

Abstract
Compensatory mutation occurs when a loss of fitness caused by a deleterious mutation is restored by its epistatic interaction with a second mutation at a different site in the genome. How many different compensatory mutations can act on a given deleterious mutation? Although this quantity is fundamentally important to understanding the evolutionary consequence of mutation and the genetic complexity of adaptation, it remains poorly understood. To determine the shape of the statistical distribution for the number of compensatory mutations per deleterious mutation, we have performed a maximum-likelihood analysis of experimental data collected from the suppressor mutation literature. Suppressor mutations are used widely to assess protein interactions and are under certain conditions equivalent to compensatory mutations. By comparing the maximum likelihood of a variety of candidate distribution functions, we established that an L-shaped gamma distribution (α = 0.564, θ = 21.01) is the most successful at explaining the collected data. This distribution predicts an average of 11.8 compensatory mutations per deleterious mutation. Furthermore, the success of the L-shaped gamma distribution is robust to variation in mutation rates among sites. We have detected significant differences among viral, prokaryotic, and eukaryotic data subsets in the number of compensatory mutations and also in the proportion of compensatory mutations that are intragenic. This is the first attempt to characterize the overall diversity of compensatory mutations, identifying a consistent and accurate prior distribution of compensatory mutation diversity for theoretical evolutionary models.
THE number of compensatory mutations that will affect a specific deleterious mutation is a fundamental evolutionary quantity of which little is known. A mutation is compensatory if it has a beneficial effect on fitness that is conditional on the presence of a deleterious mutation at a different site in the genome (Kimura 1985). Hence, a compensatory effect is the outcome of a strong epistatic interaction between two mutations. Each compensatory mutation represents an alternate genetic solution to adaptation; thus, fitness recovery from the accumulation of deleterious mutations becomes less likely to occur by back mutation when compensatory mutations are increasingly common (Whitlock and Otto 1999). Indeed, one must invoke a compensatory mutation to explain the rapid, stepwise recovery of fitness observed in mutationally degraded experimental populations (Burch and Chao 1999; Moore et al. 2000; Estes and Lynch 2003). Compensatory mutations are also implicated in the persistence of alleles that confer resistance in populations. The fitness cost associated with retaining a resistance-conferring allele in the absence of the selective agent, such as an antibiotic or pesticide, can often be reduced by compensatory mutations (Schrag and Perrot 1996; Maisnier-Patin and Andersson 2004). Finally, the number of compensatory mutations informs us about the ruggedness of the genotype-to-fitness landscape (Wilke et al. 2003).
Because we expect the number of different compensatory mutations (C) to vary among deleterious mutations in different genes and organisms, our aim is to characterize the shape of a statistical distribution of C that represents the overall diversity of compensatory mutation abundance. Presently, almost no direct empirical information on compensatory mutations can be applied to this problem. Fortunately, experimental screens for suppressor mutations (Hartman and Roth 1973) are sufficiently abundant to be used for an indirect measure of compensatory mutation diversity. A suppressor mutation literally suppresses the expression of the phenotypic effect of another mutation. When the phenotype being suppressed is closely associated with fitness, suppressor mutations can be equivalent to compensatory mutations. Suppressor mutations are widely employed in a variety of model organisms to locate and identify functional interactions within or among gene products (Sujatha and Chatterji 2000). We have surveyed a large number of studies in which a debilitating mutant phenotype is suppressed by subsequent mutations in one or more evolving populations. Each study represents a random sample of the diversity of compensatory mutations. We focused specifically on studies that report compensatory interactions within and among proteins, between mutations caused by nucleotide substitution, insertion, or deletion. Consequently, we excluded cases of compensatory mutations that occur within mRNAs (Stephan and Kirby 1993) and tRNAs (Kern and Kondrashov 2004) or involve transposable element insertions (Moore et al. 2000). Furthermore, we required that compensatory interactions were mapped to specific mutations by sequencing, so that multiple occurrences of a compensatory mutation could be detected. The frequency of multiple occurrences in a random sample allows us to infer the total number of compensatory mutations, denoted by C. As C increases, the probability that a specific mutation occurs multiple times in a random sample declines.
An explicit formula for this probability is obtained from a solution of the coupon collector's problem (Read 1998), which describes the random sampling of items that each belong to one of a finite number of classes. This formula allows us to calculate the likelihood that a hypothetical distribution of C explains the observed frequencies of compensatory mutations in our collection of studies. However, this solution of the coupon collector's problem requires that classes are equally frequent, which is equivalent to assuming that mutation rates are uniform across sites in the genome. Site-specific variation in mutation rates (Berg 1999) will inevitably cause an analysis based on the coupon collector's problem to underestimate the true number of sites in the genome at which compensatory mutations can occur. Accordingly, we have also developed an alternative to the uniform-rates model, in which mutation rates to different compensatory mutations are each drawn from a prespecified statistical distribution. Our likelihood analysis reveals that the distribution of C can be consistently and accurately represented by a particular function, irrespective of site-specific variation in mutation rates. We have also detected differences among viruses, prokaryotes, and eukaryotes, not only in the diversity of compensatory mutations, but also in the intragenic localization of compensatory interactions. Taken together, these analyses represent the first attempt to characterize the overall diversity and nature of compensatory mutations and thereby begin to quantify the evolutionary importance of compensatory mutation.
MATERIALS AND METHODS
Data collection from mutation studies:
We collected research articles from the peer-reviewed literature by performing searches in the ISI Web of the Science database, based on the key words “second-site,” “suppressor mutation,” “compensatory mutation,” and “pseudo-revertant,” and also from citations of review articles on suppressor mutation (e.g., Hartman and Roth 1973) and pioneering experimental studies (e.g., Jarvik and Botstein 1975). Articles were selected on the basis of the following criteria:
One or more initial mutations being studied produce a debilitating phenotype that either is known to reduce fitness or is likely to cause a loss of fitness in the selective environment. For example, mutations that confer resistance to an antibiotic agent but are costly to growth in the antibiotic-free environment meet this criterion.
