

Abstract
This article presents an efficient importance-sampling method for computing the likelihood of the effective size of a population under the coalescent model of Berthier et al. Previous computational approaches, using Markov chain Monte Carlo, required many minutes to several hours to analyze small data sets. The approach presented here is orders of magnitude faster and can provide an approximation to the likelihood curve, even for large data sets, in a matter of seconds. Additionally, confidence intervals on the estimated likelihood curve provide a useful estimate of the Monte Carlo error. Simulations show the importance sampling to be stable across a wide range of scenarios and show that the Ne estimator itself performs well. Further simulations show that the 95% confidence intervals around the Ne estimate are accurate. User-friendly software implementing the algorithm for Mac, Windows, and Unix/Linux is available for download. Applications of this computational framework to other problems are discussed.
THE effective size Ne of a population is an important parameter determining the rate at which genetic drift and inbreeding occur in the population, as well as the population's capacity to respond to natural selection and to purge itself of deleterious mutations. It is consequently a parameter of great interest. However, it is difficult to estimate Ne using demographic data alone, especially for organisms with high fecundity and high juvenile mortality. For this reason, a variety of methods have been developed for estimating Ne from genetic data, including the “temporal methods” in which a population's effective size is estimated using data on the change of allele frequencies observed in two or more temporally spaced genetic samples.
The first temporal methods used moment-based estimators (Krimbas and Tsakas 1971; Nei and Tajima 1981; Pollak 1983; Waples 1989; Jorde and Ryman 1995). These estimators suffer from upward bias when low-frequency alleles are present. Williamson and Slatkin (1999) introduced a likelihood-based estimator of Ne by modeling the genetic samples as observations of the hidden Markov chain that arises from the Wright-Fisher population model. They showed the likelihood-based estimator to be less biased than the moment-based estimators, but their formulation allowed only for the analysis of loci with two alleles. Anderson et al. (2000) extended that work to loci with more than two alleles, using a computationally intensive Monte Carlo likelihood scheme. Using the same hidden Markov model, Wang (2001) developed a faster method for approximating the likelihood and conducted numerous simulations demonstrating the superiority of the likelihood-based method over moment-based estimators.
Berthier et al. (2002) introduced a likelihood method for two temporally spaced samples based on a different underlying model—they derive the likelihood using the coalescent (Kingman 1982; Hudson 1990). This provides a computational advantage when a large number of generations separate the samples. Additionally, it is easier to understand how this model applies to a continuously reproducing population rather than the likelihood models based on the discrete-generation Wright-Fisher population. Beaumont (2003) extended Berthier et al.'s (2002) model to multiple samples in time and developed several computational improvements. He also provided a general formula for using importance sampling within Markov chain Monte Carlo (MCMC) in difficult problems. Unfortunately, the approaches of both Berthier et al. (2002) and Beaumont (2003) are computationally intensive, requiring computation on the order of hours to analyze a small data set of 30 individuals per sample with 10–20 loci (Berthier et al. 2002). Further, since the posterior density curves for Ne are obtained by performing density estimation on values of Ne generated from a Markov chain, it is difficult to assess their accuracy.
The purpose of this article is to present a more efficient Monte Carlo approximation of the two-sample likelihood of Berthier et al. (2002). This new method is an importance sampling approach that is upward of 1000 times faster than the MCMC method. Excellent approximations to the likelihood curve for Ne are achieved in several seconds. Further, since the method is not based on MCMC, assessing the accuracy of the Monte Carlo estimate is easy and robust. In the following I review the likelihood introduced by Berthier et al. (2002). Then I present the new importance sampling method for computing the likelihood. Finally, I conduct simulations to verify that the estimates obtained are comparable to those in Berthier et al. (2002), to explore the accuracy of the importance sampling method, to assess the behavior of the estimator in the presence of many alleles, to show that the confidence intervals for estimates of Ne using the genealogical model are reliable, and to determine how much effect random mutations have on the estimate of Ne.
PROBABILITY MODEL
I first consider the probability model for a single locus. The extension to multiple loci is straightforward and is described later. The data are two genetic samples, one of n0 codominant gene copies (n0/2 diploid individuals) assumed sampled without replacement from the population at time 0, and another sample of nT codominant gene copies assumed sampled with replacement from the population T generations before time 0, at time T. The sample at time 0 is assumed to be sampled without replacement because we will be modeling the sample using the neutral coalescent, which assumes that the sample consists of n0 distinct gene copies sampled from the population. In contrast, the sample at time T is assumed to be sampled with replacement because that allows us to model it as a multinomial sample, which, as described below, leads to further simplifications. In practice, samples are typically drawn without replacement because distinct individuals are seldom multiply sampled, and, if they are, then the duplicates are identified by allelic identity at multiple loci, and one of the individuals is removed. The model of sampling with replacement, however, is a good approximation of sampling without replacement as long as the actual size (and not necessarily the effective size) of the population from which the sample is taken is much larger than the sample itself.
The number of distinct allelic types observed in the samples at times 0 and T is denoted by K, and the observed counts of different allelic types in the samples are denoted a0 = (a0,1, … , a0,K) and aT = (aT,1, … , aT,K), respectively. We denote by K(0) and K(T) the number of distinct allelic types found in the sample at time 0 or in the sample at time T, respectively. What constitutes an allelic type will depend on the genetic marker system being used. For example, if one is using microsatellites, then alleles correspond to different numbers of repeats observable on a gel; with allozymes the alleles correspond to proteins with different electrophoretic mobilities; with single-nucleotide polymorphisms the alleles correspond to different nucleotide bases, etc. The probability model of Berthier et al. (2002) arises by considering the genealogy of the n0 gene copies sampled at time 0 and assuming that the genealogy follows the neutral coalescent process for a population of size Ne between time T and time 0.
At time 0 the n0 gene copies represent n0 separate lineages; however, if we were to trace each of those lineages back in time, some lineages may merge (“coalesce”) so that the number of lineages extant in the population at time T and ancestral to the n0 gene copies will be a number smaller than or equal to n0. We let nf denote the (unknown) number of lineages extant at time T that are ancestral to the n0 sampled genes. If the effective size of the population is small, coalescences will occur rapidly and nf will typically be smaller than it would be if Ne were large. The probability that n0 lineages at time 0 are the descendants of nf lineages at time T in a population of effective size Ne can be computed analytically (Tavaré 1984) as described below.
The nf extant lineages at time T can be considered nf gene copies that existed in the population at time T and that represent all of the ancestors at time T of the n0 genes sampled at time 0. We denote the (unknown) numbers of different allelic types carried among those nf ancestors by af = (af,1, … , af,K). It is assumed that no mutation occurs between time T and time 0. As discussed later, this assumption means the method is suitable for samples that are taken a moderate number of generations apart. It follows from this that only allelic types appearing in the sample at time 0 appear among the nf ancestors (i.e., a0,k = 0 implies af,k = 0 for all k = 1, … , K). It also follows that each allelic type observed in the n0 genes at time 0 must occur at least once among the nf ancestors (i.e., a0,k > 0 ⇒ af,k > 0, k = 1, … , K), which implies that K(0) ≤ nf ≤ n0. Just as the sample of nT gene copies was assumed to be sampled with replacement from the population at time T, the nf gene copies are assumed to be a separate, independent sample, with replacement, of nf gene copies from the population at time T. The unknown frequencies of the K alleles in the population at time T are denoted by p = (p1, … , pK). My notation differs here from that of Berthier et al. (2002) who used x to denote the allele frequencies. The vector p is a nuisance parameter that may be integrated out by assuming a prior distribution for it. The prior is taken to be a K − 1-dimensional Dirichlet distribution with parameter λ = (λ1, … , λK). Such a distribution arises as the equilibrium distribution of allele frequencies under a K-allele model with reversible mutation (Wright 1937). Often each λk is set equal to 1, giving a uniform prior for p, although, especially for large K, another sensible prior would be λk = 1/K, k = 1, … , K (Kass and Wasserman 1995).
