

Abstract
Protein kinases represent promising drug targets for a number of human and animal diseases. The recent completion of the sequenced genomes of three human-infective trypanosomatid protozoa, Leishmania major, Trypanosoma brucei and Trypanosoma cruzi, has allowed the kinome for each parasite to be defined as 179, 156 and 171 eukaryotic protein kinases respectively, that is about one third of the human complement. The analysis revealed that the trypanosomatids lack members of the receptor-linked or cytosolic tyrosine kinase families, but have an abundance of STE and CMGC family protein kinases likely to be involved in regulating cell cycle control, differentiation and response to stress during their complex life-cycles. In this review we examine the prospects for exploiting differences between parasite and mammalian protein kinases to develop novel anti-parasitic chemotherapeutic agents.
Keywords: Protein kinase, cell cycle, chemotherapy, Trypanosoma, Leishmania
Abbreviations: CAK, cdc2-activating kinase; CDK, cyclin-dependent kinase; CKI, casein kinase I; CRK, cdc2-related kinase gene; CRK, cdc2-related kinase protein; ePK, eukaryotic protein kinase; HAT; human African trypanosomiasis
1. Diseases caused by trypanosomatid parasitic protozoa
African trypanosomiasis is a vector-borne parasitic disease caused by protozoan parasites of the Trypanosoma genus. Trypanosoma brucei species can infect both humans and animals, causing human African trypanosomiasis (HAT, also known as African sleeping sickness) in man and Nagana in cattle. The disease threatens over 60 million people and uncounted numbers of cattle in 36 countries of sub-Saharan Africa and has a devastating impact on human health and the economy in affected areas. Unless treated, HAT is always fatal. Political instability and economic problems are leading factors for reduced efficacy in vector and disease control, resulting in a resurgence of disease that continues to this day (http://www.who.int/tdr).
Trypanosoma cruzi is responsible in South America for Chagas disease, which can cause acute illness and death, especially in young children. More commonly, patients develop a chronic form of the disease which affects most organs of the body, often causing fatal damage to the heart and digestive tract. Transmission occurs via bloodsucking triatomine bugs and congenitally from mother to unborn child, but can also occur through contaminated blood transfusions (http://www.who.int/en/).
The leishmaniases are caused by 20 species pathogenic for humans belonging to the genus Leishmania, transmitted by the bite of phlebotomine sandflies. Leishmaniasis currently threatens 350 million people in 88 countries around the world. Clinical symptoms range from cutaneous, mucocutaneous to visceral, depending on the Leishmania species. Cutaneous forms of the disease produce skin ulcers on exposed parts of the body causing serious disability and scarring. In mucocutaneous forms of leishmaniasis, lesions can lead to partial or total destruction of the mucous membranes of the nose, mouth and throat cavities and surrounding tissues. Visceral leishmaniasis (kala azar) is characterized by irregular bouts of fever, substantial weight loss, swelling of the spleen and liver, and anaemia. If left untreated, the fatality rate for kala azar in developing countries can be as high as 100% within 2 years (http://www.who.int/en/).
2. Complex life cycle of trypanosomatid parasites
All three trypanosomatid species discussed exhibit complicated life cycles, and are transmitted between mammalian hosts by hematophagous insects. In each host, the parasites traverse many life cycle stages with different morphologies and proliferation properties, each of which is adapted to a particular compartment within the host. These developmental stages are tightly regulated and complex control mechanisms are in place to ensure completion of the life cycle. There are both proliferative life cycle stages to establish infection and colonisation and cell cycle arrested stages that are pre-adapted for the transmission to the next host. Thus, life and cell cycle control must be intricately linked.
By way of example, a simplified biphasic life cycle of T. brucei is illustrated in Figure 1. The free-living long slender form trypanosome in the bloodstream of the mammalian host and the procyclic form in the midgut of the tsetse fly are the proliferative forms that establish infection. The long slender form trypanosome differentiates into the cell cycle arrested short stumpy form that is pre-adapted for transmission into the tsetse fly. Similarly, the procyclic form trypanosome differentiates into the cell cycle arrested metacyclic form trypanosome (via several intermediate stages), pre-adapted for transmission into the mammalian host.
Figure 1.
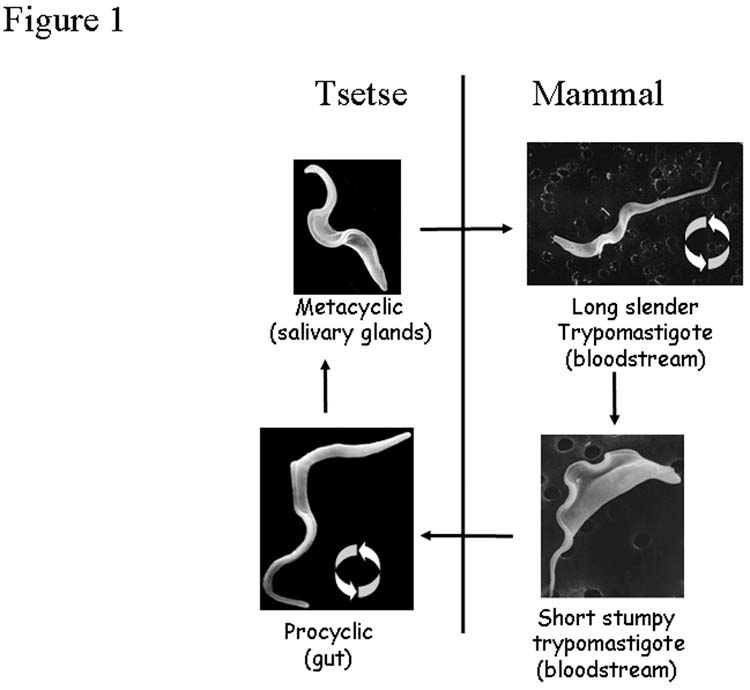
Abbreviated life-cycle of Trypanosoma brucei, highlighting replicative and non-replicative life-cycle stages of the parasite (circular arrows). Micrographs (L. Tetley, University of Glasgow).
The life cycle of Leishmania is reminiscent of that of T. brucei in that the parasite requires transition between proliferative and cell cycle arrested stages to complete the life cycle, but in contrast to T. brucei, it includes an intracellular stage. Upon infection of the mammalian host, Leishmania species invade macrophages and differentiate into the proliferative amastigote form, surviving and multiplying in a parasitophorous vacuole. After transmission into the sand fly vector, the amastigote differentiates into the flagellated proliferative promastigote form, which colonizes the gut of the sandfly. After migrating to the sandfly mouthparts, they differentiate into cell cycle arrested metacyclic promastigotes pre-adapted for the transmission into the mammalian host.
T. cruzi parasites are transmitted through the faeces of triatomine bugs and enter the mammalian host via damage to the skin. The cell cycle arrested metacyclic trypomastigotes invade a wide variety of host cells and transform into replicating amastigotes. These then differentiate into trypomastigotes and are released for another round of invasion or for transmission into the insect vector. The trypomastigote differentiates into the epimastigote form in the midgut of the vector to establish infection. They migrate to the rectal gland where differentiation into the infective metacyclic trypomastigote takes place.
