

Abstract
Signaling by the mammalian target of rapamycin (mTOR) has been reported to be necessary for mechanical load-induced growth of skeletal muscle. The mechanisms involved in the mechanical activation of mTOR signaling are not known, but several studies indicate that a unique [phosphotidylinositol-3-kinase (PI3K)- and nutrient-independent] mechanism is involved. In this study, we have demonstrated that a regulatory pathway for mTOR signaling that involves phospholipase D (PLD) and the lipid second messenger phosphatidic acid (PA) plays a critical role in the mechanical activation of mTOR signaling. First, an elevation in PA concentration was sufficient for the activation of mTOR signaling. Second, the isozymes of PLD (PLD1 and PLD2) are localized to the z-band in skeletal muscle (a critical site of mechanical force transmission). Third, mechanical stimulation of skeletal muscle with intermittent passive stretch ex vivo induced PLD activation, PA accumulation, and mTOR signaling. Finally, pharmacological inhibition of PLD blocked the mechanically induced increase in PA and the activation of mTOR signaling. Combined, these results indicate that mechanical stimuli activate mTOR signaling through a PLD-dependent increase in PA. Furthermore, we showed that mTOR signaling was partially resistant to rapamycin in muscles subjected to mechanical stimulation. Because rapamycin and PA compete for binding to the FRB domain on mTOR, these results suggest that mechanical stimuli activate mTOR signaling through an enhanced binding of PA to the FRB domain on mTOR.
Keywords: mechanical load, skeletal muscle growth, exercise, stretch, atrophy
Mechanical loads play a central role in the regulation of skeletal muscle mass; however, the mechanisms involved in converting mechanical signals into the molecular events that control this process have not been defined (1, 2). Recent studies on this topic have focused on a signaling network that is regulated by a protein kinase called the mammalian target of rapamycin (mTOR) (3–5). Interest in mTOR signaling was initially motivated by studies that suggested that this protein was the master regulator of a signaling network that controls cell growth (6, 7). One of the most intensely studied proteins in the mTOR signaling network is the ribosomal S6 kinase (p70S6k), and measurements of p70S6k phosphorylation on the threonine 389 residue [p70S6k (389)] are commonly used as a readout of mTOR signaling (6, 8, 9).
Mechanical loading of skeletal muscle has been shown to induce p70S6k (389) phosphorylation through a rapamycin-sensitive mechanism, implicating a role for mTOR in this pathway (5, 10). Furthermore, the mechanical activation of p70S6k phosphorylation correlates with the extent of the concomitant growth response, and inhibiting mTOR with rapamycin prevents the mechanical-load-induced growth (3–5, 11) Taken together, these observations indicate that the activation of mTOR signaling is an essential step for converting mechanical signals into a growth response.
To date, the mechanisms involved in the mechanical activation of mTOR signaling have not been defined. However, the mechanisms for the activation of mTOR signaling in response to other forms of growth-promoting stimuli, such as nutrients and growth factors, have been fairly well established to involve a wortmannin-sensitive mechanism [i.e., phosphotidylinositol-3-kinase (PI3K)-dependent] (8, 12). Furthermore, the presence of exogenous nutrients is required for the activation of mTOR signaling by growth factors (12). Conversely, mechanical stimuli have been shown to activate mTOR signaling through a wortmannin-independent mechanism that does not require exogenous nutrients (5, 13). These findings indicate that mechanical stimuli use a unique (PI3K- and nutrient-independent) mechanism to activate mTOR signaling.
Recently, a regulatory mechanism of mTOR signaling involving phospholipase D (PLD) and the lipid second messenger phosphatidic acid (PA) was described (8, 14–16). In this study, we examined the potential role of this pathway in the mechanical activation of mTOR signaling, and our results indicate that a PLD-dependent increase in PA is an essential step in the unique mechanism through which mechanical stimuli activate mTOR signaling.
Results
PA Is Sufficient for the Activation of mTOR Signaling.
To determine whether an elevation in PA was sufficient for the induction of mTOR signaling in skeletal muscle, mouse extensor digitorum longus (EDL) muscles were incubated in an ex vivo organ culture system with a PA phosphatase inhibitor propranolol (17). As shown in Fig. 1, incubating muscles with propranolol promoted an increase in the concentration of PA ([PA]) and an increase in p70S6k (389) phosphorylation. When muscles were incubated with rapamycin, p70S6k (389) phosphorylation was abolished in both control and propranolol-treated muscles (Fig. 1B). These results indicate that an elevation in [PA] leads to an activation of mTOR signaling in skeletal muscle.
Fig. 1.