The above sampling scheme implies a set of conditional probability densities involving the parameters and variables a0, aT, nf, af, p, n0, nT, T, Ne, and λ. These conditional densities are derived as follows. Both aT and af are independent multinomial samples from a population with allele frequencies p. Thus, P(aT|nT, p) ≡ MultK(nT, p) and P(af|nf, p) ≡ MultK(nf, p), where MultK(n, p) denotes the probability mass function of a multinomial random variable of n trials with K categories having cell probabilities p. Conditional on af, the counts of different alleles a0 among the n0 descendants sampled at time 0 follow a distribution having the form of a Dirichlet-compound multinomial distribution (Johnson et al. 1997) defined by a product of binomial coefficients,
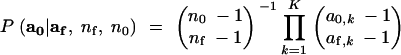 |
1 |
for values of a0 satisfying a0,k ≥ af,k, k = 1, … , K, and where the binomial coefficient
| −1 |
| −1 |
is defined as 1 (to easily deal with values of k for which a0,k = af,k = 0). This follows from the fact that forward in time, the bifurcations of a neutral coalescent starting with labeled lineages can be interpreted as steps in a Pólya-Eggenberger urn scheme (Hoppe 1984) in which each round of sampling involves taking a ball from the urn and placing it in a separate sample, and then returning two balls of like color to the urn. Under this interpretation, the nf lineages at time T are like nf balls in an urn, each one colored according to the allelic type it carries, so af counts the numbers of balls of K different colors in an urn before the onset of Pólya-Eggenberger sampling. Then, a0 represents the number of balls of each color in the urn after n0 − nf rounds of sampling. This is equivalent to collecting a sample in n0 − nf rounds of sampling in which the number of different colors of balls is given by the vector a0 − af, which follows the probability given in (1).
The probability that n0 lineages at time 0 have nf extant ancestral lineages T generations in the past in a neutral coalescent process, given an effective population size of Ne, can be computed following Tavaré (1984). Letting t = T/(2Ne) we have
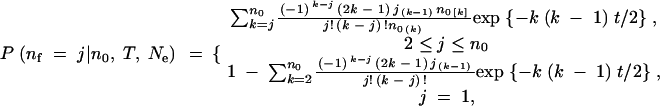 |
2 |
where i[k] = i(i − 1) · · · (i − k + 1) and i(k) = i(i + 1) · · · (i + k − 1) are notations for the falling and rising factorial functions, respectively.
With the component conditional densities specified as above, the joint probability density of all the variables may be written
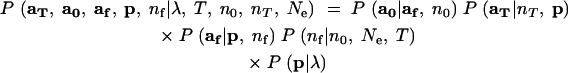 |
3 |
The factorization of the probability model above is depicted in the acyclic directed graph of Figure 1.
Figure 1.—

A directed graph showing the relationship of the observed and latent variables in the probability model arising from the genealogical perspective. Each node represents a variable in the model. Solid nodes represent observed quantities, the shaded node represents a variable whose value is assumed to provide a prior distribution, and open nodes represent unobserved variables or, in the case of Ne, the unknown parameter of interest.
The likelihood of Ne is obtained by integrating the nuisance parameter p and the unknown, latent variables af and nf out of the joint density:
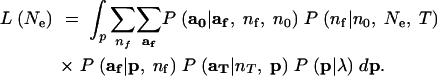 |
4 |
The sum over af in (4) has a great many terms in it, especially if n0, nf, and K are large, so an approximation to that sum is desirable. The next section will explain how that sum can be efficiently approximated using an importance-sampling algorithm.
Before describing my own importance-sampling algorithm, I briefly describe the computational approach taken by Berthier et al. (2002) and Beaumont (2003). They use the Metropolis-Hastings algorithm to define a Markov chain of values of Ne (and of p in Berthier et al. 2002), having a limiting distribution proportional (or almost proportional) to
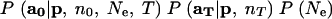 |
5 |
in the case of in Berthier et al. (2002) and
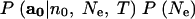 |
6 |
in the case of Beaumont (2003), where P(Ne) is a prior distribution assumed for Ne. Samples from the Markov chain are used to make a density estimate of the posterior density for Ne. Note that the first two terms of (5) are what would remain of the integrand in (4) after the sum over nf and af was performed. Similarly, the first term in (6) is what would remain of the integrand in (4) after integrating out p and then summing over nf and af. Thus, the MCMC method requires approximating the two sums in (4) for every step in the Markov chain to approximate (5) or (6) for use in a Metropolis-Hastings ratio. Berthier et al. (2002) and Beaumont (2003) do this approximation by Monte Carlo, using P(af, nf|a0, Ne, T) as an importance sampling distribution. As Beaumont (2003) notes, this importance sampling distribution could be improved by accounting for the dependence (which is apparent in Equation 4 and in the directed graph of Figure 1) of af on p. That improvement and others are demonstrated in the next section.
EFFICIENT APPROXIMATION OF THE LIKELIHOOD
The computation of (4) presented here is made efficient by: (i) avoiding the use of MCMC altogether; (ii) integrating over p in (4) analytically; (iii) recognizing that the difficult steps in calculating (4) involve neither Ne nor T, so that a number of quantities may be computed only once for any a0 and aT and then used to quickly calculate L(Ne) for any value of Ne; and (iv) choosing a suitable importance-sampling distribution.
