

Abstract
Following replication arrest, multiple cellular responses are triggered to maintain genomic integrity. In fission yeast, the RecQ helicase, Rqh1, plays a critical role in this process. This is demonstrated in Δrqh1 cells that, following treatment with hydroxyurea (HU), undergo an aberrant mitosis leading to cell death. Previous data suggest that Rqh1 functions with homologous recombination (HR) in recovery from replication arrest. We have found that loss of the HR genes rhp55+ or rhp57+, but not rhp51+ or rhp54+, suppresses the HU sensitivity of Δrqh1 cells. Much of this suppression requires Rhp51 and Rhp54. In addition, this suppression is partially dependent on swi5+. In budding yeast, overexpressing Rad51 (the Rhp51 homolog) minimized the need for Rad55/57 (Rhp55/57) in nucleoprotein filament formation. We overexpressed Rhp51 in Schizosaccharomyces pombe and found that it greatly reduced the requirement for Rhp55/57 in recovery from DNA damage. However, overexpressing Rhp51 did not change the Δrhp55 suppression of the HU sensitivity of Δrqh1, supporting an Rhp55/57 function during HR independent of nucleoprotein filament formation. These results are consistent with Rqh1 playing a role late in HR following replication arrest and provide evidence for a postsynaptic function for Rhp55/57.
REPLICATION arrest is a common occurrence even in unperturbed cells. Studies in Escherichia coli have shown that spontaneous replication arrest occurs in 18% of cells and could be as high as 50% (Cox et al. 2000; Maisnier-Patin et al. 2001; McGlynn and Lloyd 2002). We can assume that this problem is even greater in eukaryotic cells where the genomes are generally much larger and multiple origins of replication are used. When the replication machinery encounters DNA damage, the S-phase checkpoint is induced, allowing time for the cell to repair or bypass the DNA damage prior to entry into mitosis (Diffley et al. 2000; Michel 2000; Carr 2002; Nyberg et al. 2002). What has become increasingly evident is the need for homologous recombination (HR) in the recovery and restart of replication following arrest (Michel et al. 2001; Saintigny et al. 2001; Lundin et al. 2002). It remains unclear how HR functions in replication restart but several models have been proposed (Cox et al. 2000; McGlynn and Lloyd 2002; Helleday 2003). One model favors branch migration of the stalled fork, leading to the formation of a pseudo-Holliday junction (HJ) known as a chicken foot structure (Cox et al. 2000; McGlynn and Lloyd 2002; Helleday 2003; Heyer et al. 2003). Alternatively HR can act in the process of template switching (Liberi et al. 2000). The structure recognized as a substrate for HR following replication arrest has not been definitively established although it has been shown that double-strand breaks (DSBs) form during replication arrest (Michel et al. 1997; Rogakou et al. 1999). However, in at least one study, replication restart by HR was shown to occur in the absence of detectable DSBs (Lundin et al. 2002).
In Saccharomyces cerevisiae, HR proteins were initially identified as conferring resistance to ionizing radiation (IR), although increasingly their main function appears to be in maintaining genomic integrity during replication (Michel 2000; Michel et al. 2001; Helleday 2003). Following the formation of a DSB, a complex of three proteins, Mre11p, Rad50p, and Xrs2p (MRX complex), is thought to be recruited to the site (Nelms et al. 1998). The MRX complex participates in the production of a 3′ single-stranded end particularly during meiosis (Bressan et al. 1999; Paques and Haber 1999; D'Amours and Jackson 2002; Symington 2002; Helleday 2003; Trujillo et al. 2003). The single-strand binding protein, RPA, rapidly coats this 3′ single strand. Rad52 aids in the loading of Rad51 onto the 3′ single-strand end. Rad51 binds DNA weakly so the obligate heterodimer, Rad55/Rad57, acts to stabilize its binding, leading to Rad51 polymerization along the 3′ tail, forming a nucleoprotein filament (Johnson and Symington 1995; Sung 1997; Paques and Haber 1999; Fortin and Symington 2002; Helleday 2003). Next, aided by Rad54, the Rad51 filament invades its homologous sequence either on its sister chromatid or, in diploid cells, on its homologous chromosome, forming a heteroduplex (Van Komen et al. 2000, 2002; Solinger et al. 2001). This creates a joint molecule that either can be resolved by HJ resolvase or is simply displaced by collapse of the D-loop, restoring the original duplex (Kuzminov 1993; Sharples et al. 1999; Haber and Heyer 2001).
Rad55 and Rad57 are referred to as Rad51 paralogs because of their close sequence homology to Rad51 (Symington 2002). rad55 and rad57 mutants are only mildly sensitive to IR at 30° but are as sensitive as rad51 mutants at low temperatures (23°) (Lovett and Mortimer 1987; Johnson and Symington 1995). This, along with suppression of rad55 and rad57 by overexpression of Rad51, was the original basis for predicting their role as mediators (Hays et al. 1995; Johnson and Symington 1995). Cold-enhanced sensitivity is also seen in Schizosaccharomyces pombe Δrhp55 and Δrhp57 mutants (Tsutsui et al. 2000). A recent article showed that a rad51 mutant with increased DNA binding could also suppress a rad55 mutant (Fortin and Symington 2002). These results further support the role of rad55/57 as mediators of Rad51 function. Recent data have implicated Rad51 paralogs in post-strand invasion events. In two reports on human Rad51 paralogs, Rad51b protein was shown to preferentially bind HJ and Rad51c and Xrcc3 were shown to be necessary for HJ resolution (Yokoyama et al. 2003; Liu et al. 2004). This role for Rad51c has been shown only in cell extracts and was not demonstrated in vivo.
Homologs of all of the S. cerevisiae HR proteins have been identified in S. pombe (Muris et al. 1993, 1997; Khasanov et al. 1999; Wilson et al. 1999; Fukushima et al. 2000; Tsutsui et al. 2000; Ueno et al. 2003). While it is generally assumed that the S. pombe homologs will carry out functions similar to those of their S. cerevisiae counterparts, significant differences have been reported between HR in these two organisms. For example, while rad52 mutants are the most sensitive of the HR mutants to DSBs in S. cerevisiae, the equivalent mutation in S. pombe, rad22, has only a slight sensitivity to IR (Muris et al. 1997; Suto et al. 1999; van den Bosch et al. 2001). This discrepancy may be due to the existence of a second Rad52 homolog in S. pombe known as Rti1/Rad22B (Suto et al. 1999; van den Bosch et al. 2001), the function of which becomes important in Δrad22 mutants. In S. cerevisiae, mutations in members of the RAD52 epistasis group (RAD51, RAD52, RAD54, RAD55, and RAD57) confer only slight sensitivity to ultraviolet (UV) radiation. By contrast, mutants of the S. pombe homologs (Δrhp51, Δrad22, Δrhp54, Δrhp55, and Δrhp57, respectively) are sensitive to UV radiation as well as to other DNA-damaging agents and hydroxyurea (HU). This suggests that in S. pombe various types of DNA damage may be converted into substrates recognized by HR proteins, such as nicks, gaps, or DSBs (Caspari et al. 2002; Laursen et al. 2003).
Rqh1, the S. pombe RecQ homolog, has been linked to homologous recombination in several studies. Evidence indicates that HR and Rqh1 respond to DSBs and replication arrest through a common process (Murray et al. 1997; Caspari et al. 2002). rqh1+ mutants are sensitive to DNA damage and replication arrest (Murray et al. 1997; Stewart et al. 1997; Davey et al. 1998). While showing a normal or near normal checkpoint response during S-phase arrest, upon release Δrqh1 cells do not properly complete mitosis (Stewart et al. 1997; Davey et al. 1998; Marchetti et al. 2002). The mitotic defect is observed as an accumulation of cells with “cut” chromosomes or with an uneven distribution of nuclear material between daughter cells. Also, Δrqh1 cells show dramatically increased rates of HR following replication arrest or DNA damage (Stewart et al. 1997; Doe et al. 2000). When the E. coli Holliday junction resolvase, RusA, was expressed in Δrqh1 cells, their UV and HU sensitivities were partially suppressed, suggesting that in the absence of Rqh1, stalled replication forks accumulate unresolved Holliday junctions (Doe et al. 2000).
Mutants of the S. cerevisiae RecQ homolog, SGS1, show synthetic lethality with mus81/mms4, which forms a complex that cleaves a 3′ flap structure that mimics a stalled replication fork (Bastin-Shanower et al. 2003). Two studies reported that the synthetic lethality between Δmus81 and Δrqh1 is conserved in S. pombe, but two different interpretations of the data were offered for the activity of Mus81/Mms4 (Eme1): it acts in the resolution of regressed forks (HJ) or it acts on stalled replication forks (Boddy et al. 2001; Doe et al. 2002). It is conceivable that both interpretations are correct. A recent article reported that loss of HR suppressed the synthetic lethality between mus81 and sgs1 (Fabre et al. 2002), suggesting that the critical functions of these proteins are downstream of HR. Mutants defective for the yeast RecQ helicases also show synthetic interaction with Δsrs2 (srs2+ encodes another DNA helicase), which is also suppressed by loss of HR genes (Gangloff et al. 2000; Fabre et al. 2002; Maftahi et al. 2002; Doe and Whitby 2004). Together these findings have led to the speculation that yeast RecQ helicases act to prevent the deleterious effects of HR following replication arrest, either by suppressing the formation of DSB (or other structures that HR acts upon) or by participating in a process that leads to the resolution of recombination intermediates. Two recent articles have supported a role for RecQ helicases in restricting crossovers at DSBs during HR by acting on joint molecules, further supporting the role of this helicase family in recombination (Ira et al. 2003; Wu and Hickson 2003).
