

Abstract
Marker-assisted backcrossing is routinely applied in breeding programs for gene introgression. While selection theory is the most important tool for the design of breeding programs for improvement of quantitative characters, no general selection theory is available for marker-assisted backcrossing. In this treatise, we develop a theory for marker-assisted selection for the proportion of the genome originating from the recurrent parent in a backcross program, carried out after preselection for the target gene(s). Our objectives were to (i) predict response to selection and (ii) give criteria for selecting the most promising backcross individuals for further backcrossing or selfing. Prediction of response to selection is based on the marker linkage map and the marker genotype of the parent(s) of the backcross population. In comparison to standard normal distribution selection theory, the main advantage of our approach is that it considers the reduction of the variance in the donor genome proportion due to selection. The developed selection criteria take into account the marker genotype of the candidates and consider whether these will be used for selfing or backcrossing. Prediction of response to selection is illustrated for model genomes of maize and sugar beet. Selection of promising individuals is illustrated with experimental data from sugar beet. The presented approach can assist geneticists and breeders in the efficient design of gene introgression programs.
MARKER-ASSISTED backcrossing is routinely applied for gene introgression in plant and animal breeding. Its efficiency depends on the experimental design, most notably on the marker density and position, population size, and selection strategy. Gene introgression programs are commonly designed using guidelines taken from studies focusing on only one of these factors (e.g., Hospital et al. 1992; Visscher 1996; Hospital and Charcosset 1997; Frisch et al. 1999a,b). In breeding for quantitative traits, prediction of response to selection with classical selection theory is by far the most important tool for the design and optimization of breeding programs (Bernardo 2002). Adopting a selection theory approach to predict response to marker-assisted selection for the genetic background of the recurrent parent promises to combine several of the factors determining the efficiency of a gene introgression program into one criterion.
In classical selection theory, the expectation, genetic variance, and heritability of the target trait are required, as well as the covariance between the target trait and the selection criterion in the case of indirect selection (Bernardo 2002). In backcrossing without selection, the expected donor genome proportion in generation BCn is 1/2n+1. In backcrossing with selection for the presence of a target gene, Stam and Zeven (1981) derived the expected donor genome proportion on the carrier chromosome of the target gene, extending earlier results of Bartlett and Haldane (1935), Fisher (1949), and Hanson (1959) on the expected length of the donor chromosome segment attached to the target gene. Their results were extended to a chromosome carrying the target gene and the recurrent parent alleles at two flanking markers (Hospital et al. 1992) and to a chromosome carrying several target genes (Ribaut et al. 2002).
Hill (1993) derived the variance of the donor genome proportion in an unselected backcross population, whereas Ribaut et al. (2002) deduced this variance for chromosomes carrying one or several target genes. The covariance of the donor genome proportion across a chromosome and the proportion of donor alleles at markers in backcrossing was given by Visscher (1996). In their derivations, these authors assumed that the donor genome proportion of different individuals in a backcross generation is stochastically independent. This applies to large BCn populations only (a) in the absence of selection in all generations BCs (1 ≤ s ≤ n) and (b) if each BCn−1 (n > 1) individual has maximally one BCn progeny (comparable to the single-seed descent method in recurrent selfing). Visscher (1999) showed with simulations that the variance of the donor genome proportion in backcross populations under marker-assisted selection is significantly smaller than that in unselected populations of stochastically independent individuals.
Hillel et al. (1990) and Markel et al. (1997) employed the binomial distribution to describe the number of homozygous chromosome segments in backcrossing. However, Visscher (1999) demonstrated with simulations that the assumption of binomially distributed chromosome segments results in an unrealistic prediction of the number of generations required for a marker-assisted backcross program. Hence, the expectations, variances, and covariances are known for backcrossing without selection, but these approximations are of limited use as a foundation of a general selection theory for marker-assisted backcrossing.
The objective of this study was to develop a theoretical framework for marker-assisted selection for the genetic background of the recurrent parent in a backcross program to (i) predict response to selection and (ii) give criteria for selecting the most promising backcross individuals for further backcrossing or selfing. Our approach deals with selection in generation n of the backcross program, taking into account (a) preselection for the presence of one or several target genes, (b) the linkage map of the target gene(s) and markers, and (c) the marker genotype of the individuals used as nonrecurrent parents for generating backcross generations BCs (s ≤ n).
THEORY
For all derivations we assume absence of interference in crossover formation such that the recombination frequency r and map distance d are related by Haldane's (1919) mapping function r(d) = (1 − e−2d)/2. An overview of the notation used throughout this treatise is given in Table 1.