As in Beaumont (2003), analytical integration of the nuisance variable p proceeds from Mosimann's (1962) result that if a has a multinomial distribution with cell probabilities p, and p has a Dirichlet distribution, then, marginally, a will follow the Dirichlet-compound multinomial distribution. Starting from (4), reversing the order of integration and summation yields line (7) in the equation below. Because P(aT|nT, p) is a multinomial distribution (which follows from the assumption that the sample at time T is drawn with replacement), the product of P(aT|nT, p) and P(p|λ) is proportional to P(p|aT, nT, λ)—the Dirichlet-distributed posterior probability density of p conditional on aT and the prior λ. Recognizing this yields (8). Then using the fact that P(p|aT, nT, λ) is a Dirichlet density and the fact that af follows a multinomial distribution (again, this is a consequence of the assumption that the allelic types of the nf lineages are a sample with replacement from the population at time T), we apply Mosimann's (1962) result to obtain (9):
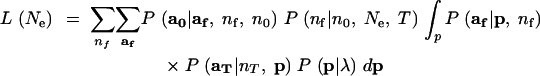 |
7 |
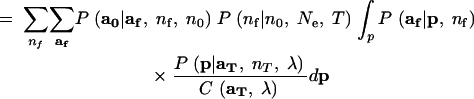 |
8 |
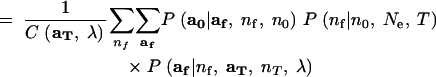 |
9 |
P(af|nf, aT, nT, λ) is a Dirichlet-compound multinomial distribution with parameters aT + λ, so that
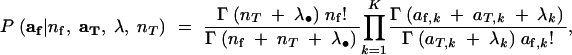 |
10 |
where λ• denotes 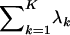 . C(aT, λ) is a constant involving multinomial coefficients and the coefficients of the Dirichlet distribution:
. C(aT, λ) is a constant involving multinomial coefficients and the coefficients of the Dirichlet distribution:
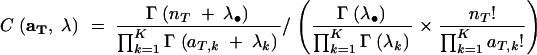 |
11 |
By rearranging the sums in (9) to obtain
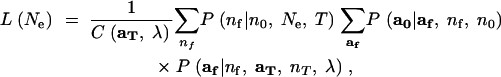 |
12 |
it can be seen that the difficult part in evaluating L(Ne) is just the sum over af. It is also clear that once the sum over af has been computed for every value of nf from K(0) to n0, then computing the likelihood for any value of Ne is achieved by a small sum over the possible values of nf. Hence, the primary task here is to develop a good approximation for
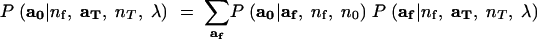 |
13 |
This is undertaken by Monte Carlo, made efficient by importance sampling. The importance-sampling formulation follows from the fact that (13) may be rewritten as
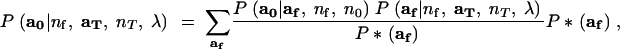 |
14 |
where P*(af) is a probability mass function for af having the property that for any value of af for which P*(af) = 0, the product P(a0|af, nf, n0)P(af|nf, aT, nT, λ) is also equal to zero. Equation 14 suggests that P(a0|nf, aT, nT, λ) may be approximated by simulating m values of af, af1, … , afm from P*(af), and computing
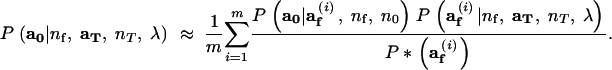 |
15 |
The variance of this Monte Carlo estimate of Ne is reduced to the extent that P*(af) can be made proportional to P(a0|af, nf, n0)P(af|nf, aT, nT, λ) (Hammersley and Handscomb 1964).
Our goal is thus to find a P*(af) that is approximately proportional to P(a0|af, nf, n0)P(af|nf, aT, nT, λ). Such an approximation may be obtained by sequentially simulating the components of af. The reasoning for this is as follows: P(a0|af, nf, n0)P(af|nf, aT, nT, λ) is the product of two Dirichlet-compound multinomial probability mass functions, and the marginal distribution of any component of a Dirichlet-compound multinomial random vector follows a beta-binomial distribution with parameters that are easily computed (see Johnson et al. 1997, p. 81). We are thus able to compute the marginal distribution of af,1, the first component of af, from a distribution exactly proportional to P(a0|af, nf, n0)P(af|nf, aT, nT, λ), as desired. That marginal distribution is proportional to the product of two beta-binomial probability mass functions, and normalizing it can be done quickly because af,1 may assume no more than nf values. We may then simulate a value, af,1i, from that distribution. After simulating af,1i, the alleles corresponding to k = 1 are conceptually “discarded” from the data set (thus reducing n0 to n0 − a0,1, nf to nf − af,1, and nT to nT − aT,1) and a similar scheme is pursued to simulate af,2. Then the alleles corresponding to k = 2 are discarded and af,3 is simulated, and so forth.
Mathematically, the probability mass function, P*(af), is defined to be
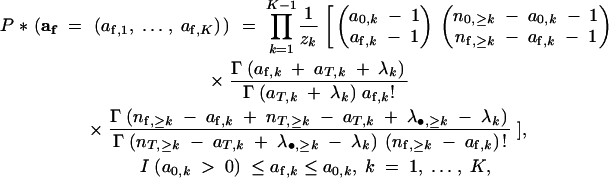 |
where I(a0,k > 0) is 1 if a0,k > 0 and 0 otherwise, and n0,≥k, nf,≥k, nT,≥k, and λ•,≥k are defined to be n0 − ∑j<ka0,j, nf − ∑j<kaf,j, nT − ∑j<kaT,j, and λ• − ∑j<kλj, respectively. The value zk is a normalizing constant equal to, for each k, the sum of the part within square brackets between the values of I(a0,k > 0) and mina0,k, nf,≥k − ∑j>kIa0,j > 0, inclusive.
While this P*(af) is not exactly proportional to P(a0|af, nf, n0)P(af|nf, aT, nT, λ), it is close enough that the Monte Carlo estimate, using (14), is quite good, even with m as small as 100. Further, by judicious use of recurrence relations for binomial coefficients and the gamma function, and by storage of frequently used quantities, values of afi may be simulated from P*(af) rapidly.
Once the Monte Carlo approximations to P(a0|nf, aT, nT, λ) are computed for all possible values of nf, they may be used in (12), to compute the likelihood for any value of Ne. In practice, the likelihood curve is computed by evaluating (12) over a fine grid of values of Ne. The maximum-likelihood estimate N̂e can then be found by parabolic interpolation of the point on the grid with highest likelihood and its two neighbors.
When data are available on multiple loci that are not in linkage disequilibrium, the overall likelihood is the product over loci of the likelihoods for each locus. The variance of the Monte Carlo estimator can be computed by standard methods, providing a direct estimate of the Monte Carlo error. For multiple loci, the calculation of the Monte Carlo variance follows that in the Appendix of Anderson et al. (2000).
The calculations described above are implemented in the computer program CoNe, which may be downloaded from santacruz.nmfs.noaa.gov/staff/eric_anderson/. CoNe computes a Monte Carlo estimate of the likelihood curve and summarizes the Monte Carlo error with upper and lower 95% confidence intervals on the estimate of the likelihood curve. It also reports the maximum-likelihood estimate (MLE), N̂e, and a 95% confidence interval around N̂e. The endpoints of the confidence interval around N̂e are the values of Ne for which the natural logarithm of the likelihood is 1.96 units smaller than log L(N̂e). Given any prior distribution for Ne, the posterior distribution may be computed from the likelihood, if desired.
SIMULATIONS AND RESULTS
It is not my goal here to undertake an exhaustive set of simulations comparing the genealogical method to other methods for estimating Ne. Such a study has been completed recently (Tallmon et al. 2004). Instead, I conduct four sets of simulations to (i) confirm that the estimator presented here estimates the same thing as Berthier et al.'s (2002) estimator, (ii) assess the reliability of the estimate of the Monte Carlo error, (iii) assess the method's behavior in the presence of many alleles, (iv) demonstrate that the 95% confidence interval on N̂e computed by CoNe is accurate, and (v) assess the effect of mutations on the estimation of Ne with CoNe.
Comparison to previous results:
First, I investigated the difference in running times between CoNe and the program TM3 presented by Berthier et al. (2002). (The program TMVP presented in Beaumont (2003) is supposed to be somewhat faster, but it gave spurious results on the data set used to test running times.) To do this comparison I used the data file supplied as an example data set in the distribution of TM3. It includes simulated data of 10 loci sampled from 50 diploids on two occasions separated by a scaled time of t = 0.05. I analyzed it assuming that the number of generations between samples was 10. Accordingly, the correct Ne is 100. The analysis of the data by CoNe using m = 250 importance-sampling repetitions required user and system time of 2.0 sec on a 2-GHz Macintosh G5 processor and had a maximum memory usage of 1.6 Mb. The likelihood curve was estimated with negligible Monte Carlo error (Figure 2, thick solid line).