3. Current state of therapy and recent developments
Control of HAT and leishmaniasis relies primarily on chemotherapy. There is a very limited arsenal of drugs and they generally have shortcomings, such as high toxicity and emerging resistance. The drugs currently available to treat HAT have been available for more than half a century. Early stages of HAT are treated with pentamidine, an aromatic diamidine, and suramin, a naphtaline derivative. Side effects for both drugs are significant and the failure rate is high, especially for suramin. Late stages of HAT can be treated with melarsoprol, a melaminophenyl arsenical compound that is able to cross the blood brain barrier. Side effects are severe and up to 5% of those treated die of drug-induced reactive encephalopathy. The only alternative to melarsoprol is eflornithine, an analogue of ornithine that acts as an inhibitor of trypanosomal ornithine decarboxylase, leading to a block in polyamine synthesis. Side effects are significant but eflornithine is much less toxic than melarsoprol [1]. However, eflornithine is not effective against the form of the disease caused by T. brucei rhodesiense in East Africa.
Treatment of leishmaniasis poses even more problems with many different Leishmania species and various infection sites. As for HAT, the main drugs currently available date back over 50 years. The first line drugs are pentavalent antimonials. However, acquired resistance against antimonials is high and common in some parts of the world, such as the Indian subcontinent. The second line drug, pentamidine, is used to treat visceral leishmaniasis refractory to antimonial treatment. Toxicity and emerging resistance are limiting factors in its use. Amphotericin B, an antifungal, is highly effective but associated with severe side effects. Newer lipid formulations of Amphothericin B remain effective with drastically reduced side effects, but their high cost prevents their utilisation in most affected countries. Miltefosine, an ether-lipid, was initially developed as an anticancer agent, but has recently been licensed for use in the treatment of visceral leishmaniasis. Miltefosine has the advantage of being orally active but again has certain drawbacks: it is expensive, teratogenic and resistance has been observed in the laboratory [2]. Drugs under development are paromomycin and sitamaquine. Little is known about their mechanism of action and widespread usefulness of those drugs remains to be shown [3,4].
As with the treatment of HAT and leishmaniasis, the treatment of Chagas disease relies on drugs discovered several decades ago. These are the nitroheterocyclic compounds nitrofuran, nifurtimox and the nitroimidazole derivative benznidazole. Nifurtimox and benznidazole are valuable drugs for the treatment of acute phase Chagas disease but are less effective against the chronic state of the disease, where the risk of significant side effects can outweigh the benefits of treatment [5].
Research aimed at the identification and validation of novel drug targets is a major focus for the kinetoplastid research community. Three recent reviews summarise the current developments [1,3,4]. In this review, we focus on the potential of protein kinases as targets for novel drugs. Protein kinases are important regulators of many different cellular processes such as transcriptional control, cell cycle progression and differentiation, and have drawn much attention as potential drug targets to treat a wide range of diseases and syndromes, such as cancer, cardiovascular disease and Alzheimer’s disease. In recent years, pharmaceutical companies have invested heavily in the development of new compounds directed against specific protein kinase targets, and there are a wide range of protein kinase inhibitors that have entered clinical trials. Two inhibitors, Gleevec (Novartis) and Iressa (Astra-Zeneca) have recently been approved to treat Chronic Myeloid Leukemia and Gastrointestinal Stromal Tumors and non-small-cell lung cancer, respectively [6] (www.gleevec.com, http://www.iressa.com/). Both drugs target tyrosine kinases; Gleevec targets the BCR-ABL tyrosine kinase, and Iressa targets the tyrosine kinase domain of the epidermal growth factor receptor.
Protein kinase inhibitors fall into four main groups: substrate-specific inhibitors, ATP-competitive inhibitors, activation inhibitors and irreversible inhibitors. The ideal protein kinase inhibitor prevents activation rather than competing with the ATP cofactor or the substrate. An example of such an inhibitor is Gleevec, which upon binding induces a conformational change that mimics substrate binding and therefore prevents activation by upstream kinases. However, most inhibitors currently in clinical trials fall into the ATP-competitive inhibitor class. The ATP-binding pocket is well suited to bind specifically designed inhibitors and, despite the relatively high degree of conservation of the ATP-binding pocket, specific inhibitors have been developed.
Progress in this field is of major importance for several reasons: understanding of function and structure of mammalian protein kinases can be used to elucidate the function of their parasite homologues, differences in function can be exploited for drug development and, most importantly, use can be made of the many libraries of compounds generated for the inhibition of mammalian protein kinases.
4. The kinomes of Trypanosoma brucei, Leishmania major and Trypanosoma cruzi
Analysis of the complement of protein kinases in the recently completed genomes of Leishmania major [7], Trypanosoma brucei [8] and Trypanosoma cruzi [9] showed they encode 179, 156 and 171 eukaryotic PKs (ePKs) that are likely to be catalytically active, as well as 17, 20 and 19 atypical protein kinase genes respectively [9], M. Parsons, P.N. Ward, E.A. Worthey, J.C. Mottram, submitted]. The majority of the ePKs reside in clusters of orthologous groups, showing synteny within the three genomes, however each species also contains distinctive protein kinases. The protein kinase complement of the trypanosomatids is about 33% larger than Saccharomyces cerevisiae, but twice that of the malaria parasite Plasmodium falciparum [10]. The trypanosomatid kinomes, which are about one third the size of the human, differ in numerous ways from their mammalian host. For comparative purposes the kinomes of the three trypanosomatids were classified into the seven major groups of ePKs (defined on the basis of sequence similarity of the catalytic domains: AGC, CAMK, CMGC, TK, TKL, STE, Other), according to the nomenclature of Manning [11]. The trypanosomatids lack members of the receptor-linked (TK) or cytosolic tyrosine kinase families (TKL), and possess only a few ePKs with predicted transmembrane domains. Certain groups of protein kinases are poorly represented, such as CAMK and AGC, while others are relatively expanded, such as CMGC and STE (see below). Also, there are many trypanosomatid ePKs that showed no strong similarity to any of the recognized ePK families, suggesting these enzymes differ in structure and function from mammalian ePKs and could be potential drug targets. However, very few of these ePKs have been studied to date. It is noteworthy that the trypanosomatid kinomes contain a number of gene products, 12 examples of which can be found in T. brucei, which are predicted to be catalytically inactive ePKs, as they lack one or more of three amino acid residues known to be essential for catalytic activity (lysine in subdomain 2, aspartic acid in subdomain 6, and aspartic acid in subdomain 7). Of the 12 catalytically inactive ePKs in T. brucei, 10 have inactive orthologues in L. major and T. cruzi, which suggests they have a conserved role, possibly involved in processes that enhance the complexity of regulatory phosphorylation networks, as proposed for inactive mammalian ePKs [12]. In humans most ePKs have accessory domains, many of which are involved in protein:protein interactions, but a search of the trypanosomatid kinome dataset showed that in general they lack additional Pfam domains. The two most numerous additional Pfam domains are the PH domain (PF00169) involved in phosphatidylinositol binding (7 examples in T. brucei) and the FHA domain (PF00498), which is a phosphopeptide binding motif (3 examples in T. brucei).
In this review we focus on the potential of the CMGC family of ePKs as drug targets. The CMGC family includes cyclin-dependent kinases, MAP kinases, GSK3 and dual specificity CLK and DYRK kinases. CMGC kinases are relatively abundant in trypanosomatids, a fact that can possibly be explained by the requirement to control the complex life cycle and cell cycle of the parasites and also by the need to ensure correct replication and segregation of organelles, such as the single mitochondrion, nucleus and flagellum, in a highly polarized cell. In addition there are fundamental differences in cell cycle control between life cycle forms of T. brucei, and key cell cycle checkpoints present in higher eukaryotes are absent from trypanosomes [13,14]. These unique features of trypanosomatid cell cycle biology may be exploitable for the development of specific inhibitors targeting parasite cell cycle transition.