Here we report on studies that support a role for Rqh1 downstream of joint molecule formation during HR. We made a series of double mutants between Δrqh1 and deletions of HR genes. We found that loss of rhp55+/57+ dramatically suppressed the HU sensitivity of Δrqh1 mutants. This suppression was largely dependent on Rhp51 and Rhp54, suggesting that the deleterious function of Rhp55/57 was acting downstream of joint molecule formation. This was further supported by our results showing that complementing the defect of Δrhp55 in the Rhp51 nucleation step did not affect the suppression of the HU sensitivity in the Δrqh1 Δrhp55 double mutant. Loss of rhp55+ decreased the number of aberrant chromosomes (showing torn nuclear material) seen in Δrqh1 cells following replication arrest, supporting the idea that these events are the result of unresolved recombination intermediates. These data imply that Rqh1 plays a late role in HR and that Rhp55/57 has a postsynaptic function.
MATERIALS AND METHODS
Media and construction of plasmids and mutant strains:
Unless indicated, cells were grown in YEA media (0.5% yeast extract, 3% glucose, and 150 mg/liter adenine). Minimal medium was EMM (QBiogene) with the appropriate supplements. G418 selection was carried out with 150 mg/liter of Geneticin (GIBCO, Grand Island, NY) in YEA. Strains containing multiple mutations were generated from crosses. Double mutants were generally isolated from tetrads and occasionally from random spores. In either case, strains containing multiple mutations were tested individually by PCR analysis and, when necessary, sequenced. Table 1 lists the strains used in this study. The Rhp51 overexpression plasmid was constructed by PCR amplification of rhp51+ from genomic DNA using primers rhp51 5′ SalI AGATCGTCGACATGGCAGATACAGAGGTGG and rhp51 3′ BamHI AGATCGGATCCTTAGACAGGTGCGATAATTTCC. The PCR product was gel purified and cloned into PCR2.1-TOPO using the TOPO TA cloning system (Invitrogen, Carlsbad, CA). The resulting plasmid pTOPO-Rhp51 was sequenced. The rhp51 fragment was then isolated from the pTOPO-Rhp51 by digestion with SalI and BamHI and ligated into SalI and BamHI or XhoI and BamHI digested pREP-3x, pREP-41x, or pREP-81x (obtained from Susan Forsburg). The resulting plasmids were designated pREP-3x-Rhp51, pREP-41x-Rhp51, and pREP-81x-Rhp51, respectively.
TABLE 1.
Yeast strains used in this study
| Strain | Genotype | Reference |
|---|---|---|
| sz472 | h+, ade6-210, ura4-D18, leu1-32 | Laboratory stock |
| sz662 | h+, ade6-210, ura4-D18, leu1-32, rqh1::kanMX4 | Maftahi et al. (2002) |
| sz215 | h+, ade6-704, ura4-D18, leu1-32, rhp51::ura4 | Jang et al. (1995) |
| sz231 | h+, ade6-210, ura4-D18, leu1-32, rhp54::ura4 | Muris et al. (1996) |
| sz844 | h+, ade6-210, ura4-D18, leu1-32, rhp55::ura4 | Khasanov et al. (1999) |
| sz664 | h−, smt-0, ura4-D18, leu1-32, his3-D, arg3-D1, swi5::his3 | Hiroshi Iwasaki |
| sz384 | h+, ade6-210, ura4-D18, leu1-32, rqh1::kanMX4, rhp51::ura4 | This study |
| sz521 | h+, ade6-210, ura4-D18, leu1-32, rqh1::kanMX4, rhp54::ura4 | This study |
| sz843 | h+, ade6-210, ura4-D18, leu1-32, rqh1::kanMX4, rhp55::ura4 | This study |
| sz638 | h+, ade6-210, ura4-D18, leu1-32, rqh1::kanMX4 rhp55::ura4, rhp51::ura4 | This study |
| sz640 | h+, ade6-210, ura4-D18, leu1-32, rqh1::kanMX4 rhp55::ura4, rhp54::ura4 | This study |
| sz694 | h−, smt-0, ura4-D18, leu1-32, his3-D, arg3-D1, rqh1::kanMX4 swi5::His3 | This study |
| sz868 | h−, smt-0, ura4-D18, leu1-32, his3-D, arg3-D1, rqh1::kanMX4, rhp55::ura4, swi5::His3 | This study |
Survival studies:
Cultures were grown overnight to midlog (106–107 cells/ml). For UV survival, cells were plated onto YEA plates and irradiated with the indicated dose of UV light. Plates were incubated at 30° for 3–4 days and colonies were counted. For HU survival, cells were counted and plated onto YEA plates containing the appropriate concentration of HU. The plates were incubated at 30° except for the cold-enhancement studies where plates were initially incubated at 22°. After 4–6 days colonies were counted.
Studies using overexpressed Rhp51:
pREP plasmids expressing Rhp51 were transformed into the various strains and selection was maintained on EMM plates with appropriate supplements including 8 μm thiamine. The presence of 8 μm thiamine suppressed the expression of Rhp51 from the nmt promoter. Previous studies of Rhp51 overexpression from the nmt promoter (Kim et al. 2001) had demonstrated that peak expression of Rhp51 occurred at 17–20 hr after the removal of thiamine. Strains containing pREP-81x-Rhp51 were grown for 20 hr in the presence or absence of 8 μm thiamine. HU (15 mm) was added to each culture and cells were collected at 3, 6, and 9 hr after addition. These cells were diluted and plated onto YEA plates and incubated at 30° for 4–6 days when colonies were counted.
Confirmation of Rhp51 overexpression:
Overnight cultures of wild-type (sz472) and Δrqh1 Δrhp55 (sz843) cells containing pREP81x-Rhp51 were grown (20 hr) in the presence of thiamine. These cells were washed and then added to media with or without thiamine. Whole-cell extracts were prepared from cells following 20 hr of growth. Cell extracts (150 μg) were separated on a 12% PAGE-SDS gel and blotted onto ECL nitrocellulose paper (Amersham, Arlington Heights, IL). Rhp51 was detected using a rabbit anti-human rad51 antibody (Santa Cruz H-92), which was previously shown to cross-react with Rhp51 (Caspari et al. 2002). The presence of antibody was detected using ECL (Amersham).
Pulse field gel electrophoresis (PFGE):
Cells were harvested at 9000 rpm in a microcentrifuge and washed in 1 ml of stop buffer (50 mm EDTA/1 mm NaN3). Cells were counted using a hemacytometer and 4.0 × 107 were resuspended in 30 μl of stop buffer. Thirty-five microliters of warm (50°) 1.5% InCert agarose in stop buffer was added to the cell suspension and the entire volume was gently transferred into a plug mold. Plugs were allowed to solidify for 20–30 min at 4° followed by incubation in spheroplasting solution (1 ml 1 m sorbitol, 40 μl 0.5 m EDTA, 10 μl 1 m Tris pH 7.5, 1 μl β-mercaptoethanol, 2 mg/ml Zymolyase, 2 mg/ml Novazyme) for 2.5 hr at 37° with gentle shaking. Spheroplasting solution was removed and plugs were incubated with 2 ml ETS (0.25 m EDTA, 50 mm Tris pH 7.5, 1% SDS) at 55° for 2 hr with one change of buffer. ETS solution was removed and plugs were incubated with 2 ml of 2 mg/ml proteinase K in SEP buffer (0.5 m EDTA, 1% lauryl sarcosine) for 1 hr at 55°. Fresh buffer was added and plugs were incubated overnight at 55°. Plugs were washed three times with 1× TE and loaded into the wells of a 0.6% agarose gel [Bio-Rad (Hercules, CA) PFGE grade] made with 1× TAE. Gels were run on a Bio-Rad CHEF-DR-II PFGE system for 72 hr at 15° at 2.0 V/cm, with switch times of 20 and 30 min. Gels were stained overnight in 1× TAE + SYBR green DNA stain (Molecular Probes, Eugene, OR) at the recommended concentration of 1:10,000 and visualized on a UV transilluminator.
RESULTS
Δrhp55 and Δrhp57 suppress HU and UV sensitivity of Δrqh1 cells:
Previous studies have reported that Δrqh1 cells are sensitive to HU treatment and, although they arrest in S phase, they undergo an aberrant mitosis where the nuclear material has a “cut” appearance and is often unevenly distributed between daughter cells (Stewart et al. 1997; Davey et al. 1998; Doe et al. 2000). One explanation for the nuclear phenotype is that HU treatment induces HR intermediates to form between sister chromatids, which are not resolved in Δrqh1 cells. We reasoned that if this were the case, loss of HR should improve viability and suppress the formation of these aberrant nuclei. We first examined the HU sensitivity of the HR mutants corresponding to the S. cerevisiae RAD52 epistasis group RAD51, RAD54, RAD55, and RAD57, which in S. pombe are rhp51+, rhp54+, rhp55+, and rhp57+, respectively. We did not pursue studies using the RAD52 homolog, rad22+, as we found Δrad22 to be synthetic lethal with Δrqh1 as was previously reported (Wilson et al. 1999). Cells were plated onto media containing various concentrations of HU and incubated for 4–6 days before colonies were counted to determine their sensitivities to replication arrest. We found that Δrhp55 and Δrhp57 mutants showed identical sensitivities to HU and DNA damage. This was expected as Rhp55 and Rhp57 act as an obligate heterodimer. Thus, for simplicity we primarily present the Δrhp55 data here. In Figure 1, a and b, Δrhp51 and Δrhp54 single mutants are shown to be sensitive to HU, particularly at higher doses. Δrhp55 cells showed essentially no sensitivity to HU exposure in the dose range examined (Figure 1c). These results show that Rhp51 and Rhp54 play a more central role in recovery from HU-induced replication arrest in rqh1+ cells than does Rhp55.
