent negative regulator of AMPAR-mediated synaptic transmission. We hypothesized that this change in function might arise from either a loss in synaptic AMPARs resulting in fewer synapses with AMPAR-mediated responses (i.e., a change in silent synapses) or a loss in the number of excitatory synapses. To test these hypotheses, we transfected GFP-SynGAP into low-density cultured neurons and subsequently labeled neurons with an antibody generated against the N terminus of the glutamate receptor subunit 1 (GluR1) of the AMPAR (14). We compared the density of GluR1 synaptic clusters in neurons expressing GFP-SynGAP with those from neighboring untransfected neurons. The surface GluR1 signal was clearly reduced in GFP-SynGAP-transfected neurons, whereas untransfected neighbors showed robust AMPAR immunolabeling (Fig. 4A). Quantification revealed a significant decrease in the density of AMPAR clusters (Fig. 4Aa2) from neurons transfected with GFP-SynGAP. There was also a significant difference in the average cluster size from all puncta when comparing transfected with untransfected neurons, indicating that clusters remaining on the surface contained fewer AMPARs (Fig. 10, which is published as supporting information on the PNAS web site). Importantly, neurons transfected with GFP-SynGAP_QTRE showed no change relative to untransfected controls (Fig. 4Aa2). We hypothesized that the reduced number of synaptic AMPARs might be due to a deficit in the total level of GluR1 expression. To test this possibility, we labeled both untransfected and GFP-SynGAP-expressing permeabilized neurons with an antibody generated against the C terminus of GluR1. There was no difference in somatic total GluR1 signal between transfected and untransfected neurons (Fig. 4 Aa3 and Aa4), indicating that reduced surface AMPAR expression was not due to changes in global GluR1 expression levels. In addition, similar results were obtained by using an antibody raised against the C terminus of GluR2 (data not shown).
Fig. 4.

SynGAP alters AMPAR trafficking. (Aa1) GFP-SynGAP and untransfected (control) neurons were labeled with N-terminal GluR1 polyclonal antibodies (GluR1-N; JH1816). Dendrites originate from neurons on the same coverslip and in close proximity to each other. (Aa2) Quantification of GluR1 surface puncta after transfection with either GFP-SynGAP (SG) or GFP-SynGAP_QTRE (SG_QTRE). Data represent numbers of detected GluR1 clusters per unit dendrite length. ∗, P < 0.001. (Aa3) Shown are neighboring somas from either a control neuron (right soma, untransfected) or GFP-SynGAP-expressing (left soma) neuron. Both are labeled with GluR1-C antibodies. (Aa4) Quantification of total GluR1 expression. (Bb1) Cy3-conjugated GluR1-N antibodies were applied to cultures in nontrafficking conditions (10°C) to label only surface receptors. (Bb2) Neurons were first labeled with unconjugated GluR1-N antibodies before being labeled with Cy3-conjugated GluR1-N antibodies (10°C). (Bb3) Neurons were blocked with unlabeled GluR1-N antibodies (10°C) and subsequently placed at 37°C with Cy3-GluR1-N for 10 min (insertion 10 min). (Bb4) Neurons were blocked with unlabeled GluR1-N antibodies (10°C) and subsequently placed at 37°C with Cy3-GluR1-N for 30 min (insertion 30 min). Images were acquired with equal parameters and were scaled identically illustrating true relative differences in signal intensity among conditions. Asterisk, location of soma. (Cc1) Dendrite from a GFP-SynGAP-transfected neuron (arrow) adjacent to a dendrite from an untransfected neuron (arrowhead). (Cc2) Insertion 30-min signal showing newly inserted AMPARs from either an untransfected neuron (arrowhead) or a GFP-SynGAP-expressing neuron (arrow). (Cc3 and Cc4) Higher-resolution images of newly inserted AMPARs from either a GFP-SynGAP-expressing neuron or an untransfected neuron (control). Dendrites were taken from neurons shown in Cc2 in the vicinity of the arrow (GFP-SynGAP) and the arrowhead (untransfected). Asterisk, location of soma. (D) Quantification of data from C (black bars, untransfected; hatched bars, GFP-SynGAP). (Dd1) Average cluster area represents the pixel area from all puncta detected in all dendrites measured. (Dd2) Cluster density represents number of detected objects from a thresholded dendrite. The average number of objects per length of dendrite was then calculated. (Dd3) Average pixel intensity represents the average gray level from a single pixel from a single cluster of newly inserted receptors. ∗∗, P < 0.01; ∗, P < 0.05.