PA is sufficient for the activation of mTOR signaling. (A) Muscles prelabeled with [3H]arachidonate were incubated with 50 μM propranolol (solid bar) or the solvent vehicle (spotted bar) for 120 min, and the concentration of 3H-labeled PA ([3H-PA]) was determined. (B) Muscles were preincubated with media containing 150 nM rapamycin or the solvent vehicle for 30 min, followed by an additional 120-min incubation with 50 μM propranolol and subjected to Western blot analysis for phosphorylated p70S6k [P-p70 (389)]. Blots are representative of at least three independent experiments. The graph represents the mean values ± SEM for each group expressed as a percentage of the vehicle control group (n = 3–8 per group). ∗, significantly different from control (P ≤ 0.05).
Mechanical Stimulation Induces PLD Activity and PA Accumulation.
To begin testing the role of PLD and PA in the mechanical activation of mTOR signaling, we used an ex vivo model for mechanically stimulating the EDL muscle (5). This model utilizes intermittent passive stretch as a source of mechanical stimulation and has been shown to activate mTOR-dependent signaling to p70S6k (389) (5). Measurements at various time points (15, 30, 60, and 90 min) after the initiation of the mechanical stimulation revealed a transient increase (15 min) in PLD activity and a sustained (15–90 min) increase in [PA] (Fig. 2). It is worth noting that the time course of the mechanically induced increase in [PA] was significantly correlated (R2 = 0.78) with the time course that we reported for mechanical activation of mTOR signaling to p70S6k (389) in this model (5).
Fig. 2.

Muscles were prelabeled with [3H]arachidonate, subjected to 15–90 min of mechanical stretch or control conditions, and measurements were made of PLD activity (A) or the concentration of 3H-labeled PA ([3H-PA]) (B) at the indicated time points. Graphs represent the mean values ± SEM for each group expressed as a percentage of the time-matched control (n = 3–4 per group). ∗, significantly different from time-matched control, (P ≤ 0.05).
Neomycin Inhibits the Mechanical Activation of mTOR Signaling but Not p38 or JNK.
To determine whether PLD was required for the mechanical activation of mTOR signaling, muscles were incubated with the PLD/phospholipase C (PLC) inhibitor neomycin. In the presence of neomycin, basal p70S6k (389) phosphorylation was elevated to a similar degree with all doses, and the mechanically induced increase in p70S6k (389) phosphorylation was inhibited in a dose-dependent manner (Fig. 3A). Specifically, 10 mM neomycin completely blocked the mechanically induced increase in p70S6k (389) phosphorylation, because there was no significant difference between control and mechanically stimulated muscles at this dose. Ten millimolar neomycin, however, did not inhibit mechanically induced signaling to the stress-activated protein kinases (SAPKs) p38 and JNK2 (Fig. 3B). These results demonstrate the specificity of neomycin and indicate that mechanical stimuli activate at least two major signaling pathways (mTOR and SAPKs) that diverge upstream of the neomycin-sensitive mechanism.
Fig. 3.

Muscles were preincubated with media containing 1–10 mM neomycin or the solvent vehicle (Neomycin −) for 30 min and then subjected to 90 min of mechanical stretch (Stretch +) or control conditions (Stretch −). Samples were subjected to Western blot analysis for phosphorylated p70S6k [P-p70 (389)] (A) or phosphorylated p38 and JNK2 (B). Blots are representative of at least three independent experiments.
Inhibitors of Intracellular (i)Ca2+, PKC, and PLC Do Not Prevent the Mechanical Activation of mTOR Signaling.
Neomycin is a polycationic aminoglycoside that binds phosphatidylinositol 4,5-bisphosphate (PIP2) with a high affinity (18). It has been reported that PIP2 is a critical cofactor in the regulation of PLD and that neomycin inhibits PLD activity by binding to PIP2 (19, 20). Binding of neomycin to PIP2 has also been reported to inhibit PLC-dependent signaling events by preventing the conversion of PIP2 to inositol 1,4,5-trisphosphate (IP3) and diacylgycerol (DAG) (21, 22). IP3 and DAG are secondary messengers that can promote the release of calcium from intracellular stores (iCa2+) and activate PKC, respectively (23). To determine whether neomycin prevented the mechanical activation of mTOR signaling by blocking these PLC-dependent signaling events, pharmacological inhibitors of iCa2+, PKC, and PLC were used.
The role of iCa2+ in the mechanical activation of mTOR signaling was assessed by using the intracellular calcium chelator 1,2-bis(2-amino-5-fluorophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl) ester (BAPTA-AM). BAPTA-AM was found to block the increases in protein kinase B (PKB) and p38 phosphorylation in muscles incubated with the calcium ionophore A23187, thus verifying that BAPTA-AM inhibited iCa2+-induced signaling events. BAPTA-AM, however, did not inhibit the mechanically induced signaling to p70S6k (389) (Fig. 4A). This observation indicates that iCa2+ is not involved in mechanical activation of mTOR signaling.