Subsequent compensatory or back mutants derived from each initial mutation are obtained as independent samples. Independence is achieved by sampling one mutant per replicate population or by immediate removal of successive mutants from a single population before their subsequent proliferation. For some cases, repeated sampling cannot ensure independence (e.g., successive clinical isolates from a patient); such cases are not included in our survey.
These mutants must be associated with specific mutations by sequencing the initial gene and other genes to which compensatory interactions have been mapped, so that the number of occurrences of each compensatory mutation can be tallied. Partitioning mutations into linkage groups alone is insufficient for our purposes.
To clarify, we use “mutant” to refer to an individual organism distinguished from its progenitor by its heritable phenotype and “mutation” to refer to a specific genetic change responsible for a phenotypic change in one or more mutant individuals. From all articles that met these criteria, we recorded the number of occurrences of the back mutation and any compensatory mutations. All records were checked by one author (A. Poon) to ensure consistency in the interpretation of each study. Of compensatory mutations involving nucleotide substitutions, only those with substitutions at sites other than the initial mutation were recorded. For example, if the phenotype of an initial mutation changing a codon from glycine (GGA) to glutamic acid (GAA) was reported as being compensated for by a second mutation at the same site, changing that codon to alanine (GCA), then that case was removed. Although a novel amino acid is being introduced in this example, there is no possibility for an epistatic interaction between the initial and compensatory mutations. Compensatory mutations were also removed if they retained the same beneficial effect when experimentally isolated from the initial mutation. In the terminology of population genetics, such a mutation would be unconditionally beneficial rather than compensatory (Wagner and Gabriel 1990). For sets of mutants that shared multiple and recurrent mutations, we recorded the minimum possible number of mutations that could explain the outcome. In addition, we recorded the following information: the study organism and gene, the type of initial mutations studied (insertion, deletion, nonsense, or missense substitution) and phenotypic effect, the location of compensatory mutations relative to the initial mutation (i.e., intragenic or extragenic), the type of amino acid substitutions involved, and the number of back mutants observed. In a few studies, not every gene implicated in compensatory interactions with an initial mutation has been sequenced by the investigators; such initial mutations are treated as involving localized mutagenesis (see below).
Uniform-rates model of mutation sampling:
For a given initial mutation, the number of different compensatory mutations (y) that occur at least once in a sample depends on the number of mutants sampled (n) and the total number of compensatory mutations (C). Note that n does not include the number of back mutants (nback), which are handled separately in our analysis. The probability distribution of y can be obtained from a general solution of the coupon collector's problem, if we assume that mutation rates are uniform across sites in the genome. Thus, the probability of having observed y out of C mutations by the nth sample can be written as
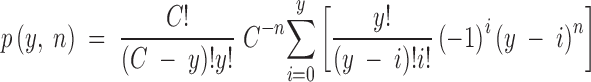 |
(Read 1998). This probability formula assumes that at least one compensatory mutation has been observed in the sample (i.e., y > 0, n > 0). Cases where only back mutations have been recovered require a separate probability formula. Assuming that the back mutation is also as likely to appear in a sample as any compensatory mutation, the probability that only the back mutation is observed in a sample of nback mutants is simply
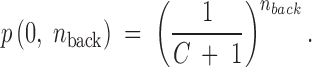 |
When the number of back mutants was not reported, we assigned as nback the total number of back and compensatory mutants derived from a different initial mutation in the same study or a conservatively high value (nback = 99) when none was available. Overall, the likelihood Li that a hypothetical distribution of C results in the sampling outcome {yi, ni} for the ith initial mutation is described by
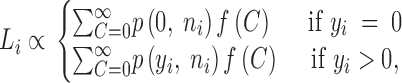 |
where f(C) is a discrete distribution function of C. Although these summations are defined over the interval (0, ∞), this is impractical in a numerical analysis and instead we use an arbitrary upper bound value of Cmax = 100. A total likelihood L for the entire data set is obtained by taking the product of individual likelihoods Li over i from 1 to N, where N is the total number of initial mutations.
Variable-rates model of mutation sampling:
Relaxing the assumption of uniform mutation rates means that we can no longer rely on the coupon collector's problem for an exact formula of the probability p(y, n). With variation in mutation rates, the probability of observing y mutations in a sample of n mutants is conditional not only on the total number of mutations (C), but also on the individual mutation rates to each of C mutations. Because it is unfeasible to calculate an exact solution of p(y, n) with continuous variation in mutation rates, we instead approximated the probability distribution with a Monte Carlo simulation procedure. The simulation was composed in the C programming language and compiled with Xcode 1.5 for Macintosh. For a given value of C, a corresponding number of mutation rates were randomly drawn from a modified gamma distribution, in which the mean is set to 1 and the variance is the reciprocal of the shape parameter (denoted by αmut, where we use a subscript to distinguish this from the parameter α of the gamma distribution function for C). Previous work has established that this one-parameter gamma distribution is an effective representation of site-specific variation in mutation rates (reviewed in Berg 1999). Next, the distribution of the sampling probability p(y, n) was estimated by drawing 1000 samples of n mutations with replacement from a normalized vector of the C mutation rates. The probability p(0, n) of sampling only back mutations was calculated simultaneously by drawing a (C + 1)th mutation rate from the gamma distribution, normalizing by the total mutation rate, and taking the nth power of the result. We reiterated the whole procedure over the range 1 ≤ C ≤ Cmax for 100 replicate samples of mutation rates, for a total of 105 replicates per value C and αmut. The upper limit Cmax was adjusted from 200 to 500 for low values of αmut to accommodate the effects of increasing variation in mutation rates. Because the entire process requires too many calculations to be incorporated into a feasible global-likelihood analysis, we approximated p(y, n) for specific values of α that were chosen to reflect the range of values reported in the literature. We confirmed that this Monte Carlo approximation of p(y, n) converges on the uniform-rates model by comparing the predicted values for increasing αmut. Calculating the likelihood L of a hypothetical distribution f(C) with variable mutation rates followed the same procedure as described above for the uniform-rates model.
