

Abstract
Among the virulence factors present in pathogenic extraintestinal Escherichia coli strains, expression of fimbrial adhesins is necessary for attachment to the host tissues and subsequent colonization. Occurrence of the sfa determinant coding for the S fimbriae is widespread among the uropathogens and meningitis isolates. The sfa operon consists of nine genes. In the biogenesis of S fimbriae, the proteins encoded by the sfa genes are presumably required in a specific stoichiometry. In the present work we studied how differential expression of the sfa operon genes occurs. Our findings indicate that a number of endoribonucleolytic cleavages occur in the mRNA from the sfa operon, and we detected the presence of different distinct transcriptional products, including sfaBA, sfaA, sfaADE, and sfaGSH. The sfaGSH transcript represents the three distal genes of the sfa operon, which code for the minor subunits of the S fimbriae. Analysis of the proteins in S fimbriae suggested that expression of the sfaGSH transcript provides equimolar amounts of the minor subunits. Furthermore, we showed that in the generation of the major sfaA transcript, the processing included RNase E endoribonuceolytic cleavage of the precursor sfaBA transcript. We suggest that posttranscriptional mRNA processing events result in differential gene expression important to achieve the stoichiometry necessary for fimbrial adhesin biogenesis.
Escherichia coli is an important human pathogen that can cause intestinal and extraintestinal infections. In the case of extraintestinal infections, the most common infections are urinary tract infections (UTI) and infections directly related to newborn meningitis. The ability of some E. coli strains to colonize the urinary tract tissues and cause further infection depends on the presence of specific virulence factors, such as adhesins, hemolysins, cytotoxic necrotizing factor 1, aerobactins, and other compounds (for a review see reference 7). Hacker et al. (13) found that the genes encoding these virulence factors are often physically linked in the large regions of the chromosome that share some characteristics with mobile elements. These regions have been termed pathogenicity islands (11).
The first step in a UTI is attachment of the bacterial cells to the urinary tract tissues; therefore, expression of specific adhesins is important. Different specific adhesins have been described for UTI and newborn meningitis isolates. For example, the S-fimbrial adhesins (Sfa) recognize and attach to receptor molecules on the cell surface that contain α-sialic acid (18). The S fimbriae are encoded by the sfa determinant, which contains nine genes, and the expression of this determinant is finely tuned. Schmoll et al. (32) demonstrated that expression of the sfa determinant is dependent on several environmental conditions, such as temperature, osmolarity, and the presence of glucose. At the molecular level, regulation of the sfa determinant is known to be mediated by two regulatory proteins, SfaB and SfaC.
A common organization of genes in bacteria is an organization in which the genes are clustered into transcriptional units termed polycistronic operons. This genetic organization ensures coordination of expression, and several interesting cases involving genes coding for virulence factors have been described. Nevertheless, the proteins encoded by these polycistronic loci are often required in nonstoichiometric amounts. Thus, the bacteria must have mechanisms to achieve differential expression at the posttranscriptional level from such polycistronic operons. Studies of the phage f1 system (17), the pap operon (25), and the atp operon (22), among others, have shown that translational control, partial termination, polarity, and mRNA processing and turnover are posttranscriptional mechanisms that may play important roles in determining the expression rates of individual genes present in polycistronic transcripts.
In the biogenesis of fimbriae, differential expression of the genes presumably must occur such that a suitable stoichiometry of the different fimbria subunits is produced (15). How such differential expression occurs has not been completely elucidated. Previous studies of an analogous fimbrial system, the pap determinant, demonstrated that processing of the mRNA and differential stability of the resulting transcripts were important for expression of the major protein subunit (1, 2, 25). Morschhäuser et al. (23) showed that expression from the sfa operon results in generation of a major transcript carrying the sequences for the major subunit, SfaA. In this study we extended the transcriptional analysis to include the whole sfa determinant. The results obtained strongly suggest that specific processing at different positions in the polycistronic mRNA is an important mechanism for achieving differential expression of the genes in this operon.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The sfa gene cluster was originally cloned from E. coli strain 536, a human urinary tract isolate (12). E. coli strains HB101 (5), N3433, and N3431 (an isogenic derivative of N3433 carrying the temperature-sensitive rne3071 mutation [9]) were the hosts of the plasmids in this study. For growth on solid media we used petri dishes containing TYS agar or CFA agar. Liquid cultures of cells were grown in Luria-Bertani (LB) medium (3) with vigorous shaking at 37°C (HB101) or at 30°C (N3431 and N3433) unless otherwise indicated. Bacterial growth was monitored by determining Klett units with a Klett-Summerson colorimeter; 50 Klett units corresponded to an optical density at 600 nm of approximately 0.4. When required, carbenicillin and isopropyl β-d-thiogalactopyranoside (IPTG) were added to final concentrations of 50 μg/ml and 1 mM, respectively.
Construction of plasmids.
The genetic organization of the plasmids used in this study is shown in Fig. 1. Plasmid pJMT1 was obtained by cloning the SmaI fragment from pANN801-13 (12) into the SmaI site of the expression vector pMMB66HE (8). pJMT2 contained the same fragment in the SmaI site of pMMB66EH in the same orientation with respect to the ptac promoter; only the direction of the polylinker was reversed to facilitate construction of the following plasmids. pJMT3 was obtained after the KpnI-BamHI fragment from pANN801-13 was inserted into KpnI/BamHI-digested pJMT2. Plasmids pJMT4 to pJMT10 were constructed by deleting the StuI-BamHI, BalI-BamHI, HpaI-BamHI, DraIII-BamHI, XmaI-BamHI, KpnI-BamHI, and ClaI-BamHI fragments, respectively, from pJMT3, blunting the recessive ends, and ligation. In the pJMT plasmids, as indicated above, the 5′ end of all the fragments cloned was the SmaI site located 25 bp downstream from the initial codon of the sfaB gene. An rrnB terminator that defined the end of the transcription was located downstream of the cloned fragments.
FIG. 1.