Fig. 4.

Muscles were preincubated with media containing a pharmacological inhibitor for 30 min, subjected to mechanical stretch (Stretch +) or control condition (Stretch −), and stimulated with the appropriate pharmacological agonist for 90 min. The pharmacological inhibitors and agonists used, respectively, are BAPTA-AM (100 μM) and A23187 (20 μM) (A), BIM (5 μM) and TPA (1 μM) (B), and U73122 (10 μM) and lysophosphatidic acid (5 μM) (C). Appropriate solvent vehicles were used in all experiments. Samples were subjected to Western blot analysis of phosphorylated PKB (P-PKB) and p38 (P-p38) (A) or phosphorylated p70S6k [P-p70 (389)] (A–C). Blots are representative of at least three independent experiments.
The role of PKCs in the mechanical activation of mTOR signaling was assessed by using the PKC-specific inhibitor bisindolylmaleimide I (BIM). BIM was found to block the increase in p70S6k (389) phosphorylation in muscles stimulated with the PKC agonist TPA, thus verifying that BIM inhibited PKC-dependent signaling. BIM, however, did not inhibit the mechanically induced signaling to p70S6k (389) (Fig. 4B). This finding indicates that PKCs are not involved in the mechanical activation of mTOR signaling.
The role of PLC in the mechanical activation of mTOR signaling was assessed by using the PLC-specific inhibitor U73122. U73122 was found to block the increase in p70S6k (389) phosphorylation in muscles stimulated with the PLC agonist lysophosphatidic acid, thus verifying that U73122 inhibited PLC-dependent signaling. U73122, however, did not inhibit the mechanically induced signaling to p70S6k (389) (Fig. 4C). Taken together, the results from the above experiments indicate that PLC-, iCa2+-, and PKC-dependent signaling events are not involved in the mechanical activation of mTOR signaling and, therefore, suggest that the effect of neomycin was attributable to the inhibition of PLD.
1-Butanol Inhibits the Mechanical Activation of mTOR Signaling.
To further assess whether the inhibition of PLD could prevent the mechanical activation of mTOR signaling, muscles were incubated with various concentrations of 1-butanol, a primary alcohol that inhibits PLD-catalyzed PA formation, or 2-butanol, a secondary alcohol that does not inhibit PLD-catalyzed PA formation (14, 24–26). These experiments demonstrate that 1-butanol produced a dose-dependent inhibition of the mechanically induced signaling to p70S6k (389), whereas 2-butanol had no effect (Fig. 5 A and B). To further confirm the specificity of 1-butanol, muscles were incubated with 1% 1-butanol and then stimulated with insulin; the results indicate that 1% 1-butanol did not inhibit insulin-induced signaling to PKB (presented in Fig. 7, which is published as supporting information on the PNAS web site). Because it has been reported that insulin-induced signaling to PKB is a PLD-independent signaling event (27), the lack of an effect of 1-butanol in this event is in concert with its specificity on PLD-dependent signaling. Taken together, these results indicate that mechanical stimuli use a PLD-dependent increase in [PA] to activate mTOR signaling.
Fig. 5.

A PLD-dependent increase in [PA] is necessary for mechanical activating of mTOR signaling. (A and B) Muscles were preincubated for 30 min with media containing the indicated concentration of 1-butanol (A) or 2-butanol (B), followed by an additional 90 min of mechanical stretch (Stretch +) or control condition (Stretch −). Samples were subjected to Western blot analysis for phosphorylated p70S6k [P-p70 (389)]. (C) Muscles were prelabeled with [3H]arachidonate and then incubated for 30 min with media containing neomycin (10 mM), 1-butanol (1%), or the solvent vehicle, followed by an additional 90 min of mechanical stretch or control conditions, and the concentration of 3H-labeled PA ([3H-PA]) was determined. (D) Muscles were preincubated for 30 min with media containing 2–50 nM rapamycin, followed by an additional 90 min of stretch or control conditions. Samples were subjected to Western blot analysis for P-p70 (389). All blots are representative of at least three independent experiments. The graph represents the mean values ± SEM for each group expressed as a percentage of the treatment control (vehicle, neomycin, or 1-butanol), (n = 3–7 per group). ∗, significantly different from treatment control (P ≤ 0.05).
Neomycin and 1-Butanol Inhibit the Mechanically Induced Increase in [PA].
To verify that PLD was required for the mechanically induced increase in [PA], muscles were incubated with PLD inhibitors (10 mM neomycin and 1% 1-butanol) and subjected to mechanical stimulation. The results indicated that both neomycin and 1-butanol blocked the mechanically induced increase in [PA] (Fig. 5C).