TABLE 1.
Notation
| c | No. of chromosomes |
|---|---|
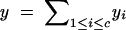 |
y, total genome length; yi, length of chromsome i. |
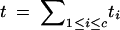 |
t, total no. of target loci; ti, no. of target loci on chromosome i. |
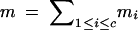 |
m, total no. of marker loci; mi, no. of marker loci on chromosome i. |
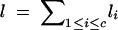 |
l, total no. of loci; li = mi + ti, no. of loci on chromosome i. |
| xi,j | Map distance between locus j on chromosome i and the telomere. |
| M | Set consisting of the indices (i, j) of marker loci M = {(i, j)|xi,j is the map position of a marker locus}. |
| T | Set consisting of the indices (i, j) of target loci T = {(i, j)|xi,j is the map position of a target locus}. |
| L = M ∪ T | Set comprising the indices (i, j) of target and marker loci. |
| di,j | Length of chromosome interval j on chromosome i in map distance. For detailed definition see Equations 16–18. |
| J | Set containing all indices (i, j) of chromosome intervals J = {(i, j)|i = 1 … c, j = 1 … li + 1}. |
| Gn,i,j, gn,i,j | Indicator variable taking the value 1 if the locus at position xi,j carries the donor allele in generation BCn and 0 otherwise. Realizations are denoted by gn,i,j. |
| Gn, gn | Random vector denoting the multilocus genotype of a BCn individual, Gn = (Gn,1,1, Gn,1,2, … , Gn,1,l1, … , Gn,c,lc). Realizations are denoted by gn. |
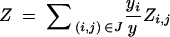 |
Zn, random variable denoting the donor genome proportion across the entire genome; Zi,j, random variable denoting the donor genome proportion in the chromosome interval corresponding to di,j.a |
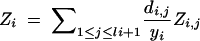 |
Zi, Random variable denoting the donor genome proportion on chromosome i.a |
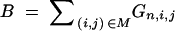 |
Random variable counting the number of donor alleles at marker loci. |
Random variables Z, Zi, and Zi,j refer to the homologous chromosomes originating from the nonrecurrent parent.
In the following we derive (1) the expected donor genome proportion of a backcross individual conditional on its multilocus genotype gn at marker and target loci, (2) the expected donor genome proportion of a backcross population generated by backcrossing an individual with multilocus genotype gn to the recurrent parent, and (3) the expected donor genome proportion of the wth-best individual of a backcross population of size u generated by backcrossing an individual with multilocus genotype gn to the recurrent parent.
Probability of multilocus genotypes:
We derive the probability that a BCn individual has multilocus genotype gn under the condition that its nonrecurrent parent has multilocus genotype gn−1. Let
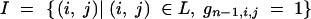 |
1 |
denote the set of indices, for which the locus at position xi,j was heterozygous in the nonrecurrent parent in generation BCn−1 (F1 = BC0). The elements of I are ordered according to
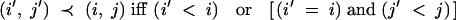 |
2 |
The conditional probability that the BCn individual has the multilocus marker genotype gn is
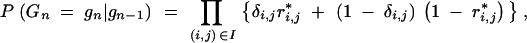 |
3 |
where
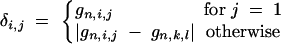 |
4 |
with
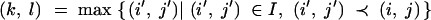 |
5 |
and
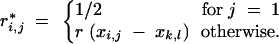 |
6 |
Distribution of donor alleles at markers:
Consider a BCn family of size u, generated by backcrossing one BCn−1 individual to the recurrent parent. Let
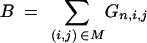 |
7 |
denote the number of donor alleles at the marker loci of a BCn individual. The probability that an individual that carries all target genes is heterozygous at exactly b loci is
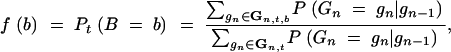 |
8 |
where
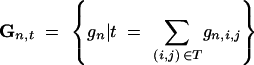 |
9 |
denotes the set of all multilocus marker genotypes carrying all target genes and
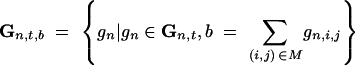 |
10 |
denotes the set of all multilocus marker genotypes carrying all target genes and the donor allele at exactly b marker loci. The respective distribution function is F(b) = Pt(B ≤ b).
Selection of individuals with a low number of donor alleles:
We determine the distribution of donor alleles in the individual carrying (1) all target genes and (2) the w smallest number of donor alleles among all carriers of the target genes (subsequently referred to as the wth best individual).