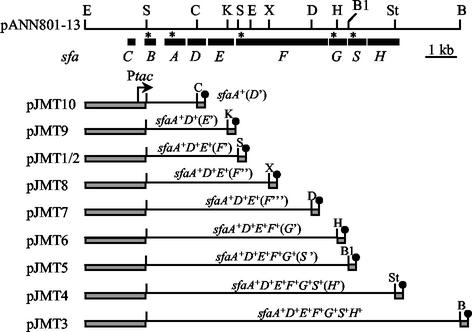
Genetic structure of the entire sfaCBADEFGSH operon cloned into plasmid pANN801-13 and deletion constructs used in this study. Plasmids pJMT1 to pJMT10 carry different parts of the sfa operon in either pMMB66HE (pJMT1) or pMMB66EH (pJMT2 to pJMT10), with the sfaB promoter replaced by the inducible tac promoter and rrnB terminators placed downstream of the sfa inserts. The solid boxes indicate the locations of the sfa genes. Relevant restriction sites are indicated as follows: B, BamHI; B1, BalI; C, ClaI; D, DraIII; E, EcoRI; H, HpaI; K, KpnI; S, SmaI; St, StuI; and X, XmaI. Asterisks indicate the binding sites of the oligonucleotides used as specific probes.
RNA isolation and Northern analysis.
RNA was extracted from strains 536 and HB101/pANN801-13 collected at the mid-log phase (50 Klett units). In the case of strains containing the pJMT plasmids, IPTG was added to the bacterial cultures when the density was 50 Klett units, and after 20 min of induction the cells were collected. To analyze rne-dependent mRNA processing, cultures were grown at 30°C (the permissive temperature for the rne mutant) to a density of 50 Klett units, divided in half, and incubated for an additional 30 min at either 30 or 44°C (the nonpermissive temperature). Transcription was then induced for 10 min, after which RNA was isolated. For transcriptional repression, 200 μg of rifampin per ml was added to the cultures 2 min after IPTG induction. In all cases, the RNA was isolated by the hot phenol method as described by von Gabain et al. (34). The RNA was separated in a 1% agarose gel in HEPES-acetate buffer containing 2.2 M formaldehyde. RNA blotting, hybridization, and washing of the membrane were performed essentially as described by Nilsson and Uhlin (25). The membranes were subjected to autoradiography. The probes used to detect expression of the different mRNA transcripts were the following [γ-32P]ATP kinase-labeled oligonucleotides: M151 (5′-CCCAAGGTCAGGGCTGAAAATACAGCC-3′) complementary to the sfaA transcript, M187 (5′-CCACTGACCAGATAATCCTTCATAGCG-3′) complementary to the sfaB transcript, CBP15 (5′-CCCCCGCCAGCAGAGCCGGTCAGT-3′) complementary to the sfaF transcript, CBP16 (5′-CCCGTCACAGTAATCGTCGTATCCACTGCC-3′) complementary to the sfaS transcript, and CBP17 (5′-CACAGAACCCGCTGCACAGAC-3′) complementary to the sfaG transcript.
Primer extension analysis.
The primers used for primer extension analysis were the [γ-32P]ATP kinase-labeled oligonucleotides M151 and M187 described above. The labeled primers were annealed with 10 μg of total RNA in 8 μl of 50 mM Tris-HCl (pH 8.3)-60 mM NaCl-10 mM dithiothreitol. The primer extension reactions were performed by using 1 h of incubation at 42°C after addition of 8 μl of extension buffer containing 1 U of avian myeloblastosis virus reverse transcriptase (Boehringer Mannheim), 25 mM Tris-HCl (pH 8.3), 30 mM NaCl, 15 mM MgCl2, 1.25 mM dithiothreitol, 1 mM dATP, 1 mM dCTP, 1 mM dGTP, and 1 mM dTTP. The reaction was stopped by addition of 16 μl of formamide sample buffer (100% formamide, 0.04% xylene cyanol, 0.04% bromophenol blue). The cDNA products were heated at 80°C for 3 min and separated by electrophoresis on a 6% polyacrylamide-urea sequencing gel.
S1 nuclease assay.
The DNA probes used in the S1 nuclease assay were the 610-bp Asp718a-EcoRI (probe I) and 324-bp Asp718a-SmaI (probe II) fragments from plasmid pANN801-13. These fragments contained the downstream part of the sfaE gene and the upstream part of the sfaF gene. The transcribed DNA strand (complementary to the RNA generated) was specifically radiolabeled by filling in the Asp718a cohesive end with the Klenow fragment and adding [α-32P]dGTP to the reaction mixture. The total RNA and the DNA probe were denatured by incubation at 75°C for 10 min in 50 μl of hybridization buffer containing 80% formamide, 40 mM piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES) (pH 6.9), and 400 mM NaCl. The hybridization mixture was incubated at 52°C for 16 h and then at 45°C for 3 h. S1 nuclease digestion was performed by incubation at 37°C for 30 min after addition of 400 μl of digestion buffer containing 100 U of S1 nuclease (Boehringer Mannheim), 30 mM sodium acetate (pH 4.5), 250 mM NaCl, 1 mM ZnCl2, and 5% glycerol. The reaction was stopped by adding 110 μl of stop buffer (4 M ammonium acetate, 50 μM EDTA) (pH 8.0). The RNA-DNA hybrids were recovered by extraction with phenol-chloroform and ethanol precipitation. The precipitate was dissolved in 80% formamide-0.04% xylene cyanol-0.04% bromophenol blue. The products were heated at 80°C for 3 min and separated by electrophoresis on a 6% polyacrylamide-urea sequencing gel.
S-fimbria purification.