Mechanical Stimulation Confers Resistance to Rapamycin.
PA has been reported to modulate mTOR signaling by binding to mTOR in its FRB domain (14). The FRB domain is also the binding site for the complex formed by the immunophilin FKBP12 and the drug rapamycin and is necessary for rapamycin to inhibit mTOR signaling (14). It has been proposed that the FKBP12–rapamycin complex and PA compete for binding to the FRB domain (14), and this hypothesis has been supported by the recent report that an elevation of PLD activity can confer resistance to rapamycin (28). To determine whether mechanical stimulation can also confer resistance to rapamycin, muscles were incubated with various concentrations of rapamycin, subjected to mechanical stimulation or control conditions, and analyzed for p70S6k (389) phosphorylation. The IC50 of rapamycin in mechanically stimulated muscles (2.32 ± 0.27 nM, R2 = 0.83) was significantly higher than the IC50 in control muscles (1.06 ± 0.24 nM, R2 = 0.95), (P < 0.01) (Fig. 5D). The higher IC50 of rapamycin in mechanically stimulated muscles demonstrates that mechanical stimulation confers resistance to rapamycin and suggests that the mechanical activation of mTOR signaling results from enhanced binding of PA to the FRB domain on mTOR.
Expression and Localization of PLD1, PLD2, and mTOR in Skeletal Muscle.
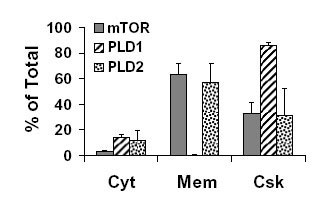
To gain insight into how mechanical stimuli might regulate PLD activity, primary antibodies generated against the two isozymes of PLD (PLD1 and PLD2) and mTOR were used to determine the subcellular localization of these proteins in skeletal muscle. Analysis of different subcellular fractions of the EDL muscle indicated that the majority of PLD1 was localized to the cytoskeletal fraction, with only a small proportion of PLD1 in the cytosolic fraction. The majority of PLD2 was detected in the membrane and cytoskeletal fractions, with a small proportion in the cytosolic fraction. Similar to PLD2, mTOR was also localized to the membrane and cytoskeletal fractions (Fig. 6A). A quantitative analysis of these experiments is presented in Fig. 8, which is published as supporting information on the PNAS web site.
Fig. 6.

Localization of PLD1, PLD2, and mTOR in skeletal muscle. (A) EDL muscles were separated into cytosol (Cyt), membrane (Mem), and cytoskeletal (Csk) fractions as described in Supporting Methods, and equal amounts of protein from each fraction were subjected to Western blot analysis for PLD1, PLD2, and mTOR. (B–G) Immunohistochemistry of cross-sections (B–D) and longitudinal sections (E–G) from the EDL muscle incubated with antibodies against PLD1 (B and E), PLD2 (C and F), or mTOR (D and G) and revealed with FITC-conjugated secondary antibodies by using an epifluorescent microscope. Longitudinal sections from the EDL muscle were also double-labeled with antibodies against α-actinin and PLD1 (H–K) or α-actinin and PLD2 (L–O). α-Actinin was revealed with TRITC-conjugated secondary antibodies (red). PLD1 and PLD2 were revealed with FITC-conjugated secondary antibodies (green). Images H and I were merged in J, which was magnified in K. Images in L and M were merged in N, which was magnified in O. Images in H–O were obtained by using a confocal microscope. All images are representative of at least three independent experiments. The boxes in J and N are the portions shown magnified in K and O.
Immunohistochemical analysis of cross-sections of the EDL muscle demonstrated that PLD1 and PLD2 were distributed throughout the interior of the muscle fibers, but PLD2 also displayed a higher intensity of staining at the sarcolemmal membrane; mTOR was localized primarily at the sarcolemmal membrane (Fig. 6 B–D). Longitudinal sections of the EDL muscle provided additional evidence that mTOR is localized to the sarcolemmal membrane, whereas PLD1 and PLD2 revealed a distinct striated pattern (Fig. 6 E–G).
To further define the striated pattern that was observed for PLD1 and PLD2, double-labeling experiments were performed with antibodies directed against the z-band protein α-actinin. Confocal images from longitudinal sections demonstrate that both PLD1 and PLD2 are localized within and immediately adjacent to the region occupied by α-actinin. Furthermore, a pattern of thin fibrous structures running perpendicular to the z-band was observed for PLD2 but not PLD1 (Fig. 6 H–O). Previous studies have demonstrated by peptide competition that the PLD2 (29) and PLD1 (30) antibodies used in this study are highly specific.