Figure 2.—

Comparison of estimated log L(Ne) between CoNe and TM3. The thick solid line shows the estimated log-likelihood curve produced by CoNe in 2 sec. Results from runs of TM3 that required 20, 200, and 2000 sec are shown by the dotted, dashed, and light solid line, respectively.
I then analyzed the same data set on the same computer using TM3 with the default settings. After 20, 200, and 2000 sec, I took the output and estimated the log-likelihood curve using the density function in the computer package R (Ikaha and Gentleman 1996). Each of these curves is plotted in Figure 2. It is apparent that after running 1000 times as long as CoNe, TM3 has obtained a good estimate for N̂e, but it has not estimated the whole likelihood curve very well. The maximum memory usage for each run of TM3 was 179 kb.
Second, I repeated the simulation experiment performed by Berthier et al. (2002), in which samples of L loci (L = 5, 10, or 20) from 30 or 60 diploid individuals, separated by one or five generations, were drawn from simulated populations of effective size 10, 20, or 50, with a variety of different initial allele frequencies (scenarios A–C, see Table 1 legend). (For brevity I omit the simulation of allele frequency scenario F from Berthier et al. 2002.) For each combination of parameters, 2000 simulated data sets were analyzed with CoNe using 1000 Monte Carlo samples (i.e., m = 1000), and the median maximum-likelihood estimate, the square root of the mean squared error, and a summary of the confidence intervals obtained were recorded. The results of these simulations are shown in italics in Table 1. The results from Berthier et al.'s (2002) simulations are shown for comparison in regular type in adjacent columns. The two methods are clearly comparable, although CoNe is much faster. The difference in results between the two methods is accounted for by the fact that this study sampled 10 times as many simulated data sets, and the distribution of estimated values of Ne is heavy tailed to the right.
TABLE 1.
Comparison of results fromCoNe and from Berthieret al.'s (2002) method
| L | n0/2 | AF | N̂e(SE) | N̂e(SE) |
|
|
C.I.'s | C.I.'s | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Ne = 10, T = 1 | ||||||||||
| 5 | 30 | A | 8.9 (1.0) | 8.4 (2.5) | 44.5 | 34.3 | 2.0–500.0 | 2.9–463 | ||
| 5 | 60 | A | 7.9 (0.5) | 7.3 (0.3) | 24.6 | 4.2 | 2.1–122.5 | 2.8–57.7 | ||
| 10 | 30 | A | 8.6 (0.1) | 7.8 (0.3) | 5.5 | 4.3 | 3.4–84.5 | 3.6–130.6 | ||
| 10 | 30 | C | 9.1 (2.3) | 12.7 (6.6) | 104.1 | 93.5 | 1.0–500.0 | 2.1–475.3 | ||
| 20 | 30 | A | 8.6 (0.1) | 7.8 (.1) | 2.9 | 2.6 | 4.0–29.4 | 4.4–21.2 | ||
| Ne = 20, T = 5 | ||||||||||
| 5 | 30 | C | 19.4 (2.9) | 19.3 (8.1) | 135.0 | 119.9 | 1.5–500.0 | 3.6–478.2 | ||
| 5 | 60 | A | 18.0 (0.2) | 17.3 (0.5) | 7.1 | 6.3 | 5.9–65.5 | 7.8–57.0 | ||
| 5 | 30 | B | 20.4 (0.2) | 19.4 (0.6) | 10.2 | 9.3 | 7.4–105.6 | 8.9–124.9 | ||
| 5 | 60 | B | 19.3 (0.2) | 18.3 (0.5) | 8.2 | 6.8 | 7.4–75.6 | 8.4–62.0 | ||
| 5 | 30 | A | 19.0 (0.2) | 18.1 (0.5) | 9.3 | 7.7 | 5.9–105.8 | 6.3–105.6 | ||
| 10 | 30 | A | 18.7 (0.1) | 18.4 (0.4) | 5.2 | 5.4 | 7.9–53.1 | 9.7–53.9 | ||
| 20 | 30 | A | 18.8 (0.1) | 17.7 (0.3) | 3.7 | 3.9 | 10.1–37.5 | 10.6–35.3 | ||
| Ne = 50, T = 5 | ||||||||||
| 10 | 60 | A | 49.5 (0.4) | 47.4 (1.0) | 16.0 | 17.3 | 21.5–159.3 | 23.3–136.7 | ||
| 10 | 30 | A | 51.9 (0.6) | 49.9 (2.1) | 29.5 | 30.0 | 18.7–408.7 | 20.4–435.5 | ||
| 20 | 30 | A | 51.9 (0.3) | 49.5 (1.3) | 15.4 | 18.4 | 24.9–161.6 | 26.4–183.0 | ||
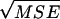
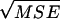
Columns in italics are results from CoNe. Columns in regular type are results from Berthier et al. (2002), taken from their Table 1. L is the number of loci, n0/2 is the number of diploid individuals sampled, AF is the allele frequency scenario [A = 5 alleles at frequencies (0.2, 0.59, 0.1, 0.07, 0.04); B = 5 alleles at uniform allele frequencies; C = 2 alleles at frequencies (0.885, 0.115)]. N̂e is the median maximum-likelihood value for Ne and SE is the standard error of the mean MLE of Ne. 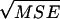 is the square root of the mean squared error. For CoNe, C.I.'s are summaries of the 95% confidence intervals for Ne computed as the ±1.96 log-likelihood unit limits. For Berthier et al.'s (2002) results, C.I.'s are summaries of the 90% credible intervals. The lower number is the 5th percentile of the lower interval limits and the higher number is the 95th percentile of the upper interval limits.
is the square root of the mean squared error. For CoNe, C.I.'s are summaries of the 95% confidence intervals for Ne computed as the ±1.96 log-likelihood unit limits. For Berthier et al.'s (2002) results, C.I.'s are summaries of the 90% credible intervals. The lower number is the 5th percentile of the lower interval limits and the higher number is the 95th percentile of the upper interval limits.
Monte Carlo error:
When estimating parameters, uncertainty is often expressed in terms of a confidence interval or a “credible set.” When the likelihood curve itself is estimated by Monte Carlo, there is another source of uncertainty due to the Monte Carlo variance in the estimate of the likelihood, and that uncertainty may also be expressed by a confidence interval. It is this uncertainty due to Monte Carlo variance that is the topic of this section, and the confidence intervals on the likelihood L(Ne), referred to here, should not be confused with confidence intervals on the estimate N̂e itself (discussed later). Ninety-five percent confidence intervals for the Monte Carlo estimate of L(Ne) may be computed by adding (for the upper limit) and subtracting (for the lower limit) 1.96 times the Monte Carlo standard error of the estimate.
In the simulations described above, 30,000 data sets were analyzed by CoNe. Of those, the data set yielding the highest Monte Carlo variance was simulated using allele frequency scenario A, with 20 loci and true Ne = 10 and T = 1. The likelihood curve for Ne given this data set and computed using m = 1000 Monte Carlo samples is shown as the solid line in Figure 3a. The dashed lines on either side of the likelihood curve are the 95% confidence intervals on the Monte Carlo estimate of L(Ne). The distance between the two dashed lines measures how much uncertainty is in the Monte Carlo estimate of L(Ne). This figure is provided as an example of how efficient this Monte Carlo scheme is. The curve in Figure 3a was achieved in <6 sec on a 2 GHz G5 processor, and, although it represents the worst Monte Carlo estimate in 30,000 data sets, the approximation is still good. By increasing the number of Monte Carlo samples to m = 50,000, an excellent approximation is achieved (Figure 3b) in only 4 min 3 sec.