5. Cyclin-dependent kinases
Over the past few years, cyclin-dependent kinases have drawn much attention as potential targets for the treatment of various human cancers. It has been shown that CDKs are frequently deregulated in many cancer forms and therefore have great potential to be selectively inhibited, leading to a reduction or a complete arrest of cell growth. There is a range of selective CDK inhibitors that all act by competing with ATP for binding in the catalytic site of CDKs [6]. Flavopiridol, for example, was the first CDK inhibitor to reach clinical trial. Flavopiridol can induce cell cycle arrest and tumor growth inhibition in many solid tumor cell lines [15]. The same general principles of cell cycle control apply to both mammalian cells and parasites, and it can be speculated that CDKs, which have a crucial role in controlling growth and cell division, pose equally potent drug targets for parasitic diseases as in tumour cell lines.
The CDK family is comparatively large in trypanosomatids, with 11 members in L. major and T. brucei, and 10 members in T. cruzi (Table 1). CDKs require an activating cyclin partner for activity, and analysis of the genome of the three trypanosomatids revealed 10 orthologous cyclins in each parasite (Table 2: named CYC2-CYC11; The T. brucei CYC1, originally proposed to be a cyclin [16] was subsequently found to lack the characteristics of a cyclin [17] and indeed has more sequence similarity to a serine peptidase [7]. L. major has an additional unique mitotic-like cyclin, CYCA. Some CDKs require phosphorylation on a conserved threonine residue (T loop, T160 in human CDK1) by a cdc2-activating kinase (CAK). Many trypanosomatid CRKs (eg CRKs 1,2,3,6,7,8,9 and 12, see Table 1) have a conserved T-loop residue, suggesting that the CRKs might be activated in vivo by a CAK activity [18]). In mammals CAK is a complex of CDK7:cyclinH:MAT1. CRK7 has the highest level of sequence identity to CDK7 of mammals, but no cyclin H or MAT1 orthologues can be identified in trypanosomatids, so it remains to be determined if CRK7 has CAK activity. Some of the trypanosomatid CRKs also have conserved Thr and Tyr residues corresponding to human CDK1 T14 and Y15 (Table 1). Human CDK1 Y15 is phosphorylated by the Wee1 tyrosine kinase to inactivate the CDK complex and Wee 1 kinases can be identified in the three trypanosomatid genomes. Thus it is likely that many trypanosomatid CDK activities are regulated by cyclin binding and phosphorylation, similar to the situation in mammals.
Table 1.
Cdc2-related kinases predicted from the genome of Leishmania major and Trypanosoma brucei. Gene identifiers can be accessed at http://www.genedb.org/. T. brucei orthologues were identified by synteny and cluster of orthologous groups (COG) analyses. * human CDK1 numbering. CRK5 was originally thought to be a CDK, but was subsequently found to be a member of the RCK family and has the highest level of sequence identity to human MOK.
| L. major | Name | Size kDa | T14, Y15* | T160* | Cyclin binding | T. bruceiorthologue | CMGC family |
|---|---|---|---|---|---|---|---|
| LMJF21.1080 | CRK1 | 34.4 | T, Y | T | PCTAIRE | Tb10.70.7040 | CDK |
| LMJF05.0550 | CRK2 | 36.4 | S, Y | T | SVSSIRE | Tb07.30D13.430 | CDK |
| LMJF36.0550 | CRK3 | 35.6 | T, Y | T | PQTALRE | Tb10.70.2210 | CDK |
| LMJF16.0990 | CRK4 | 51.7 | T, Y | S | PGAAIRE | Tb08.5H5.130 | CDK |
| LmjF35.5010 | CRK5 | 44.2 | T, F | T | QVNRLRE | Tb09.211.0960 | RCK - MOK |
| LMJF27.0560 | CRK6 | 37.3 | T, Y | T | PATTIRE | Tb11.47.0031 | CDK |
| LMJF26.0040 | CRK7 | 32.4 | Q, F | S | PHPVARE | Tb07.43M14.340 | CDK |
| LmjF11.0110 | CRK8 | 44.4 | T, F | T | HRCTFRE | Tb11.02.5010 | CDK |
| LMJF27.1940 | CRK9 | 101.8 | A, S | T | QREEARP | Tb927.2.4510 | CDK |
| LMJF29.2150 | CRK10 | 48.4 | C, G | V | RKGAFDA | Tb03.48K5.160 | CDK |
| LMJF30.1780 | CRK11 | 66.3 | G, Y | P | SATVLRE | Tb06.5F5.880 | CDK |
| LMJF09.0310 | CRK12 | 54.4 | T, Y | T | PQTSLRE | Tb11.01.4130 | CDK |
Table 2.
Cyclins and protein kinase accessory proteins predicted from the genome of Leishmania major and Trypanosoma brucei. Gene identifiers can be accessed at http://www.genedb.org/. Orthologues of the L. major genes were identified by synteny and cluster of orthologous groups (COG) analyses.
| L. major | Name | Size kDa | T. bruceiorthologue | Name | Alternative name | Products |
|---|---|---|---|---|---|---|
| LmjF25.1470 | CYCA | 34.2 | none | - | - | Mitotic-like cyclin |
| LmjF32.0820 | CYC2 | 18.8 | Tb11.01.5660 | CYC2 | CycE1 | CYC2 cyclin |
| LmjF30.0080 | CYC3 | 46.9 | Tb06.3A7.1310 | CYC3 | CycB1 | Mitotic-like cyclin |
| LmjF05.0710 | CYC4 | 149 | Tb07.21H15.170 | CYC4 | CycE3 | CYC2-like cyclin |
| LmjF33.0770 | CYC5 | 93.6 | Tb10.26.0510 | CYC5 | CycE4 | CYC2-like cyclin |
| LmjF32.3320 | CYC6 | 35.2 | Tb11.01.8460 | CYC6 | CycB2 | Mitotic cyclin |
| LmjF30.3630 | CYC7 | 27.6 | Tb06.30P15.430 | CYC7 | CycE2 | CYC2-like cyclin |
| LmjF26.0330 | CYC8 | 50.6 | Tb07.27M11.950 | CYC8 | CycB3 | Mitotic-like cyclin |
| LmjF32.0760 | CYC9 | 32.9 | Tb11.01.5600 | CYC9 | Cyclin C-like | |
| LmjF24.1890 | CYC10 | 68.7 | Tb08.11J15.340 | CYC10 | CYC2-like cyclin | |
| LmjF24.1880 | CYC11 | 101.6 | Tb08.11J15.300 | CYC11 | CYC2-like cyclin | |
| Tb07.10C21.430 | MOB1A | MOB1 | ||||
| LmjF06.0960 | MOB1 | Tb07.10C21.440 | MOB1B | MOB1 | ||
| LmjF32.3790 | CKS1 | Tb11.01.8085 | CKS1 | CKS1 |
The function of two putative CDKs in Leishmania mexicana, LmexCRK1 and LmexCRK3, have been analysed in some detail [19–24]. LmexCRK1 is present in all life cycle stages but its histone H1 kinase activity is restricted to the insect stages of the parasite, both proliferative promastigotes and cell cycle arrested metacyclic promastigotes [24]. LmexCRK1 is essential in Leishmania promastigotes, but its role in amastigotes has not been investigated, so it is not known if it could be a useful drug target [24]. Since analysis of the function of essential genes by RNA interference is not possible in Leishmania, it is a challenge to further determine the role of LmexCRK1 in cell cycle control. However, since the T. brucei CRK1 was able to complement LmexCRK1, it can be speculated that both proteins fulfil a similar function in the two parasites (see below) [21,24].