Maximum-likelihood estimation:
The shape of the distribution f(C) is determined by one or more parameters (e.g., the exponential distribution is defined by a single rate parameter λ). It follows that the likelihood L is a function of these parameters. For any well-behaved distribution function assigned to f(C), specific parameter values that maximize L should exist. Moreover, we expect that a particular distribution function will have a greater maximum likelihood assigned to it than to other functions. We have tested several candidate distribution functions (e.g., gamma, lognormal, negative binomial, Cauchy, Weibull, exponential, and Poisson) to approximate the actual distribution of C. Continuous distribution functions were discretized by numerical integration of the function over the closed interval [c, c + 1], where c is a nonnegative integer value. Each distribution function was renormalized to account for the arbitrary upper bound of summation Cmax = 100. For evaluating maximum likelihood, we applied the Davidson-Fletcher-Powell optimization algorithm (dfp; Fletcher 1970) within the R-statistical environment for Macintosh (R Development Core Team 2003). We verified that our model converged on accurate parameter estimates by employing an artificial data set of y and n values generated from a prespecified distribution of C. To discriminate between alternative candidates of f(C), we applied Akaike's information criterion (AIC; Akaike 1973), −2 log(Lmax) + 2p, which penalizes the maximum likelihood (Lmax) of a model for the number of parameters (p) that it specifies. The distribution function that minimizes AIC is favored for fitting best the data with the fewest parameters. To determine the rate of convergence to our parameter estimates, we evaluated likelihood for random subsamples of the data set with replacement.
RESULTS
Summary of data collection:
After discounting many articles on the basis of our selection criteria (see materials and methods), we recorded the experimental results from 67 research articles, for a total of 129 different initial mutations (see supplementary Table S1 at http://www.genetics.org/supplemental/). Although a variety of taxa were sampled (20 studies of viruses, 31 of prokaryotes, and 16 of eukaryotes), many studies were specifically carried out in model systems such as the lactose permease gene in Escherichia coli. In such cases, there is frequent overlap between articles in the initial mutations being studied. We combined these overlapping samples, assuming that the total number of compensatory mutations (C) does not vary among studies of the same initial mutation. While the majority of initial mutations involve nonsynonymous substitutions, there are also 10 cases of deletion in seven different genes. Removal of these deletion cases from the data has a negligible effect on our estimate of the distribution of C (data not shown). The overall relationship between the observed number of compensatory mutations and the sample size is illustrated in Figure 1. On average, ∼4.5 different compensatory mutations occurred at least once in a sample of 11 mutants (not including back mutants). In the most extensive study (Maisnier-Patin et al. 2002), a total of 35 compensatory mutations occurred at least once in a sample of 77 mutants. Of 129 initial mutations studied, in 27 cases no compensatory mutations occurred in the sample.
Figure 1.—

Relationship of the number of different compensatory mutations (y) observed at least once in a random sample of n mutants. Each point corresponds to a unique initial mutation from which compensatory mutations are subsequently derived in one or more studies. All points have been displaced by a small random amount so that multiple overlapping points can be discerned. A solid line at y = n is drawn to emphasize the increasing rate of resampling particular compensatory mutations with larger sample sizes. Five initial mutations for which no compensatory mutants were recovered despite extensive sampling are omitted, having no sample number of mutants specified.
Intragenic vs. extragenic mutation:
We examined the frequency of mutant individuals in which compensatory interactions were mapped to mutations within the same gene as the initial mutation (i.e., intragenic), instead of another gene (i.e., extragenic). To obtain an unbiased measure of the frequency of intragenic mutants, we excluded cases in which the occurrence of intragenic mutations in a sample is artificially skewed by an experimental treatment. For example, 33 of 129 initial mutations were subjected to localized mutagenesis (e.g., a gene or segment of the gene containing the initial mutation is transferred to a plasmid, which is subsequently mutagenized; Table S1), creating a bias for intragenic mutations. Five other initial mutations involved the partial or complete deletion of the gene, making intragenic interactions almost impossible. After removing these cases, ∼78% (796/1021; Table 1) of the remaining compensatory mutants were caused by intragenic mutations. By treating the number of intragenic or extragenic mutants in a sample as a binomial outcome, we can estimate the probability of intragenic compensatory mutation (herein referred to as pintra) for a given initial mutation. We employed a beta-distribution function to approximate the overall distribution of pintra and estimated the parameter values of the distribution by maximum likelihood. The relative frequency of intragenic mutants in a random sample is best predicted by a U-shaped beta-distribution function of pintra with an average E[pintra] = 0.83 (Table 1). In other words, on average, 83% of independently isolated compensatory mutants represent a genetic change within the same gene as the initial mutation. The scarcity of intermediate values of pintra suggests that sites that participate in compensatory interactions either within or among gene products tend to be mutually exclusive.
TABLE 1 .
Relative frequency of intragenic and extragenic mutants
| N | No. intragenic |
No. extragenic |
α | β | E[pintra] | −log Lmax | |
|---|---|---|---|---|---|---|---|
| Viruses | 27 | 125 | 70 | 0.212 | 0.096 | 0.689 | 32.37 |
| Prokaryotes | 33 | 285 | 115 | 0.242 | 0.022 | 0.916 | 21.16 |
| Eukaryotes | 16 | 388 | 40 | 1.795 | 0.200 | 0.900 | 23.07 |
| Total | 76 | 796 | 225 | 76.60 | |||
| All data | 0.320 | 0.066 | 0.829 | 83.09 |
No. intragenic and no. extragenic columns list the respective frequencies of compensatory mutant individuals, derived from one of N initial mutations in viruses, prokaryotes, or eukaryotes. Cases that involve localized mutagenesis or partial to complete deletion of the initial gene are excluded. Parameter estimates (e.g., α and β) for a beta-distribution function, used to represent the distribution among initial mutations of the probability of intragenic mutation (pintra), were obtained by maximum likelihood. The mean and negative log-likelihood for each beta distribution are listed in the E[pintra] and −log Lmax columns, respectively. Significance of the taxonomic partition was determined by a likelihood-ratio test (χ2 = 12.98, d.f. = 4, P < 0.05).