Figure 3.—


The worst Monte Carlo error from 30,000 data sets. (a and b) Solid lines are the estimated likelihood curves and dashed lines are the 95% confidence intervals around the estimated likelihood curves. The data set analyzed here had the widest confidence intervals of all 30,000 data sets analyzed for Table 1. It had 20 loci with five alleles. (a) Results for a CoNe run with m = 1000 Monte Carlo replicates, requiring <6 sec on a 2-GHz G5 processor. (b) Results for m = 50,000 Monte Carlo replicates, requiring 4 min 3 sec on the same processor. The dashed lines are difficult to see in b since the confidence interval around the likelihood curve is very narrow. Clearly the Monte Carlo error is minimal, and it is easily reduced by using more Monte Carlo replicates.
It should be apparent that the lower and upper confidence intervals on the Monte Carlo estimate (Figure 3, dashed lines) provide a convenient summary of the accuracy of the approximation. This is a more useful measure of uncertainty than is available when MCMC and density estimation are used to estimate the likelihood curve.
A series of simulations was undertaken to determine how reliable the Monte Carlo confidence intervals are. This was done by analyzing 70 separate data sets [two samples, separated by 20 generations, of 100 individuals typed at 12 loci with seven alleles having frequencies drawn from a uniform Dirichlet (1, … , 1) distribution] from simulated populations of 1000 individuals. For each data set an estimate of the likelihood curve was computed with CoNe using m = 100,000 replicates. This estimate was taken to be close to the exact likelihood. Then the same data set was reanalyzed 500 more times, each time with only m = 100 replicates, and it was recorded whether the “exact” likelihood curve estimated with m = 100,000 fell within the Monte Carlo confidence intervals given by analyzing the data with m = 100. Even with as few as m = 100 replicates, the average width of the Monte Carlo confidence intervals over all the simulations was 0.07 units of log-likelihood. Sixty percent of the time, the exact likelihood curve fell entirely within the confidence intervals at all points within 4 log-likelihood units of the maximum. When confidence intervals failed to contain the exact likelihood, the average distance between the exact likelihood and the edge of the confidence interval was only 0.013. Thus, while the confidence intervals for the estimate of L(Ne) are not strictly 95% confidence intervals, they do provide a very good measure of the uncertainty in the Monte Carlo estimation. Regardless, in all simulated data sets I have analyzed, the Monte Carlo error can be reduced to negligible levels with never more than a few minutes of computation and typically in a matter of seconds.
Behavior with many alleles:
Some computational methods for estimating Ne become unstable or converge slowly when applied to data sets with many alleles (cf. Anderson et al. 2000). Therefore, I investigated the performance of the importance-sampling method and of the coalescent-based Ne estimation procedure, with loci having many alleles. CoNe was applied to simulated data of 100 individuals genotyped at 10 loci, each with K alleles and a uniform initial allele frequency. K was set equal to 5, 8, 13, 20, 30, 50, 75, or 100 in each simulation for all 10 of the loci. These simulations were done using a coalescent method. In one set of simulations the genetic drift between samples was set to that of 20 generations in a population of size 100 [i.e., a scaled time of t = T/(2Ne) = 0.1] and in another set of simulations it was set to that of 2 generations in a population of size 100 (t = 0.01). For each combination of number of alleles and amount of genetic drift, 250 data sets were simulated and analyzed. For all data sets the importance-sampling procedure remained stable. Even with as many as 100 alleles, the likelihood curve was reliably estimated with as few as m = 1000 Monte Carlo replicates. For the scaled time of 0.1 (corresponding to 20 generations in a population of size 100), the performance of the coalescent-based estimator of Ne was not greatly affected by the number of alleles. At all numbers of alleles, the estimator had a slight upward bias, with the mean maximum-likelihood estimate of Ne being ∼103—only slightly greater than the true value of 100 (Figure 4a). With a scaled time of 0.01 (corresponding to 2 generations in a population of size 100), the importance-sampling procedure was once again stable. However, the estimator itself shows an upward bias, particularly as the number of alleles increases beyond 20. Additionally, with very large numbers of alleles (75 and 100), on average, the 95% confidence interval around the maximum-likelihood estimate of Ne does not overlap the true value of 100 (Figure 4b).
Figure 4.—


Summary of simulations using loci with different numbers of alleles. Points on solid lines show mean maximum-likelihood estimates from 250 simulated data sets as a function of number of alleles. See text for full explanation of simulations. Points on dashed lines show mean of upper and lower 95% confidence intervals for Ne. Vertical bars are two times the standard error of the mean in all cases. The thin dotted line shows the true value of Ne = 100. (a) Scaled time = 0.1, corresponding to 20 generations of drift with Ne = 100. (b) Scaled time = 0.01, corresponding to 2 generations of drift with Ne = 100. Note the upward bias in b.
It is important to note that such pathological data sets would rarely, if ever, be encountered from natural populations having an Ne low enough that it might be reliably estimated. The appearance of so many alleles in such a population would suggest either that the alleles were under some sort of balancing selection or that the mutation rate at the locus was quite high, violating the assumptions of the Ne estimation method.
Accuracy of confidence intervals for Ne:
CoNe reports a 95% confidence interval around the MLE, N̂e. The true value of Ne ought to be contained in that confidence interval in 95% of data sets (simulated under the model) analyzed by CoNe. Tallmon et al. (2004) report that the credible intervals (these are like confidence intervals, but are computed from a Bayesian perspective) computed by a related program, TMVP (Beaumont 2003), are grossly inaccurate. TMVP is based on the same likelihood model as CoNe, but it approximates the likelihood by MCMC instead of using the efficient importance sampling algorithm presented here. In Tallmon et al.'s (2004) simulations, data sets of n = 20 or 60 diploid individuals were drawn from populations of Ne = 20, 50, or 100, separated by 1, 3, 5, or 10 generations. The individuals were genotyped at 5 or 15 loci, which were initialized with eight alleles having frequencies drawn from a uniform Dirichlet distribution. Over all the simulation conditions, TMVP's credible intervals failed to contain the true value of Ne 24.4% of the time. In one instance, with Ne = 20, n = 60, T = 3, and with 15 loci, TMVP's credible intervals failed to contain the true Ne >78% of the time.
Since CoNe is based on the same likelihood model as TMVP, I performed simulations like those of Tallmon et al. (2004) to investigate whether CoNe's confidence intervals suffer similar degrees of inaccuracy. For each set of simulation parameters, I applied CoNe to 1000 simulated data sets, recording how often the true Ne was not contained within CoNe's 95% confidence intervals for Ne (Table 2).
TABLE 2.