L. major CRK3 was able to complement a temperature-sensitive Schizosaccharomyces pombe cdc2 mutant. Furthermore, CRK3 is essential for the growth of the parasite [20]. A screen to test 634 chemically diverse antimitotic compounds revealed 27 inhibitors of CRK3, 16 of which also inhibited growth and replication of L. donovani amastigotes in infected peritoneal macrophages. The effects of some of the compounds on L. mexicana promastigotes were analysed by flow cytometry and microscopy. The analysis revealed a high proportion of cells with aberrant DNA content and abnormal morphology, indicating a disruption of normal cell cycle control mechanisms [25]. This study, the only one to date that systematically analysed the effects of CDK inhibitors in kinetoplastid parasites, proves the validity of these ePKs as potential drug targets.
In contrast to Leishmania, T. brucei gene function can be analysed by RNAi. However, of the large family of putative CDKs and cyclins in T. brucei, only a few have been characterised in detail [13,26–29]. RNAi studies of CRK1, 2, 3, 4 and 6 have shed some light on potential roles of CRKs in the trypanosome cell cycle [27,28]. When CRK1 was downregulated by RNAi in both procyclic and bloodstream form trypanosomes, there was a reduced growth and an increase in G1 cells, indicating a role in controlling the G1/S transition in both procyclic and bloodstream form trypanosomes [27,28]. A simultaneous downregulation of CRK1 and either CRK2, 4 and 6 enhanced this phenotype, suggesting an auxiliary role for these three CRKs. Neither CRK2 nor CRK6 are essential for cell cycle control progression individually, but CRK4 may have a non-essential role in both G1/S and G2/M transitions. Downregulation of CRK3 in both procyclic and bloodstream form trypanosomes resulted in a mitotic block and growth arrest. The nuclei of the arrested cells were enlarged and bromodeoxyuridine (BrdU) incorporation showed continuous DNA synthesis without subsequent mitosis [28]. Kinetoplast (mitochondrial DNA) segregation was not impaired. In procyclic form trypanosomes, kinetoplast segregation appeared to be able to drive cytokinesis and cell division when the cells were arrested in G2/M, resulting in the formation of zoids (anucleate cells). This did not appear to be the case for RNAi of CRK3 in bloodstream form trypanosomes. In this case there was a significant increase in highly abnormal cells containing multiple kinetoplasts and a grossly enlarged nucleus, indicating that cytokinesis and cell division were blocked but not kinetoplast replication and segregation [27,28]. It has been shown that CRK3 interacts with CYC2 and CYC6 [13,26,29,30] and that down regulation of these two cyclins by RNA interference correlates with the CRK3 data. CYC6 down regulation resulted in a mitotic block both in procylic and bloodstream form trypanosomes [13], in agreement with bioinformatic predictions that CYC6 is a mitotic cyclin (Table 2). Thus, the CRK3:CYC6 complex has the properties of a G2/M phase kinase required for regulation of entry into mitosis (Figure 2).
Figure 2.
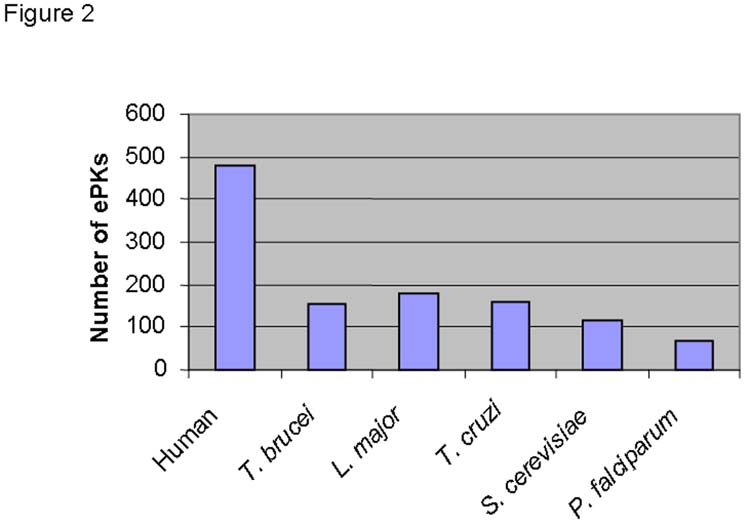
Comparison of the ePK complement in the genomes of selected eukaryotes
Two papers describe the function of CYC2 using RNAi [26,29]. Downregulation of CYC2 resulted in an irreversible growth arrest in procyclic and bloodstream form trypanosomes with a slightly decreased penetrance of the phenotype in the bloodstream form trypanosomes [26,29]). One study [29] described the formation of zoids upon downregulation of CYC2, similar to the phenotype observed when CRK3 was down regulated in the same life cycle stage. However, zoid formation was not observed by Hammarton et al [26]. This discrepancy might be explained by the use of different RNA interference systems, resulting in different penetrance of the phenotype. Taken together, however, the data of both studies suggest that the CRK3:CYC2 complex is required for progression through G1 (Figure 2).
A few reports describe the identification and characterisation of two CRKs, TzCRK1 and TzCRK3 in T. cruzi [31–34]. TzCRK3 is a possible CDK1 homologue whose activity in synchronised T. cruzi cells was found to peak at the boundary of G2/M, whereas TzCRK1 was active throughout G1 and S [34]. Both kinases were capable of interacting with mammalian cyclins, confirming that they function as cyclin-dependent kinases. However, TzCRK1 was active as a recombinant protein suggesting cyclin binding might not be required for activity and that there might be other different mechanisms of activation [32]. TzCRK3 and to a lesser extent TzCRK1 was capable of phosphorylating T. cruzi histone H1 and this phosphorylation was highest at the transitions of G2/M and G1/S. There is evidence that H1 phosphorylation varies during the life cycle of T. cruzi [31], but the possible relevance of phosphorylation of histone H1 by TzCRK3 has yet to be elucidated.
6. MAP kinases
Mitogen-activated protein (MAP) kinases are important regulators of differentiation and cell proliferation in many eukaryotes [35]. External stimuli and other signals are transmitted via three MAP kinases receiver molecules, ultimately leading to a change in transcription and a change of the proliferative status or the state of differentiation of a cell. Malfunction of MAP kinases is implicated in many forms of cancer and over the past few years MAP kinases have become important targets for the development of novel drugs to treat malignancies. Small-molecule inhibitors designed to target various steps of the MAPK pathway have entered clinical trials [35,36]
Several reports describe the identification of MAP kinases (MAPKs) and their activators, the MAPK kinases (MAPKK), from T. brucei and Leishmania [37–41]. However, despite the completion of the genome sequencing projects in the three kinetoplastid parasites discussed, it has proven impossible to predict a complete MAP kinase signalling pathway for these parasites based on sequence analysis alone.