Furthermore, we partitioned the data set into viruses, prokaryotes, and eukaryotes to determine whether pintra varies among these taxonomic groups (Table 1). There is a significant improvement in the fit of the beta-distribution model with this partition (likelihood-ratio test χ2 = 12.98, d.f. = 4, P < 0.05). The average probability that a compensatory mutation is intragenic is substantially lower in viruses than in prokaryotes or eukaryotes (E[pintra]virus = 0.69; Table 1). In fact, this observed viral average is significantly lower than that expected by chance (P < 0.01), using randomized subsampling of N = 27 outcomes from the entire data set with replacement. There is, however, little difference in E[pintra] between prokaryotes and eukaryotes (Table 1). Divergence in the model between prokaryotes and eukaryotes is instead largely caused by differences in variation in pintra.
Estimating the distribution of C by likelihood:
We applied a model of random sampling—derived from the coupon collector's problem—to calculate the likelihood of a hypothetical distribution of C, based on the number of mutations that occur in the random samples of mutant individuals in our data set. This model assumes that mutation rates are uniform across sites; in a following section, we examine the effect of site-specific variation in mutation rates. On the basis of maximum-likelihood values calculated from our data set according to the uniform-rates model, we were able to discriminate among several hypothetical distribution functions for C. In Table 2, we report maximum-likelihood values and the associated parameter estimates for the five most successful distribution functions (i.e., gamma, Weibull, negative binomial, lognormal, and exponential). These distributions are plotted against C in Figure 2. Overall, all five distributions have a very similar shape with a mode at zero and a long right tail (Figure 2). The highest value of maximum likelihood was obtained for the gamma-distribution function (Table 2). After adjusting maximum likelihoods for the number of parameters in each distribution function (Akaike 1973), the gamma-distribution function remained the most successful. This distribution predicts that on average E[C] ≃ 11.8 compensatory mutations are associated with a deleterious mutation. Using a Markov chain Monte Carlo procedure, we obtained estimates for the gamma distribution of the Bayesian 95% confidence intervals for E[C] = (8.61, 15.25). Note that this interval is substantially narrower than what would be predicted from the confidence intervals provided for the individual parameters of the gamma distribution (Table 2). This occurs because the likelihood surface is shallower along the axis α = 1/θ, and the expectation of the gamma distribution is the product αθ. According to the gamma distribution, only 20% of all deleterious mutations cannot be compensated for by mutations at any other site in the genome. Moreover, it predicts that over one-third (37%) of all deleterious mutations are associated with at least 10 compensatory mutations.
TABLE 2 .
Likelihood results from uniform-rates model
| Distribution function | Parameter estimates | E[C] | σC | −log Lmax | AIC | |
|---|---|---|---|---|---|---|
| Gamma | α: 0.564 (0.418, 0.737) | 11.84 | 15.77 | 246.96 | 497.92 | |
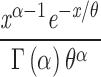
|
θ: 21.01 (12.66, 35.34) | |||||
| Weibull | α: 0.684 (0.555, 0.843) | 12.68 | 19.05 | 247.19 | 498.38 | |
| αβ−αxα−1e−x/βα | β: 9.789 (6.887, 13.73) | |||||
| Negative binomial | r: 0.544 (0.404, 0.723) | 10.89 | 15.13 | 247.55 | 499.10 | |
|
p: 0.048 (0.030, 0.074) | |||||
| r−1 | ||||||
| Lognormal | m: 1.822 (1.337, 2.460) | 39.90a | 254.3 | 248.96 | 501.92 | |
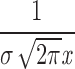 e−ln x−m2/2σ2 e−ln x−m2/2σ2
|
σ: 1.931 (1.455, 2.541) | |||||
| Exponential | ||||||
| λe−λx | λ: 0.100 (0.073, 0.137) | 9.95 | Same | 255.89 | 513.78 |
Parameter estimates for each distribution function are followed by their 95% confidence limits (lower, upper). Estimates were obtained by a likelihood analysis of the entire data set under the uniform-rates model, which assumes that mutation rates are uniform across sites in the genome. The mean and variance of the resulting distribution are given under the headings E[C] and σC. Negative log-likelihood (−log Lmax) is minimized by the most successful model, after being penalized for the number of parameters according to Akaike's information criterion (AIC).
The lognormal distribution exhibits properties of a broad distribution, in that the tail of the distribution exerts such a disproportionate influence on the mean and variance that they assume spurious values (Romeo et al. 2003).
Figure 2.—

Maximum-likelihood estimates of various probability density functions (exponential, gamma, negative binomial, Weibull, and lognormal), each representing a hypothetical distribution of the number of different compensatory mutations, C. The functions shown are the five best distributions among all those tested. All functions possess a similar L-shape, with a mode located at zero and a long right tail. Estimates of maximum likelihood are obtained from a uniform-rates model of mutation sampling.
We approximated the likelihood surface above the two parameters of the gamma-distribution function by evaluating the likelihood for a 50-by-50 grid of parameter values. A contour plot of these likelihood estimates defines a smooth unimodal likelihood surface centered around our parameter estimates. This implies that our maximum-likelihood estimate of the shape of the gamma distribution is a global maximum for our data set. To determine how rapidly the maximum-likelihood analysis converges on the final parameter estimates, we randomly subsampled with replacement from our data set and reevaluated maximum likelihood. We found rapid convergence to the final parameter estimates with less than half of the data incorporated (data not shown). From this result, we infer that our data set is sufficiently comprehensive so that our estimate of the distribution of C does not rely on a disproportionate influence of a few outcomes.
Partitioning data by taxonomic group for C:
We reapplied the taxonomic partition of our data—previously used to detect differences in the frequency of intragenic mutations—to detect differences between viruses, prokaryotes, and eukaryotes in the distribution of C. For each data subset, we reevaluated the maximum likelihood for all distribution functions under the uniform-rates model. Our results from this partitioned analysis are presented in Table 3. Partitioning the data produces a significant improvement in the overall fit of the gamma-distribution function (likelihood-ratio test χ2 = 15.70, d.f. = 4, P < 0.01); the performances of other distribution functions are also significantly improved (Table 3).