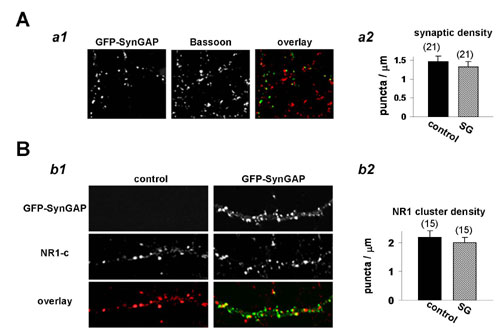
It is possible that a reduction in the frequency of mEPSCs can occur from changes in the number of excitatory synapses. To test whether SynGAP overexpression regulates synapses in general, we transfected cultured neurons with GFP-SynGAP and subsequently labeled neurons for Bassoon or NR1. Bassoon has been shown to be a component of nearly all synapses in the forebrain, rendering it an ideal marker for changes in synapse number (15). We observed no changes in total synaptic density or excitatory synapse number as measured by Bassoon and NMDA receptor immunolabeling (Fig. 11, which is published as supporting information on the PNAS web site), suggesting that acute SynGAP overexpression specifically regulates AMPARs at existing synapses.
Our data show that SynGAP overexpression results in a decrease in the number of AMPARs found at excitatory synapses and reduces synaptic strength. The steady state level of synaptic receptors is a balance of exocytosis, endocytosis, and recycling processes. To examine the exocytosis of AMPARs, we developed an assay to measure the rate of newly inserted endogenous AMPARs in cultured neurons (Fig. 4B). Receptor insertion in control untransfected neurons was robust, suggesting a high level of basal insertion of AMPARs, whereas receptor insertion in neurons expressing GFP-SynGAP was low (Fig. 4C). Quantification of these data revealed that the average cluster density, cluster size, and single-pixel intensity of newly inserted receptors were all significantly decreased in GFP-SynGAP-expressing neurons (Fig. 4D). These data strongly suggest that GFP-SynGAP regulates plasma membrane insertion of AMPARs in cultured neurons and provide a mechanism for decreased AMPAR synaptic expression.
SynGAP Negatively Regulates ERK Signaling.
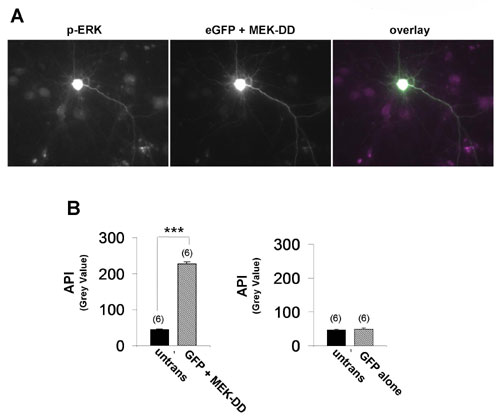
SynGAP contains a RasGAP domain, and mutation of this domain disrupts its ability to regulate AMPAR-mediated synaptic transmission (Fig. 2). To test whether SynGAP may regulate AMPAR trafficking through the MAPK pathway, we examined the regulation of ERK activity in neurons using an antibody that specifically detects phosphorylated, or activated, ERK (16). To observe the maximal range of activation of ERK in cultured neurons, it was necessary to first inhibit the high basal Ras/ERK activity due to spontaneous synaptic activity in the cultures. Cultures were treated with tetrodotoxin, 2-amino-5-phosphonovaleric acid, and 2,3-dihydroxy-6-nitro-7-sulfamoylbenzo[f]quinoxaline to inhibit synaptic transmission and glutamate receptor function for at least 8 h (“blocked” neurons). This treatment significantly suppressed the level of phospho-ERK staining in the cultures (Fig. 5Aa1). The ERK pathway was then acutely activated by applying media containing low magnesium and high glycine to facilitate synaptic NMDA receptor activation (“activated” neurons). Indeed, this treatment resulted in significant ERK activation, as shown by the robust increase in phospho-ERK antibody labeling (Fig. 5Aa2). Because this activation was completely abolished by the MAPK kinase (MEK) inhibitor U0126 (Fig. 5Aa3) and a constitutively active form of MAPK kinase (MEK-DD) resulted in a significant increase in phospho signal (Fig. 12, which is published as supporting information on the PNAS web site), we are confident that this antibody preferentially detects activated ERK1/2 kinase. To test the possibility that SynGAP suppresses NMDA-receptor-dependent ERK activation, we compared phosphorylated ERK signal in neurons expressing GFP-SynGAP to untransfected neighboring neurons in activated cultures (Fig. 5 Aa7–Aa9). GFP-SynGAP significantly reduced the level of detectable phosphorylated ERK signal, indicating that SynGAP can depress this pathway (Fig. 5B). Interestingly, GFP-SynGAP with a mutated GAP domain did not significantly reduce phosphorylated ERK, implicating GAP activity as a mechanism responsible for this effect (Fig. 5B). eGFP expression did not affect ERK activation because transfected and untransfected neurons had equally robust phospho-Erk signal (Fig. 5 Aa4–Aa6 and B). Finally, we observed no change in phospho-ERK signal with these constructs in blocked neurons (data not shown), indicating that our synaptic blocking paradigm largely inhibits ERK activity.
Fig. 5.