In order to detect the different subunits of the S fimbriae by electrophoretic analysis, a purification protocol was developed. Cultures (500 ml) of HB101/pBR322 and HB101/pANN801-13 were grown until the density was 50 Klett units. Cells were harvested by centrifugation (7,000 × g, 10 min, 4°C), washed in phosphate-buffered saline, and concentrated 200-fold in phosphate-buffered saline. The fimbriae were sheared from the cells with a tissue homogenizer operated at 20,000 rpm for 5 min on ice. The cell debris was centrifuged for 20 min at 16,000 × g at 4°C. The presence of S fimbriae in the resulting supernatant was analyzed by Tricine-sodium dodecyl sulfate (SDS)-15% polyacrylamide gel electrophoresis (PAGE) (27), and proteins were stained with silver nitrate. When immunodetection of S-fimbria subunits was carried out, samples obtained after PAGE were blotted onto a polyvinylidene difluoride membrane. Polyclonal rabbit antiserum raised against isolated S fimbriae was used as the primary antibody, followed by a horseradish peroxidase-conjugated antibody. The membrane was developed with the ECL-Plus reagent (Amersham Pharmacia Biotech) according to the method specified by the manufacturer and was scanned with the STORM system (Molecular Dynamics).
AFM analysis.
Bacterial cultures that were analyzed by atomic force microscopy (AFM) were grown either on solid media (TYS agar and CFA agar) for 16 h at 37°C or in LB liquid medium at 37°C with vigorous shaking to the mid-log phase. Bacterial cells were washed with 10 mM Tris-HCl (pH 8.0). Washed bacterial cells were briefly resuspended in water before 5 to 10 μl was placed onto freshly cleaved ruby red mica (Goodfellow Cambridge Ltd., Cambridge, United Kingdom). The cells were incubated for 2 to 5 min at room temperature, washed with water, and blotted dry before they were placed into a desiccator for a minimum of 1 h. Images were collected with a Nanoscope IIIa (Digital Instruments, Santa Barbara, Calif.) AFM by using TappingMode with standard silicon cantilevers oscillated at resonant frequency (270 to 305 kHz). Images were collected in air at a scan rate of approximately 0.5 to 1.5 Hz. The final images were flattened and/or plane fitted in both axes by using DI software and were presented in either height or amplitude (error) mode.
RESULTS
Three proximal genes of the sfa operon were expressed from a single polycistronic mRNA.
In a previous study, two transcripts containing the sfaA sequences were identified by Northern hybridization of total RNA from strain HB101/pANN801-13 (23). A major 700-nucleotide transcript corresponding to the transcript containing the coding sequences of the sfaA gene and a less abundant 1.4-kb transcript that represented the sfaB and sfaA genes were detected. In order to carry out a transcriptional analysis of all of the sfa genes, the plasmids shown in Fig. 1 were constructed (see Materials and Methods). The pJMT plasmids carried various lengths of the sfa operon, and expression of the different cloned sfa genes could be induced from the ptac promoter. As a first approach to analyze transcription of the proximal genes, we determined the hybridization pattern of an sfaA-specific probe (M151) in total RNA of the strains containing the pJMT plasmids. As shown in the Fig. 2A, the pattern obtained was consistent with the previous results; one major transcript consisting of 700 nucleotides corresponding to the sfaA transcript was detected. Interestingly, some larger bands were also detected. The bands shown in Fig. 2A, lanes 1, 2, and 3 (the sizes were estimated to be approximately 1,200, 2,200, and 2,400 nucleotides, respectively) corresponded to the transcripts starting in front of the sfaA gene and ending at the rrnB terminator located downstream of the cloning site. These transcripts were designated the sfaAD(Δ), sfaADE, and sfaAF(Δ) transcripts, respectively. In lanes 4 to 9 corresponding to the RNA samples from strains containing plasmids with regions downstream of the sfaE gene, only a low-intensity band was detected. This band corresponded to a transcript that was designated sfaADE (see below). These results indicated that in addition to formation of the transcripts consisting of 700 and 1,400 nucleotides, a longer transcript consisting of about 2,200 nucleotides was produced; this longer transcript started upstream of the sfaA gene and covered the three proximal genes, sfaA, sfaD, and sfaE. Similar transcripts were detected with total RNA from strains in which expression of the sfa operon was under control of the native promoter. Transcriptional analysis of clinical isolate 536 confirmed that the most abundant transcript containing the sfaA sequences corresponded to a 700-base transcript (Fig. 2A, lane 10) that was identified previously by analyzing samples from HB101/pANN801-13 (23) (Fig. 2A, lane 11a). Furthermore, when samples from HB101/pANN801-13 were used, it was possible to detect the minor sfaBA transcript and the newly described sfaADE transcript (Fig. 2A, lane 11b).
FIG. 2.
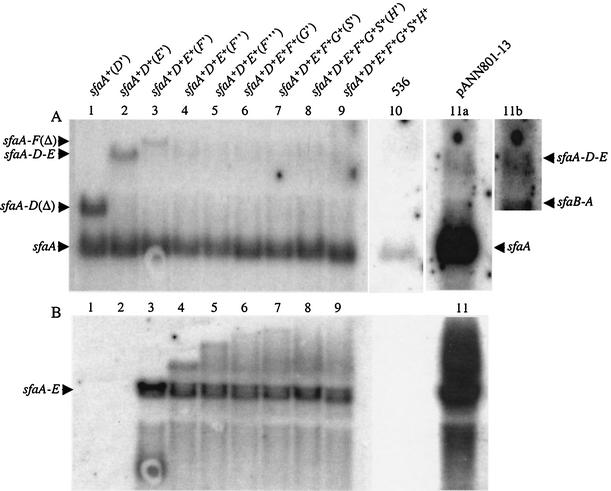
Northern hybridization of total RNA from strain HB101 carrying different sfa plasmids and from strain 536. Either an sfaA-specific probe (A) or an sfaF-specific probe (B) was used. Lane 1, HB101/pJMT10; lane 2, HB101/pJMT9; lane 3, HB101/pJMT2; lane 4, HB101/pJMT8; lane 5, HB101/pJMT7; lane 6, HB101/pJMT6; lane 7, HB101/pJMT5; lane 8, HB101/pJMT4; lane 9, HB101/pJMT3; lane 10, 536; lane 11a, HB101/pANN801-13; lane 11b, overexposure of part of the blot in lane 11a in order to visualize the small amount of sfaBA and sfaADE transcripts detected. The positions of the different transcripts are indicated on the left and right.
sfaA transcript was produced by RNase E endonucleolytic processing from precursor transcript sfaBA.