TABLE 3 .
Likelihood results with taxonomic partition
| N | Distribution function |
E[C] | σC | −log Lmax |
|---|---|---|---|---|
| Virus | ||||
| 44 | Gamma | 8.49 | 11.25 | 70.69 |
| Negative binomial | 7.97 | 11.50 | 70.92 | |
| Weibull | 9.06 | 13.27 | 71.04 | |
| Lognormal | 30.88a | 206.79 | 72.06 | |
| Geometric | 6.32 | 6.80 | 72.94 | |
| Prokaryote | ||||
| 51 | Lognormal | 21.65a | 69.69 | 109.25 |
| Cauchy | NAb | NA | 109.41 | |
| Weibull | 13.31 | 17.88 | 109.80 | |
| Negative binomial | 12.16 | 15.58 | 110.21 | |
| Gamma | 13.01 | 15.93 | 110.22 | |
| Eukaryote | ||||
| 34 | Gamma | 13.59 | 19.41 | 58.19 |
| Weibull | 16.93 | 29.55 | 58.53 | |
| Negative binomial | 9.74 | 14.24 | 58.59 | |
| Lognormal | 376.28a | 12281.72 | 59.05 | |
| Geometric | 9.33 | 9.82 | 61.25 | |
Maximum-likelihood results from the uniform-rates model are given for the best five distributions in viruses, prokaryotes, or eukaryotes. N, the number of initial (deleterious) mutations screened for compensatory mutations within each taxonomic group; E[C], distribution mean; and σC, standard deviation.
The lognormal distribution exhibits properties of a broad distribution, in that the tail of the distribution exerts such a disproportionate influence on the mean and variance that they assume spurious values (Romeo et al. 2003).
Moments of the Cauchy distribution are undefined.
We can distinguish two general patterns in the partitioned analysis. First, there are apparently fewer compensatory mutations on average in viruses than in prokaryotes or eukaryotes, regardless of which distribution function is used to approximate the distribution of C (Table 3). This trend suggests that broad differences between viruses and higher organisms in genome size and gene lengths can constrain the number of possible interactions within and among gene products. However, our statistical power in a partitioned analysis is limited by the availability of mutational data. A randomized “viral” subset of only N = 44 (Table 3) initial mutations, sampled with replacement from the entire data set, assumes a mean of E[C] ≤ 8.49 with a probability of P ≈ 0.15. Second, maximum likelihood favors an L-shaped gamma distribution for describing mutation data from both viral (α = 0.570, θ = 14.89) and eukaryotic (α = 0.490, θ = 27.72) systems (Table 3). The overall rank of the alternative distributions by maximum likelihood is also fairly congruent between viruses and eukaryotes. However, this pattern is not carried over to the analysis of prokaryotic data, for which maximum likelihood favors a lognormal distribution (m = 1.860, σ = 1.559; Table 3). In randomized “prokaryotic” subsets of N = 51 initial mutations, maximum likelihood was greater for the lognormal than for the gamma-distribution function with a probability of P ≈ 0.1. Therefore, there is no reason to believe that the diversity of compensatory mutations in prokaryotes actually follows a different distribution.
Effect of variation in mutation rates:
The preceding analyses for the distribution of C assume that mutation rates to different compensatory mutations are equal. Site-specific variation in mutation rates will inevitably cause this uniform-rates model to underestimate the number of compensatory mutations. To evaluate the effects of relaxing this assumption, we have taken two different approaches: (1) removing known sources of site-specific mutation rate variation from the data set and (2) reanalyzing the entire data set using a variable-rates model. First, we removed any cases from the data in which a mutagenic treatment had been used to generate compensatory mutations. Because a mutational bias is inherent in most mutagenic treatments, they can potentially represent a major source of variation in mutation rates among sites. We recorded use of a chemical mutagen or UV irradiation in 16 studies and localized mutagenesis in 20 studies. In total, 49 initial mutations were affected by one or more of these treatments and were removed from our data set. Removal of these mutations had a small effect on the shape of the gamma distribution (α = 0.69, θ = 16.97) determined by maximum likelihood under the uniform-rates model, with an ∼1% decrease in the predicted mean number of compensatory mutations. The apparent robustness of the distributional estimate to the removal of mutagenic treatments implies that either: (1) the effect of variation in mutation rates caused by mutagenesis in 49 cases is small relative to the overall effect of naturally occurring rate variation or (2) estimating the distribution of C by maximum likelihood is, for some reason, robust to variation in mutation rates. In the following section, we demonstrate that the latter explanation is unrealistic.
For a more direct approach to relaxing the assumption of uniform mutation rates, we replaced the uniform-rates model with a Monte Carlo procedure that approximates the distribution p(y, n) (i.e., the probability of observing y mutations in a random sample of n mutants) when mutation rates to C different compensatory mutations are each drawn from a prespecified distribution. Previous work has established that site-specific variation in mutation rates can be accurately represented by a modified gamma distribution (reviewed in Berg 1999), in which the mean is set to 1 and the variance equals 1/αmut. We approximated p(y, n) for several values of αmut, reflecting the range of empirical estimates reported in the literature (αmut = 0.3–1.6; Berg 1999). As a result, we were able to reevaluate our likelihood analysis of the data for different levels of mutation rate variation. Maximum likelihood consistently favors an L-shaped gamma distribution of C over the alternative distribution functions, irrespective of what value of αmut is employed in the variable-rates model. In Figure 3, these gamma distributions are plotted against C for different values of αmut. As predicted, increasing site-specific variation in mutation rates causes an increase in the average number of compensatory mutations predicted from the data. In fact, the average E[C] increases linearly with the variance of the mutation rate distribution, 1/αmut. In Figure 4, we have plotted the mean values E[C] corresponding to the family of gamma distributions shown in Figure 3, as a function of αmut. According to this linear relationship, the range of values of αmut = 0.3–1.6 that have been reported in the literature (Oller and Schaaper 1994; Yang 1996; Berg 1999) correspond to an average number of compensatory mutations E[C] = 16.7–38.0 (Figure 4).