SynGAP suppresses ERK activity. (Aa1–Aa3) Neurons were transferred to a media containing 1 μM tetrodotoxin, 10 μM 2,3-dihydroxy-6-nitro-7-sulfamoylbenzo[f]quinoxaline, and 100 μM 2-amino-5-phosphonovaleric acid for 5 min (blocked). (Aa2) Neurons undergoing activation (5 min) were placed in artificial cerebrospinal fluid free of synaptic blockers and MgCl2 that included 100 μM glycine. (Aa3) Neurons were first pretreated with 10 μM U0126 for 30 min and then treated as in Aa2. All neurons were then labeled with antibodies that detect phosphorylated ERK. (Aa4–Aa9) Neurons were first transfected with either eGFP (Aa4–Aa6) or GFP-SynGAP (Aa7–Aa9), then activated (as in Aa2), and finally labeled with phospho-ERK antibodies. (B) Quantification of phosphorylated ERK immunofluorescence from neurons expressing eGFP, GFP-SynGAP (SG), GFP-SynGAP_AL (GAP**), SynGAP-targeted siRNA (si_PAN), or control siRNA. The intensity of transfected neurons was normalized to untransfected neighboring neurons. U0, ratio of neurons pretreated with U0126 to neurons pretreated with DMSO. Filled bars, activated (as in Aa2); open bars, blocked (as in Aa1). ∗∗∗, P < 0.001. (Cc1) WT (+/+) neurons were either blocked or activated (see Aa1 and Aa2) and then labeled with both phospho-ERK (p-ERK) and pan-ERK (total ERK) antibodies. (Cc2) Neurons from SynGAP-null mice (−/−) were blocked or activated and then labeled with both phospho-ERK (p-ERK) and pan-ERK (total ERK) antibodies. (D) Quantification of data from phosphorylated ERK immunocytochemical labeling in neurons from WT and SynGAP KO mice. Normalized values were derived from dividing p-ERK integrated intensity by total Erk integrated intensity from either WT or KO neurons (see Materials and Methods). Open bars, blocked; filled bars, activated. ∗∗∗, P < 0.01. (E) Normalized AMPAR mEPSCs from neurons expressing a dominant negative (dn) or WT (wt) form of ERK2. Black bars, mEPSC frequency; gray bars, mEPSC amplitude. ∗, P < 0.05.
Overexpression of SynGAP protein inhibits ERK activation in neurons, suggesting that neurons derived from SynGAP KO mice may exhibit enhanced ERK activation. In WT mice, blocking synaptic transmission had similar effects to those seen in rat neurons because there is little phosphorylated ERK signal detected (Fig. 5Cc1). After activating WT neurons by promoting NMDA receptor activation, a modest phospho-ERK signal was seen in dendrites, whereas a pronounced signal was observed in the soma. Interestingly, when we analyzed ERK activity in SynGAP KO mice, we observed substantial ERK activation even when synaptic transmission and glutamate receptors were inhibited (Fig. 5 Cc2 and D). However, activation of the cultures resulted in a further increase in ERK activation, demonstrating that signaling via this pathway was not saturated (Fig. 5 Cc2 and D).
To determine whether acute down-regulation of SynGAP had the same effect on ERK activity, we transfected siRNAs targeted against SynGAP into primary neurons and measured phosphorylated ERK immunofluorescence. siRNA knockdown of SynGAP protein resulted in a significant increase in phosphorylated ERK from activity-inhibited neurons, whereas control siRNAs had no effect on ERK activity (Fig. 5B). We observed no change in ERK signal from activated neurons with either control or SynGAP siRNA (data not shown).
These data suggest that ERK lies downstream of SynGAP in neurons and may mediate the effect of SynGAP on AMPAR function. To test whether ERK is a positive regulator of AMPAR function, we transfected either WT or dominant negative ERK2 into cultured neurons and recorded AMPAR-mediated mEPSCs (Fig. 5E). Dominant negative ERK2 significantly suppressed the amplitude of AMPAR-mediated mEPSCs while also depressing the frequency of events, although this effect was not significant. In contrast, WT ERK2 exhibited a trend toward enhanced frequency and amplitude of AMPAR-mediated mEPSCs indicating that ERK signaling positively regulates AMPAR function in cultured neurons. These data are consistent with the hypothesis that SynGAP mediates its effects on AMPAR function, at least in part, through ERK activation.
SynGAP Positively Regulates p38 MAPK Signaling.
ERK and P38 kinases are thought to have opposing functions on AMPAR trafficking in neurons (17). Because our data indicate that SynGAP normally suppresses ERK function, we designed experiments to test the effect of SynGAP on P38 signaling. Neurons that had been blocked overnight (no synaptic transmission) displayed minimal, although detectable, levels of phospho-specific P38 labeling (Fig. 6A). Stimulation of these cultures, which promoted synaptic NMDA receptor activation, resulted in a robust increase in phospho-P38 signal especially in the nuclear region (Fig. 6 A and B). Interestingly, blocked neurons transfected with GFP-SynGAP exhibited significantly enhanced P38 activity (Fig. 6D) reminiscent of a pattern seen with synaptic activation (Fig. 6A), although we observed no effect of SynGAP on P38 activity in activated neurons (data not shown). In contrast, GFP-SynGAP with compromised GAP function had no effect on P38 activity (Fig. 6D). We then examined P38 function in SynGAP KO mice. The phosphorylated P38 signal in blocked neurons from SynGAP KO mice was significantly lower than that measured in WT neurons (Fig. 6C), although there was no detectable difference between WT and KO mice in activated cultures. These data suggest that the presence of SynGAP protein increases P38 MAPK activity in activity-deficient cultures. We also examined P38 activity in neurons transfected with siRNAs directed against SynGAP into neurons and measured P38 activity. siRNA knockdown of SynGAP protein did not affect P38 phospho-signal in either blocked or activated neurons (Fig. 6D).