Previously, it was suggested that expression of the genes present in the sfa determinant might be under control of three promoters (31), divergent promoters pB and pC located in the intercistronic region between sfaB and sfaC and presumed promoter pA located upstream of the sfaA gene. However, it was evident that the activity of the pA promoter was very low and that most of the sfa transcription was initiated at pB under the conditions used (23). It was proposed previously that the sfaA transcript could be produced primarily by endoribonucleic processing of mRNA precursors that initiated at pB, similar to what was described in the transcriptional studies of the sfaA analog papA (1, 24, 26).
To investigate generation of the sfaA transcript in more detail, RNA samples were prepared from strain HB101 carrying pJMT1 (sfaA+D+E+[F′]) at different times after induction by addition of IPTG. The samples were analyzed by primer extension by using the sfaA-specific probe M151 and the sfaB-specific probe M187. Immediately after IPTG induction, a strong signal was detected that corresponded to the transcriptional start site at the ptac promoter in front of sfaB (Fig. 3A). This signal became weaker after 5 min and then remained at a constant level in spite of continuing transcriptional activation. Moreover, after 30 s of induction with IPTG, a transcript starting at positions −165 and −164 in front of sfaA became visible, and after a little delay, another transcript starting at position −83 and the neighboring nucleotides appeared (Fig. 3B). The latter transcript, which accumulated over time, corresponded to the major sfaA mRNA detected previously (23) (Fig. 2A). The time course of appearance of the different transcripts suggested that the sfaA mRNA was generated from the sfaBA transcript by endoribonucleolytic processing at position −165/−164, yielding a precursor transcript which was subsequently processed further at position −83 to produce the mature sfaA mRNA. To confirm this two-step sfaBA transcript processing, we determined the levels of the two transcripts that exclusively contained the sequences of sfaA (identified as pre-sfaA with a transcription start site at position −165 and as mature sfaA with a transcription start site at position −84) after transcription was inhibited by addition of rifampin. As shown in Fig. 3C, the mature sfaA transcript remained stable after inhibition of transcription, while a gradual decrease in the amount of the pre-sfaA transcript was detected. Ten minutes after inhibition, the pre-sfaA transcript was barely detected. Together, these results strongly indicated that generation of the stable sfaA transcript was due to two-step RNA processing of the sfaBA mRNA precursor.
FIG. 3.
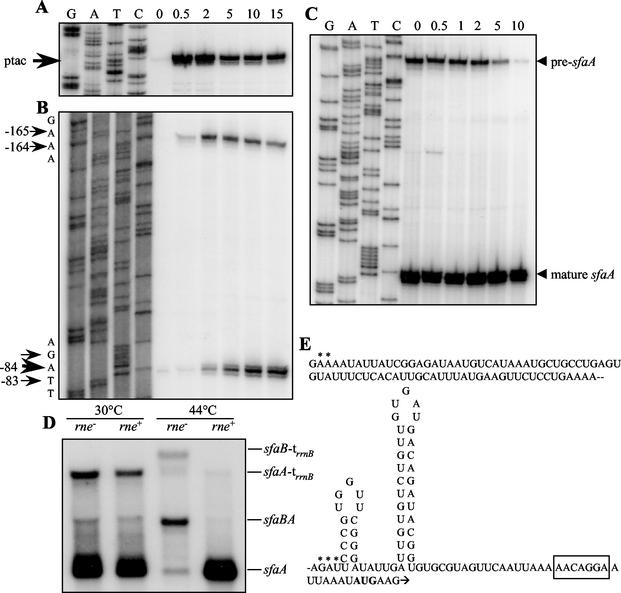
The sfaA transcript was generated by RNase E-dependent endoribonucleolytic cleavage. (A and B) Primer extension analysis with total RNA from strain HB101/pJMT1 and either sfaB-specific probe M187 (A) or sfaA-specific probe M151 (B). The time (in minutes) after induction with IPTG at which RNA was isolated is indicated at the top. The arrows indicate the positions of mRNA start points. Sequencing reaction mixtures containing the same primers and plasmid pANN801-13 as the template were used as size markers. In panel A the estimated location of the transcriptional start from the ptac promoter is indicated. (C) Primer extension analysis of sfaA transcript stability. Total RNA was isolated from strain N3433 (rne+) carrying plasmid pJMT1 before rifampin addition (lane 0) and at different times after rifampin addition (in minutes, as indicated at the top). sfaA-specific primer M151 was used. A sequencing reaction mixture containing the same primer and plasmid pANN801-13 as the template was used as the size marker. The arrowheads indicate the mRNA start points of the pre-sfaA and mature sfaA transcripts. (D) Northern hybridization of total RNA isolated from strains N3431 (rne) and N3433 (rne+) carrying plasmid pJMT1 and grown under permissive (30°C) or nonpermissive (44°C) conditions with sfaA-specific probe M151. The positions of the transcripts are indicated on the right. (E) Structural analysis of the UTR of the sfaA transcript. The secondary structure was predicted by using the genetic algorithm of the program STAR 4.4. Asterisks indicate the positions of the transcription start sites mapped by primer extension analysis (see panel A). The box indicates the position of the putative Shine-Dalgarno sequence. The first codon of sfaA is indicated by boldface type.
In the case of the pap operon, it was shown that the processing that generated a stable papA transcript was RNase E dependent. We tested whether this was also the case in the processing of the sfaBA transcript. The transcription products obtained from pJMT1 (sfaA+D+E+[F′]) were analyzed in E. coli strain N3431 carrying the temperature-sensitive rne3071 mutation and its isogenic rne+ parent, N3433. Northern hybridization performed with probe M151 demonstrated that at the permissive temperature, both strains produced low levels of the sfaBA transcript, large amounts of the mature sfaA mRNA, and significant amounts of the sfaA-(trrnB) transcript (Fig. 3D). In contrast, only low levels of the processed sfaA and sfaA-(trrnB) transcripts were detected in the rne mutant grown at the nonpermissive temperature. Instead, the corresponding precursors starting at the ptac promoter, sfaBA and sfaB-(trrnB), accumulated at much higher levels. In the rne+ control strain the longer transcripts were barely detected, probably because of accelerated decay at the higher temperature. These results demonstrated that processing of the sfa transcripts in front of sfaA was indeed rne dependent. Sequence analysis of the upstream untranslated region (UTR) of the sfaA transcript showed that the sequences upstream of the processing sites mapped by primer extension at positions −165 and −84 were A-U rich (Fig. 3E) and may be considered potential RNase E cleavage sites. Furthermore, computational analysis of the UTR downstream of the 5′ end of the mature sfaA transcript was performed, and two putative stem-loop structures were detected. The appearance of these stem-loop structures was determined by using the principles of a genetic algorithm of the program STAR 4.4 (10, 33). The secondary structure shown in Fig. 3E had a calculated ΔG value of −16.9 kcal/mol. The presence of this kind of secondary structure with similar free energy has been described in the UTR of other stable transcripts (6).