Assume that v out of u individuals of a backcross family carry all target genes. Then, the distribution of donor alleles in the wth best individual among the v carriers of the target gene is described by the wth order statistic of v independent random variables with distribution function F(b). Its distribution function is
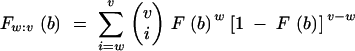 |
11 |
(David 1981). Weighing with the probability that exactly v individuals carry the target gene yields the distribution function of donor alleles in the wth best carrier of all target genes in a BCn family of size u,
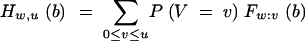 |
12 |
with
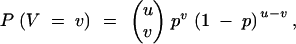 |
13 |
where the probability p that an individual carries all target genes is
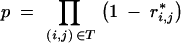 |
14 |
and r*i,j is calculated analogously to Equations 5 and 6 but replacing I with T.
The probability that the wth best individual carries b donor alleles is
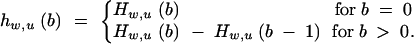 |
15 |
Distribution of the donor genome proportion:
In the following, we investigate the homologous chromosomes of backcross individuals that originate from the nonrecurrent parent. We divide the chromosomes into nonoverlapping intervals,
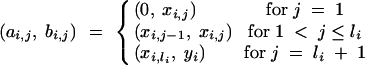 |
16 |
with length
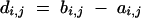 |
17 |
for each
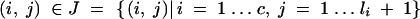 |
18 |
Consider a BCn individual with genotype gn of which the genotype of the nonrecurrent parent in generations BCs (1 ≤ s < n) was gs. We first derive the expected donor genome proportion E(Zi,j) of a chromosome interval delimited by (ai,j, bi,j). Assume at first a finite number e of loci equidistantly distributed on the chromosome interval at positions x*i,1, … , x*i,e; the corresponding random variables indicating the presence of the donor allele are G*n,i,1, … , G*n,i,e. The expected donor genome proportion in the interval is then
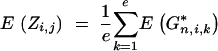 |
19 |
According to Hill (1993), who used results of Franklin (1977), Equation 19 can be extended to an infinite number of loci at positions x*i,k:
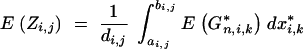 |
20 |
with
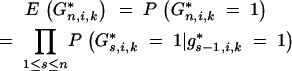 |
21 |
The probability 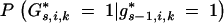 depends on the genotypes of the loci flanking the interval (i, j) in generations BCs−1 and BCs. For telomere chromosome segments (j = 1, j = li + 1)
depends on the genotypes of the loci flanking the interval (i, j) in generations BCs−1 and BCs. For telomere chromosome segments (j = 1, j = li + 1)
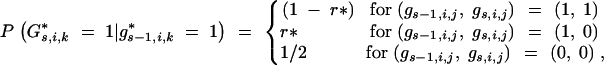 |
22 |
where
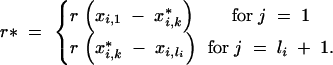 |
23 |
For nontelomere chromosome segments (1 < j < li + 1) the probability 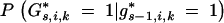 can be calculated with the equations in Table 2.
can be calculated with the equations in Table 2.
TABLE 2.
Probability 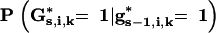 depending on flanking marker genotypesgs−1,i,j−1,gs−1,i,j,gs,i,j−1, andgs,i,j for 1 <j <l + 1
depending on flanking marker genotypesgs−1,i,j−1,gs−1,i,j,gs,i,j−1, andgs,i,j for 1 <j <l + 1
|
gs,i,j−1, gs,i,j
|
||||||||
|---|---|---|---|---|---|---|---|---|
| gs−1,i,j−1, gs−1,i,j | 1, 1 | 1, 0 | 0, 1 | 0, 0 | ||||
| 1, 1 |
|
|
|
|
||||
| 1, 0 | 0 | 1 − r*1 | 0 | r*1 | ||||
| 0, 1 | 0 | 0 | 1 − r*2 | r*2 | ||||
| 0, 0 | 0 | 0 | 0 | 1/2 | ||||
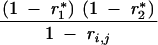
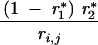
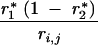
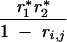
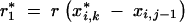 and
and 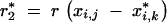 .
.