Fig. 6.

SynGAP enhances P38 signaling. (A) Neurons were either blocked or activated as above, except that the stimulation duration was increased to 10 min. Neurons were then immunolabeled with phospho-P38 (p-P38) and microtubule-associated protein 2 (MAP2) antibodies. (B) Quantification of phospho-P38 signal [average pixel intensity (API)] in neurons. ∗∗∗, P < 0.001. (C) Quantification of data (average pixel intensity) from phosphorylated P38 labeling of neurons derived from WT or SynGAP KO mice. Open bars, blocked; filled bars, activated. ∗, P < 0.01. (D) Quantification of phosphorylated P38 immunofluorescence from either blocked or activated neurons expressing eGFP, GFP-SynGAP, GFP-SynGAP_AL (GAP**), or siRNA (si_PAN) constructs. The intensity of transfected neurons was normalized to untransfected neighboring neurons. ∗∗∗, P < 0.001.
We examined phospho-P38 labeling before and after sorbitol stimulation of HEK293 or primary neurons to determine the specificity of this antibody. Osmotic shock through sorbitol stimulation is known to activate the P38 pathway in many cell types (18). This treatment resulted in a significant increase of P38 activity in both HEK293 cells and neurons (Fig. 13, which is published as supporting information on the PNAS web site), confirming the specificity of phospho-P38 immunolabeling.
Discussion
We report here that SynGAP expression level in cultured neurons has a dramatic effect on synaptic strength. When SynGAP protein is present at high levels, AMPAR-mediated synaptic transmission was significantly depressed. We further show that this change in function is a consequence of the presence of fewer AMPARs at excitatory synapses. Because we observed little change in NMDA receptor synaptic clustering, or total synaptic density, these results strongly suggest that SynGAP specifically regulates AMPARs and therefore silent synapse number. This idea is strengthened by data showing a potentiation of AMPAR-mediated synaptic transmission in neurons with low-to-absent SynGAP expression. Previously, we and others have shown that SynGAP hetero (+/−) mice have decreased LTP (8, 9). Recent studies have suggested that LTP expression in CA1 hippocampus can arise from insertion of AMPARs into previously silent excitatory synapses (5). If loss of SynGAP function results in fewer silent synapses, then the magnitude of LTP expression may be significantly reduced. Therefore, it is possible that the mechanism for reduced LTP in SynGAP mutant mice results from a decrease in silent synapse number. In support of this idea, overexpression of proteins that potentiate AMPAR transmission, such as PSD-95 (19, 20) or CaMKII (21), have also been shown to occlude LTP induction. In addition, overexpression of PSD-95 decreases the number of functionally silent excitatory synapses (19), further suggesting that proteins involved in regulating silent synapses also regulate neuronal plasticity.
Our data show that the activity of two different MAPK signaling pathways are differentially regulated by SynGAP expression levels. SynGAP inhibits ERK activity, most likely by direct inhibition of Ras signaling, and activates P38, most likely through indirect pathways. Interestingly, in nonneuronal cells, P38 and ERK have been shown to have opposing functions (22, 23), and stimuli that activate one pathway can lead to inhibition of the other (24). In neurons overexpressing SynGAP, we report that ERK activation is suppressed after NMDA receptor activation, a stimulation known to potently activate this pathway (17, 25). We also show that P38 signaling is enhanced in neurons overexpressing SynGAP. In contrast, basal ERK signaling is potentiated in SynGAP-null mice, and the P38 pathway is suppressed. Furthermore, in situations where synaptic activity and glutamate receptor function is inhibited, robust ERK activation persists in neurons derived from SynGAP KO mice. This finding suggests that SynGAP suppresses the ERK pathway under normal synaptic signaling. It has been shown previously that SynGAP mutant mice exhibit enhanced ERK activity in vivo (8), although it was unclear whether changes in this pathway were an indirect consequence of deficient neuronal plasticity or some other long-term change caused by loss of SynGAP during development. Our data show that SynGAP is likely a direct regulator of this pathway as acute gain-of-function and loss-of-function experiments result in reciprocal changes in the level of activated ERK. These data raise the question of how SynGAP-ERK-P38 signaling could alter synaptic AMPAR surface expression and vesicle trafficking. Our data show that endogenous SynGAP activity promotes a condition that favors ERK suppression and P38 activation. We also show that conditions that favor ERK activation (SynGAP siRNA or SynGAP KO) result in an enhancement of synaptic strength, whereas conditions that favor P38 activation (SynGAP overexpression) lead to a depression in synaptic strength. It has been shown previously that ERK activation is necessary for insertion of AMPARs during LTP, whereas P38 activity is necessary for removal of AMPARs after long-term depression (LTD) (17). Taken together, these data implicate SynGAP as a central regulator of synaptic signaling modules necessary for certain types of neuronal plasticity.