3′ end of the sfaADE transcript maps to the coding sequence of sfaF.
The results shown in Fig. 2A provided evidence for generation of a transcript covering the three proximal genes (sfaA, sfaD, and sfaE). Presumably, this involved the hypothesis that the transcript representing the sfaF gene should have a 5′ end located downstream of the sfaE gene. In order to test this hypothesis, we hybridized total RNA isolated from strains carrying the pJMT plasmids with an sfaF-specific probe (CBP15) in a Northern blot analysis. The results obtained are shown in Fig. 2B. Surprisingly, one band at approximately 2.2 kb was detected in the sample from strain HB101/pJMT2 (sfaA+D+E+[F′]) (lane 3). Plasmid pJMT2 carried the SmaI fragment of the sfa determinant that contained the four proximal genes (sfaB′ADE) and a short region (133 bp) of the sfaF gene. Therefore, the data indicated that the band detected corresponded to the transcript containing the sequences of the three proximal genes (sfaA, sfaD and sfaE) and that the 3′ end of this transcript maps to the sfaF gene. This 2.2-kb transcript was also detected in all the RNA samples from strains harboring plasmids with sequences downstream of the SmaI site located in the sfaF gene (Fig. 2B, lanes 4 to 9). As a control, no signal was detected with samples from HB101/pJMT10 (sfaA+[D′]) and HB101/pJMT9 (sfaA+D+[E′]) (Fig. 2B, lanes 1 and 2). In fact, the 2.2-kb transcript was also detected when the sfaA-specific probe was used (Fig. 2) and corresponded to the transcript designated sfaADE. In the samples from HB101/pJMT2 (Fig. 2B, lane 3) a transcript consisting of about 500 bases was also detected, and this transcript corresponded to a small mRNA fragment with the 5′ end in the sfaE sequence and the 3′ end at the rrnB terminator. Taking into consideration the length of the transcript detected, we mapped as a possible 5′ end a putative RNase E cleavage site located 360 bases upstream of the AUG codon of the sfaF gene. As this transcript was not detected in the other samples, we considered it an artifact due to the possible anomalous structure of the mRNA produced from plasmid pJMT2.
As shown in lanes 4 and 5 of Fig. 2B, we detected some weak larger bands that on the basis of their estimated sizes should correspond to transcripts covering increasing portions of the sfaF gene. Such bands were not clearly present in lanes 6 to 9 containing samples from strains carrying plasmids with downstream sequences of the sfaF gene. Another sfaF-specific probe that hybridized with the central region of the sfaF gene was used to perform Northern hybridization to detect the transcript that should represent this gene. However, after several trials no clear bands were detected, suggesting that this putative transcript was very rare and/or quickly degraded.
The 3′ end of the proximal transcript was mapped by S1 nuclease analysis by using simultaneously two different double-stranded probes (Fig. 4A) and total RNA samples from strains HB101/pJMT2 and HB101/pJMT8. The results are shown in Fig. 4B. When total RNA from HB101/pJMT8 was used, a band at 296 bases was detected by using either of the probes. In addition, an extra band at 334 bases was detected only with probe II. The exact positions of the 3′ ends were mapped, and the locations are indicated in Fig. 4B. The results suggested that there were two different 3′ ends of the sfaAF transcript spaced 39 bases apart, flanking the SmaI site. The SmaI site was the cloning site for plasmid pJMT2. Therefore, corroborating these results, only the band at 296 bases was detected with both probes when total RNA from strain HB101/pJMT2 was analyzed. As a control for S1 nuclease digestion, probe II was used, and a band with a migration position similar to that of the band observed with probe I was generated. When lanes 4 and 6 of Fig. 4B were compared, we observed an important difference in the efficiency of S1 nuclease digestion when probe II was used with RNA samples from strains HB101/pJMT8 and HB101/pJMT2. Most of probe II was digested with S1 nuclease in the presence of RNA from strain HB101/pJMT2. This could be explained by the fact that in the total RNA from strain HB101/pJMT2, the amount of the sfaADE transcript was greater than the amounts in the rest of the samples analyzed (Fig. 2B, lane 3).
FIG. 4.
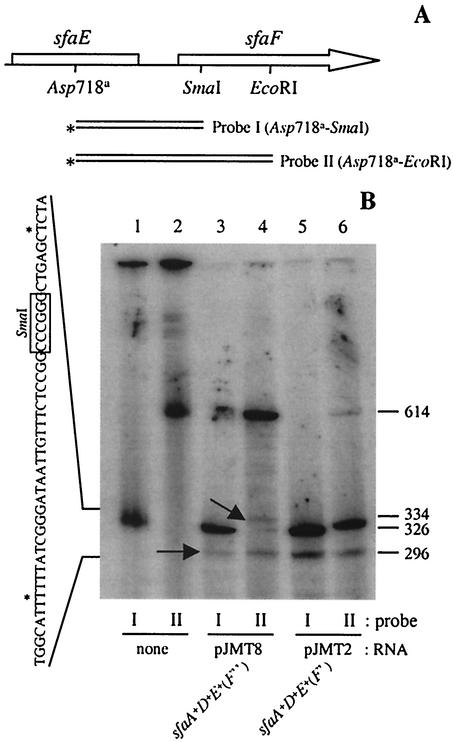
The 3′ end of the sfaADE transcript mapped in the coding sequence of sfaF. (A) Genetic map of the DNA fragments used as probes for S1 nuclease digestion. The asterisks indicate the 3′end that was radiolabeled. (B) S1 nuclease digestion of total RNA from strains HB101/pJMT8 (lanes 3 and 4) and HB101/pJMT2 (lanes 5 and 6) by using probe I (lanes 1, 3, and 5) and probe II (lanes 2, 4, and 6). The arrows indicate the positions of two different kinds of products obtained. On the left side the exact positions of the two 3′ ends of the sfaADE transcript are indicated (asterisks) in the sequence. The boxed nucleotides indicate the position of the SmaI restriction site. The exact size (number of nucleotides, shown on the right) of the S1 products was determined by using sequencing reaction mixtures as molecular mass markers (data not shown).