Proportion of 95% confidence intervals that do not contain trueNe
|
Ne = 20
|
Ne = 50
|
Ne = 100
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Conditions | T | L | U | Tot | N̂ȯe | L | U | Tot | N̂ȯe | L | U | Tot | N̂ȯe |
| 5 loci | |||||||||||||
| n = 20 | 1 | 0.0 | 2.1 | 2.1 | 8.9 | 0.0 | 1.0 | 1.0 | 37.8 | ||||
| 3 | 4.5 | 1.5 | 6.0 | 0.1 | 0.5 | 1.2 | 1.7 | 7.2 | |||||
| 5 | 3.9 | 1.0 | 4.9 | 0.0 | 4.5 | 1.1 | 5.6 | 0.7 | |||||
| 10 | 2.9 | 3.0 | 5.9 | 0.0 | 7.2 | 1.0 | 8.2 | 0.0 | |||||
| n = 60 | 1 | 1.6 | 4.2 | 5.8 | 0.1 | 0.0 | 1.3 | 1.3 | 6.2 | 0.0 | 1.2 | 1.2 | 26.8 |
| 3 | 1.9 | 4.6 | 6.5 | 0.0 | 3.8 | 2.1 | 5.9 | 0.1 | 2.7 | 1.4 | 4.2 | 1.6 | |
| 5 | 1.6 | 4.5 | 6.1 | 0.0 | 3.8 | 2.8 | 6.6 | 0.0 | 4.2 | 1.9 | 6.1 | 0.4 | |
| 10 | 1.1 | 4.8 | 5.9 | 0.0 | 3.3 | 3.3 | 6.6 | 0.0 | 4.1 | 2.2 | 6.3 | 0.0 | |
| 15 loci | |||||||||||||
| n = 20 | 1 | 6.9 | 0.6 | 7.5 | 1.4 | 0.0 | 0.3 | 0.3 | 29.6 | ||||
| 3 | 5.6 | 1.2 | 6.8 | 0.0 | 10.8 | 0.3 | 11.1 | 0.8 | |||||
| 5 | 4.6 | 1.1 | 5.7 | 0.0 | 9.4 | 0.7 | 10.1 | 0.0 | |||||
| 10 | 3.6 | 2.1 | 5.7 | 0.0 | 9.0 | 0.8 | 9.8 | 0.0 | |||||
| n = 60 | 1 | 1.4 | 7.2 | 8.6 | 0.0 | 3.2 | 1.3 | 4.5 | 0.9 | 0.0 | 1.1 | 1.1 | 12.9 |
| 3 | 0.3 | 7.6 | 7.9 | 0.0 | 3.8 | 2.8 | 6.6 | 0.0 | 5.4 | 1.1 | 6.5 | 0.0 | |
| 5 | 0.5 | 7.0 | 7.5 | 0.0 | 3.1 | 2.7 | 5.8 | 0.0 | 4.9 | 0.9 | 5.8 | 0.0 | |
| 10 | 0.8 | 7.6 | 8.4 | 0.0 | 2.2 | 2.6 | 4.8 | 0.0 | 4.8 | 2.2 | 7.0 | 0.0 | |
L(U) is the proportion of lower (upper) endpoints of 95% confidence intervals that are greater (less) than the true Ne of 20, 50, or 100. “Tot” is the proportion of all confidence intervals that do not contain the true Ne. N̂ȯe is the proportion of simulated data sets for which the maximum-likelihood estimate of Ne was >400 (and for which the confidence interval was not considered). Values are from 1000 simulated data sets of n diploids sampled T generations apart. Following Tallmon et al. (2004) simulations of n = 20 when Ne = 100 were not done.
CoNe's confidence intervals appear to be more accurate than TMVP's credible intervals as reported by Tallmon et al. (2004). Over all simulation conditions, true Ne was contained in the 95% confidence interval 94.3% of the time, just as expected. The worst performance of CoNe's 95% confidence intervals was on the data simulated with Ne = 50, n = 20, T = 3, using 15 loci, when 11.1% of the time the true Ne was not contained in the confidence interval.
It is not clear why Tallmon et al. (2004) found TMVP's credible intervals to perform so poorly when evaluated from a frequentist perspective on simulated data. It is possible that the Markov chain of Ne values produced by TMVP was not run long enough to achieve a good estimate of the posterior density near the tails of the posterior distribution. At any rate, the confidence intervals around Ne computed by CoNe seem to be reliable, and it is straightforward to assess how well the Monte Carlo estimate of the likelihood has converged.
The effect of mutations:
Methods for estimating Ne from temporally spaced samples have traditionally been applied to samples separated by a small number of generations in populations of plants or animals. In such situations, the assumption of no mutation is quite reasonable. However, increasingly the ability to extract and amplify genetic markers from archived samples, as well as the investigation of short-lived organisms like bacteria and viruses, makes it more likely that two samples will be separated by enough time that the assumption of no mutation may be violated. An apparent mutation can be caused by any heritable alteration that changes the observed allelic state of an allelic type. Depending on the type of marker system used these alterations could be point mutations, insertions, deletions, recombinations, or gene conversions, etc. I undertook a short simulation to determine under what conditions (of mutation rate and time between samples) mutation can appreciably affect the inference of Ne with a program like CoNe. I initialized a simulated Wright-Fisher population of Ne = 1000 diploids at time T with allele frequencies drawn from a uniform Dirichlet distribution with eight alleles and simulated the population forward in time until time 0, under both an infinite-alleles model (IAM) of mutation and a symmetric K-allele model (KAM) of mutation, with mutation rate u per gamete per generation. Samples of size 100 were drawn at times T and 0, and CoNe was used to estimate Ne and the scaled time t = T/(2Ne). This was done for all combinations of u ∈ {0, 10−3, 10−4, 10−5, 10−6} and T ∈ {100, 200, 400, 600, 1000} generations. For each T and u and mutation model (IAM or KAM) 300 data sets were simulated and analyzed. The mean estimated Ne and the mean estimated scaled time t for each condition were recorded.
Mutation biases the estimate of Ne upward. This makes sense—it tends to counteract the effects of drift (i.e., the fixation and loss of alleles), so it makes it appear that the population is larger than it is. In investigating the effect of mutation it is more convenient to express its effect on the estimates of the scaled time t = T/(2Ne). Obviously mutation biases the estimate of the scaled time t between samples downward. Figure 5 plots the results of the simulations.
Figure 5.—

The effect of mutation on estimates of t = T/(2Ne) under the infinite-alleles model (A) and the K-allele model (B). In A and B the x-axis plots the mean from 300 simulated data sets of the estimate of t, when the true value of t was 0.05, 0.1, 0.2, 0.3, or 0.5 and the mutation rate is zero. The y-axis shows the mean estimated t from 300 data sets simulated with mutation at a rate u as indicated by the text to the right of each line. If mutation is causing no bias in the estimate, then the points will fall along the y = x line, which is indicated by the dotted line. Higher values of the mutation rate and higher values of the true t between sampling episodes increase the amount of bias that mutation causes. A downward bias in the estimate of t means an upward bias in the estimate of Ne.
On Figure 5's x-axis are the mean estimates of t = T/(2Ne) for each of the five values of T with no mutation (u = 0). On Figure 5's y-axis are the mean estimates of t under either the IAM or the KAM models for the different values of u. It is clear from the figure that the effect of mutation becomes more pronounced as t increases. This is expected—as more time elapses between the samples, there is more opportunity for mutations to occur. However, it is also clear that the mutation rate must be quite high for it to have a substantial effect, especially at values of t < 0.2.
Because mutations will have an effect only if they occur on lineages ancestral to the sample at time 0, and because the probability that such mutations will occur depends on the scaled mutation rate θ = 4Neu and only weakly on sample size, some rough generalizations may be drawn from the above results. When θ < 4 × 1000 × 10−5 = 0.04 the effect of mutation for all t < 0.5 is not overwhelming. Further, for the KAM, scaled mutation rates of θ < 4 × 1000 × 10−4 = 0.4 are unlikely to bias estimates of Ne for t ≤ 0.1. Conversely, if you are using markers that mutate according to an IAM, then even with θ = 0.4 your estimates of t may be substantially biased downward and hence your estimates of Ne biased upward.