It is not surprising that SynGAP regulates MAPK signaling because it contains a putative RasGAP domain that is highly homologous to known RasGAPs, and Ras is thought to be upstream of ERK in neurons (17, 26). Indeed, we show that disruption of residues that are known to be crucial for RasGAP function in similar proteins (12) completely abolishes SynGAP function with respect to its effect on synaptic AMPARs and MAPK regulation. This finding suggests that SynGAP may regulate Ras in neurons. However, a recent report suggested that SynGAP may not regulate Ras, because of persistent Erk activation in H-Ras-null, SynGAP-hetero mice (8). The Ras-like superfamily of small GTPases is quite extensive, although Ras genes themselves are comprised of three closely related proteins, H-, N-, and K-Ras (27). It is possible that H-Ras is not the major postsynaptic isoform, and that K- or N-Ras may mediate signaling in this compartment. Indeed, K-Ras has been implicated in regulation of LTP expression as well as learning and memory (28). Alternatively, it is possible that SynGAP functions to catalyze inactivation of Ras related GTPases such as Rin and Rap, or even distinct GTPases including members of the Rab family. Indeed, there is precedent for promiscuity among classes of GAPs and small G proteins (29), and SynGAP has recently been shown to regulate Rap1 in vitro (13). Interestingly, calcium influx stimulates a Rap1/BRAF signaling pathway that leads to ERK activation in neurons (30), and the expression of an inhibitory Rap1 isoform in the forebrain results in suppression of ERK1/2 (31), suggesting multiple independent pathways for this type of signaling.
GTP-bound Ras is thought to be the principle activator of ERK in neurons (26), and this pathway has been shown to regulate synaptic function and LTP expression in hippocampal slices (17). In addition, Erk signaling is necessary for LTP induction as well as some forms of spatial learning (32–35). A recent report has shown that overexpression of activated Ras leads to AMPAR insertion that results in LTP occlusion in CA1 pyramidal neurons from hippocampal slice cultures (17). This report also suggested that Ras lies downstream from CaMKII activation, because neurons expressing both dominant negative Ras and constitutively active CaMKII-α do not have potentiated AMPA-mediated synaptic transmission. It is unclear how CaMKII activation and the Ras/MAP kinase pathway, which are both necessary for LTP induction and AMPAR delivery to synapses, are coupled. One possibility is that SynGAP activity is negatively regulated after NMDA receptor activation of CaMKII, resulting in synaptic potentiation and LTP induction through stimulation of the Ras/ERK pathway. Our data supports this scenario because SynGAP acts to potentiate ERK and depress P38 signaling in silent or resting cultures with little or no SynGAP protein expression (KO or siRNA knockdown), an experimental condition analogous to disruption of SynGAP activity. Mechanistically, this could occur through phosphorylation of SynGAP directly by CaMKII, resulting in a change in GAP activity. Indeed, SynGAP is phosphorylated by CaMKII in cultured neurons, although the effect on GAP activity is controversial (7, 13, 36). Alternatively, calcium influx through NMDA receptors, as a result of synaptic activation, could act to induce the activity of GEFs leading to GTPase activation by overriding SynGAP function.
During the preparation of this article, a report was published suggesting that SynGAP positively regulates AMPAR function and synaptic strength through inhibition of the Rap1/P38 pathway, while demonstrating no changes in ERK function (13). This study investigated the functional consequences of disrupting endogenous SynGAP-PDZ interactions as well as disrupting SynGAP protein expression using RNAi. These results are quite different from those reported here. In these experiments, Krapivinsky et al. (13) used peptide and RNAi reagents that should only disrupt the function or expression of the SynGAP-alpha isoform. However, we observed identical effects with regards to AMPAR function at synapses whether we disrupted all SynGAP isoforms or only SynGAP-alpha by siRNA transfection. It is unlikely that these discrepancies can be attributed to differences in culture systems that result in SynGAP coupling to distinct downstream signaling pathways because a recent report suggests that SynGAP regulates the timing of ERK activation in synapses (37). Clearly, more work is necessary to better understand the regulation of signaling events at synapses and their impact on AMPAR function.
In conclusion, we provide strong evidence that SynGAP modifies synaptic strength by altering membrane trafficking of AMPARs through GTPase regulation. Indeed, small GTPase regulation in neurons is emerging as an important process in neuronal physiology. Elucidating the interplay between small GTPase(s), their regulators, and downstream effectors should provide valuable insight into mechanisms of synaptic plasticity, potentially strengthening the idea of a molecular basis for information storage.
Materials and Methods
Molecular Biology and Cloning.