10 MAPK (LmxMPK – LmxMPK9) and 1 MAPKK (LmxMKK) have been identified and partially characterised in Leishmania mexicana [37–41]. Of these, LmxMPK, LmxMPK9 and LmxMKK [37,39,41] have been characterised in detail. Both LmxMKK and LmxMPK9 are exclusively expressed in promastigotes and are involved in the maintenance of flagellar length [37,39]. Whereas LmxMPK9 null mutants display elongated flagella, LmxMKK null mutants have a very short flagellum and are not motile. Moreover they show defects in the paraflagellar rod (PFR) and an accumulation of membrane fragments in the flagellar pocket. In BALB/c mice infected with LmxMKK null mutants, lesion development is delayed, but later the infection progresses as in wild type infections. There is no morphological defect in the vestigial amastigote flagellum, indicating that retrograde transport in the flagellum remains unaffected. LmxMKK is possibly involved in the anterograde transport. Because both LmxMKK and LmxMPK9 null mutants are viable throughout the entire life cycle, neither of them can be classified as a potential drug target. LmxMPK, however, shows potential as a drug target [41], since null mutants can infect peritoneal macrophages and differentiate into amastigotes, but are unable to proliferate within the parsitophorous vacuole. It seems, therefore, that LmxMPK is essential for parasite growth in the clinically relevant amastigote form. Moreover, the null mutant is unable to establish infection and causes footpad lesions in BALB/c mice.
Three MAPKs have been described in some detail in T. brucei [42–45]. KFR1 can be classified into the ERK subfamily of MAP kinases. It has been proposed that KFR1 is involved in the proliferation of bloodstream form trypanosomes, based on the finding that KFR1 could be activated by IFN-γ in bloodstream form trypanosomes, but not the procyclic form trypanosomes, leading to slightly faster growth of the parasites. However, the increase in growth was modest and this needs to be further investigated [43]. TbMAPK2, also classified as an ERK subfamily kinase, is not essential for the bloodstream form trypanosome, but is important for differentiation from the bloodstream form to the procyclic form. Mutants lacking TbMAPK2 could differentiate, but were unable to proliferate in the insect stage. Moreover, this proliferation was only possible in the presence of active TbMAPK2 [42]. TbECK1, which has characteristics of both MAPKs and CDKs, and was named T. brucei ERK-like, CDK-like protein kinase [45], falls into the CDK-like family by phylogenetic analysis [18]. The kinase appears to be essential in all life cycle stages analysed [45]. TbECK1 has an unusual C-terminal extension that can also be found in several Leishmania MAP kinases (LmxMPK2, 3, 6, 7,8 and 9) and several human ERK kinases. Overexpression of TbECK1 lacking the C-terminal extension in procyclic trypanosomes leads to a significant reduction in growth, and an accumulation of cells with aberrant nucleus/kinetoplast configurations, suggesting a role in the cell cycle of T. brucei. The C-terminal extension appears to be acting as a cis-acting negative regulator of TbECK1. The kinase could not be ectopically expressed in the bloodstream form trypanosome, suggesting that its activity needs to be tightly regulated in this stage [45]. Of the three T. brucei MAP type kinases described, TbECK1 has the most potential as a drug target. It remains to be seen if any of the multiple MAP kinases identified through the L. major and T. brucei genome projects are possible drug targets.
7. Other kinases
Several other kinase families are worth noting as potential drug targets in trypanosomatids. The NEK family is particularly interesting, as there is a significant expansion of NEK kinases in trypanosomatids (~20) when compared to humans (15), suggesting that some of the parasite enzymes will have unique functions. Not much is known about the function of NEK during the life cycle of trypanosomatids [46–48], although one is known to be expressed at a higher level in the bloodstream than the procyclic form [46] and another has an essential role in basal body duplication (D. Robinson, personal communication).
Members of the Casein kinase I (CK1) family are also of potential interest. In a search for intracellular targets of purvalanols, a group of well characterised CDK inhibitors, by affinity chromatography with an immobilised inhibitor (purvalanol B), one CKI was identified as a major putative target in L. mexicana and T. cruzi [49]. In contrast, mammalian CK1 did not bind to purvalanol B from any of the tissues tested [49], indicating that the parasite CK1s are significantly different from their mammalian counterparts. Purvalanols have relatively broad inhibitory specificities, as they inhibit mammalian CK1, ERK1, ERK2 and other kinases, but with a 1000-fold lower potency than CDK1, hence would normally count as selective. It remains to be shown whether the binding of leishmanial CK1 to purvalanol B is due to higher abundance of CK1 than of CDKs in the parasite extract. Furthermore, it needs to be shown whether growth inhibition of trypanosomatid parasites is due to inhibition of CK1 or CDKs [49].
A member of the AGC family, which is under-represented in trypanosomatids, PK50, has been shown to interact with MOB1 and form an active kinase complex in vivo [50]. Knockdown of MOB1 results in growth arrest and a block of cytokinesis in bloodstream forms [50] (Figure 2). There are no syntenic orthologues of PK50 in L. major or T. cruzi, but the finding that the PK50:MOB1 complex is essential for cytokinesis makes it a promising potential drug target in trypanosomes.
8. Conclusions and perspectives
Several lines of evidence suggest that the protein kinase family is worth pursuing to identify drug targets for trypanosomes and Leishmania. Firstly, several protein kinases have been shown to be essential for proliferation and/or viability of parasite life-cycle stages that are clinically relevant. These include examples from several families (eg CRK3 of the CMGC family and PK50 of the AGC family). Secondly, protein kinases of trypanosomatids have significant sequence differences from mammalian homologues. Indeed, a recent analysis of human protein kinases described the relationship between sequence similarity and structure activity relationships for inhibitors of 78 kinases [51]. The authors propose that, as a general rule, protein kinases having more than 60% sequence identity over the core catalytic domain have a high probability of being inhibited by the same group of low molecular mass compounds. The corollary is that there is a higher level of confidence that specific inhibitors can be designed to target protein kinases with <60% sequence identity, which is the case for the vast majority of the parasite kinomes when compared to that of humans. This concept has only been validated in one case for trypanosomatids, where CDK inhibitors of the indirubin, paullone and 2,6,9-trisubstituted purine families were found to have up to 1000-fold difference in IC50s between the L. mexicana CRK3 and human cdc2 [25]. These two CDKs have homologous functions in the G2/M phase transition, but only 54% sequence identity [19]. A third point for consideration is the concern that resistance to highly specific inhibitors might arise in the target protein kinase, making a drug ineffective. This has already been observed in some chronic myelogenous leukaemia patients, who have been treated with the highly specific Bcr-Abl kinase inhibitor Gleevec. A major mechanism of resistance has been the appearance of point mutations in the kinase that affects residues involved in binding of the inhibitor [52]. Thus, it might be an advantage to develop a less specific inhibitor that targets several closely related protein kinases within a family.
Figure 3.