Figure 3.—

Gamma-distribution functions evaluated by maximum likelihood with the variable-rates model for different values of αmut (indicated in inset), chosen to represent the empirically observed range. The solid line represents the maximum-likelihood estimate from the uniform-rates model (αmut = ∞). As αmut decreases, there is greater site-specific variation in mutation rates, and the gamma-distribution function becomes steeper. Parameter estimates for gamma-distribution functions are α = 0.505, θ = 31.15 (αmut = 2); α = 0.474, θ = 41.19 (αmut = 1); α = 0.434, θ = 62.53 (αmut = 0.5); and α = 0.392, θ = 110.9 (αmut = 0.25).
Figure 4.—

Average number of compensatory mutations plotted against the variance of the distribution of mutation rates (1/αmut, one-parameter gamma). Average values are determined from gamma distributions estimated by maximum likelihood in the variable-rates model. The solid line represents a least-squares linear fit that yields the approximate formula E[C] = 7.88/αmut + 11.76.
DISCUSSION
Suppressor mutations have a long history of use in molecular biology to reveal functional interactions within and among proteins (Hartman and Roth 1973; Sujatha and Chatterji 2000). In this study, we have employed data from this literature to infer the overall diversity in the abundance of compensatory mutations, expressed as a statistical distribution in the number of compensatory mutations per deleterious mutation (C). Under a uniform-rates model of random sampling, a gamma-distribution function was the most successful representation of the distribution of C and predicted a mean of E[C] = 11.84 mutations. The success of the gamma-distribution function is robust to site-specific variation in mutation rates, which makes this function the best overall representation of compensatory mutation diversity to employ in theoretical models of evolution. Furthermore, the mean E[C] varies predictably with the level of site-specific variation in mutation rates in a linear fashion. For typical levels of mutation rate variation (Berg 1999), E[C] increases about twofold.
What does the shape of this gamma distribution predict about the evolutionary consequences of compensatory mutation? Although it is evident from empirical work that compensatory mutation plays an important role in several aspects of evolution (Schrag and Perrot 1996; Burch and Chao 1999; Kondrashov et al. 2002), there are as yet few theoretical models in evolutionary biology in which it is explicitly incorporated. However, in the following section we apply results from a previous investigation of the compensatory evolution of resistance (Levin et al. 2000) to identify the range of C where the evolutionary influence of compensatory mutation eclipses that of reversion.
Compensatory evolution of resistance:
Under strong selective pressure from an antibiotic or pesticide, pathogens can rapidly acquire alleles that confer resistance. However, these resistance-conferring alleles are often accompanied by a reduction in fitness relative to the nonresistant wild type that is exposed in the absence of the selective agent (Andersson and Levin 1999). To minimize this cost of resistance in an antibiotic- or pesticide-free environment, some pathogens will acquire compensatory mutations instead of reverting the resistance-conferring allele to the wild type (Maisnier-Patin and Andersson 2004). However, these compensatory mutations will not always restore fitness completely (Andersson and Levin 1999). Why would a population preferentially retain a costly resistance-conferring allele by fixing a compensatory mutation that only partially restores fitness? Levin et al. (2000) postulate that a sufficiently high rate of compensatory mutation, combined with recurrent bottlenecks in population size, can produce this outcome. If the rate of compensatory mutation is greater than the rate of reversion, then compensatory mutations benefit disproportionately on average from the momentary period of exponential growth following a population bottleneck. This process is named the bottleneck-mutation hypothesis (Levin et al. 2000).
In computer simulations of the evolution of streptomycin resistance in E. coli (Schrag and Perrot 1996), compensatory mutations fixed or rose to near fixation in >55% of mildly bottlenecked populations (Ne ≈ 106), despite having a substantial selective disadvantage (8%) relative to the wild-type reversion (Levin et al. 2000). This outcome requires that the rate of compensatory mutation is at least 10-fold greater than that of reversion. Having measured the distribution of C, we can extrapolate from this study to make general predictions about the evolution and maintenance of resistance-conferring alleles. The rate of compensatory mutation will exceed the rate of reversion by 10-fold when C ≥ 10, on average. Given our results, this condition is met by at least 37% of all resistance-conferring alleles with a fitness cost comparable to the streptomycin resistance mutation. Thus, pathogenic populations are able to retain a substantial proportion of resistance-conferring alleles despite their selective disadvantage in the wild-type environment.
The bottleneck-mutation hypothesis can also be generalized to assessing the role of compensatory mutations in the recovery of fitness from the accumulation of deleterious mutations in severely bottlenecked populations. There are several examples of compensatory mutations outcompeting the reversion of deleterious mutations in the empirical literature. For example, two populations of T7 bacteriophage that had been degraded by the accumulation of hundreds of mutations recovered fitness almost entirely by compensatory mutations, upon return to large population size (N ≈ 105; Bull et al. 2003). Similarly, very few of the mutations acquired in the fitness-recovery phase (N ≈ 107) of mutationally degraded populations of foot-and-mouth disease virus (FMDV) represented reversions (Escarmís et al. 1999). In these examples, the population size during fitness recovery is consistent with the size employed in the preceding example of the bottleneck-mutation hypothesis. However, reducing the population size during this recovery phase lowers the threshold amount by which the rate of compensatory mutation must exceed the rate of reversion. Thus, all four experimental populations of bacteriophage Φ6 that were maintained at a small size (Ne < 500) recovered fitness by compensatory mutation rather than by reversion (Burch and Chao 1999), even though the reversion was selectively superior. Not only do we expect compensatory mutations to occur more often, but also we expect the overall rate of fitness recovery to be improved by compensatory mutation. This motivates an extension of the bottleneck-mutation hypothesis to determine the degree to which compensatory mutation may reduce the extinction risk of small populations (Whitlock and Otto 1999).