N-terminally tagged GFP and Myc-tagged SynGAP constructs were created by PCR amplification of the original SynGAPα1 clone (6). GFP-SynGAP_AL was generated by PCR mutagenesis. The codons corresponding to amino acids 311–312 of SynGAP (6) were mutated from F → A and R → L with a QuikChange reaction (Stratagene). GFP-SynGAP_QTRE was generated by PCR amplification from the original SynGAPα1 clone. Because this strategy called for mutating only the final amino acid in the coding sequence, we designed a reverse primer that switched the terminal V to E followed by a stop codon. Mouse ERK2 cDNA was generated by PCR inserted into the pRK5 mammalian expression vector downstream of an N-terminal myc epitope tag. The previously described K52R kinase-dead mutation (38) was generated by a QuikChange reaction.
Cell Culture and Neuronal Transfection.
Medium-density forebrain cultures from embryonic day 18 rat pups were prepared as reported in ref. 14 with minor alterations. Neurons (0.75 × 106) were added to a 60-mm culture dish containing poly(l-lysine)-coated coverslips. Growth media consisted of NeuroBasal (Invitrogen) supplemented with equine serum (HyClone, Logan, UT), 2% B27, 1% Glutamax, and 1% Penn/Strep. Neurons were fed twice per week with glia conditioned growth medium. Mouse cultures were prepared in an identical manner except that pups were postnatal day 1. Postnatal day 1 pups were used because SynGAP −/− mice die shortly after birth (9). Therefore, we prepared SynGAP +/− matings and genotyped newborns to identify WT from homozygous mice. Low-density hippocampal neurons were prepared as described in ref. 39. Neuronal transfections were performed with Lipofectamine 2000 (Invitrogen).
All neurons in this study were used between 14 and 21 days in vitro. The equivalent ages for postnatal mouse cultures were 12–19 days in vitro. Experiments were always performed within 24 h of transfection with the exception of siRNA experiments. For knockdown experiments, we originally purchased SynGAP siRNAs and control siRNAs (ON-TARGET siRNA; Dharmacon). In addition, we purchased siRNAs previously shown to knock down SynGAP-α expression [Ambion, Austin, TX (13)]. To transfer oligonucleotides into neurons, we double-transfected 1 μg/ml siRNA (75–80 nM) and 0.5 μg of pEGFP-C3 plasmid (Clontech-Invitrogen) with Lipofectamine 2000 and performed experiments after 3 days in vitro to ensure maximal effects.
Electrophysiology and mEPSC Analysis.
Whole-cell patch-clamp recordings were performed from forebrain cultures at the in vitro day indicated. To isolate AMPA-mediated mEPSCs, neurons were continuously perfused with artificial cerebrospinal fluid (aCSF) at a flow rate of 1 ml/min. The composition of the artificial cerebrospinal fluid was as follows: 150 mM NaCl, 3.1 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM Hepes, 0.1 mM dl-2-amino-5-phosphonovaleric acid, 0.005 mM strychnine, 0.1 mM picrotoxin, and 0.001 mM tetrodotoxin. The osmolarity of the artificial cerebrospinal fluid was adjusted to 305–310, and the pH was adjusted to 7.3–7.4. Intracellular saline consisted of 135 mM Cs-MeSO4, 10 mM CsCl, 10 mM Hepes, 5 mM EGTA, 2 mM MgCl2, 4 mM Na-ATP, and 0.1 mM Na-GTP. This saline was adjusted to 290–295 mosM, and the pH was adjusted to 7.2.
Transfected neurons were selected based on fluorescent (eGFP) signal. Once the whole-cell recording configuration was achieved, neurons were voltage-clamped, and passive properties were monitored throughout. In the event of a change in series resistance or input resistance >15% during the course of a recording, the data were excluded from the set. mEPSCs were acquired through a MultiClamp 700A amplifier (Axon Instruments), filtered at 2 kHz, and digitized at 5 kHz. Data were recorded continuously only after a period of 2 min, during which the cell was allowed to stabilize. mEPSCs were detected manually with minianalysis software (Synaptosoft, Decatur, GA) by setting the amplitude threshold to × 3 (usually 4 pA). A minimum of 100 events were collected from each neuron. In all electrophysiological experiments, a similar amount of data was acquired from both transfected and untransfected neurons on the same day. Data from each group were then averaged, and statistical significance was determined by using Student's t test (unless noted otherwise). All electrophysiological experiments were performed from at least two individual platings of neurons from three different transfections.
Immunocytochemistry.
In general, primary neurons were fixed with 4% paraformaldehyde/4% sucrose in PBS for 10 min. For efficient NMDA receptor or phosphorylated MAPK immunolabeling, an additional methanol fixation step was necessary. To label surface GluR1-containing AMPARs, we first added 5 μg/ml Cy3-conjugated JH1816 (GluR1-N) into neuronal growth media and incubated neurons at 10°C for 20 min. All antibodies were previously described or were acquired commercially: SynGAP (6), NR1 [C-terminal, mAb (40)], PSD-95 (mAb; Upstate, Charlottesville, VA), Bassoon (Stressgen Biotechnologies), GluR1-N [pAb, JH1816 (14)], GluR1-C [JH1710 (40)], microtubule-associated protein 2 (mAb; Sigma), phospho-specific Erk1/2 [Sigma (16) or Promega (Anti-Active MAPK)], pan-Erk1/2 (Cell Signaling Technology), and phospho-specific P38 MAPK (Anti-Active P38).