The expected donor genome proportion on the homologous chromosomes originating from the nonrecurrent parent of a BCn individual with genotype gn can then be determined as
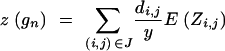 |
24 |
Response to selection:
We define response to selection R as the difference between the expected donor genome proportion μ in the selected fraction of a BCn population and the expected donor genome portion μ′ in the unselected BCn population:
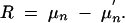 |
25 |
We consider a BCn family of size uq generated by backcrossing one BCn−1 individual of genotype gn−1,q. With respect to this family
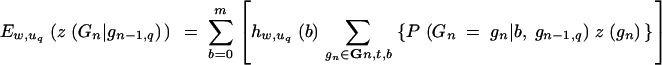 |
26 |
denotes the expected donor genome proportion of the wth best individual, where
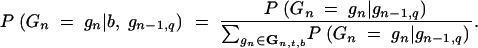 |
27 |
We now consider p BCn−1 individuals with genotypes gn−1,q (q = 1, … , p)that are backcrossed to the recurrent parent. Family size of family q is uq such that the size of the BCn population is u = ∑quq. From family q, the wq best individuals are selected such that the selected fraction consists of w = ∑qwq individuals. We then have
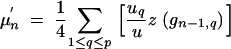 |
28 |
and
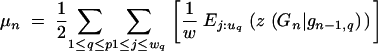 |
29 |
Note that z(gn) refers to one set of homologous chromosomes, whereas μn and μ′n refer to both homologous chromosome sets. This results in the factors 1/4 and 1/2 in Equations 28 and 29.
Numerical implementation:
Calculations for Equations 8 and 26 require enumeration of all realizations of the random vector Gn. For a large number of markers, a Monte Carlo method can be used to limit the necessary calculations. Instead of enumerating all realizations of Gn, a random sample of realizations, determined with a random-walk procedure from the probability of occurrence of multilocus genotypes (Equation 3), can be used as basis for the calculations. The routines developed for implementing our theory are available in the software Plabsoft (Maurer et al. 2004).
DISCUSSION
Comparison to normal distribution selection theory:
Normal distribution selection theory can be applied to marker-assisted backcrossing by considering a BCn population in which indirect selection for low donor genome proportion Z is carried out by selecting individuals with a low count B of donor alleles at markers. Assuming a heritabiltiy of h2 = 1 for the marker score B, response to selection R can be predicted (Bernardo 2002, p. 264) as
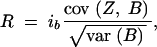 |
30 |
where ib is the selection intensity.
Under the assumptions of (i) no selection in generations BCs (s < n) and (ii) no preselection for the presence of target genes in generation BCn, we have (appendix A, using results of Hill 1993 and Visscher 1996)
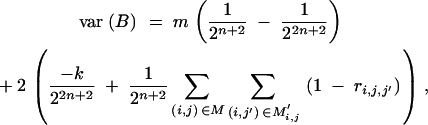 |
31 |
where
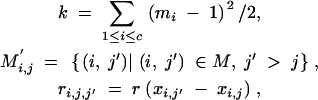 |
32 |
and (appendix A)
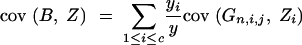 |
33 |
with (Visscher 1996)
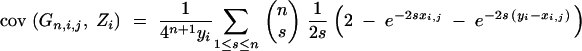 |
34 |
From a mathematical point of view, applying normal distribution selection theory to marker-assisted backcrossing has the following shortcomings:
The distribution of marker scores is discrete, but the normal approximation is continuous.
The distribution of the marker scores is limited, but the normal approximation is unlimited.
The relationship between marker score and donor genome proportion of an individual is nonlinear (this can be shown by using Equation 20), but normal distribution selection theory assumes a linear relationship.
From a genetic point of view, the derivations (appendix A) of variance and covariance presented for the normal approximation (Equation 30) are based on the following assumptions:
The BCn population is generated by recurrent backcrossing of unselected BCs (1 ≤ s < n) populations of large size.
No preselection for the presence of target genes was carried out in the BCn population under consideration.
We illustrate the effects of these shortcomings and assumptions with a model close to the maize genome with 10 chromosomes of length 2 M, markers evenly distributed across the genome, and two target genes located in the center of chromosomes 1 and 2.
For unselected BC1 populations and large numbers of markers (e.g., 200), the normal approximation of the distribution of donor alleles fits very well the exact distribution (Figure 1A). However, if only few markers are employed, the discretization of the probability density function of the normal distribution approximates only roughly the exact distribution (Figure 1B). In particular, for donor genome proportions <0.2, where selection will most likely take place, a considerable underestimation of the exact distribution is observed. This results in an underestimation of the response to selection when normal distribution selection theory is employed. The underestimation is even more severe if an order statistics approach for normal distribution selection theory is applied (Hill 1976), which takes the finite population size into account.