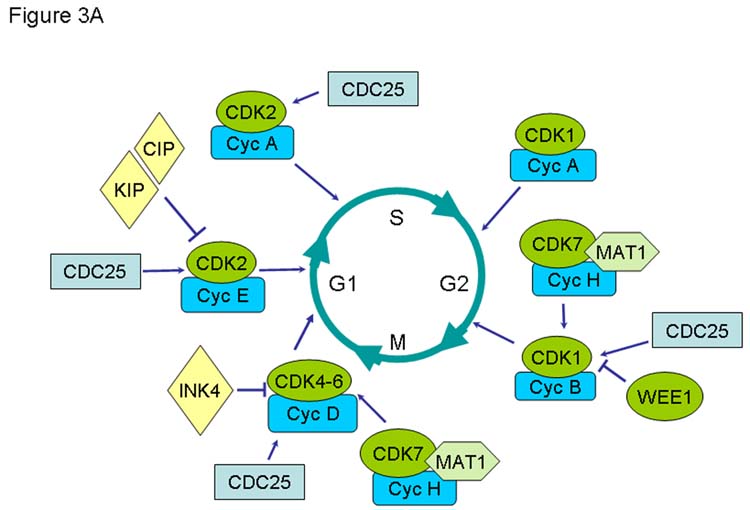
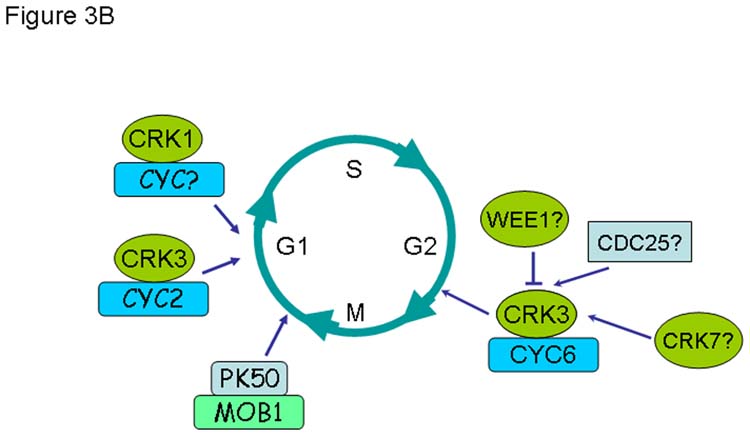
Comparison of known functions of cyclin-dependent kinases and associated regulatory molecules during the cell cycle of human (A) and T. brucei (B).
Acknowledgments
We thank Karen Grant and Tansy Hammarton for critical comments on the manuscript and participants at the 4th international conference on inhibitors of protein kinases, Warsaw June 25–29th 2005 for useful discussions. JCM is supported by the Medical Research Council and the Wellcome Trust. This work was supported in part by NIH AI31077 (MP).
References
- 1.Naula C, Burchmore R. A plethora of targets, a paucity of drugs: progress towards the development of novel chemotherapies for human African trypanosomiasis. Expert Rev Anti Infect Ther. 2003;1:157–165. doi: 10.1586/14787210.1.1.157. [DOI] [PubMed] [Google Scholar]
- 2.Perez-Victoria FJ, Castanys S, Gamarro F. Leishmania donovani resistance to miltefosine involves a defective inward translocation of the drug. Antimicrob Agents Chemother. 2003;47:2397–2403. doi: 10.1128/AAC.47.8.2397-2403.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Croft SL, Coombs GH. Leishmaniasis--current chemotherapy and recent advances in the search for novel drugs. Trends Parasitol. 2003;19:502–508. doi: 10.1016/j.pt.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 4.Ouellette M, Drummelsmith J, Papadopoulou B. Leishmaniasis: drugs in the clinic, resistance and new developments. Drug Resist Updat. 2004;7:257–266. doi: 10.1016/j.drup.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 5.Urbina JA, Docampo R. Specific chemotherapy of Chagas disease: controversies and advances. Trends Parasitol. 2003;19:495–501. doi: 10.1016/j.pt.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 6.R.Buijsman, Structural Aspects of Kinases and Their Inhibitors, in: H.Kubinyi and G.M¨ller (Eds.), Chemogenomics in Drug Discovery: A Medicinal Chemistry Perspective, Wiley Publishers, 2004, pp.191–219.
- 7.Ivens AC, Peacock CS, Worthey EA, Murphy L, Aggarwal G, Berriman M, Sisk E, Rajandream MA, Adlem E, Aert R, Anupama A, Apostolou Z, Attipoe P, Bason N, Bauser C, Beck A, Beverley SM, Bianchettin G, Borzym K, Bothe G, Bruschi CV, Collins M, Cadag E, Ciarloni L, Clayton C, Coulson RMR, Cronin A, Cruz AK, Davies RM, De Gaudenzi J, Dobson DE, Duesterhoeft A, Fazelina G, Fosker N, Frasch AC, Fraser A, Fuchs M, Gabel C, Goble A, Goffeau A, Harris D, Hertz-Fowler C, Hilbert H, Horn D, Huang Y, Klages S, Knights A, Kube M, Larke N, Litvin L, Lord A, Louie T, Marra M, Masuy D, Matthews K, Michaeli S, Mottram JC, Muller-Auer S, Munden H, Nelson S, Norbertczak H, Oliver K, O’Neil S, Pentony M, Pohl TM, Price C, Purnelle B, Quail MA, Rabbinowitsch E, Reinhardt R, Rieger M, Rinta J, Robben J, Robertson L, Ruiz JC, Rutter S, Saunders D, Schafer M, Schein J, Schwartz DC, Seeger K, Seyler A, Sharp S, Shin H, Sivam D, Squares R, Squares S, Tosato V, Vogt C, Volckaert G, Wambutt R, Warren T, Wedler H, Woodward J, Zhou S, Zimmermann W, Smith DF, Blackwell JM, Stuart KD, Barrell B, Myler PJ. The genome of the kinetoplastid parasite, Leishmania major. Science. 2005;309:436–442. doi: 10.1126/science.1112680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berriman M, Ghedin E, Hertz-Fowler C, Blandin G, Renauld H, Bartholomeu DC, Lennard NJ, Caler E, Hamlin NE, Haas B, Bohme U, Hannick L, Aslett MA, Shallom L, Marcello L, Hou L, Wickstead B, Alsmark UC, Arrowsmith C, Atkin RJ, Barron AJ, Bringaud F, Brooks K, Carrington M, Cherevach I, Chillingworth TJ, Churcher C, Clark LN, Corton CH, Cronin A, Davies RM, Doggett J, Djikeng A, Feldblyum T, Field MC, Fraser A, Goodhead I, Hance Z, Harper D, Harris BR, Hauser H, Hostetler J, Ivens A, Jagels K, Johnson D, Johnson J, Jones K, Kerhornou AX, Koo H, Larke N, Landfear S, Larkin C, Leech V, Line A, Lord A, MacLeod A, Mooney PJ, Moule S, Martin DMA, Morgan GW, Mungall K, Norbertczak H, Ormond D, Pai G, Peacock CS, Peterson J, Quail MA, Rabbinowitsch E, Rajandream MA, Reitter C, Salzberg SL, Sanders M, Schobel S, Sharp S, Simmonds M, Simpson AJ, Tallon L, Turner CM, Tait A, Tivey AR, Van Aken S, Walker D, Wanless D, Wang S, White B, White O, Whitehead S, Woodward J, Wortman J, Adams MD, Embley TM, Gull K, Ullu E, Barry JD, Fairlamb AH, Opperdoes F, Barrell BG, Donelson JE, Hall N, Fraser CM, Melville SE, El Sayed NM. The genome of the African trypanosome Trypanosoma brucei. Science. 2005;309:416–422. doi: 10.1126/science.1112642. [DOI] [PubMed] [Google Scholar]
- 9.ElSayed NM, Myler PJ, Bartholomeu DC, Nilsson D, Aggarwal G, Tran AN, Ghedin E, Worthey EA, Delcher AL, Blandin G, Westenberger SJ, Caler E, Cerqueira GC, Branche C, Haas B, Anupama A, Arner E, Aslund L, Attipoe P, Bontempi E, Bringaud F, Burton P, Cadag E, Campbell DA, Carrington M, Crabtree J, Darban H, da Silveira JF, de Jong P, Edwards K, Englund PT, Fazelina G, Feldblyum T, Ferella M, Frasch AC, Gull K, Horn D, Hou L, Huang Y, Kindlund E, Klingbeil M, Kluge S, Koo H, Lacerda D, Levin MJ, Lorenzi H, Louie T, Machado CR, McCulloch R, McKenna A, Mizuno Y, Mottram JC, Nelson S, Ochaya S, Osoegawa K, Pai G, Parsons M, Pentony M, Pettersson U, Pop M, Ramirez JL, Rinta J, Robertson L, Salzberg SL, Sanchez DO, Seyler A, Sharma R, Shetty J, Simpson AJ, Sisk E, Tammi MT, Tarleton R, Teixeira S, Van Aken S, Vogt C, Ward PN, Wickstead B, Wortman J, White O, Fraser CM, Stuart KD, Andersson B. The genome sequence of Trypanosoma cruzi, etiologic agent of chagas disease. Science. 2005;309:409–415. doi: 10.1126/science.1112631. [DOI] [PubMed] [Google Scholar]