Transcriptional analysis of the distal genes of the sfa operon.
The results obtained by using the sfaF-specific probe showed larger bands only when we analyzed samples from strains containing plasmids with fragments covering the sfaF gene but not the downstream region (Fig. 2B). This strongly indicated that another processing event occurred downstream of the sfaF gene. The patterns of hybridization obtained by using an sfaG-specific probe (CBP17) were determined for RNA samples from the strains carrying plasmids with the distal genes. The results obtained are shown in Fig. 5. When we analyzed total RNA from strains containing the whole sfa determinant (e.g., HB101/pJMT3 [sfaA+D+E+F+G+S+H+] and HB101/pANN801-13), one transcript that was approximately 2 kb long was detected. A similar transcript was detected when an sfaS-specific probe (CBP18) was used (data not shown). Based on its size, this transcript should correspond to a transcript containing the sequences for the three distal genes (sfaG, sfaS, and sfaH). The results obtained when total RNA from strain HB101/pJMT4 (sfaA+D+E+F+G+S+[H′]) was analyzed supported our conclusions (Fig. 5, lane 3). In the latter case, we detected a slightly shorter transcript since plasmid pJMT4 did not contain the entire sfa determinant and the rrnB terminator was located inside the sfaH gene. Therefore, our results indicated that the three distal genes of the sfa operon were represented in a single polycistronic transcript that resulted from processing of the larger precursor, sfaAH.
FIG. 5.

Northern hybridization of total RNA isolated from strains HB101/pANN801-13 (lane 1), HB101/pJMT7 (lane 2), HB101/pJMT4 (lane 3), and HB101/pJMT3 (lane 4) with sfaS-specific probe CBP16.
Analysis of S-fimbria proteins and morphology.
The following question about the role of RNA processing in the differential expression from the sfa operon arose from the previous findings: are the distal genes of the sfa determinant represented by the sfaGSH transcriptional product expressed at similar levels? Therefore, the level of protein expression from distal genes sfaG, sfaS, and sfaH was studied. These three genes encode proteins that are involved in S-fimbria biogenesis as structural proteins located at the tips of the fimbriae. S fimbriae were isolated from HB101/pANN801-13 grown under the conditions used in the expression studies, and the different structural subunits were resolved by SDS-PAGE and Western blot analysis. The three minor subunits, SfaG, SfaS, and SfaH, were identified by their relative migration positions, as previously described (29). The results (Fig. 6) indicated that there was not a significant difference in the amounts of the three minor subunits in the S fimbriae under the conditions used.
FIG. 6.
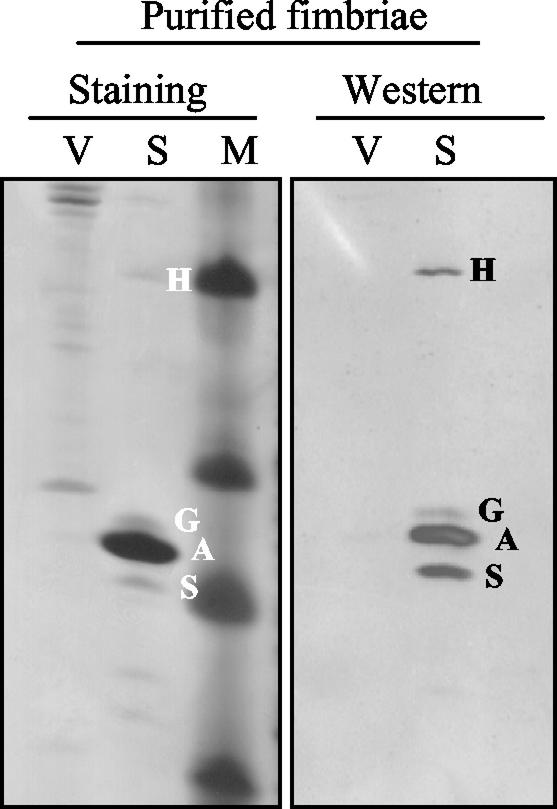
Analysis of the Sfa fimbrial proteins: Tricine-SDS-PAGE of purified fimbria samples from cultures of strains HB101/pBR322 (lanes V) (vector control) and HB101/pANN801-13 (lanes S) (S-fimbria operon cloned). Lane M contained molecular markers. The samples were analyzed by silver staining and Western blotting by using antisera raised against S fimbriae. The bands corresponding to the different fimbrial subunits are indicated as follows: S, SfaS; A, SfaA; G, SfaG; and H, SfaH.
Most of the studies on the assembly and structure of fimbriae have been performed by using P fimbriae as a model (19). Ultramicroscopy studies performed by using transmission electron microscopy of P fimbriae have shown that two different parts can be differentiated morphologically: the pilus rod and the tip fibrilla. The pilus rod is composed of PapA subunits packed in a helical conformation, and the tip fibrilla is mainly composed of PapE, one of the minor subunits of the P fimbriae (19). The results of the protein analysis described above indicated that none of the SfaG, SfaS, and SfaH minor subunits of the S fimbriae was present at a higher level than the other subunits. This suggested that under the conditions used, the S fimbriae might not have a tip fibrilla structure. AFM analyses by using TappingMode were performed in order to observe the morphological features of the fimbriae. As a control, cells expressing P fimbriae were analyzed by AFM, and tip fibrilla structures, as previously described in transmission electron microscopy studies, were indeed visualized (Fig. 7A and B). When S-fimbriated cells were analyzed, we did not observe tip fibrillae similar to those detected in the case of P pili (Fig. 7C to F) either when bacteria were grown on solid media or when they were grown in LB medium under the conditions used in the transcriptional studies described above. Thus, the results of the AFM studies were in agreement with the results obtained in the analysis of the amounts of the minor subunit proteins present in fimbriae. There was apparently no exceptionally abundant minor subunit that could be identified as a major tip fibrillar subunit.