Figure 1.—

Distribution of the donor genome proportion at markers throughout the entire genome (comprising homologous chromosomes originating from the nonrecurrent parent and the recurrent parent) calculated with a normal approximation (solid line) and the exact approach presented (histogram) for a model of the maize genome. Diagrams are shown for a BC1 population without preselection for the presence of target genes employing (A) 200 markers and (B) 20 markers, for a BC1 population after preselection for the presence of two target genes located in the center of chromosomes 1 and 2 employing (C) 80 markers, and for a BC2 population after preselection for the presence of two target genes employing 80 markers (D). The BC2 population was generated by backcrossing one BC1 individual with donor genome content 0.25.
Due to the donor chromosome segments attached to the target genes, the donor genome proportion in backcross populations preselected for the presence of target genes is greater than that in unselected backcross populations. This can result in an overestimation of the response to selection, when employing the normal distribution selection theory and using 1/2n+1 as the population mean of the donor genome proportion (Figure 1C). Note, however, that an adaptation of the normal selection approach should be possible by adjusting the population mean with the expected length of the attached donor segment using results of Hanson (1959).
In marker-assisted backcross programs, usually a high selection intensity is employed and only one or few individuals of a backcross population are used as nonrecurrent parents for the next backcross generation. This results in a smaller variance in the donor genome proportion at markers compared with backcrossing the entire unselected population that is assumed by the normal distribution approach (Figure 1D). The result can be a severe overestimation of the response to selection.
The suggested exact approach overcomes the shortcomings and assumptions listed under a–e. In conclusion, it can be applied to a much larger range of situations than the normal distribution approach.
Comparison to simulation studies:
Simulation studies were successfully applied for obtaining guidelines for the design of marker-assisted backcrossing (Hospital et al. 1992; Frisch et al. 1999b; Visscher 1999; Ribaut et al. 2002). According to Visscher (1999), one of the most important advantages of simulation studies is that selection is taken into account, whereas previous theoretical approaches yielded only reliable estimates for backcrossing without selection.
Our approach solves the problem of using selected individuals as nonrecurrent parents. With respect to two areas, however, simulation studies cover a broader range of scenarios than the selection theory approach presented: (i) Simulations allow the comparison of alternative selection strategies, while in this study we developed the selection theory approach for using the marker score B as a selection index, and (ii) simulations allow coverage of an entire backcross program, while we developed our approach only for one backcross generation. Both issues are promising areas for further research.
Prediction of response to selection:
Prediction of response to selection with Equation 25 can be employed to compare alternative scenarios with respect to population size and required number of markers. We illustrate this application by the example of a BC1 population using model genomes close to maize (10 chromosomes of length 2 M) and sugar beet (9 chromosomes of length 1 M). Markers are evenly distributed across all chromosomes and a target gene is located 66 cM from the telomere on chromosome 1. The donor of the target gene and the recurrent parent are completely homozygous. One individual is selected as the nonrecurrent parent of generation BC2.
The expected response to selection for maize ranges from ∼5% of the donor genome (20 markers, 20 plants) to 12% (120 markers, 1000 plants), and for sugar beet it ranges from ∼7 to 15% (Figure 2). To obtain a response to selection of ∼10% with 60 markers, a population size of 180 is required in maize, corresponding to ∼180/2 × 60 = 5400 marker data points (MDP). By comparison, in sugar beet a population size of 60 is sufficient, resulting in only 30% of the MDP required for maize. This result indicates that the efficiency of marker-assisted backcrossing in crops with smaller genomes is much higher than that in crops with larger genomes. Stam (2003) obtained similar results in a simulation study.
Figure 2.—

Expected response to selection throughout the entire genome (comprising homologous chromosomes originating from the nonrecurrent parent and the recurrent parent) and expected number of required marker data points (MDP) when selecting the best out of u = 20, 40, 60, 80, 100, 200, 500, and 1000 BC1 individuals. The values depend on the number of markers (20–120) and on the number and length of the chromosomes. Left, model of the maize genome with 10 chromosomes of length 2 M. Right, model of the sugar beet genome with 9 chromosomes of length 1 M.
Using >80 markers in maize (corresponding to a marker density of 25 cM) or >60 markers in sugar beet (marker density 15 cM) resulted only in a marginal increase of the response to selection, irrespective of the population size employed (Figure 2). Increasing the population size up to 100 plants results in substantial increase in response to selection in both crops, and using even larger populations still improves the expected response to selection. In conclusion, increasing the response to selection by increasing the number of markers employed is possible only up to an upper limit that depends on the number and length of chromosomes. In contrast, increasing response to selection by increasing the population size is possible up to population sizes that exceed the reproduction coefficient of most crop and animal species.