Figure 1.—
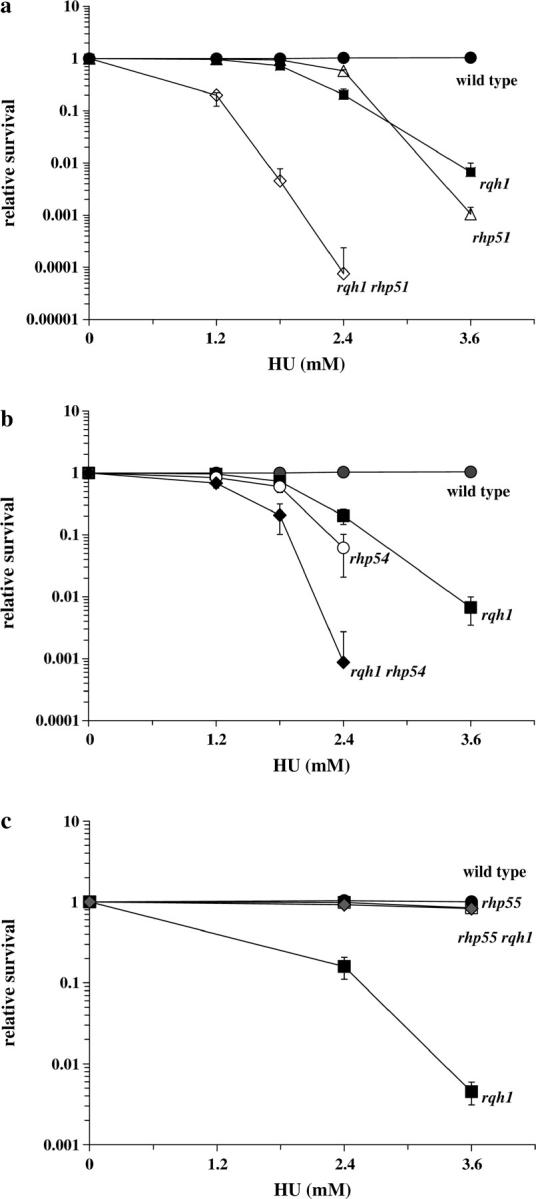

HU and UV sensitivity of HR mutants alone and combined with Δrqh1. Double mutants between Δrqh1 and mutants of the RAD52 epistasis group were created. To measure HU sensitivity, cells were grown to midlog and then each single and double mutant was plated onto plates containing HU of varying concentrations and colonies were counted after 4–6 days of incubation at 30°. To measure UV sensitivity, midlog cultures were grown and cells were spread onto YEA plates at varying concentrations and irradiated with the indicated dose of 254 nm UV light. The results are shown. •, wild type; ▪, Δrqh1. (a) ▵, Δrhp51; ⋄, Δrqh1 Δrhp51. (b) ○, Δrhp54; ♦, Δrqh1 Δrhp54. (c) ▵, Δrhp55; ♦, Δrqh1 Δrhp55. (d) ▵, Δrhp51; ⋄, Δrqh1 Δrhp51; ○, Δrhp54; ▴, Δrqh1 Δrhp54. (e) ▵, Δrhp55; ♦, Δrqh1 Δrhp55. (Note that some error bars are smaller than the symbols.)
Next we tested the HU sensitivity of double mutants made between the HR mutants and Δrqh1 (Figure 1, a–c). We found that Δrqh1 Δrhp51 and Δrqh1 Δrhp54 double mutants were actually more sensitive to HU than the single Δrqh1 mutant (Figure 1, a and b). However, loss of rhp55+ significantly suppressed the HU sensitivity of Δrqh1 cells, to essentially the levels seen in the single Δrhp55 mutant (Figure 1c). These data suggest that the action of Rhp55/57 leads to the sensitivity in replication-arrested cells lacking Rqh1. To make certain that the losses of Rhp55 and Rhp57 were equivalent, we created a triple mutant, Δrqh1 Δrhp55 Δrhp57, and tested its sensitivity to HU. As expected, the triple mutant showed levels of sensitivity identical to those seen in the Δrqh1 Δrhp55 and Δrqh1 Δrhp57 double mutants (data not shown).
Δrqh1 and HR mutants are sensitive to exposure to UV radiation (Muris et al. 1993, 1996; Ostermann et al. 1993; Murray et al. 1997; Davey et al. 1998; Gangloff et al. 2000; Fabre et al. 2002; Maftahi et al. 2002; Doe and Whitby 2004). When we tested the UV sensitivity of double mutants between Δrqh1 and genes of the HR pathway we found a pattern of suppression similar to that seen with HU treatment. Δrqh1 Δrhp51 and Δrqh1 Δrhp54 double mutants showed sensitivities to UV damage identical to those of the Δrhp51 and Δrhp54 single mutants, which are more sensitive than the Δrqh1 single mutant (Figure 1d). By contrast, deletion of rhp55+ in a Δrqh1 background significantly suppressed the sensitivity of Δrqh1 mutants (Figure 1e). These data are consistent with our findings with HU treatment and suggest that Rqh1 has a role in recovery from DNA damage and replication arrest that acts downstream of Rhp55/57 function. A recent study by Doe and Whitby (2004) also showed that loss of rhp55+ suppressed both the HU and the DNA damage sensitivity of Δrqh1 mutants.
Δrhp55 partially suppresses the presence of torn nuclear material and speeds the formation of intact chromosomes in HU-treated Δrqh1 cells:
Since the loss of rhp55+ improved the HU resistance of Δrqh1 cells, we speculated that its loss would also suppress the cut phenotype of Δrqh1 cells following replication arrest, supporting the hypothesis that these could represent unresolved recombination intermediates. To test this hypothesis, wild-type, Δrqh1, Δrhp55, and Δrqh1 Δrhp55 strains were incubated in HU for 5 hr, sufficient time to achieve 100% arrest of cells in S phase, based on PFGE results shown in Figure 2c and FACS analysis (not shown).
Figure 2.—


Evidence that HR intermediates accumulate in HU-treated Δrqh1 cells that are suppressed by Δrhp55. Previous studies had shown that aberrant mitosis occurs in Δrqh1 cells following replication arrest. We speculated that torn and unevenly distributed nuclear material was due to unresolved recombinant intermediates. We tested this hypothesis by determining if loss of rhp55+ could suppress this phenotype. (a) Examples of DAPI-stained cells visualized by fluorescent microscopy are shown following HU treatment and a 3-hr recovery. Arrowheads point to septa of dividing cells. Asterisks indicate cut chromosomes. The double asterisk indicate cells where all DNA segregated into one daughter cell. (b) Quantitation of the number of cells with aberrant chromosomes visible following DAPI staining after a 3-hr release from replication block. (c) PFGE was used as another way of monitoring the fate of chromosomes following HU treatment. Replication fork structures and recombination intermediates are inhibited from exiting the well. We compare the chromosomes from Δrqh1 cells with those from wild type, Δrhp55, and Δrqh1 Δrhp55. Lanes 1, 6, 11, and 16, chromosomes from cycling cells; lanes 2, 7, 12, and 17, chromosomes from cells exposed to 15 mm HU for 5 hr; lanes 3, 8, 13, and 18, chromosomes from cells 2 hr after release; lanes 4, 9, 14, and 19, chromosomes from cells 4 hr after release; lanes 5, 10, 15, and 20, chromosomes from cells 6 hr after release.
Cells were allowed to recover for various times from the HU block and then stained with 4′,6-diamidino-2-phenylindole (DAPI) to examine their nuclear material by fluorescence microscopy. Dividing cells were observed at times from 2 to 5 hr after HU release. The 3-hr time point had the greatest number of cells in the process of cell division, so we picked this time point for quantitative analysis. Photographs depicting representative examples of the four strains from the 3-hr time point are shown in Figure 2a. The presence of cells with cut nuclei and unevenly distributed chromosomal material is evident in dividing Δrqh1 cells.
For quantitative analysis, we counted only cells that had clearly undergone mitosis, where either a septum was present or daughter cells were still attached following cell division. Dividing cells were grouped into three categories: (1) cells undergoing normal cell division where nuclear material appeared normal and was equally distributed between daughter cells, (2) cells with torn nuclei and an unequal distribution of nuclear material, and (3) cells where torn chromosomes were not evident but where there was clearly an unequal distribution of nuclear material. These results, obtained from scoring >200 dividing cells from each strain, are summarized in Figure 2b. The data indicate that while 95% of dividing wild-type cells showed normal cell division, only 20% of Δrqh1 cells showed normal segregation of nuclear material. By comparison, 76% of Δrhp55 cells appeared normal, indicating that Δrhp55 cells are somewhat compromised in their ability to recover from HU. As predicted, loss of rhp55+ significantly improved the ability of Δrqh1 cells to undergo normal mitosis; 52% of Δrqh1 Δrhp55 cells were found to divide normally. These results demonstrate that there is a correlation between the cut phenotype seen in HU-treated Δrqh1 cells and HR and, while not conclusive, are consistent with these nuclear aberrations representing HR intermediates. If these are recombination intermediates, these data cannot distinguish whether their formation is suppressed in a Δrhp55 background or their resolution is improved in this background.