The genotype-to-fitness landscape:
What does our estimate of the distribution of C imply about the ruggedness of the genotype-to-fitness landscape (Kauffman 1993)? By intuition, one often equates ruggedness with modality and thus with the number of distinct fitness optima in the landscape. As we demonstrate, however, this concept of ruggedness is difficult to apply in practice (Horn and Goldberg 1995). Our distribution of C describes the local ruggedness adjacent to a fitness optimum, in that the first deleterious mutation leads to at least 11.8 secondary mutations that each ascend a separate fitness optimum. This interpretation, however, assumes the absence of neutral mutations that may connect separate fitness optima. Furthermore, the total number of possible mutations is often so large that this local ruggedness represents a negligible fraction of mutations at a genetic distance of 2. Indeed, we calculate that for a genome of G = 106 sites, a random walk from a fitness optimum (in the absence of selection) will first encounter a compensatory mutation at an average genetic distance of at least 200 deleterious mutations.
For genomes of short lengths, however, this average distance is sufficiently short to explain why an average excess of antagonistic epistasis is repeatedly observed in cases of experimental evolution. Such is the case for short RNA sequences (G = 76) studied in silico by Wilke et al. (2003), where fitness is determined by the conformation of their secondary structure. They were able to detect a significant excess of antagonistic epistasis in random walks in the sequence space over the interval of zero to eight mutations (Wilke et al. 2003). Furthermore, they determined that this effect was caused by acquiring compensatory mutations in a rugged landscape. Similar results have been obtained in the experimental evolution of proteins. For example, in mutagenized libraries of a single-chain Fv antibody gene, the fraction of clones losing gene function decayed linearly with a small number of mutations (Daugherty et al. 2000). However, a significantly greater fraction of highly mutagenized clones, containing 8–22.5 mutations, retain function than that predicted from the effects of fewer mutations. Daugherty et al. (2000) also attribute this phenomenon to encountering alternative fitness optima in a rugged landscape. If we assume that ∼500 nucleotide sites in the Fv antibody gene affect function, then our results predict that a mutagenized clone acquires a compensatory mutation at an average distance of 8 mutations.
The biological basis of C:
What determines the number of compensatory mutations that can affect a given deleterious mutation? One possible factor is the number of protein interactions encountered by the mutation of a gene product. However, the overall majority of compensatory mutations are intragenic, which suggests that the number of interactions among different proteins may be not as generally important as the complexity of the protein itself. In addition, our analysis suggests that there are fewer compensatory mutations for a deleterious mutation in a virus, on average, than in a prokaryote or eukaryote. Viral genes tend to be shorter, which implies that the size of the gene product constrains the number of possible interactions. We also find that a significantly greater proportion of compensatory mutations in viruses than in prokaryotes or eukaryotes are extragenic. This pattern is counterintuitive because many more genes are in prokaryotic and eukaryotic genomes and consequently a greater number of potential interactions among gene products. However, the unique constraints on viral genomes require viruses to accomplish many biological functions using a limited number of genes. Prokaryotes and eukaryotes, on the other hand, may compartmentalize biological functions into specialized genes through a proliferation of gene number and subsequent divergence in function or expression (Lynch and Force 2000). Finally, mutations that interact with both intragenic and extragenic mutations with equal frequency are underrepresented in our data. Whether this pattern reflects a real biological constraint on the range of functional interactions or an observational bias will require further experimental work.
Our ability to determine how C varies in response to biological factors is limited by difficulties in measuring C itself. All empirical investigations are affected by at least one of two detection thresholds, which originate respectively from variation in the compensatory mutation rate and the mutation effect size. For example, experiments that rely on the influx of spontaneous mutations can easily overlook mutations that occur at a very low relative rate. We have corrected our estimates of C for this mutation rate threshold by implementing a variable-rates model, in which individual mutation rates are random deviates from a one-parameter gamma distribution. Good empirical evidence supports the use of this distribution to represent site-specific variation in mutation rates (Berg 1999). It is also difficult to detect compensatory mutations that have only a marginal effect on fitness. Although it is conceivably possible to correct for this detection threshold as well (Otto and Jones 2000), there is currently not enough information on the effect-size distribution of compensatory mutations. Thus, our estimate of the distribution of C is the best that can be achieved with the available empirical data.
Many issues remain to be resolved in our understanding of the diversity of compensatory mutations. We have so far determined that the statistical distribution of the number of compensatory mutations per deleterious mutation consistently conforms to a gamma-distribution function. However, it is unclear whether this distribution is arrived at by adaptation or simply represents a biological constraint in the number of functional interactions. By identifying this distribution, we have facilitated the incorporation of compensatory mutation into theoretical models of evolution, which in turn will enable us to address whether the distribution of C itself represents an adaptation. Another interesting direction of research is to evaluate the extent that compensatory amino acid substitutions tend to restore biochemical properties of the wild type (B. Davis and A. Poon, unpublished data). Finally, a great deal more of the evolutionary consequences of compensatory mutation will be revealed when we have obtained estimates of the effect-size distribution of compensatory mutations.
Acknowledgments
The authors thank the Schluter, Otto, Whitlock and Doebeli (SOWD) lab groups at University of British Columbia, Doris Bachtrog, Hopi Hoekstra, Peter Andolfatto, and two anonymous reviewers for their insightful comments on earlier versions of this article; Jon Bollback for advice on our likelihood analysis; and Daniel M. Weinreich for helpful discussions on sign epistasis and the fitness landscape. This work was supported by a grant from the National Institutes of Health (GM-60916) to L.C. and a Natural Sciences and Engineering Research Council of Canada postgraduate scholarship (NSERC PGS-B) to B.D. We also acknowledge Michael C. Whitlock for providing financial support from an NSERC discovery grant.
References
- Akaike, H., 1973 Information theory and an extension of the maximum likelihood principle, pp. 267–291 in 2nd International Symposium on Information Theory, edited by B. N. Petrov and F. Csaki. Publishing House of the Hungarian Academy of Sciences, Budapest.