For additional methods, see Supporting Methods: Microscopy and Data Analysis, which is published as supporting information on the PNAS web site.
Supplementary Material
Acknowledgments
We thank Richard Johnson and Dr. Gareth Thomas for technical assistance and Drs. Jean-Claude Béïque and Radhika Reddy for helpful discussions during the preparation of this work. This work was supported National Institute of Mental Health Grant R01 MH64856 (to R.L.H.) and National Institute of Neurological Disorders and Stroke Grant F32 NS43071 (to G.R.).
Abbreviations
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- AMPAR
AMPA receptor
- CaMKII
calcium/calmodulin-dependent protein kinase II
- ERK
extracellular signal-regulated kinase
- GluR
glutamate receptor
- LTP
long-term potentiation
- MAPK
mitogen-activation protein kinase
- mEPSC
miniature excitatory postsynaptic current
- siRNA
small interfering RNA.
Conflict of interest statement: Under a licensing agreement between Upstate Group, Inc. and the Johns Hopkins University, R.L.H. is entitled to a share of royalty received by the University on sales of products described in this article. R.L.H. is a paid consultant to Upstate Group, Inc. The terms of this arrangement are being managed by the Johns Hopkins University in accordance with its conflict of interest policies.
See accompanying Profile on page 4341.
References
- 1.McBain C., Dingledine R. J. Neurophysiol. 1992;68:16–27. doi: 10.1152/jn.1992.68.1.16. [DOI] [PubMed] [Google Scholar]
- 2.Forsythe I. D., Westbrook G. L. J. Physiol. (London) 1988;396:515–533. doi: 10.1113/jphysiol.1988.sp016975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hestrin S., Nicoll R. A., Perkel D. J., Sah P. J. Physiol. (London) 1990;422:203–225. doi: 10.1113/jphysiol.1990.sp017980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Song I., Huganir R. L. Trends Neurosci. 2002;25:578–588. doi: 10.1016/s0166-2236(02)02270-1. [DOI] [PubMed] [Google Scholar]
- 5.Malinow R. Philos. Trans. R. Soc. London B. 2003;358:707–714. doi: 10.1098/rstb.2002.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim J. H., Liao D., Lau L. F., Huganir R. L. Neuron. 1998;20:683–691. doi: 10.1016/s0896-6273(00)81008-9. [DOI] [PubMed] [Google Scholar]
- 7.Chen H. J., Rojas-Soto M., Oguni A., Kennedy M. B. Neuron. 1998;20:895–904. doi: 10.1016/s0896-6273(00)80471-7. [DOI] [PubMed] [Google Scholar]
- 8.Komiyama N. H., Watabe A. M., Carlisle H. J., Porter K., Charlesworth P., Monti J., Strathdee D. J., O’Carroll C. M., Martin S. J., Morris R. G., et al. J. Neurosci. 2002;22:9721–9732. doi: 10.1523/JNEUROSCI.22-22-09721.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim J. H., Lee H. K., Takamiya K., Huganir R. L. J. Neurosci. 2003;23:1119–1124. doi: 10.1523/JNEUROSCI.23-04-01119.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pei L., Teves R. L., Wallace M. C., Gurd J. W. J. Cereb. Blood Flow Metab. 2001;21:955–963. doi: 10.1097/00004647-200108000-00008. [DOI] [PubMed] [Google Scholar]
- 11.Song B., Meng F., Yan X., Guo J., Zhang G. Neurosci. Lett. 2003;349:183–186. doi: 10.1016/s0304-3940(03)00830-9. [DOI] [PubMed] [Google Scholar]
- 12.Scheffzek K., Ahmadian M. R., Kabsch W., Wiesmuller L., Lautwein A., Schmitz F., Wittinghofer A. Science. 1997;277:333–338. doi: 10.1126/science.277.5324.333. [DOI] [PubMed] [Google Scholar]
- 13.Krapivinsky G., Medina I., Krapivinsky L., Gapon S., Clapham D. E. Neuron. 2004;43:563–574. doi: 10.1016/j.neuron.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 14.Rumbaugh G., Sia G. M., Garner C. C., Huganir R. L. J. Neurosci. 2003;23:4567–4576. doi: 10.1523/JNEUROSCI.23-11-04567.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Richter K., Langnaese K., Kreutz M. R., Olias G., Zhai R., Scheich H., Garner C. C., Gundelfinger E. D. J. Comp. Neurol. 1999;408:437–448. doi: 10.1002/(sici)1096-9861(19990607)408:3<437::aid-cne9>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 16.Goldin M., Segal M. Eur. J. Neurosci. 2003;17:2529–2539. doi: 10.1046/j.1460-9568.2003.02694.x. [DOI] [PubMed] [Google Scholar]