FIG. 7.
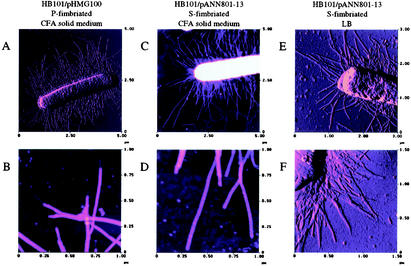
AFM images of fimbriated E. coli. (A) P-fimbriated bacterium (HB101/pHMG100) grown on CFA solid medium. (B) Higher magnification of P fimbriae shown in panel A. (C) S-fimbriated bacterium (HB101/pANN801-13) grown on CFA solid medium. (D) Higher magnification of S fimbriae shown in panel C. (E and F) S-fimbriated bacteria grown in LB medium.
DISCUSSION
The fimbrial adhesins expressed by extraintestinal E. coli pathogens are encoded by loci that contain a large number of genes. Fimbria biogenesis studies have shown that the different subunits are clearly required in nonstoichiometric amounts. Thus, the data imply that the bacteria should have mechanisms to achieve differential expression at the posttranscriptional level. Early experiments on expression of the pap determinant demonstrated that RNase E-dependent endoribonucleolytic cleavages of the polycistronic mRNA generated a stable transcript containing the sequences of the gene coding for the major subunit of the P fimbriae (1, 2, 25). Extensive studies have been performed on the regulation of biogenesis of the F1845 fimbriae, which are encoded by six genes present in the daa determinant. It has been shown that endoribonucleotytic cleavages and the subsequent different stabilities of the cleavage fragments result in the formation of a stable transcript coding only for DaaE, the major subunit of the F1845 fimbriae. The endoribonucleotide activity responsible for these processing events is independent of RNase E and RNase III (4, 21).
In the present study, using the sfa determinant, we obtained data indicating that the polycistronic sfaBADEFGSH transcript is subject to several mRNA processing events that generate different transcriptional products, as summarized in Fig. 8. The results obtained seem to be in total agreement with the notion that the gene products are expressed to optimize biogenesis of this kind of fimbriae. We used different approaches to analyze the posttranscriptional events in the sfa operon. Use of the pJMT plasmids allowed us to induce expression of different parts of the sfa operon, which facilitated detection of the different transcripts. Furthermore, the results obtained by overexpression of the sfa operon were confirmed by comparison with the sfa expression from plasmid pANN801-13, in which the sfa determinant is expressed under control of the natural promoter sequences.
FIG. 8.
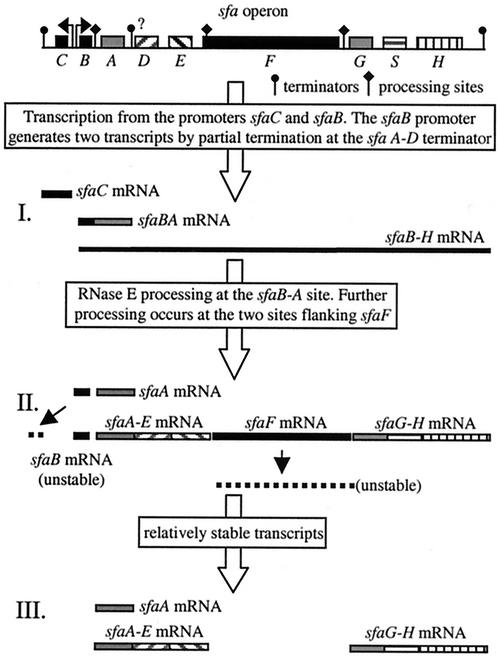
Summary of transcriptional events occurring in the sfa operon.
Taken together, the results led us to conclude that seven different transcripts were generated from the sfa determinant. The greatest complexity was found in the promoter-proximal region, where five different transcripts were generated, and three of these transcripts (sfaBA, sfaA, and sfaADE) contained the sfaA sequences. The existence of the sfaBA and sfaA transcripts and the existence of the sfaB and sfaC transcripts were described previously (23). The major transcript generated from the sfa determinant represented the sfaA gene, and this is consistent with the fact that the SfaA protein is the major subunit of the S fimbriae (30). The high steady-state level of the sfaA transcript seems to be achieved by mechanisms similar to those described previously for other stable transcripts. For the gene analogous to sfaA, papA, it was determined that a stable transcript corresponding to only this gene is generated by RNase E-mediated cleavage at the intercistronic region between the papB and papA genes (24, 26). Recently, it was demonstrated that the secondary structure of the resulting stable transcript after the RNase E cleavage includes a stem-loop near the 5′ end that stabilizes the transcript by protecting it from degradation (6). A similar mechanism of protection was described for the rather stable OmpA transcript (14). In our study, we demonstrated that generation of the major sfaA transcript was the result of RNase E cleavage (Fig. 3). Determination of the 5′ end of this stable transcript revealed two mRNA species that differ in size by about 82 nucleotides. The shorter transcript (sfaA) appeared after generation of the larger transcript (pre-sfaA) (Fig. 3). Interestingly, when the secondary structure of the sfaA transcript was determined by computer prediction, two putative stem-loop structures were detected in the UTR. The free energy value coincided exactly with the value obtained when, using the same program, we calculated the free energy of the secondary structure of the UTR of the papA transcript described by Bricker and Belasco (6). The resemblance to the secondary structure of the papA transcript and the fact that the 5′ end of the sfaA transcript maps exactly just upstream of the first stem-loop (Fig. 3E) led us to speculate that the presence of these two stem-loop structures is important in defining the high level of stability of this transcript. It will be of interest to determine the biological significance of these stem-loop structures by, for example, altering the nucleotide sequence. Figure 3D shows that in the presence of RNase E (at the permissive temperature) two major transcripts, sfaA and sfaA-trrnB, were generated. In contrast, when RNase E was not active (at the nonpermissive temperature), the sfaBA and sfaB-trrnB transcripts were detected. These results strongly indicated that the 3′ end of sfaA was not generated by RNase E-mediated processing. These results support the hypothesis that the major transcript, the sfaA transcript, is generated by partial termination at a putative terminator located downstream of the sfaA gene. Consistent with this hypothesis, a stable stem-loop followed by a U-rich region was detected downstream of sfaA in the intercistronic region sfaA-sfaD (data not shown). Further studies are needed to confirm the in vivo significance of such a structure as a rho-independent terminator.