An optimum criterion for the design of marker-assisted selection in a backcross population can be defined by the expected response to selection reached with a fixed number of MDP. For fixed numbers of MDP in sugar beet, designs with large populations and few markers always reached larger values of response to selection than designs with small populations and many markers (Figure 2). For maize, the same trend was observed for 500 and 1000 MDP, while for a larger number of MDP the optimum design ranged between 40 and 50 markers. In conclusion, in BC1 populations of maize and sugar beet and a fixed number of MDP, marker-assisted selection is, within certain limits, more efficient for larger populations than for higher marker densities.
Selecting backcross individuals:
Selection of backcross individuals can be carried out by using the number of donor alleles at markers B as a selection index. However, when employing markers not evenly distributed across the genome, the donor genome proportion at markers reflects only poorly the donor genome proportion across the entire genome.
The selection theory presented provides two alternative criteria that can be used as a selection index for evaluation of each backcross individual: (1) the expected donor genome proportion z(gn) (Equation 24) of the backcross individual and (2) the expected donor genome proportion E1,u(z(Gn+1|gn)) (Equation 26) of the best of the progenies obtained when using the backcross individual as nonrecurrent parent of the next backcross generation. Employing z(gn) is recommended when selecting plants for selfing from the final generation of a backcross program, because the ultimate goal of a backcross program is to generate an individual (carrying the target genes) with a low donor genome proportion. In contrast, employing E1,u(z(Gn+1|gn)) is recommended for selecting individuals as parents for subsequent backcross generations, because here the donor genome proportion in the progenies is more important than the donor genome in the selected individual itself. Both criteria take into account the position of the markers and are, therefore, more suitable than B, if unequally distributed markers are employed.
Comparison of B, z(gn), and E1,u(z(Gn+1|gn)) is demonstrated with experimental data from a gene introgression program in sugar beet. The target gene was located on chromosome 1 with map distance 6 cM from the telomere, and 25 codominant polymorphic markers were employed for background selection. The map positions of the markers were (chromosome number/distance from the telomere in centimorgans): 1/12, 1/28, 1/32, 1/40, 1/46, 1/75, 2/1, 2/16, 2/96, 3/0, 3/55, 3/78, 4/36, 4/64, 4/67, 5/33, 5/65, 6/42, 6/57, 7/4, 7/67, 8/14, 8/74, 9/0, and 9/12. The lengths of chromsomes 1–9 were 90, 102, 78, 84, 102, 89, 75, 94, and 94 cM. After producing the BC1 generation, 89 plants carrying the target gene were preselected and analyzed for the 25 markers. The criteria B, z(gn), and E1,u(z(Gn+1|gn)) for u = 20, 40, and 80 were calculated and presented for the 25 plants with the smallest marker scores B (Table 3).
TABLE 3.
Selection criteria for the 25 BC1 plants with highest marker scoreB in the sample data set for sugar beet consisting of 89 plants
|
E1,u(z(Gn+1|gn)) × 100
|
|||||
|---|---|---|---|---|---|
| Plant no. | B | z(gn) × 100 | u = 20 | u = 40 | u = 80 |
| 1 | 2 | 7.4 | 3.4 | 3.2 | 3.0 |
| 2 | 4 | 18.0 | 6.7 | 6.1 | 5.6 |
| 3 | 4 | 11.5 | 3.9 | 3.5 | 3.2 |
| 4 | 5 | 14.8 | 5.4 | 4.9 | 4.5 |
| 5 | 5 | 14.9 | 5.5 | 5.0 | 4.6 |
| 6 | 6 | 9.0 | 3.8 | 3.4 | 3.1 |
| 7 | 6 | 16.2 | 5.9 | 5.3 | 4.9 |
| 8 | 6 | 15.4 | 5.0 | 4.4 | 3.9 |
| 9 | 6 | 14.8 | 4.8 | 4.2 | 3.7 |
| 10 | 6 | 17.0 | 5.6 | 5.0 | 4.4 |
| 11 | 7 | 17.2 | 6.4 | 5.7 | 5.2 |
| 12 | 7 | 20.2 | 6.3 | 5.5 | 4.9 |
| 13 | 8 | 19.6 | 6.5 | 5.7 | 5.1 |
| 14 | 8 | 23.2 | 8.0 | 7.1 | 6.4 |
| 15 | 8 | 14.5 | 5.6 | 4.9 | 4.3 |
| 16 | 8 | 17.1 | 6.0 | 5.4 | 4.9 |
| 17 | 8 | 12.1 | 4.9 | 4.3 | 3.8 |
| 18 | 9 | 16.7 | 6.4 | 5.5 | 4.7 |
| 19 | 9 | 17.0 | 6.7 | 5.9 | 5.3 |
| 20 | 9 | 18.0 | 7.0 | 6.4 | 5.8 |
| 21 | 9 | 21.4 | 8.4 | 7.7 | 7.1 |
| 22 | 9 | 27.3 | 10.7 | 9.7 | 8.6 |
| 23 | 9 | 14.8 | 5.6 | 4.9 | 4.3 |
| 24 | 9 | 18.3 | 5.6 | 4.9 | 4.3 |
| 25 | 9 | 18.3 | 6.1 | 5.4 | 4.9 |
For details and explanation of z(gn) and E1,u(z(Gn+1|gn)) see text.