Discussion
Previous reports have demonstrated that signaling by mTOR is required for mechanical-load-induced growth of skeletal muscle (3–5, 11). To date, the mechanisms involved in the mechanical activation of mTOR signaling have not been defined, but recent studies indicate that an unique (PI3K- and nutrient-independent) pathway is involved (2, 5, 13). This finding is surprising, because PI3K is generally considered to be indispensable for the activation of mTOR signaling (12, 31, 32). In fact, we have found only one other report of a PI3K-independent activation of mTOR signaling, where Wang and Proud (33) demonstrated that the Gq protein-coupled receptor agonist phenylephrine (PE) activated mTOR signaling through a wortmannin-insensitive (i.e., PI3K-independent) pathway. Later studies by Ballou et al. (34) implicated a role for PLD in this pathway, when it was shown that 1-butanol could inhibit the activation of mTOR signaling by PE. Therefore, this study was undertaken to test the hypothesis that mechanical stimuli activate mTOR signaling through a PLD-dependent mechanism.
The results from this study provide several lines of evidence to support the hypothesis that mechanical stimuli activate mTOR signaling through a PLD-dependent increase in [PA]. First, an elevation in [PA] was sufficient for the activation of mTOR signaling (Fig. 1). Second, the isozymes of PLD (PLD1 and PLD2) are localized to the z-band in skeletal muscle (a critical site of mechanical force transmission) (Fig. 6). Third, mechanical stimulation induced PLD activation, PA accumulation, and mTOR signaling (Figs. 2–5). Fourth, pharmacological inhibition of PLD blocked the mechanically induced increase in [PA] and activation of mTOR signaling (Figs. 3 and 5). Finally, mTOR signaling was partially resistant to rapamycin in mechanically stimulated muscles (Fig. 5), suggesting that the mechanical activation of mTOR signaling resulted from enhanced binding of PA to the FRB domain on mTOR.
Although our results indicate that PLD is the enzyme responsible for the mechanically induced increase in [PA], we cannot rule out the potential contribution of additional PA regulatory enzymes. Such enzymes might include the PA phosphatases (PAP), which dephosphorylate PA to DAG, and the DAG kinases (DAGK), which generate PA through the phosphorylation of DAG (35, 36). In fact, previous studies have shown that PLD-derived PA can be rapidly converted to DAG by PAP and that DAG can, in turn, be transformed back to PA by DAGK (37). Such a mechanism might explain why mechanical stimulation produced a transient increase in PLD activity, whereas the elevation in [PA] was sustained. Additional studies will be needed to fully define the mechanisms by which PLD contributes to the mechanically induced increase in [PA].
Our finding that mechanical stimulation promoted an increase in PLD activity raises questions about the mechanism(s) involved in this process. Previous studies have shown that PLD activity can be regulated by various protein kinases such as PKC, protein-tyrosine kinases, and the MAP kinase family as well as the small G proteins from the ARF, Rho, and Ras families (38, 39). Furthermore, the activity of PLD has been shown to be regulated through interactions with various cytoskeletal proteins such as tubulin, actin, and α-actinin (40–42). For example, Park et al. (41) demonstrated that PLD can bind to α-actinin and that this interaction inhibits the activity of PLD. This observation is of particular interest, because our results demonstrated that both isozymes of PLD are colocalized with α-actinin in skeletal muscle. Because α-actinin is a vital component of the z-band in skeletal muscle, and the z-band is a focal point for mechanical force transmission (43), it is tempting to speculate that mechanical stimuli induce a physical dissociation of PLD from α-actinin and relieve PLD from the inhibitory effect of the interaction. Additional interest in the cytoskeletal network comes from the observation that disruption of the actin cytoskeleton with cytochalasin D can prevent the mechanical activation of signaling to p70S6k in skeletal muscle cells in vitro (44). Clearly, the mechanisms responsible for the mechanical activation of PLD activity remain to be more fully defined, and the role of the cytoskeletal network in this process will be an area worthy of further investigations.
In summary, the results of this study indicate that mechanical stimuli activate mTOR signaling through a PLD-dependent increase in [PA]. Because the activation of mTOR signaling is required for mechanically induced growth of skeletal muscle, these findings contribute significantly to our understanding of how mechanical signals are transduced into the molecular events that regulate skeletal muscle growth and remodeling.
Materials and Methods
Materials.
Details regarding the specific materials used in this study are described in Supporting Methods, which is published as supporting information on the PNAS website.
Animal Care and Use.
All experimental procedures were approved by the University of California at San Diego Animal Care and Use Committee. Male C57BL6 mice (The Jackson Laboratories and Harlan Laboratories), 8–14 weeks of age, were randomly assigned to different experimental groups. All animals were allowed free access to food and water.
Organ Culture and Mechanical Stimulation.