As a practical example, suppose you have sampled microsatellites in a fish population. Assume that the mutation rate at each locus is u = 10−4 and that the KAM provides a reasonable approximation over these timescales to the mode of mutation of microsatellites. If the estimate of the scaled time between samples is 0.1, then, if your samples are separated by no more than 200 generations, it is unlikely that mutations are biasing your estimate.
DISCUSSION
I have presented a computational method for fast and precise approximation of the likelihood for Ne under a coalescent model. This method is an importance sampling algorithm implemented in the computer program CoNe. Previous approaches used MCMC and were considerably slower. The performance of my importance-sampling algorithm demonstrates that, although MCMC is broadly applicable and is typically easy to implement, a much faster solution may be available if the problem can be decomposed in such a manner as to avoid MCMC. In addition to being faster, it is often also easier to assess the Monte Carlo error if one is using independently simulated samples (as in CoNe) rather than correlated samples from a Markov chain (as in MCMC).
The program CoNe is based upon the same underlying model as the programs TM3 (Berthier et al. 2002) and TMVP, in the case with only two samples (Beaumont 2003). Thus, the coalescent-based estimator implemented in CoNe is expected to perform similarly to TM3 and TMVP. In this article, I used computer simulations to show that estimates made by CoNe and TM3 are similar.
Some approximations to the likelihood for Ne become unstable when the data include many alleles (e.g., Anderson et al. 2000). This is not the case with CoNe. I subjected CoNe to a number of tests involving samples with very large numbers of alleles. The importance-sampling algorithm always performed well in approximating the likelihood. The maximum-likelihood estimator, however, seems to be upwardly biased for Ne when the amount of genetic drift is small, i.e., when T/(2Ne) is on the order of 0.01. This upward bias is exacerbated when more alleles at low frequency are present. Interestingly, this bias is of a different nature than the bias shown by moment-based estimators of Ne applied to data with low-frequency alleles. Moment-based estimators show more bias when drift is relatively strong and the low-frequency alleles have been lost from the population (Waples 1989). With CoNe the situation is exactly the opposite—the bias is very small (≈3%) when t = T/(2Ne) = 0.1 but the bias is more severe with t = 0.01.
I have shown how to compute the likelihood for Ne in what is essentially a frequentist analysis. However, two points must be made. First, should one desire a Bayesian posterior distribution for Ne, it can easily be computed from the likelihood. Second, the likelihood is an integrated likelihood: a prior for the allele frequencies at time T must be assumed. I have used a Dirichlet prior with parameter λ. This corresponds to the equilibrium frequencies of a K-allele model with reversible mutation or to the equilibrium distribution for allele frequencies under drift and recurrent migration from a large population (Wright 1937). In simulations (not shown) I found that changing the value of λ from (1, … , 1) to (1/K, … , 1/K) had little effect on the inference of Ne. It is accordingly unlikely that the use of other diffuse priors would greatly influence the estimates of Ne.
The importance-sampling algorithm presented here is quite efficient for the case where only two temporally spaced samples are taken; however, it is worth asking if the importance sampling could be extended to multiple samples in time (Beaumont 2003). Such a task could be challenging. The algorithm presented here works well because it is possible to compute the probability of the observed data given that there were nf lineages at time T for all n0 − K0 + 1 possible values of nf. Those probabilities are then used in (13) to compute the likelihood for Ne. Naively taking the same approach with more than two samples could lead to computational demands that are exponential in the number of samples, but it might be possible to make the problem linear in the number of samples by using an algorithm like that described in Baum (1971). Unfortunately, the conditional probabilities that would have to be calculated and normalized for each sampling episode would require considerably more (on the order of n times more, where n is the sample size) computation than they do with only two samples, and there is no guarantee that the resulting importance-sampling distribution would be as effective as it is in the two-sample case. Extending this importance sampling approach to more than two samples remains an open problem.
It is important to point out that, although the likelihood for Ne used here is based on the coalescent, the calculation of the likelihood is easier than in many other coalescent-based inference problems. By making the assumption of no mutation between the samples, it is possible to treat the different allelic types separately, without considering the number of mutational steps between alleles. This simplification makes it unnecessary to consider different topologies of coalescent trees. In effect, the formulation of (1) follows from an implicit sum over all possible topologies—without having to actually perform that sum. Thus, although the importance-sampling algorithm presented here offers dramatic improvements for calculations involving the coalescent without mutation, it is not a solution that applies equally well to other difficult problems such as computing the likelihood for θ = 4Neu from a single sample of sequences (Griffiths and Tavaré 1994a; Kuhner et al. 1995; Stephens and Donnelly 2000) or computing the likelihood of recombination rates from a single sample of sequences (Griffiths and Marjoram 1996) or of migration rates from a single sample of sequences or microsatellites (Beerli and Felsenstein 1999). In those cases, not only is it necessary to explicitly sum over different genealogical trees and their branch lengths, but also it is necessary to sum over the unknown ancestral state of the progenitor of all alleles in the sample and over locations in the tree where mutations might have occurred.
The program CoNe is intended to provide estimates of contemporary Ne of well-circumscribed populations. The Ne that is estimated is the effective size of the population that prevailed over the time interval between the samples. This contrasts with the methods that estimate θ = 4Neu from a single sample. The Ne referred to there is the effective size of the population over the entire coalescent history of the sample, which typically represents far more time than just the interval between two samples taken from the population. Recently, a method that allows the separate estimation of Ne and u (rather than estimation only of the composite parameter θ) from temporally spaced samples of sequences was developed by Drummond et al. (2002). Their program, Bayesian Evolutionary Analysis Sampling Trees (BEAST), has been used in a number of instances involving genetic sequences sampled at short time intervals from rapidly mutating and short-lived organisms like viruses or sampled at long time intervals (by obtaining DNA from subfossil material) from longer-lived organisms (reviewed in Drummond et al. 2003). BEAST is designed for use with temporally spaced samples of sequences, and the temporally spaced element of the samples is useful to the program only if the samples are separated by enough time that mutations are expected to have accumulated in the lineages. This is very different from CoNe, which uses codominant allelic count data (which may come from sequences, microsatellites, SNPs, etc.) and performs best when enough time has elapsed between the samples for a substantial amount of drift to have occurred, but not so much time that many mutations have occurred between the two sampling episodes.
In the absence of mutation the importance-sampling algorithm presented here applies directly to the general problem of computing the likelihood of the number of coalescences that have occurred during the time between two sampling episodes. Accordingly, there are a number of related inference problems to which the algorithm could be applied. First, estimating NeT, the effective size of the population at time T, and a growth rate r of the population until the sample at time zero would be straightforward. This is because (4) can be expressed as a likelihood for the scaled time t, and any pair of NeT and r implies a single scaled time t by the results of Griffiths and Tavaré (1994b) for rates of coalescence in populations of varying size; hence (4) implies a likelihood for pairs NeT, r. Also, with some modifications, the method could be used to estimate the number of lineages founding small, colonized populations. Such a problem, like the estimation of effective size, is similar to the problem of estimating the degree of inbreeding accumulated in a population between two time points. Laval et al. (2003) approach the problem of estimating inbreeding using MCMC and “several levels of approximation … to circumvent the combinatorial problems raised by the exact coalescent approach when the number of alleles increases” (p. 1199). The Monte Carlo approach taken in this article could be applied to the problem of estimating inbreeding using the exact coalescent approach, dealing with the combinatorial problem by efficient importance sampling. It is likely this would lead to a better estimator that could be computed many times more quickly and without the need to “tune” various parameters for MCMC.