We refer here only to the most interesting results:
Plant 6 had z(gn) = 9.0% and plant 10 had z(gn) = 17.0%, in spite of an identical marker score of B = 6.
Plant 1 was the best with respect to all three criteria. However, plant 6 was second best with respect to the expected donor genome proportion but had only rank 6 with respect to the marker score B.
Plant 9 had a considerably larger expected donor genome proportion [z(gn) = 14.8%] than plant 17 [z(gn) = 12.1%], but the expected donor genome proportion in the best progeny of plant 9 was lower than that of plant 17 for all three populations sizes.
These results demonstrate that the criteria B, z(gn), and E1,u(z(Gn+1|gn)) can result in different rankings of individuals. In conclusion, if markers are not evenly distributed, calculating the proposed selection criteria in addition to the marker score B provides additional information to assess the value of backcross individuals and can assist geneticists and breeders in their selection decision.
QTL introgression:
Marker-assisted selection in introgression of favorable alleles at quantitative trait loci (QTL) usually comprises selection for (1) presence of the donor allele at two markers delimiting the interval in which the putative QTL was detected and (2) the recurrent parent allele at markers outside the QTL interval. Our results can be applied for the latter purpose in exactly the same way as previously described for the transfer of a single target gene. Hence, our approach is applicable to many scenarios in application of marker-assisted backcrossing for qualitative and quantitative traits.
Acknowledgments
We thank Dietrich Borchardt for critical reading and helpful comments on the manuscript. We are indebted to KWS Saat AG, 75555 Einbeck, Germany, for providing the experimental data on sugar beet. We greatly appreciate the helpful comments and suggestions of an anonymous reviewer.
APPENDIX A
We use here an abbreviated notation: Gi,j (i = 1 … c, j = 1 … mi) is a random variable taking 1 if the jth marker on the ith chromosome is heterozygous and 0 otherwise.
We derive the variance of B in a BCn population under the assumptions of (1) no selection in generations BCs (s < n) and (2) no preselection for presence of target genes in generation BCn (i.e., the entire BCn population is considered, comprising individuals carrying the target genes as well as individuals not carrying the target gene). We have
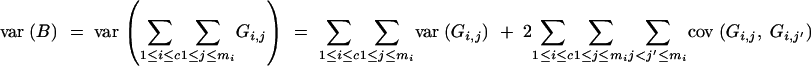 |
Under assumptions (1) and (2) we have for any Gn,i,j
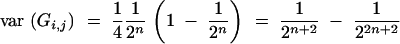 |
(Hill 1993) and further
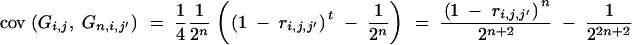 |
(Visscher 1996) with
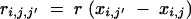 |
Therefore,
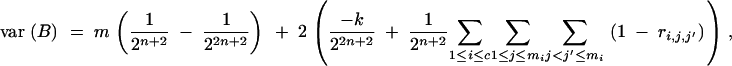 |
where
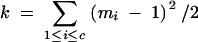 |
is the number of covariance terms.