Mice were anesthetized with sodium pentobarbital (150 mg/kg of body weight), and the EDL muscle of the hind limb was placed in the ex vivo organ culture system at optimal length (Lo) as described in ref. 13. Details regarding the protocol for mechanical stimulation have been described in ref. 5. Briefly, 15% intermittent passive stretch was used as a source of mechanical stimulation; in addition, muscles were maintained at Lo as a control condition.
Pharmacological Inhibitors.
Muscles were preincubated with pharmacological inhibitors or the solvent vehicle for 30 min before stimulation with pharmacological agonists or mechanical stretch. All pharmacological inhibitors were present throughout the entire stimulation period. Stock solutions of the inhibitors BAPTA-AM, BIM, U73122, and rapamycin were dissolved in DMSO, neomycin was dissolved in distilled (d)H2O, and 1-butanol and 2-butanol were added directly to the culture media.
Pharmacological Agonists.
Muscles were incubated with the pharmacological agonists or solvent vehicles for 90–120 min as indicated in the figure legends. Stock solutions of the pharmacological agonists TPA and A23187 were dissolved in DMSO and lysophosphatidic acid was dissolved in PBS with 0.1% (wt/vol) BSA. Insulin and propranolol were dissolved in dH2O.
Western Blot Analysis and Sample Preparation.
Unless otherwise noted, muscles were removed from the organ culture system, immediately frozen in liquid nitrogen, homogenized, and subjected to Western blot analysis as described in ref. 13. Details regarding the procedure for separating the cytosolic, membrane, and cytoskeletal muscle fractions are provided in Supporting Methods.
Immunohistochemistry.
EDL muscles were mounted at resting length in optimal cutting temperature compound and frozen in isopentane chilled with liquid nitrogen. Longitudinal and cross-sections (10 μm) were generated with a cryostat and immediately fixed in −20°C acetone or −20°C methanol. Fixed sections were incubated with PLD1, PLD2, mTOR, or α-actinin primary antibodies. Primary antibodies were detected with FITC- or tetramethylrhodamine isothiocyanate (TRITC)-conjugated secondary antibodies and visualized with an epifluorescent or confocal microscope. Additional details regarding these procedures are provided in Supporting Methods.
Analysis of PLD Activity and [PA].
PLD activity was measured with the transphosphatidylation assay described by Facchinetti et al. (24). Briefly, muscles were prelabeled in the organ culture system with media containing [3H]arachidonate (1 μCi/ml) (1 Ci = 37 GBq) for 2 h and then subjected to experimental treatments. PLD activity was measured during the final 15 min before the indicated treatment time by washing the muscle with fresh media for 5 min and then incubating with media containing 0.5% 1-butanol for 15 min. The same procedure was used for measurements of 3H-labeled PA, except 1-butanol was not added to the culture media. Samples were homogenized in chloroform–methanol 2:1 (vol/vol) with a polytron, and lipids were extracted according to Folch et al. (45). Phosphotidylbutanol (PtdBut) and PA standards (10 μg) were added to the extracted lipids, and aliquots were used for the measurement of radioactivity in the total lipids or spotted on LK5D silica gel plates for separation of PtdBut and PA by TLC. The plates were developed with a solvent system consisting of ethyl acetate–isooctane–acetic acid–water 13:2:3:10 (vol/vol). This system provided efficient separation of PtdBut (Rf = 0.41) and PA (Rf = 0.14), which were visualized by iodine staining. Iodine-stained spots containing the 3H-labeled PtdBut and PA were scraped off the TLC plate, and the radioactivity was measured by liquid scintillation spectrometry. Final calculations for PLD activity and [PA] were made by dividing the amount of radioactivity in the Ptdbut or PA spot by the amount of radioactivity in the total lipid extract.
Statistical Analysis.
All values are expressed as means ± SEM. Statistical significance was determined by using ANOVA, followed by Student–Newman–Keuls post hoc analysis. Differences between groups were considered significant if P ≤ 0.05.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Grants R01HL64382, R01HL80518, and F32AR052240.
Abbreviations
- BAPTA-AM
1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl) ester
- DAG
diacylglycerol
- EDL
extensor digitorum longus
- iCa2+
intracellular Ca2+
- mTOR
mammalian target of rapamycin
- PA
phosphatidic acid
- [PA]
PA concentration
- PLC
phospholipase C
- PLD
phospholipase D
- PI3K
phosphotidylinositol-3-kinase
- PKB
protein kinase B
- p70S6k
ribosomal S6 kinase.
Footnotes
Conflict of interest statement: No conflicts declared.