- Andersson, D. I., and B. R. Levin, 1999. The biological cost of antibiotic resistance. Curr. Opin. Microbiol. 2 489–493. [DOI] [PubMed] [Google Scholar]
- Berg, O. G., 1999. Synonymous nucleotide divergence and saturation: effects of site-specific variations in codon bias and mutation rates. J. Mol. Evol. 48 398–407. [DOI] [PubMed] [Google Scholar]
- Bull, J. J., M. R. Badgett, D. Rokyta and I. J. Molineux, 2003. Experimental evolution yields hundreds of mutations in a functional viral genome. J. Mol. Evol. 57 241–248. [DOI] [PubMed] [Google Scholar]
- Burch, C., and L. Chao, 1999. Evolution by small steps and rugged landscapes in the RNA virus Φ6. Genetics 151 921–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daugherty, P., G. Chen, B. L. Iverson and G. Georgiou, 2000. Quantitative analysis of the effect of the mutation frequency on the affinity maturation of single chain Fv antibodies. Proc. Natl. Acad. Sci. USA 97 2029–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escarmís, C., M. Dávila and E. Domingo, 1999. Multiple molecular pathways for fitness recovery of an RNA virus debilitated by operation of Muller's ratchet. J. Mol. Biol. 285 495–505. [DOI] [PubMed] [Google Scholar]
- Estes, S., and M. Lynch, 2003. Rapid recovery of mutation-accumulation lines by compensatory mutation. Evolution 57 1022–1030. [DOI] [PubMed] [Google Scholar]
- Fletcher, R., 1970. A new approach to variable metric algorithms. Comp. J. 13 317–322. [Google Scholar]
- Hartman, P. E., and J. R. Roth, 1973. Mechanisms of suppression. Adv. Genet. 17 1–105. [DOI] [PubMed] [Google Scholar]
- Horn, J., and D. E. Goldberg, 1995 Genetic algorithm difficulty and the modality of fitness landscapes, pp. 243–269 in Foundations of Genetic Algorithms 3, edited by L. Darrell Whitley and M. D. Vose. Morgan Kauffman, San Francisco.
- Jarvik, J., and D. Botstein, 1975. Conditional-lethal mutations that suppress genetic defects in morphogenesis by altering structural proteins. Proc. Natl. Acad. Sci. USA 72 2738–2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauffman, S. A., 1993 The Origins of Order. Oxford University Press, New York.
- Kern, A. D., and F. A. Kondrashov, 2004. Mechanisms and convergence of compensatory evolution in mammalian tRNAs. Nat. Genet. 36 1207–1212. [DOI] [PubMed] [Google Scholar]
- Kimura, M., 1985. The role of compensatory neutral mutations in molecular evolution. J. Genet. 64 7–19. [Google Scholar]
- Kondrashov, A. S., S. Sunyaev and F. A. Kondrashov, 2002. Dobzhansky-Muller incompatibilities in protein evolution. Proc. Natl. Acad. Sci. USA 99 14878–14883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin, B., V. Perrot and N. Walker, 2000. Compensatory mutation, antibiotic resistance and the population genetics of adaptive evolution in bacteria. Genetics 154 985–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch, M., and A. Force, 2000. The probability of duplicate gene preservation by subfunctionalization. Genetics 154 459–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maisnier-Patin, S., and D. I. Andersson, 2004. Adaptation to the deleterious effects of antimicrobial drug resistance mutations by compensatory evolution. Res. Microbiol. 155 360–369. [DOI] [PubMed] [Google Scholar]
- Maisnier-Patin, S., O. G. Berg, L. Liljas and D. I. Andersson, 2002. Compensatory adaptation to the deleterious effect of antibiotic resistance in Salmonella typhimurium. Mol. Microbiol. 2 355–366. [DOI] [PubMed] [Google Scholar]
- Moore, F. B. G., D. E. Rozen and R. E. Lenski, 2000. Pervasive compensatory adaptation in Escherichia coli. Proc. R. Soc. Lond. Ser. B 267 515–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oller, A. R., and R. M. Schaaper, 1994. Spontaneous mutation in Escherichia coli containing dnaE911 DNA polymerase antimutator allele. Genetics 138 263–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto, S. P., and C. D. Jones, 2000. Detecting the undetected: estimating the number of loci underlying a trait in QTL analysis. Genetics 156 2093–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team, 2003 R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna.
- Read, K. L. Q., 1998. A lognormal approximation for the collector's problem. Am. Stat. 52 175–180. [Google Scholar]
- Romeo, M., V. Da Costa and F. Bardou, 2003. Broad distribution effects in sums of lognormal random variables. Eur. Phys. J. B 32 513–525. [Google Scholar]
- Schrag, S. J., and V. Perrot, 1996. Reducing antibiotic resistance. Nature 381 120–121. [DOI] [PubMed] [Google Scholar]
- Stephan, W., and D. A. Kirby, 1993. RNA folding in Drosophila shows a distance effect for compensatory fitness interactions. Genetics 135 97–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sujatha, S., and D. Chatterji, 2000. Understanding protein-protein interactions by genetic suppression. J. Genet. 79 125–129. [Google Scholar]
- Wagner, G. P., and W. Gabriel, 1990. Quantitative variation in finite populations: What stops Muller's ratchet in the absence of recombination? Evolution 44 715–731. [DOI] [PubMed] [Google Scholar]
- Whitlock, M., and S. P. Otto, 1999. The panda and the phage: compensatory mutations and the persistence of small populations. Trends Ecol. Evol. 14 295–296. [DOI] [PubMed] [Google Scholar]
- Wilke, C. O., R. E. Lenski and C. Adami, 2003. Compensatory mutations cause excess of antagonistic epistasis in RNA secondary structure folding. BMC Evol. Biol. 3 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Z., 1996. Maximum-likelihood models for combined analyses of multiple sequence data. J. Mol. Evol. 42 587–596. [DOI] [PubMed] [Google Scholar]