Finally, the problem of estimating admixture proportions in populations subject to drift, first addressed by Thompson (1973), could be treated using the importance-sampling scheme presented here. In this scenario, an admixed population is formed a known time in the past with an unknown proportion π of the colonizers coming from population A and 1 − π from population B. Using present-day genetic samples from populations A and B and the admixed population, the goal is to estimate π while taking account of the genetic drift that has occurred in all three populations since the time of colonization. Chikhi et al. (2001) develop a coalescent-based likelihood for a two-population admixture model and estimate the admixture proportion using MCMC. Runs of the chain required ∼1 week on a 500-Mhz Pentium computer. Being able to compute the likelihood more efficiently using importance sampling would clearly be desirable, and, with some adjustments and approximation, the importance-sampling scheme presented here could deliver substantial improvements in computation time. Further, since the importance-sampling algorithm would yield an unnormalized likelihood, it would be useful for conducting tests of the null hypothesis that the admixed population contains ancestry only from the two putative source populations. This would permit a way of dealing with the possibility that the admixture contains ancestry from another, unsampled population.
The genealogical perspective provides a powerful framework for formulating likelihoods in a number of problems; however, its use in estimating Ne and admixture proportions has not been rapidly adopted, in part because of the computational burden of currently available methods. The algorithm presented in this article reduces that computational burden and should make genealogical approaches even more practical in the future.
Acknowledgments
I thank Dave Tallmon, Mark Beaumont, Montgomery Slatkin, and Carlos Garza for helpful discussions on this article; Kevin Dunham for help with testing and releasing the software; and the editor and two anonymous referees for their insightful comments. This article developed out of work initiated while E.C.A. was supported by National Institutes of Health grant GM-40282 to M. Slatkin.
References
- Anderson, E. C., E. G. Williamson and E. A. Thompson, 2000. Monte Carlo evaluation of the likelihood for Ne from temporally spaced samples. Genetics 156: 2109–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum, L. E., 1971. Statistical inference for probabilistic functions of finite state Markov chains. Ann. Math. Stat. 37: 1554–1563. [Google Scholar]
- Beaumont, M. A., 2003. Estimation of population growth or decline in genetically monitored populations. Genetics 164: 1139–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beerli, P., and J. Felsenstein, 1999. Maximum-likelihood estimation of migration rates and effective population numbers in two populations using a coalescent approach. Genetics 152: 763–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthier, P., M. A. Beaumont, J. M. Cornuet and G. Luikart, 2002. Likelihood-based estimation of the effective population size using temporal changes in allele frequencies: a genealogical approach. Genetics 160: 741–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chikhi, L., M. W. Bruford and M. A. Beaumont, 2001. Estimation of admixture proportions: a likelihood-based approach using Markov chain Monte Carlo. Genetics 158: 1347–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond, A. J., G. K. Nicholls, A. G. Rodrigo and W. Solomon, 2002. Estimating mutation parameters, population history and genealogy simultaneously from temporally spaced sequence data. Genetics 161: 1307–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond, A. J., O. G. Pybus, A. Rambaut, R. Forsberg and A. G. Rodrigo, 2003. Measurably evolving populations. Trends Ecol. Evol. 18: 481–488. [Google Scholar]
- Griffiths, R. C., and P. Marjoram, 1996. Ancestral inference from samples of DNA sequences with recombination. J. Comput. Biol. 3: 479–502. [DOI] [PubMed] [Google Scholar]
- Griffiths, R. C., and S. Tavaré, 1994. a Ancestral inference in population genetics. Stat. Sci. 9: 307–319. [Google Scholar]
- Griffiths, R. C., and S. Tavaré, 1994. b Sampling theory for neutral alleles in a varying environment. Philos. Trans. R. Soc. Lond. Ser. B 344: 403–410. [DOI] [PubMed] [Google Scholar]
- Hammersley, J. M., and D. C. Handscomb, 1964 Monte Carlo Methods. Methuen & Co., London.
- Hoppe, F., 1984. Polya-like urns and the Ewen's sampling formula. J. Math. Biol. 20: 91–94. [Google Scholar]
- Hudson, R. R., 1990 Gene genealogies and the coalescent process, pp. 1–44 in Oxford Surveys in Evolutionary Biology, edited by D. Futuyma and J. Antonovics. Oxford University Press, Oxford.
- Ikaha, R., and R. Gentleman, 1996. R: a language for data analysis and graphics. J. Comput. Graph. Stat. 5: 299–314. [Google Scholar]
- Johnson, N. L., Z. Kotz and N. Balakrishnan, 1997 Discrete Multivariate Distributions. Wiley & Sons, New York.
- Jorde, P. E., and N. Ryman, 1995. Temporal allele frequency change and estimation of effective size in populations with overlapping generations. Genetics 139: 1077–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kass, R. E., and L. Wasserman, 1995. A reference Bayesian test for nested hypotheses and its relationship to the Schwarz criterion. J. Am. Stat. Assoc. 90: 928–934. [Google Scholar]
- Kingman, J. F. C., 1982. On the genealogy of large populations. J. Appl. Probab. 19A: 27–43. [Google Scholar]
- Krimbas, C. B., and S. Tsakas, 1971. The genetics of Dacus oleae. V. Changes of esterase polymorphism in a natural population following insecticide control—selection or drift? Evolution 25: 454–460. [DOI] [PubMed] [Google Scholar]
- Kuhner, M. K., J. Yamato and J. Felsenstein, 1995. Estimating effective population size and mutation rate from sequence data using Metropolis-Hastings sampling. Genetics 140: 1421–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laval, G., M. SanCristobal and C. Chevalet, 2003. Maximum-likelihood and Markov chain Monte Carlo approaches to estimate inbreeding and effective size from allele frequency changes. Genetics 164: 1189–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosimann, J. E., 1962. On the compound multinomial distribution, the multivariate β-distribution and correlations among proportions. Biometrika 49: 65–82. [Google Scholar]
- Nei, M., and F. Tajima, 1981. Genetic drift and estimation of effective population size. Genetics 98: 625–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollak, E., 1983. A new method for estimating the effective population size from allele frequency changes. Genetics 104: 531–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens, M., and P. Donnelly, 2000. Inference in molecular population genetics (with discussion). J. R. Stat. Soc. Ser. B 62: 605–655. [Google Scholar]
- Tallmon, D. A., G. Luikart and M. Beaumont, 2004. Comparative evaluation of a new effective population size estimator based on approximate Bayesian computation. Genetics 167: 977–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavaré, S., 1984. Lines of descent and genealogical processes, and their applications in population genetics models. Theor. Popul. Biol. 26: 119–164. [DOI] [PubMed] [Google Scholar]
- Thompson, E. A., 1973. The Icelandic admixture problem. Ann. Hum. Genet. 37: 69–80. [DOI] [PubMed] [Google Scholar]
- Wang, J., 2001. A pseudo-likelihood method for estimating effective population size from temporally spaced samples. Genet. Res. 78: 243–257. [DOI] [PubMed] [Google Scholar]
- Waples, R. S., 1989. A generalized approach for estimating effective population size from temporal changes in allele frequency. Genetics 121: 379–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson, E. G., and M. Slatkin, 1999. Using maximum likelihood to estimate population size from temporal changes in allele frequencies. Genetics 152: 755–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, S., 1937. The distribution of gene frequencies in populations. Proc. Natl. Acad. Sci. USA 23: 307–320. [DOI] [PMC free article] [PubMed] [Google Scholar]