In complementary experiments the fate of chromosomes in cells following HU treatment was examined directly by PFGE. Incompletely replicated DNA containing replication forks cannot migrate out of the wells of PFGs due to their branched structures (Cha and Kleckner 2002). Recombination intermediates presumably would behave likewise. In these studies, cells were collected at 2-hr time points following release from a 5-hr HU block. An example of one experiment is shown in Figure 2c. All three chromosomes are visible in the gel in samples prepared from unsynchronized cells (lanes 1, 6, 11, and 16). After 5 hr in HU all of the chromosomal material was found in the wells with no distinct chromosomes detected in the gel for any strain (lanes 2, 7, 12, and 17). By 2 hr after release, DNA synthesis appears to be complete in wild-type and Δrhp55 cells on the basis of the intensity of the chromosomal bands seen in the gel (lanes 3 and 13); compare with unsynchronized cells in lanes 1 and 11. Also no further increase in chromosome intensity is seen after 2 hr (compare lanes 3 and 13 to lanes 4 and 5 and lanes 14 and 15). By contrast chromosomal staining in the Δrqh1 cells is significantly less intense at 2 hr (lane 8). Even after 6 hr of recovery, the staining intensity of the chromosomes from Δrqh1 cells did not reach those of the unsynchronized cells (compare lane 10 to lane 6). Previous studies using FACS analysis showed that Δrqh1 cells are not delayed in completion of DNA synthesis following release from an HU block (Marchetti et al. 2002). This suggests that the DNA retained in the wells in the Δrqh1 cells is due to the presence of unresolved recombination intermediates. The intensity of chromosomal bands present in Δrqh1Δrhp55 cells by 2 hr after release from HU (lane 18) is comparable with wild type or the Δrhp55 single mutant at this time point (lanes 3 and 13, respectively). Also the intensity of chromosome staining does not further intensify at later time points (compare lane 18 with lanes 19 and 20). We suggest that these results are further evidence that loss of rhp55+ suppresses the accumulation of recombination intermediates in replication-arrested Δrqh1 cells but acknowledge that we cannot absolutely rule out the possibility that the retardation of chromosomal migration is due to residual replication intermediates.
Rhp51 and Rhp54 activities are required for the suppression of Δrqh1 HU sensitivity:
We next asked if the suppression of Δrqh1 sensitivity to HU by Δrhp55/Δrhp57 depends on the functions of Rhp51 and Rhp54. Figure 1, a and b, shows that both are critical in recovery of cells from replication arrest. To address this we created Δrqh1 Δrhp55 Δrhp51 and Δrqh1 Δrhp55 Δrhp54 triple mutants. We compared the HU sensitivities of these mutants to wild type and to single and double mutants. The Δrqh1 Δrhp55 Δrhp51 and Δrqh1 Δrhp55 Δrhp54 triple mutants are much more sensitive than the Δrqh1 Δrhp55 double mutant, showing that suppression by Δrhp55 is dependent on the presence of Rhp51 and Rhp54 (Figure 3, a and b). The growth of the Δrqh1 Δrhp55 Δrhp51 triple mutant on HU-containing plates did slightly improve over that of the Δrqh1 Δrhp51 double mutant. One interpretation of these findings is that two lines of suppression of Δrqh1 cells exist, one that is Rhp51 dependent and another that is Rhp51 independent. A comparable change is not seen in the Δrqh1 Δrhp55 Δrhp54 triple mutant where the sensitivity was the same as in the Δrqh1 Δrhp54 double mutant.
Figure 3.—


Suppression of Δrqh1 HU sensitivity depends on the function of Δrhp51 and Δrhp54. (a and b) Tenfold serial dilutions of each strain were spotted onto plates containing either 0 mm or 3.6 mm HU. Plates were incubated for 5 days at 30° and photographed. (c) At reduced temperatures Δrhp55 mutants show reduced resistance to HU but maintain their suppression of Δrhq1. The plates contain 2.4 mm HU and were incubated at 30° for 5 days or 22° for 8 days.
One note concerning these experiments is that the HU sensitivity of Δrhp51 cells appears to be less than that of Δrqh1 cells. This is in contrast to the results seen in Figure 1, where Δrqh1 and Δrhp51 mutants show similar sensitivities to HU. We have repeated both experiments multiple times, with identical results. Our only explanation is based on our observation that, in addition to forming a few visible colonies, Δrhp51 cells form microscopic colonies (∼15–50 cells) on 3.6 mm HU plates after ≥5 days of incubation. By contrast Δrqh1 cells either die immediately, seen as single cells, or grow a very few generations, seen as colonies of 2–10 cells, when plated on 3.6 mm HU and incubated for 5 days. Thus, when Δrhp51 cells are spotted onto 3.6 mm HU plates, microcolonies form. These microcolonies are not visible individually but collectively form a visible spot when viewed in a spot test assay.
The Rhp55/57 activity responsible for Δrqh1 sensitivity to HU treatment is independent of Rhp51 filament formation:
In S. cerevisiae several studies have contributed to developing a profile of Rad55/57 functioning in stimulating Rad51 filament formation. The evidence is threefold. First, while rad55 and rad57 are much less sensitive to IR damage at 30° compared to rad51, their sensitivities are much greater at lower temperatures (Lovett and Mortimer 1987; Johnson and Symington 1995). The argument for this phenomenon is that at lower temperatures the Rad51 filament is less stable and so depends more on rad55/57. Second, in vitro studies by P. Sung demonstrated that Rad51 filament formation on ssDNA is stimulated by the presence of Rad55/57 (Sung 1997). Finally, the IR sensitivity of rad55/57 mutants was significantly reduced in strains overexpressing Rad51 or containing a Rad51 mutant with increased DNA binding capacity (Johnson and Symington 1995; Fortin and Symington 2002).
On the basis of these results, we sought to test whether the role of Rhp55/57 in nucleoprotein filament formation was separate from its role in suppressing the HU sensitivity in Δrqh1 cells. In S. pombe, rhp55 and rhp57 mutants also show cold-enhanced sensitivity; at 30° these mutants are much less sensitive to γ-ray damage than a Δrhp51 mutant, but at lower temperatures Δrhp55 and Δrhp57 mutants are as sensitive as a Δrhp51 mutant (Khasanov et al. 1999). We reasoned that if the Δrhp55/57 suppression of Δrqh1 sensitivity to replication arrest was lost at low temperatures it would be consistent with this suppression being associated with its role in Rhp51 nucleoprotein filament formation. If on the other hand we found that Δrhp55/57 suppression was maintained at lower temperatures this would support the conclusion that that suppression was due to loss of a function that is independent of filament formation. Figure 3c shows that when spotted onto plates containing 2.4 mm HU followed by incubation at 22°, the suppression of the HU sensitivity of Δrqh1 cells by Δrhp55 is maintained.
To further test for an Rhp55/57 activity independent of nucleoprotein filament formation, we cloned rhp51+ into a series of thiamine-suppressible plasmids, pREP-3x, pREP-41x, and pREP-81x. These same plasmids were previously used to create Rhp51-overexpressing plasmids that were able to complement Δrhp51 in DNA damage assays (Kim et al. 2001). We confirmed that each plasmid was able to suppress the IR sensitivity of Δrhp51 (data not shown). We picked the plasmid that produced the lowest level of Rhp51 protein, pREP81x-Rhp51, for the remaining studies. Wild-type and Δrhp55 cells were transformed with pREP81X-Rhp51 and their sensitivity to IR was tested. Cells transformed with pREP81x-Rhp51 were incubated for 17 hr in the absence of thiamine to induce maximal Rhp51 expression. These cells were irradiated with varying doses of γ-rays, plated on YEA plus thiamine plates, and incubated at 30° for 5 days when colonies were counted. Control strains included Δrhp55 containing the vector alone and Δrhp55 with pREP81x-Rhp51 but grown with thiamine prior to irradiation. The results shown in Figure 4a demonstrate that overexpression of Rhp51 reduced the sensitivity of Δrhp55 cells to near wild-type levels. These findings are consistent with Rhp55/57 playing an early role in nucleoprotein filament formation and, as seen in S. cerevisiae, overexpression of Rhp51 largely circumvents this need. This provides a mechanism of potentially separating the role of Rhp55/57 in nucleoprotein filament formation from other functions.