References
- 1.Tidball J. G. J. Appl. Physiol. 2005;98:1900–1908. doi: 10.1152/japplphysiol.01178.2004. [DOI] [PubMed] [Google Scholar]
- 2.Hornberger T. A., Esser K. A. Proc. Nutr. Soc; 2004. pp. 331–335. [DOI] [PubMed] [Google Scholar]
- 3.Bodine S. C., Stitt T. N., Gonzalez M., Kline W. O., Stover G. L., Bauerlein R., Zlotchenko E., Scrimgeour A., Lawrence J. C., Glass D. J., Yancopoulos G. D. Nat. Cell Biol. 2001;3:1014–1019. doi: 10.1038/ncb1101-1014. [DOI] [PubMed] [Google Scholar]
- 4.Kubica N., Bolster D. R., Farrell P. A., Kimball S. R., Jefferson L. S. J. Biol. Chem. 2005;280:7570–7580. doi: 10.1074/jbc.M413732200. [DOI] [PubMed] [Google Scholar]
- 5.Hornberger T. A., Stuppard R., Conley K. E., Fedele M. J., Fiorotto M. L., Chin E. R., Esser K. A. Biochem. J. 2004;380:795–804. doi: 10.1042/BJ20040274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jacinto E., Hall M. N. Nat. Rev. Mol. Cell Biol. 2003;4:117–126. doi: 10.1038/nrm1018. [DOI] [PubMed] [Google Scholar]
- 7.Thomas G., Hall M. N. Curr. Opin. Cell Biol. 1997;9:782–787. doi: 10.1016/s0955-0674(97)80078-6. [DOI] [PubMed] [Google Scholar]
- 8.Chen J. Curr. Top Microbiol. Immunol. 2004;279:245–257. doi: 10.1007/978-3-642-18930-2_14. [DOI] [PubMed] [Google Scholar]
- 9.Sarbassov dos D., Ali S. M., Sabatini D. M. Curr. Opin. Cell Biol. 2005;17:596–603. doi: 10.1016/j.ceb.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 10.Baar K., Esser K. Am. J. Physiol. 1999;276:C120–C127. doi: 10.1152/ajpcell.1999.276.1.C120. [DOI] [PubMed] [Google Scholar]
- 11.Hornberger T. A., McLoughlin T. J., Leszczynski J. K., Armstrong D. D., Jameson R. R., Bowen P. E., Hwang E. S., Hou H., Moustafa M. E., Carlson B. A., et al. J. Nutr. 2003;133:3091–3097. doi: 10.1093/jn/133.10.3091. [DOI] [PubMed] [Google Scholar]
- 12.Harris T. E., Lawrence J. C., Jr. Sci. STKE. 2003;2003:re15. doi: 10.1126/stke.2122003re15. [DOI] [PubMed] [Google Scholar]
- 13.Hornberger T. A., Chien S. J. Cell. Biochem. 2005 Nov 28; doi: 10.1002/jcb.20671. [DOI] [PubMed] [Google Scholar]
- 14.Fang Y., Vilella-Bach M., Bachmann R., Flanigan A., Chen J. Science. 2001;294:1942–1945. doi: 10.1126/science.1066015. [DOI] [PubMed] [Google Scholar]
- 15.Kam Y., Exton J. H. FASEB J. 2004;18:311–319. doi: 10.1096/fj.03-0731com. [DOI] [PubMed] [Google Scholar]
- 16.Fang Y., Park I. H., Wu A. L., Du G., Huang P., Frohman M. A., Walker S. J., Brown H. A., Chen J. Curr. Biol. 2003;13:2037–2044. doi: 10.1016/j.cub.2003.11.021. [DOI] [PubMed] [Google Scholar]
- 17.Murthy K. S., Makhlouf G. M. Mol. Pharmacol. 1995;48:293–304. [PubMed] [Google Scholar]
- 18.Gabev E., Kasianowicz J., Abbott T., McLaughlin S. Biochim. Biophys. Acta. 1989;979:105–112. doi: 10.1016/0005-2736(89)90529-4. [DOI] [PubMed] [Google Scholar]
- 19.Kurz T., Kemken D., Mier K., Weber I., Richardt G. J. Mol. Cell. Cardiol. 2004;36:225–232. doi: 10.1016/j.yjmcc.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 20.Liscovitch M., Chalifa V., Pertile P., Chen C. S., Cantley L. C. J. Biol. Chem. 1994;269:21403–21406. [PubMed] [Google Scholar]
- 21.Facchinetti M. M., Boland R., de Boland A. R. Mol. Cell. Endocrinol. 1998;136:131–138. doi: 10.1016/s0303-7207(97)00221-9. [DOI] [PubMed] [Google Scholar]
- 22.Neri L. M., Borgatti P., Capitani S., Martelli A. M. J. Biol. Chem. 1998;273:29738–29744. doi: 10.1074/jbc.273.45.29738. [DOI] [PubMed] [Google Scholar]