- 10.Ward P, Equinet L, Packer J, Doerig C. Protein kinases of the human malaria parasite Plasmodium falciparum: the kinome of a divergent eukaryote. BMC Genomics. 2004;5:79. doi: 10.1186/1471-2164-5-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 12.Pils B, Schultz J. Inactive enzyme-homologues find new function in regulatory processes. J Mol Biol. 2004;340:399–404. doi: 10.1016/j.jmb.2004.04.063. [DOI] [PubMed] [Google Scholar]
- 13.Hammarton TC, Clark J, Douglas F, Boshart M, Mottram JC. Stage-specific differences in cell cycle control in Trypansoma brucei revealed by RNA interference of a mitotic cyclin. J Biol Chem. 2003;278:22877–22886. doi: 10.1074/jbc.M300813200. [DOI] [PubMed] [Google Scholar]
- 14.Hammarton TC, Mottram JC, Doerig C. The cell cycle of parasitic protozoa: potential for chemotherapeutic exploitation. Prog Cell Cycle Res. 2003;5:91–101. [PubMed] [Google Scholar]
- 15.Shapiro GI. Preclinical and clinical development of the cyclin-dependent kinase inhibitor flavopiridol. Clin Cancer Res. 2004;10:4270s–4275s. doi: 10.1158/1078-0432.CCR-040020. [DOI] [PubMed] [Google Scholar]
- 16.Affranchino JL, Gonzalez SA, Pays E. Isolation of a mitotic-like cyclin homologue from the protozoan Trypanosoma brucei. Gene. 1993;132:75–82. doi: 10.1016/0378-1119(93)90516-6. [DOI] [PubMed] [Google Scholar]
- 17.Hammarton TC, Ford JR, Mottram JC. Trypanosoma brucei CYC1 does not have characteristics of a mitotic cyclin. Mol Biochem Parasitol. 2000;111:229–233. doi: 10.1016/s0166-6851(00)00308-x. [DOI] [PubMed] [Google Scholar]
- 18.M. Parsons, P.N. Ward, E.A. Worthey, J.C. Mottram, Comparative analysis of the kinomes of three pathogenic trypanosomatids; Leishmania major, Trypanosoma brucei and Trypanosoma cruzi, BMC. Genomics submitted (2005). [DOI] [PMC free article] [PubMed]
- 19.Grant KM, Hassan P, Anderson JS, Mottram JC. The CRK3 gene of Leishmania mexicana encodes a stage-regulated cdc2-related histone H1 kinase that associates with p12. J Biol Chem. 1998;273:10153–10159. doi: 10.1074/jbc.273.17.10153. [DOI] [PubMed] [Google Scholar]
- 20.Hassan P, Fergusson D, Grant KM, Mottram JC. The CRK3 protein kinase is essential for cell cycle progression of Leishmania mexicana. Mol Biochem Parasitol. 2001;113:189–198. doi: 10.1016/s0166-6851(01)00220-1. [DOI] [PubMed] [Google Scholar]
- 21.Mottram JC, McCready BP, Brown KG, Grant KM. Gene disruptions indicate an essential function for the LmmCRK1 cdc2-related kinase of Leishmania mexicana. Mol Microbiol. 1996;22:573–583. doi: 10.1046/j.1365-2958.1996.00136.x. [DOI] [PubMed] [Google Scholar]
- 22.Mottram JC, Grant KM. Leishmania mexicana p12cks1, a homologue of fission yeast p13suc1, associates with a stage-regulated histone H1 kinase. Biochem J. 1996;316(Pt3):833–839. doi: 10.1042/bj3160833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Y, Dimitrov K, Garrity LK, Sazer S, Beverley SM. Stage-specific activity of the Leishmania major CRK3 kinase and functional rescue of a Schizosaccharomyces pombe cdc2 mutant. Mol Biochem Parasitol. 1998;96:139–150. doi: 10.1016/s0166-6851(98)00121-2. [DOI] [PubMed] [Google Scholar]
- 24.Mottram JC, Kinnaird JH, Shiels BR, Tait A, Barry JD. A novel CDC2-related protein kinase from Leishmania mexicana, LmmCRK1, is post-translationally regulated during the life cycle. J Biol Chem. 1993;268:21044–21052. [PubMed] [Google Scholar]
- 25.Grant KM, Dunion MH, Yardley V, Skaltsounis AL, Marko D, Eisenbrand G, Croft SL, Meijer L, Mottram JC. Inhibitors of Leishmania mexicana CRK3 cyclin-dependent kinase: chemical library screen and antileishmanial activity. Antimicrob Agents Chemother. 2004;48:3033–3042. doi: 10.1128/AAC.48.8.3033-3042.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hammarton TC, Engstler M, Mottram JC. The Trypanosoma brucei cyclin, CYC2, is required for cell cycle progression through G1 phase and for maintenance of procyclic form cell morphology. J Biol Chem. 2004;279:24757–24764. doi: 10.1074/jbc.M401276200. [DOI] [PubMed] [Google Scholar]
- 27.Tu X, Wang CC. Pairwise Knockdowns of cdc2-Related Kinases (CRKs) in Trypanosoma brucei Identified the CRKs for G1/S and G2/M Transitions and Demonstrated Distinctive Cytokinetic Regulations between Two Developmental Stages of the Organism. Eukaryot Cell. 2005;4:755–764. doi: 10.1128/EC.4.4.755-764.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tu X, Wang CC. The involvement of two cdc2-related kinases (CRKs) in Trypanosoma brucei cell cycle regulation and the distinctive stage-specific phenotypes caused by CRK3 depletion. J Biol Chem. 2004;279:20519–20528. doi: 10.1074/jbc.M312862200. [DOI] [PubMed] [Google Scholar]
- 29.Li Z, Wang CC. A PHO80-like cyclin and a B-type cyclin control the cell cycle of the procyclic form of Trypanosoma brucei. J Biol Chem. 2003;278:20652–20658. doi: 10.1074/jbc.M301635200. [DOI] [PubMed] [Google Scholar]
- 30.Van Hellemond JJ, Neuville P, Schwarz RT, Matthews KR, Mottram JC. Isolation of Trypanosoma brucei CYC2 and CYC3 cyclin genes by rescue of a yeast G(1) cyclin mutant. Functional characterization of CYC2. J Biol Chem. 2000;275:8315–8323. doi: 10.1074/jbc.275.12.8315. [DOI] [PubMed] [Google Scholar]