Figure 4.—
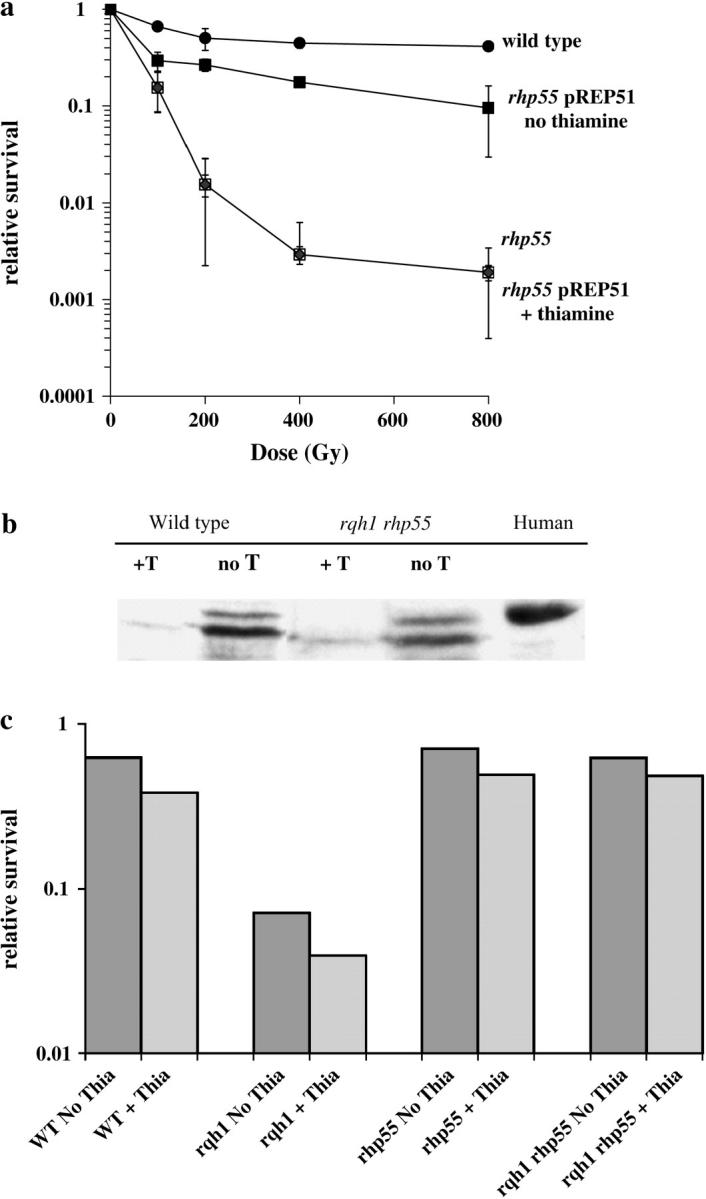
Overexpression of Rhp51. Full-length rhp51+ was cloned into the pREP81x vector (pREP 81x-Rhp51), transformed into various strains, and tested for HU sensitivity. (a) Wild-type and Δrhp55 cells containing pREP 81x-Rhp51 were grown to midlog in media either containing 8 mm thiamine or lacking thiamine. These cells were then irradiated with varying doses of γ-rays and subsequently plated onto YEA plates containing 8 mm thiamine. (b) Wild-type (WT), Δrhp55, Δrqh1, and Δrhp55 Δrqh1 strains were transformed with pREP 81x-Rhp51. Cells were grown to midlog in media either containing 8 mm thiamine or lacking thiamine. Then 15 mm HU was added to each culture and allowed to incubate for an additional 9 hr. Samples were then collected and plated onto YEA plates with 8 mm thiamine. The plates were incubated for 4 days and colonies were counted. (c) Extracts were prepared from wild-type and Δrhp55 Δrqh1 cells all containing pREP 81x-Rhp51 with cultures grown in the presence or absence of thiamine. A total of 150 μg of each extract was loaded onto a 12% PAGE SDS gel and the samples were separated by electrophoresis. As a control, 100 μg of nuclear extract prepared from HeLa cells was also loaded onto the gel. The gel was blotted and Rhp51 was detected using an antibody against human Rad51, which cross-reacts with S. pombe Rhp51. Antibody binding was detected by chemiluminescence.
Δrqh1 and Δrqh1 Δrhp55 strains were transformed with pREP81x-Rhp51. We then tested whether inducing Rhp51 expression would influence the sensitivity of these strains to HU treatment. Cells were incubated for 17 hr in the presence or absence of thiamine. HU was then added to the cultures at a concentration of 15 mm. The cultures were incubated for 0, 3, 6, or 9 hr in HU before washing and plating onto YES plates. Plates were incubated for 5 days and colonies were counted. As seen in Figure 4b, the overexpression of Rhp51 did not reduce the Δrhp55 suppression of the HU sensitivity of Δrqh1 cells, arguing that the Rhp55/57 function responsible for this sensitivity is independent of nucleoprotein formation. For completion we confirmed that Rhp51 was overexpressed in these cells. Whole-cell extracts were prepared from wild type and Δrqh1 Δrhp55 cells grown in the presence or absence of thiamine for 17 hr. Western blot analysis (Figure 4c) shows that Rhp51 levels are significantly elevated in strains grown in the absence of thiamine.
The HU sensitivity of Δrqh1 cells can also be suppressed by Δswi5 and suppression by Δrhp55 is partially dependent on swi5+:
It has recently been reported that S. pombe has an Rhp55/57-independent recombination repair pathway that requires Rhp51 (Akamatsu et al. 2003). This pathway is defined by swi5+, a gene originally identified in a screen for mating-type switching mutants (Egel et al. 1984). We considered the possibility that swi5+ was required for the improved resistance of Δrqh1 Δrhp55 mutants. We first created a Δswi5 Δrhp55 double mutant that we tested for HU sensitivity. We found that while Δswi5 showed wild-type levels of resistance to HU the Δswi5 Δrhp55 double mutant was more sensitive than the Δrhp55 single mutant (Figure 5a). We found that the double mutant was not as sensitive to HU as a Δrhp51 mutant. This differs from the results reported for IR sensitivity of the double mutant, which was shown to be comparable to that of a Δrhp51 mutant, as we also found to be the case (Akamatsu et al. 2003; data not shown). We next examined the effect of Δswi5 on the HU sensitivity of Δrqh1. We found that loss of swi5+ partially suppressed the HU sensitivity of Δrqh1 cells although not back to the level of a Δswi5 single mutant (Figure 5b). Next we created a Δrqh1 Δrhp55 Δswi5 triple mutant and compared its HU sensitivity to that of the Δrqh1 Δrhp55 strain. The addition of the Δswi5 mutation to Δrqh1 Δrhp55 increased the HU sensitivity to an intermediate level between a Δrqh1 and a Δrqh1 Δrhp55 (Figure 5c). These data demonstrate that part of the suppression by Δrhp55 depends on a Swi5 function. However, the suppression of the HU sensitivity of Δrqh1 cells by Δswi5 shows that the situation is more complicated than Swi5 simply acting in an alternative pathway in the absence of Rhp55/57.
Figure 5.—
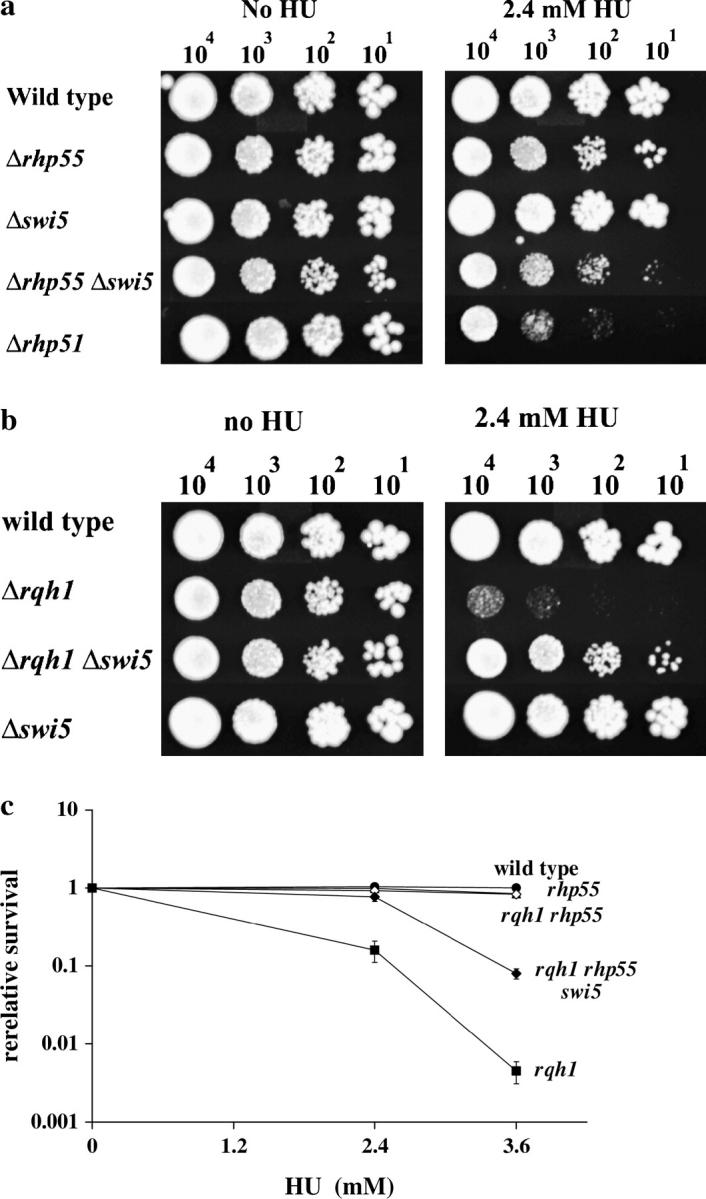
Suppression of Δrqh1 HU sensitivity by Δrhp55 is partially dependent on Swi5. We investigated the possibility that Swi5 was necessary for the suppression of the HU sensitivity of Δrqh1 by Δrhp55. (a) Serial dilutions of midlog cultures of wild-type, Δrhp55, Δswi5, Δrhp51, and Δrhp55 Δswi5 cells were plated onto YEA or YEA containing 2.4 mm HU followed by incubation for 5 days. (b) Serial dilutions of midlog cultures of wild-type, Δrqh1, Δswi5, and Δrqh1 Δswi5 cells were plated onto YEA or YEA containing 2.4 mm HU followed by incubation for 5 days. (c) Midlog cultures of wild type, Δrhp55, Δswi5, Δrqh1, Δrqh1 Δrhp55, and Δrqh1 Δrhp55 Δswi5 were plated onto YEA plates containing varying concentrations of HU and incubated for 4–6 days before colonies were counted.