- 23.Becker K. P., Hannun Y. A. Cell. Mol. Life Sci. 2005;62:1448–1461. doi: 10.1007/s00018-005-4531-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Facchinetti M. M., Boland R., de Boland A. R. J. Lipid Res. 1998;39:197–204. [PubMed] [Google Scholar]
- 25.Wakelam M. J., Hodgkin M., Martin A. Methods Mol. Biol. 1995;41:271–278. doi: 10.1385/0-89603-298-1:271. [DOI] [PubMed] [Google Scholar]
- 26.Shen Y., Xu L., Foster D. A. Mol. Cell. Biol. 2001;21:595–602. doi: 10.1128/MCB.21.2.595-602.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Farese R. V. Exp. Biol. Med. (Maywood) 2001;226:283–295. doi: 10.1177/153537020122600404. [DOI] [PubMed] [Google Scholar]
- 28.Chen Y., Zheng Y., Foster D. A. Oncogene. 2003;22:3937–3942. doi: 10.1038/sj.onc.1206565. [DOI] [PubMed] [Google Scholar]
- 29.Freyberg Z., Bourgoin S., Shields D. Mol. Biol. Cell. 2002;13:3930–3942. doi: 10.1091/mbc.02-04-0059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Freyberg Z., Sweeney D., Siddhanta A., Bourgoin S., Frohman M., Shields D. Mol. Biol. Cell. 2001;12:943–955. doi: 10.1091/mbc.12.4.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen J., Fang Y. Biochem. Pharmacol. 2002;64:1071–1077. doi: 10.1016/s0006-2952(02)01263-7. [DOI] [PubMed] [Google Scholar]
- 32.Gingras A. C., Raught B., Sonenberg N. Genes Dev. 2001;15:807–826. doi: 10.1101/gad.887201. [DOI] [PubMed] [Google Scholar]
- 33.Wang L., Proud C. G. Circ. Res. 2002;91:821–829. doi: 10.1161/01.res.0000041029.97988.e9. [DOI] [PubMed] [Google Scholar]
- 34.Ballou L. M., Jiang Y. P., Du G., Frohman M. A., Lin R. Z. FEBS Lett. 2003;550:51–56. doi: 10.1016/s0014-5793(03)00816-0. [DOI] [PubMed] [Google Scholar]
- 35.Luo B., Regier D. S., Prescott S. M., Topham M. K. Cell. Signaling. 2004;16:983–989. doi: 10.1016/j.cellsig.2004.03.016. [DOI] [PubMed] [Google Scholar]
- 36.Brindley D. N., English D., Pilquil C., Buri K., Ling Z. C. Biochim. Biophys. Acta. 2002;1582:33–44. doi: 10.1016/s1388-1981(02)00135-x. [DOI] [PubMed] [Google Scholar]
- 37.Maestre N., Bezombes C., Plo I., Levade T., Lavelle F., Laurent G., Jaffrezou J. P. J. Exp. Ther. Oncol. 2003;3:36–46. doi: 10.1046/j.1359-4117.2003.01065.x. [DOI] [PubMed] [Google Scholar]
- 38.Foster D. A., Xu L. Mol. Cancer Res. 2003;1:789–800. [PubMed] [Google Scholar]
- 39.Exton J. H. FEBS Lett. 2002;531:58–61. doi: 10.1016/s0014-5793(02)03405-1. [DOI] [PubMed] [Google Scholar]
- 40.Chae Y. C., Lee S., Lee H. Y., Heo K., Kim J. H., Suh P. G., Ryu S. H. J. Biol. Chem. 2005;280:3723–3730. doi: 10.1074/jbc.M406987200. [DOI] [PubMed] [Google Scholar]
- 41.Park J. B., Kim J. H., Kim Y., Ha S. H., Yoo J. S., Du G., Frohman M. A., Suh P. G., Ryu S. H. J. Biol. Chem. 2000;275:21295–21301. doi: 10.1074/jbc.M002463200. [DOI] [PubMed] [Google Scholar]
- 42.Lee S., Park J. B., Kim J. H., Kim Y., Shin K. J., Lee J. S., Ha S. H., Suh P. G., Ryu S. H. J. Biol. Chem. 2001;276:28252–28260. doi: 10.1074/jbc.M008521200. [DOI] [PubMed] [Google Scholar]
- 43.Patel T. J., Lieber R. L. Exerc. Sport Sci. Rev. 1997;25:321–363. [PubMed] [Google Scholar]
- 44.Hornberger T. A., Armstrong D. D., Koh T. J., Burkholder T. J., Esser K. A. Am. J. Physiol. 2005;288:C185–C194. doi: 10.1152/ajpcell.00207.2004. [DOI] [PubMed] [Google Scholar]
- 45.Folch J., Lees M., Sloane Stanley G. H. J. Biol. Chem. 1957;226:497–509. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}