- 31.da Cunha JP, Nakayasu ES, Elias MC, Pimenta DC, Tellez-Inon MT, Rojas F, Manuel M, Almeida IC, Schenkman S. Trypanosoma cruzi histone H1 is phosphorylated in a typical cyclin dependent kinase site accordingly to the cell cycle. Mol Biochem Parasitol. 2005;140:75–86. doi: 10.1016/j.molbiopara.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 32.Gomez EB, Kornblihtt AR, Tellez-Inon MT. Cloning of a cdc2-related protein kinase from Trypanosoma cruzi that interacts with mammalian cyclins. Mol Biochem Parasitol. 1998;91:337–351. doi: 10.1016/s0166-6851(97)00218-1. [DOI] [PubMed] [Google Scholar]
- 33.Gomez EB, Santori MI, Laria S, Engel JC, Swindle J, Eisen H, Szankasi P, Tellez-Inon MT. Characterization of the Trypanosoma cruzi Cdc2p-related protein kinase 1 and identification of three novel associating cyclins. Mol Biochem Parasitol. 2001;113:97–108. doi: 10.1016/s0166-6851(00)00382-0. [DOI] [PubMed] [Google Scholar]
- 34.Santori MI, Laria S, Gomez EB, Espinosa I, Galanti N, Tellez-Inon MT. Evidence for CRK3 participation in the cell division cycle of Trypanosoma cruzi. Mol Biochem Parasitol. 2002;121:225–232. doi: 10.1016/s0166-6851(02)00039-7. [DOI] [PubMed] [Google Scholar]
- 35.Sebolt-Leopold JS, Herrera R. Targeting the mitogen-activated protein kinase cascade to treat cancer. Nat Rev Cancer. 2004;4:937–947. doi: 10.1038/nrc1503. [DOI] [PubMed] [Google Scholar]
- 36.Strumberg D, Seeber S. Raf kinase inhibitors in oncology. Onkologie. 2005;28:101–107. doi: 10.1159/000083373. [DOI] [PubMed] [Google Scholar]
- 37.Bengs F, Scholz A, Kuhn D, Wiese M. LmxMPK9, a mitogen-activated protein kinase homologue affects flagellar length in Leishmania mexicana. Mol Microbiol. 2005;55:1606–1615. doi: 10.1111/j.1365-2958.2005.04498.x. [DOI] [PubMed] [Google Scholar]
- 38.Wiese M, Wang Q, Gorcke I. Identification of mitogen-activated protein kinase homologues from Leishmania mexicana. Int J Parasitol. 2003;33:1577–1587. doi: 10.1016/s0020-7519(03)00252-2. [DOI] [PubMed] [Google Scholar]
- 39.Wiese M, Kuhn D, Grunfelder CG. Protein kinase involved in flagellar-length control. Eukaryot Cell. 2003;2:769–777. doi: 10.1128/EC.2.4.769-777.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wiese M, Gorcke I. Homologues of LMPK, a mitogen-activated protein kinase from Leishmania mexicana, in different Leishmania species. Med Microbiol Immunol (Berl) 2001;190:19–22. doi: 10.1007/s004300100072. [DOI] [PubMed] [Google Scholar]
- 41.Wiese M. A mitogen-activated protein (MAP) kinase homologue of Leishmania mexicana is essential for parasite survival in the infected host. EMBO J. 1998;17:2619–2628. doi: 10.1093/emboj/17.9.2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Muller IB, Domenicali-Pfister D, Roditi I, Vassella E. Stage-specific requirement of a mitogen-activated protein kinase by Trypanosoma brucei. Mol Biol Cell. 2002;13:3787–3799. doi: 10.1091/mbc.E02-02-0093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hua SB, Wang CC. Interferon-gamma activation of a mitogen-activated protein kinase, KFR1, in the bloodstream form of Trypanosoma brucei. J Biol Chem. 1997;272:10797–10803. doi: 10.1074/jbc.272.16.10797. [DOI] [PubMed] [Google Scholar]
- 44.Hua SB, Wang CC. Differential accumulation of a protein kinase homolog in Trypanosoma brucei. J Cell Biochem. 1994;54:20–31. doi: 10.1002/jcb.240540104. [DOI] [PubMed] [Google Scholar]
- 45.Ellis J, Sarkar M, Hendriks E, Matthews K. A novel ERK-like, CRK-like protein kinase that modulates growth in Trypanosoma brucei via an autoregulatory C-terminal extension. Mol Microbiol. 2004;53:1487–1499. doi: 10.1111/j.1365-2958.2004.04218.x. [DOI] [PubMed] [Google Scholar]
- 46.Gale M, Jr, Carter V, Parsons M. Translational control mediates the developmental regulation of the Trypanosoma brucei Nrk protein kinase. J Biol Chem. 1994;269:31659–31665. [PubMed] [Google Scholar]
- 47.Gale M, Jr, Carter V, Parsons M. Cell cycle-specific induction of an 89 kDa serine/threonine protein kinase activity in Trypanosoma brucei. J Cell Sci. 1994;107(Pt7):1825–1832. doi: 10.1242/jcs.107.7.1825. [DOI] [PubMed] [Google Scholar]
- 48.Gale M, Jr, Parsons M. A Trypanosoma brucei gene family encoding protein kinases with catalytic domains structurally related to Nek1 and NIMA. Mol Biochem Parasitol. 1993;59:111–121. doi: 10.1016/0166-6851(93)90012-m. [DOI] [PubMed] [Google Scholar]
- 49.Knockaert M, Gray N, Damiens E, Chang YT, Grellier P, Grant K, Fergusson D, Mottram J, Soete M, Dubremetz JF, Le Roch K, Doerig C, Schultz P, Meijer L. Intracellular targets of cyclin-dependent kinase inhibitors: identification by affinity chromatography using immobilised inhibitors. Chem Biol. 2000;7:411–422. doi: 10.1016/s1074-5521(00)00124-1. [DOI] [PubMed] [Google Scholar]
- 50.Hammarton TC, Lillico SG, Welburn SC, Mottram JC. Trypanosoma brucei MOB1 is required for accurate and efficient cytokinesis but not for exit from mitosis. Mol Microbiol. 2005;56:104–116. doi: 10.1111/j.1365-2958.2005.04542.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vieth M, Higgs RE, Robertson DH, Shapiro M, Gragg EA, Hemmerle H. Kinomics-structural biology and chemogenomics of kinase inhibitors and targets. Biochim Biophys Acta. 2004;1697:243–257. doi: 10.1016/j.bbapap.2003.11.028. [DOI] [PubMed] [Google Scholar]
- 52.Shah NP, Tran C, Lee FY, Chen P, Norris D, Sawyers CL. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science. 2004;305:399–401. doi: 10.1126/science.1099480. [DOI] [PubMed] [Google Scholar]