Rqh1 and HR share a common response to IR-induced DSBs:
We also analyzed the sensitivity of our mutants to IR. IR creates DSBs that must be repaired by HR or NHEJ. The observation that Δrqh1 cells are sensitive to IR indicates that Rqh1 functions in the repair of DSBs (Figure 6a). Δrhp51 cells were the most γ-ray sensitive of the HR mutants tested (Figure 6b). The Δrqh1 Δrhp51 double mutant has sensitivity identical to that of the Δrhp51 single mutant, showing that these proteins are in the same epistasis group for repair of DSBs (Figure 6b; also see Caspari et al. 2002). The Δrhp55 single mutant showed sensitivity to IR that was very similar to that of Δrqh1 (Figure 6a). Moreover, the Δrqh1 Δrhp55 double mutant was indistinguishable from either single mutant with regard to its IR sensitivity (Figure 6b). We also examined the γ-ray sensitivity of the Δrqh1 Δrhp55 Δrhp51 triple mutant to determine if the strong IR sensitivity of the Δrqh1 Δrhp51 mutant would be suppressed. Δrhp55 did not improve the IR resistance of a Δrqh1 Δrhp51 double mutant (Figure 6b). Comparable results were seen for Δrhp54 (data not shown). We did not see the mild improvement in survival of this triple mutant over the double mutant that we found with HU treatment (Figure 3a), showing that no Rhp51-independent repair of these DSBs takes place.
Figure 6.—

The IR sensitivities of double mutants between Δrqh1 and HR genes show survival patterns similar to their HU sensitivities. The IR sensitivities of Δrhp55, Δrhp51, Δrqh1, Δrqh1 Δrhp55, and Δrqh1 Δrhp51 were tested. Cells from midlog cultures were plated onto YEA plates and irradiated with the indicated dose. Colonies were counted after 5 days. •, wild type; ▪, Δrqh1. (a) ▴, Δrhp55; ▵, Δrqh1 Δrhp55. (b) ▴, Δrhp51; ▵, Δrqh1 Δrhp51; ⋄, Δrqh1 Δrhp55 Δrhp51.
DISCUSSION
While Rqh1 plays an important role in recovery from replication arrest, it is unclear how it participates in this process. Several lines of evidence have suggested that it carries out its function with HR (Murray et al. 1997; Stewart et al. 1997; Davey et al. 1998). One possible role for Rqh1 helicase is to act at a late step in HR by unwinding the heteroduplex formed by strand invasion, although it has also been proposed to function in an earlier step of HR (Caspari et al. 2002). In these studies we sought to investigate the role of Rqh1 in recovery from replication arrest.
Rhp51, Rhp54, and Rqh1 are critical in recovery from replication arrest:
HU treatment leads to an S-phase arrest as replication is inhibited. Wild-type cells eventually recover from this arrest without loss of viability or obvious accumulation of mutations despite a dramatic increase in HR rates. Δrqh1 mutants show low survival and high rates of chromosomal loss following HU treatment (Stewart et al. 1997). In addition, Δrhp51 and Δrhp54 mutants are also quite sensitive to HU, demonstrating that HR plays a vital role in recovery from replication arrest. The need for HR in recovery from HU treatment can be explained in two ways: replication arrest ultimately leads to formation of DSBs, which would require HR or NHEJ for repair, or HR acts on a DNA structure other than a DSB, possibly protecting stalled forks from collapse and promoting replication restart. Support for the former explanation comes from data showing that replication arrest leads to formation of DSBs (Michel et al. 1997; Lundin et al. 2002), although this issue is far from resolved.
The relatively severe sensitivity of Δrqh1 cells to HU demonstrates that Rqh1 also plays an important role in replication arrest recovery. Δrhp55 and Δrhp57 mutants show mild sensitivity and would appear to play a minor role in this process. The interpretation of the role of Rhp55/57 is complicated by the reported backup role of Swi5 in repair of DSBs (see below) (Akamatsu et al. 2003).
Loss of rhp55+ or rhp57+ suppresses the HU sensitivity of Δrqh1 cells:
We found that double mutants between Δrqh1 and various HR genes showed very different sensitivities to HU. While Δrqh1 Δrhp51 and Δrqh1 Δrhp54 double mutants were more sensitive to HU than the single mutants, we found that the additional loss of either rhp55+ or rhp57+ suppressed the HU as well as the UV sensitivity of Δrqh1 cells. A recent article by Doe and Whitby (2004) also reported that loss of rhp55+ suppressed the HU and UV as well as MMS sensitivity of Δrqh1 mutants, although they did not describe studies beyond this point. They also stated that loss of rhp51+ had a similar effect, which would be in conflict with our data. However, no data were shown for this statement, making it difficult to evaluate this claim. The suppression of the HU and UV sensitivity of Δrqh1 cells by Δrhp55 or Δrhp57 implies that, in response to these agents, Rhp55/57 functions in a process that is deleterious to cells lacking Rqh1. But what is this process? Previous studies in S. cerevisiae have characterized Rad55/57 as a mediator, aiding in the early step of HR by helping to stabilize Rad51 loading onto single-stranded DNA (Symington 2002). Our results show that suppression of Δrqh1 by Δrhp55 largely depends on the presence of Rhp51 and Rhp54 (Figure 3, a and b). On the basis of the early roles of Rhp51 and Rhp54 in HR, these results suggest that this Rhp55/57 activity acts downstream of joint molecule formation.
To investigate this further we carried out two studies aimed at determining whether we could separate the role of Rhp55/57 in nucleoprotein filament formation from a late function in HR. First, S. cerevisiae rad55/57 mutants are less sensitive to IR at 30° than are other members of the RAD52 epistasis group. However, at lower temperatures (20°), they become more sensitive, approaching the level of rad51 mutants (Lovett and Mortimer 1987; Johnson and Symington 1995). The same phenotype has been reported for Δrhp55/57 mutants in S. pombe (Khasanov et al. 1999). The explanation for this cold-enhanced sensitivity is that Rad55/57-independent Rad51 nucleation on DNA is inhibited at lower temperatures, increasing the requirement for Rad55/57 mediator function. We found that Δrhp55 is also more sensitive to HU at 22° than at 30°. However, even at 22° Δrhp55 suppressed the HU sensitivity of Δrqh1 (Figure 3c).
Second, in a separate experiment we overexpressed Rhp51 and showed that it suppressed the IR sensitivity of Δrhp55 mutants. This same result has been described in S. cerevisiae and is interpreted as further evidence of the role of Rad55/57 in helping to establish the Rhp51 nucleoprotein filament. The explanation is that having more Rhp51 on hand alters the kinetics of nucleoprotein filament formation, largely eliminating the need for Rhp55/57 in this process. We next tested whether overexpression of Rhp51 would alter the suppressor effect of Δrhp55 on the HU sensitivity of Δrqh1 cells. We reasoned that if the increase in resistance involved the role of Rhp55 in the nucleoprotein filament formation, then overexpressing Rhp51 should make a Δrqh1 Δrhp55 double mutant sensitive to HU. Overexpression of Rhp51 had no effect on the ability of Δrhp55 to suppress the HU sensitivity of Δrqh1 mutants. Together these results suggest that Rhp55/57 has a function that is independent of its role in Rhp51 nucleoprotein filament formation.
This raises the question of what is this second Rhp55/57 function. One clue may come from studies in human cells where Rad51 paralogs have been implicated in playing a late function in HR (Brenneman et al. 2002; Yokoyama et al. 2003; Liu et al. 2004). In one study it was shown that the human Rad51 paralog Rad51B binds to HJs (Yokoyama et al. 2003). Using cell free extracts, Liu et al. (2004) provided data suggesting that Rad51C and XRCC3 play a role in HJ resolution. And finally, Brenneman et al. (2002) carried out studies on XRCC3 and suggested that Rad51 paralogs were likely acting to stabilize the heteroduplex following strand invasion. Our data provide genetic evidence that Rhp55/57 is acting late in HR, likely downstream of joint molecule formation. If Rhp55/57 were acting to stabilize HJs, as suggested in human studies, then loss of rhp55+ or rhp57+ should destabilized HJs. How could this suppress the loss of Rqh1 activity? It is well established that RecQ helicases function to suppress recombination and crossovers. This could be accomplished either by blocking the initiation of HR or by eliminating recombination intermediates via a mechanism that yields noncrossover products (Ira et al. 2003; Wu and Hickson 2003). If it were the latter, then the likely role for RecQ helicases would be to resolve the joint molecule formed during HR. If this were the case then in the absence of RecQ, heteroduplex DNA would persist. If eliminating Rhp55/57 destabilized the HJ, this could lead to branch migration, leading to resolution of the joint molecule, minimizing the need for Rqh1. We recognize that any model proposed for RecQ helicases needs to include a role for Top3. We imagine that Top3 strand passage activity could act to allow the displaced strand to reform the original duplex DNA, although admittedly we have no direct evidence to support this. A study in S. cerevisiae proposed a model in which Sgs1 and Top3 function late in HR, but to resolve double HJ (Ira et al. 2003). An in vitro study using human Blm and Topo IIIα showed that these proteins could resolve a synthetic double HJ (Wu and Hickson 2003). Needless to say, the actual roles of RecQ and Top3 in HR remain uncertain.