We derive cov(B, Z) under assumptions (1) and (2). Because
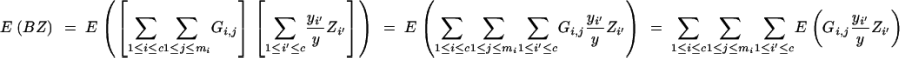 |
and
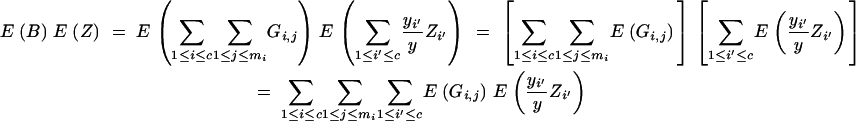 |
we have
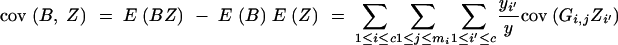 |
and from
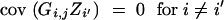 |
follows
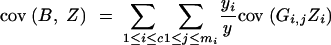 |
This article is dedicated to Professor Dr. H. F. Utz on the occasion of his 65th birthday. His teaching of selection theory was most instrumental to the authors.
References
- Bartlett, M. S., and J. B. S. Haldane, 1935. The theory of inbreeding with forced heterozygosity. J. Genet. 31: 327–340. [Google Scholar]
- Bernardo, R., 2002 Breeding for Quantitative Traits in Plants. Stemma Press, Woodbury, MN.
- David, H. A., 1981 Order Statistics. Wiley, New York.
- Franklin, I. R., 1977. The distribution of the proportion of genome which is homozygous by descent in inbred individuals. Theor. Popul. Biol. 11: 60–80. [DOI] [PubMed] [Google Scholar]
- Fisher, R. A., 1949 The Theory of Inbreeding. Oliver & Boyd, Edinburgh.
- Frisch, M., M. Bohn and A. E. Melchinger, 1999. a Minimum sample size and optimal positioning of flanking markers in marker-assisted backcrossing for transfer of a target gene. Crop Sci. 39: 967–975 (erratum: Crop Sci. 39: 1913). [Google Scholar]
- Frisch, M., M. Bohn and A. E. Melchinger, 1999. b Comparison of selection strategies for marker-assisted backcrossing of a gene. Crop Sci. 39: 1295–1301. [Google Scholar]
- Haldane, J. B. S., 1919. The combination of linkage values and the calculation of distance between the loci of linkage factors. J. Genet. 8: 299–309. [Google Scholar]
- Hanson, W. D., 1959. Early generation analysis of lengths of heterozygous chromosome segments around a locus held heterozygous with backcrossing or selfing. Genetics 44: 833–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill, W. G., 1976. Order statistics of correlated variables and implications in genetic selection programmes. Biometrics 32: 889–902. [PubMed] [Google Scholar]
- Hill, W. G., 1993. Variation in genetic composition in backcrossing programs. J. Hered. 84: 212–213. [Google Scholar]
- Hillel, J., T. Schaap, A. Haberfeld, A. J. Jeffreys, Y. Plotzky et al., 1990. DNA fingerprints applied to gene introgression in breeding programs. Genetics 124: 783–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hospital, F., and A. Charcosset, 1997. Marker-assisted introgression of quantitative trait loci. Genetics 147: 1469–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hospital, F., C. Chevalet and P. Mulsant, 1992. Using markers in gene introgression breeding programs. Genetics 132: 1199–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markel, P., P. Shu, C. Ebeling, G. A. Carlson, D. L. Nagle et al., 1997. Theoretical and empirical issues for marker-assisted breeding of congenic mouse strains. Nat. Genet. 17: 280–284. [DOI] [PubMed] [Google Scholar]
- Maurer, H. P., A. E. Melchinger and M. Frisch, 2004 Plabsoft: software for simulation and data analysis in plant breeding. Proceedings of the 17th EUCARPIA General Congress, September 8–11, 2004, Tulln, Austria, pp. 359–362.
- Ribaut, J.-M., C. Jiang and D. Hoisington, 2002. Simulation experiments on efficiencies of gene introgression by backcrossing. Crop Sci. 42: 557–565. [Google Scholar]
- Stam, P., 2003 Marker-assisted introgression: Speed at any cost?, pp. 117–124 in EUCARPIA Leafy Vegetables 2003, edited by Th. J. L. van Hintum, A. Lebeda, D. Pink and J. W. Schut. EUCARPIA, Valencia, Spain.
- Stam, P., and A. C. Zeven, 1981. The theoretical proportion of the donor genome in near-isogenic lines of self-fertilizers bred by backcrossing. Euphytica 30: 227–238. [Google Scholar]
- Visscher, P. M, 1996. Proportion of the variation in genetic composition in backcrossing programs explained by genetic markers. J. Hered. 87: 136–138. [Google Scholar]
- Visscher, P. M, 1999. Speed congenics: accelerated genome recovery using genetic markers. Genet. Res. 74: 81–85. [DOI] [PubMed] [Google Scholar]