- 17.Zhu J. J., Qin Y., Zhao M., Van Aelst L., Malinow R. Cell. 2002;110:443–455. doi: 10.1016/s0092-8674(02)00897-8. [DOI] [PubMed] [Google Scholar]
- 18.Tibbles L. A., Woodgett J. R. Cell Mol. Life Sci. 1999;55:1230–1254. doi: 10.1007/s000180050369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stein V., House D. R., Bredt D. S., Nicoll R. A. J. Neurosci. 2003;23:5503–5506. doi: 10.1523/JNEUROSCI.23-13-05503.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ehrlich I., Malinow R. J. Neurosci. 2004;24:916–927. doi: 10.1523/JNEUROSCI.4733-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hayashi Y., Shi S. H., Esteban J. A., Piccini A., Poncer J. C., Malinow R. Science. 2000;287:2262–2267. doi: 10.1126/science.287.5461.2262. [DOI] [PubMed] [Google Scholar]
- 22.Oh C. D., Chang S. H., Yoon Y. M., Lee S. J., Lee Y. S., Kang S. S., Chun J. S. J. Biol. Chem. 2000;275:5613–5619. doi: 10.1074/jbc.275.8.5613. [DOI] [PubMed] [Google Scholar]
- 23.Heffron D. S., Mandell J. W. Mol. Cell. Neurosci. 2005;28:779–790. doi: 10.1016/j.mcn.2004.12.010. [DOI] [PubMed] [Google Scholar]
- 24.Westermarck J., Li S. P., Kallunki T., Han J., Kahari V. M. Mol. Cell. Biol. 2001;21:2373–2383. doi: 10.1128/MCB.21.7.2373-2383.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yun H. Y., Gonzalez-Zulueta M., Dawson V. L., Dawson T. M. Proc. Natl. Acad. Sci. USA. 1998;95:5773–5778. doi: 10.1073/pnas.95.10.5773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thomas G. M., Huganir R. L. Nat. Rev. Neurosci. 2004;5:173–183. doi: 10.1038/nrn1346. [DOI] [PubMed] [Google Scholar]
- 27.Cullen P. J., Lockyer P. J. Nat. Rev. Mol. Cell Biol. 2002;3:339–348. doi: 10.1038/nrm808. [DOI] [PubMed] [Google Scholar]
- 28.Ohno M., Frankland P. W., Chen A. P., Costa R. M., Silva A. J. Nat. Neurosci. 2001;4:1238–1243. doi: 10.1038/nn771. [DOI] [PubMed] [Google Scholar]
- 29.Bernards A. Biochim. Biophys. Acta. 2003;1603:47–82. doi: 10.1016/s0304-419x(02)00082-3. [DOI] [PubMed] [Google Scholar]
- 30.Grewal S. S., Horgan A. M., York R. D., Withers G. S., Banker G. A., Stork P. J. J. Biol. Chem. 2000;275:3722–3728. doi: 10.1074/jbc.275.5.3722. [DOI] [PubMed] [Google Scholar]
- 31.Morozov A., Muzzio I. A., Bourtchouladze R., Van-Strien N., Lapidus K., Yin D., Winder D. G., Adams J. P., Sweatt J. D., Kandel E. R. Neuron. 2003;39:309–325. doi: 10.1016/s0896-6273(03)00404-5. [DOI] [PubMed] [Google Scholar]
- 32.English J. D., Sweatt J. D. J. Biol. Chem. 1997;272:19103–19106. doi: 10.1074/jbc.272.31.19103. [DOI] [PubMed] [Google Scholar]
- 33.Selcher J. C., Atkins C. M., Trzaskos J. M., Paylor R., Sweatt J. D. Learn. Mem. 1999;6:478–490. doi: 10.1101/lm.6.5.478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Blum S., Moore A. N., Adams F., Dash P. K. J. Neurosci. 1999;19:3535–3544. doi: 10.1523/JNEUROSCI.19-09-03535.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Adams J. P., Sweatt J. D. Annu. Rev. Pharmacol. Toxicol. 2002;42:135–163. doi: 10.1146/annurev.pharmtox.42.082701.145401. [DOI] [PubMed] [Google Scholar]
- 36.Oh J. S., Manzerra P., Kennedy M. B. J. Biol. Chem. 2004;279:17980–17988. doi: 10.1074/jbc.M314109200. [DOI] [PubMed] [Google Scholar]
- 37.Kim M. J., Dunah A. W., Wang Y. T., Sheng M. Neuron. 2005;46:745–760. doi: 10.1016/j.neuron.2005.04.031. [DOI] [PubMed] [Google Scholar]
- 38.Robbins D. J., Zhen E., Owaki H., Vanderbilt C. A., Ebert D., Geppert T. D., Cobb M. H. J. Biol. Chem. 1993;268:5097–5106. [PubMed] [Google Scholar]
- 39.Banker G. A., Cowan W. M. Brain Res. 1977;126:397–425. doi: 10.1016/0006-8993(77)90594-7. [DOI] [PubMed] [Google Scholar]
- 40.Liao D., Zhang X., O’Brien R., Ehlers M. D., Huganir R. L. Nat. Neurosci. 1999;2:37–43. doi: 10.1038/4540. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}