Further evidence that Swi5 functions in a process similar to Rhp55/57:
A recent article described results suggesting that Swi5 functions in a process parallel to Rhp55/57 that depends on Rhp51 (Akamatsu et al. 2003). On their own, Δswi5 mutants show little sensitivity to DNA damage, including IR, UV, or MMS treatment. However, when combined with Δrhp55, the double mutant reaches a level of sensitivity to DNA damage that is comparable to the more sensitive Δrhp51 mutant. These data have been interpreted as showing that Swi5 acts as an alternative to Rhp55/57 (Akamatsu et al. 2003). We wanted to test whether Δrqh1 Δrhp55 mutants depended on Swi5 for recovery from HU. For this we created a Δrqh1 Δrhp55 Δswi5 triple mutant and compared its HU sensitivity to Δrqh1 Δrhp55. As we suspected, the triple mutant showed increased sensitivity to replication arrest over the Δrqh1 Δrhp55 double mutant. The sensitivity is intermediate between Δrqh1 and Δrqh1 Δrhp55 mutants, implying that some of the recovery from arrest is dependent on a function of Swi5 in the absence of rhp55+. However, the story is not simply that Swi5 acts in a parallel pathway in the absence of Rhp55/57. We also found that in an rhp55+ background Δswi5 suppressed the HU sensitivity of Δrqh1. This shows that Swi5 is functioning even in the presence of Rhp55/57. Further, we found that the Δrhp55 Δswi5 double mutant does not become nearly as sensitive to HU as a Δrhp51 mutant. These results suggest a slightly more complex function for Swi5 than simply acting in a parallel pathway to Rhp55/57 during recovery from replication arrest. The suppression of the HU sensitivity of Δrqh1 mutants by loss of swi5+ is qualitatively different from the suppression by loss of rhp55+ (compare the colony morphologies of Figures 3a and 5b and the level of suppression). This and the intermediate phenotype of the triple mutant imply that the mechanisms of suppression are different. Further experiments are necessary to better understand the roles that Rhp55 and Swi5 play in recovery from replication arrest.
Acknowledgments
The authors thank Gloria Osorio and Sarah Mense for invaluable technical assistance and Lorraine Symington, Lance Langston, and Steve Brill for helpful discussions and critical reading of this manuscript. J.C.H. is a Ruth L. Kischstein Fellow (GM20376). This work was supported by National Institutes of Health grant CA072647.
References
- Akamatsu, Y., D. Dziadkowiec, M. Ikeguchi, H. Shinagawa and H. Iwasaki, 2003. Two different Swi5-containing protein complexes are involved in mating-type switching and recombination repair in fission yeast. Proc. Natl. Acad. Sci. USA 100: 15770–15775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastin-Shanower, S. A., W. M. Fricke, J. R. Mullen and S. J. Brill, 2003. The mechanism of Mus81-Mms4 cleavage site selection distinguishes it from the homologous endonuclease Rad1-Rad10. Mol. Cell. Biol. 23: 3487–3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boddy, M. N., P. H. Gaillard, W. H. McDonald, P. Shanahan, J. R. Yates, III et al., 2001. Mus81-Eme1 are essential components of a Holliday junction resolvase. Cell 107: 537–548. [DOI] [PubMed] [Google Scholar]
- Brenneman, M. A., B. M. Wagener, C. A. Miller, C. Allen and J. A. Nickoloff, 2002. XRCC3 controls the fidelity of homologous recombination: roles for XRCC3 in late stages of recombination. Mol. Cell 10: 387–395. [DOI] [PubMed] [Google Scholar]
- Bressan, D. A., B. K. Baxter and J. H. Petrini, 1999. The Mre11-Rad50-Xrs2 protein complex facilitates homologous recombination-based double-strand break repair in Saccharomyces cerevisiae. Mol. Cell. Biol. 19: 7681–7687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr, A. M., 2002. DNA structure dependent checkpoints as regulators of DNA repair. DNA Repair 1: 983–994. [DOI] [PubMed] [Google Scholar]
- Caspari, T., J. M. Murray and A. M. Carr, 2002. Cdc2-cyclin B kinase activity links Crb2 and Rqh1-topoisomerase III. Genes Dev. 16: 1195–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha, R. S., and N. Kleckner, 2002. ATR homolog Mec1 promotes fork progression, thus averting breaks in replication slow zones. Science 297: 602–606. [DOI] [PubMed] [Google Scholar]
- Cox, M. M., M. F. Goodman, K. N. Kreuzer, D. J. Sherratt, S. J. Sandler et al., 2000. The importance of repairing stalled replication forks. Nature 404: 37–41. [DOI] [PubMed] [Google Scholar]
- D'Amours, D., and S. P. Jackson, 2002. The Mre11 complex: at the crossroads of DNA repair and checkpoint signalling. Nat. Rev. Mol. Cell. Biol. 3: 317–327. [DOI] [PubMed] [Google Scholar]
- Davey, S., C. S. Han, S. A. Ramer, J. C. Klassen, A. Jacobson et al., 1998. Fission yeast rad12+ regulates cell cycle checkpoint control and is homologous to the Bloom's syndrome disease gene. Mol. Cell. Biol. 18: 2721–2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diffley, J. F., K. Bousset, K. Labib, E. A. Noton, C. Santocanale et al., 2000. Coping with and recovering from hydroxyurea-induced replication fork arrest in budding yeast. Cold Spring Harbor Symp. Quant. Biol. 65: 333–342. [DOI] [PubMed] [Google Scholar]
- Doe, C. L., and M. C. Whitby, 2004. The involvement of Srs2 in post-replication repair and homologous recombination in fission yeast. Nucleic Acids Res. 32: 1480–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doe, C. L., J. Dixon, F. Osman and M. C. Whitby, 2000. Partial suppression of the fission yeast rqh1(-) phenotype by expression of a bacterial Holliday junction resolvase. EMBO J. 19: 2751–2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doe, C. L., J. S. Ahn, J. Dixon and M. C. Whitby, 2002. Mus81-Eme1 and Rqh1 involvement in processing stalled and collapsed replication forks. J. Biol. Chem. 277: 32753–32759. [DOI] [PubMed] [Google Scholar]
- Egel, R., D. H. Beach and A. J. Klar, 1984. Genes required for initiation and resolution steps of mating-type switching in fission yeast. Proc. Natl. Acad. Sci. USA 81: 3481–3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabre, F., A. Chan, W. D. Heyer and S. Gangloff, 2002. Alternate pathways involving Sgs1/Top3, Mus81/ Mms4, and Srs2 prevent formation of toxic recombination intermediates from single-stranded gaps created by DNA replication. Proc. Natl. Acad. Sci. USA 99: 16887–16892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin, G. S., and L. S. Symington, 2002. Mutations in yeast Rad51 that partially bypass the requirement for Rad55 and Rad57 in DNA repair by increasing the stability of Rad51-DNA complexes. EMBO J. 21: 3160–3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukushima, K., Y. Tanaka, K. Nabeshima, T. Yoneki, T. Tougan et al., 2000. Dmc1 of Schizosaccharomyces pombe plays a role in meiotic recombination. Nucleic Acids Res. 28: 2709–2716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangloff, S., C. Soustelle and F. Fabre, 2000. Homologous recombination is responsible for cell death in the absence of the Sgs1 and Srs2 helicases. Nat. Genet. 25: 192–194. [DOI] [PubMed] [Google Scholar]
- Haber, J. E., and W. D. Heyer, 2001. The fuss about Mus81. Cell 107: 551–554. [DOI] [PubMed] [Google Scholar]
- Hays, S. L., A. A. Firmenich and P. Berg, 1995. Complex formation in yeast double-strand break repair: participation of Rad51, Rad52, Rad55, and Rad57 proteins. Proc. Natl. Acad. Sci. USA 92: 6925–6929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helleday, T., 2003. Pathways for mitotic homologous recombination in mammalian cells. Mutat. Res. 532: 103–115. [DOI] [PubMed] [Google Scholar]
- Heyer, W. D., K. T. Ehmsen and J. A. Solinger, 2003. Holliday junctions in the eukaryotic nucleus: resolution in sight? Trends Biochem. Sci. 28: 548–557. [DOI] [PubMed] [Google Scholar]
- Ira, G., A. Malkova, G. Liberi, M. Foiani and J. E. Haber, 2003. Srs2 and Sgs1-Top3 suppress crossovers during double-strand break repair in yeast. Cell 115: 401–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang, Y. K., Y. H. Jin, Y. S. Shim, M. J. Kim, E. J. Yoo et al., 1995. Evidences for possible involvement of Rhp51 protein in mitotic events including chromosome segregation. Biochem. Mol. Biol. Int. 37: 329–337. [PubMed] [Google Scholar]
- Johnson, R. D., and L. S. Symington, 1995. Functional differences and interactions among the putative RecA homologs Rad51, Rad55, and Rad57. Mol. Cell. Biol. 15: 4843–4850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khasanov, F. K., G. V. Savchenko, E. V. Bashkirova, V. G. Korolev, W. D. Heyer et al., 1999. A new recombinational DNA repair gene from Schizosaccharomyces pombe with homology to Escherichia coli RecA. Genetics 152: 1557–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, W. J., H. Lee, E. J. Park, J. K. Park and S. D. Park, 2001. Gain- and loss-of-function of Rhp51, a Rad51 homolog in fission yeast, reveals dissimilarities in chromosome integrity. Nucleic Acids Res. 29: 1724–1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzminov, A., 1993. RuvA, RuvB and RuvC proteins: cleaning-up after recombinational repairs in E. coli. BioEssays 15: 355–358. [DOI] [PubMed] [Google Scholar]
- Laursen, L. V., E. Ampatzidou, A. H. Andersen and J. M. Murray, 2003. Role for the fission yeast RecQ helicase in DNA repair in G2. Mol. Cell. Biol. 23: 3692–3705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberi, G., I. Chiolo, A. Pellicioli, M. Lopes, P. Plevani et al., 2000. Srs2 DNA helicase is involved in checkpoint response and its regulation requires a functional Mec1-dependent pathway and Cdk1 activity. EMBO J. 19: 5027–5038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y., J. Y. Masson, R. Shah, P. O'Regan and S. C. West, 2004. RAD51C is required for Holliday junction processing in mammalian cells. Science 303: 243–246. [DOI] [PubMed] [Google Scholar]
- Lovett, S. T., and R. K. Mortimer, 1987. Characterization of null mutants of the RAD55 gene of Saccharomyces cerevisiae: effects of temperature, osmotic strength and mating type. Genetics 116: 547–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundin, C., K. Erixon, C. Arnaudeau, N. Schultz, D. Jenssen et al., 2002. Different roles for nonhomologous end joining and homologous recombination following replication arrest in mammalian cells. Mol. Cell. Biol. 22: 5869–5878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maftahi, M., J. C. Hope, L. Delgado-Cruzata, C. S. Han and G. A. Freyer, 2002. The severe slow growth of Deltasrs2 Deltarqh1 in Schizosaccharomyces pombe is suppressed by loss of recombination and checkpoint genes. Nucleic Acids Res. 30: 4781–4792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maisnier-Patin, S., K. Nordstrom and S. Dasgupta, 2001. Replication arrests during a single round of replication of the Escherichia coli chromosome in the absence of DnaC activity. Mol. Microbiol. 42: 1371–1382. [DOI] [PubMed] [Google Scholar]
- Marchetti, M. A., S. Kumar, E. Hartsuiker, M. Maftahi, A. M. Carr et al., 2002. A single unbranched S-phase DNA damage and replication fork blockage checkpoint pathway. Proc. Natl. Acad. Sci. USA 99: 7472–7477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGlynn, P., and R. G. Lloyd, 2002. Recombinational repair and restart of damaged replication forks. Nat. Rev. Mol. Cell. Biol. 3: 859–870. [DOI] [PubMed] [Google Scholar]
- Michel, B., 2000. Replication fork arrest and DNA recombination. Trends Biochem. Sci. 25: 173–178. [DOI] [PubMed] [Google Scholar]
- Michel, B., S. D. Ehrlich and M. Uzest, 1997. DNA double-strand breaks caused by replication arrest. EMBO J. 16: 430–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel, B., M. J. Flores, E. Viguera, G. Grompone, M. Seigneur et al., 2001. Rescue of arrested replication forks by homologous recombination. Proc. Natl. Acad. Sci. USA 98: 8181–8188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muris, D. F., K. Vreeken, A. M. Carr, B. C. Broughton, A. R. Lehmann et al., 1993. Cloning the RAD51 homologue of Schizosaccharomyces pombe. Nucleic Acids Res. 21: 4586–4591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muris, D. F., K. Vreeken, A. M. Carr, J. M. Murray, C. Smit et al., 1996. Isolation of the Schizosaccharomyces pombe RAD54 homologue, rhp54+, a gene involved in the repair of radiation damage and replication fidelity. J. Cell Sci. 109(Pt. 1): 73–81. [DOI] [PubMed] [Google Scholar]
- Muris, D. F., K. Vreeken, H. Schmidt, K. Ostermann, B. Clever et al., 1997. Homologous recombination in the fission yeast Schizosaccharomyces pombe: different requirements for the rhp51+, rhp54+ and rad22+ genes. Curr. Genet. 31: 248–254. [DOI] [PubMed] [Google Scholar]
- Murray, J. M., H. D. Lindsay, C. A. Munday and A. M. Carr, 1997. Role of Schizosaccharomyces pombe RecQ homolog, recombination, and checkpoint genes in UV damage tolerance. Mol. Cell. Biol 17: 6868–6875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelms, B. E., R. S. Maser, J. F. MacKay, M. G. Lagally and J. H. Petrini, 1998. In situ visualization of DNA double-strand break repair in human fibroblasts. Science 280: 590–592. [DOI] [PubMed] [Google Scholar]
- Nyberg, K. A., R. J. Michelson, C. W. Putnam and T. A. Weinert, 2002. Toward maintaining the genome: DNA damage and replication checkpoints. Annu. Rev. Genet. 36: 617–656. [DOI] [PubMed] [Google Scholar]
- Ostermann, K., A. Lorentz and H. Schmidt, 1993. The fission yeast rad22 gene, having a function in mating-type switching and repair of DNA damages, encodes a protein homolog to Rad52 of Saccharomyces cerevisiae. Nucleic Acids Res. 21: 5940–5944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paques, F., and J. E. Haber, 1999. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 63: 349–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogakou, E. P., C. Boon, C. Redon and W. M. Bonner, 1999. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell Biol. 146: 905–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saintigny, Y., F. Delacote, G. Vares, F. Petitot, S. Lambert et al., 2001. Characterization of homologous recombination induced by replication inhibition in mammalian cells. EMBO J. 20: 3861–3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharples, G. J., S. M. Ingleston and R. G. Lloyd, 1999. Holliday junction processing in bacteria: insights from the evolutionary conservation of RuvABC, RecG, and RusA. J. Bacteriol. 181: 5543–5550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solinger, J. A., G. Lutz, T. Sugiyama, S. C. Kowalczykowski and W. D. Heyer, 2001. Rad54 protein stimulates heteroduplex DNA formation in the synaptic phase of DNA strand exchange via specific interactions with the presynaptic Rad51 nucleoprotein filament. J. Mol. Biol. 307: 1207–1221. [DOI] [PubMed] [Google Scholar]
- Stewart, E., C. R. Chapman, F. Al-Khodairy, A. M. Carr and T. Enoch, 1997. rqh1+, a fission yeast gene related to the Bloom's and Werner's syndrome genes, is required for reversible S phase arrest. EMBO J. 16: 2682–2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung, P., 1997. Yeast Rad55 and Rad57 proteins form a heterodimer that functions with replication protein A to promote DNA strand exchange by Rad51 recombinase. Genes Dev. 11: 1111–1121. [DOI] [PubMed] [Google Scholar]
- Suto, K., A. Nagata, H. Murakami and H. Okayama, 1999. A double-strand break repair component is essential for S phase completion in fission yeast cell cycling. Mol. Biol. Cell 10: 3331–3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symington, L. S., 2002. Role of RAD52 epistasis group genes in homologous recombination and double-strand break repair. Microbiol. Mol. Biol. Rev. 66: 630–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trujillo, K. M., D. H. Roh, L. Chen, S. Van Komen, A. Tomkinson et al., 2003. Yeast xrs2 binds DNA and helps target rad50 and mre11 to DNA ends. J. Biol. Chem. 278: 48957–48964. [DOI] [PubMed] [Google Scholar]
- Tsutsui, Y., T. Morishita, H. Iwasaki, H. Toh and H. Shinagawa, 2000. A recombination repair gene of Schizosaccharomyces pombe, rhp57, is a functional homolog of the Saccharomyces cerevisiae RAD57 gene and is phylogenetically related to the human XRCC3 gene. Genetics 154: 1451–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno, M., T. Nakazaki, Y. Akamatsu, K. Watanabe, K. Tomita et al., 2003. Molecular characterization of the Schizosaccharomyces pombe nbs1+ gene involved in DNA repair and telomere maintenance. Mol. Cell. Biol. 23: 6553–6563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Bosch, M., K. Vreeken, J. B. Zonneveld, J. A. Brandsma, M. Lombaerts et al., 2001. Characterization of RAD52 homologs in the fission yeast Schizosaccharomyces pombe. Mutat. Res. 461: 311–323. [DOI] [PubMed] [Google Scholar]
- Van Komen, S., G. Petukhova, S. Sigurdsson, S. Stratton and P. Sung, 2000. Superhelicity-driven homologous DNA pairing by yeast recombination factors Rad51 and Rad54. Mol. Cell 6: 563–572. [DOI] [PubMed] [Google Scholar]
- Van Komen, S., G. Petukhova, S. Sigurdsson and P. Sung, 2002. Functional cross-talk among Rad51, Rad54, and replication protein A in heteroduplex DNA joint formation. J. Biol. Chem. 277: 43578–43587. [DOI] [PubMed] [Google Scholar]
- Wilson, S., N. Warr, D. L. Taylor and F. Z. Watts, 1999. The role of Schizosaccharomyces pombe Rad32, the Mre11 homologue, and other DNA damage response proteins in non-homologous end joining and telomere length maintenance. Nucleic Acids Res. 27: 2655–2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, L., and I. D. Hickson, 2003. The Bloom's syndrome helicase suppresses crossing over during homologous recombination. Nature 426: 870–874. [DOI] [PubMed] [Google Scholar]
- Yokoyama, H., H. Kurumizaka, S. Ikawa, S. Yokoyama and T. Shibata, 2003. Holliday junction binding activity of the human Rad51B protein. J. Biol. Chem. 278: 2767–2772. [DOI] [PubMed] [Google Scholar]