We described the existence of the transcript spanning the three proximal genes, the sfaADE transcript. This transcript resembles the papAH transcript produced in the pap determinant (2). Similar to the sfaADE transcript, it contains the sequences from the major subunit gene to the sequence coding for the outer membrane translocation assembly proteins (papC and sfaF in the pap and sfa determinants, respectively). The fact that the sfaADE transcript hybridized with one sfaF-specific probe for the upstream part of the gene was an unexpected result. However, possible endoribonucleolytic cleavage(s) within coding sequences could be an efficient system for the cell to quickly silence expression from a transcript. The outer membrane translocation protein encoded by the sfaF gene is one protein that theoretically should be necessary in very few copies for correct biogenesis of the fimbriae. Therefore, the putative cleavage within the coding sequence of the sfaF gene could have important biological significance since this type of processing could be a general mechanism for regulation of differential expression in polycistronic loci.
The distal part of the sfa operon contains only three genes instead of six genes, as found in the pap operon located downstream of papC. The smaller number of minor subunit genes could be related to the lower complexity of the fimbrial structure. Moreover, when isolated P fimbriae were analyzed, the relatively abundant minor subunit, PapE, was detected, and this was consistent with the fact that PapE is the major component of the fibrilla tip (20, 21). When purified S fimbriae were analyzed, our results indicated that all three minor subunits were present at similar levels. Furthermore, we found that a unique transcript represented the three distal genes of the sfa operon. The equimolarity of the minor subunits is in agreement with the absence in the S fimbriae of a long fibrilla tip of the type described for the P fimbriae (Fig. 7). However, we cannot rule out the possibility that the adhesin of the S fimbriae, SfaS, is organized in the S fimbriae in short fibrilla tips resembling the tips described for the type 1 fimbriae (16). The genetic loci coding for Sfa and type 1 fimbriae contain the same number of fimbrial structural genes, and they are organized similarly. In both cases, there are three distal genes coding for the minor subunits of the fimbriae. Studies of expression of the fim operon at the protein level (16) and at the transcriptional level (28) indicated that the fim genes coding for the minor subunits are expressed at a similar ratio. Consistent with this observation, it has been found that the type 1 tip fibrilla is a stubby structure compared with the P tip fibrilla. Therefore, we can speculate that the genetic similarities between S fimbriae and type 1 fimbriae give rise to analogous structures.
The final stoichiometry of the fimbrial organelle may depend on factors such as the amounts and stabilities of individual mRNA species, the quality of ribosomal binding sites, codon usage, protein-protein interactions between structural components, and/or members of the transport machinery. In the present study we established that several posttranscriptional mRNA processing events occur in the polycistronic sfaBADEFGSH transcript. We suggest that generation of several transcripts is a mechanism for achieving the differential expression from the sfa genes necessary for S-fimbria biogenesis.
Acknowledgments
This work was supported by grants from The Swedish Research Council and from The Deutsche Forschungsgemeinschaft. The Knut and Alice Wallenberg Foundation is acknowledged for a grant in support of AFM equipment.
REFERENCES
- 1.Båga, M., M. Göransson, S. Normark, and B. E. Uhlin. 1988. Processed mRNA with differential stability in the regulation of the E. coli pilin gene expression. Cell 52:197-206. [DOI] [PubMed] [Google Scholar]
- 2.Båga, M., M. Norgren, and S. Normark. 1987. Biogenesis of E. coli Pap pili: PapH, a minor pilin subunit involved in cell anchoring and length modulation. Cell 49:241-251. [DOI] [PubMed] [Google Scholar]
- 3.Bertani, G. 1951. Studies on lysogenesis. I. The mode of phage liberation by lysogenic Escherichia coli. J. Bacteriol. 62:293-300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bilge, S. S., J. M. Apostol, Jr., M. A. Aldape, and S. L. Moseley. 1993. mRNA processing independent of RNase III and RNase E in the expression of the F1845 fimbrial adhesin of Escherichia coli. Proc. Natl. Acad. Sci. USA 90:1455-1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boyer, H. W., and D. Roulland-Dussoix. 1969. A complementation analysis of the restriction and modification of DNA in Escherichia coli. J. Mol. Biol. 41:459-472. [DOI] [PubMed] [Google Scholar]
- 6.Bricker, A. L., and J. G. Belasco. 1999. Importance of a 5′ stem-loop for longevity of papA mRNA in Escherichia coli. J. Bacteriol. 181:3587-3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Donnenberg, M. S., and R. A. Welch. 1996. Virulence determinants of uropathogenic Escherichia coli, p. 135-174. In H. L. T. Mobley and J. W. Warren (ed.), Urinary tract infections: molecular pathogenesis and clinical management. American Society for Microbiology, Washington, D.C.
- 8.Fürste, J. P., W. Pansegrau, R. Frank, H. Blöcker, P. Scholz, M. Bagdasarian, and E. Lanka. 1986. Molecular cloning of the plasmid RP4 primase region in a multi-host-range tacP expression vector. Gene 48:119-131. [DOI] [PubMed] [Google Scholar]
- 9.Goldblum, K., and D. Apirion. 1981. Inactivation of the ribonucleic acid processing enzyme ribonuclease E blocks cell division. J. Bacteriol. 146:128-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gultyaev, A. P., F. H. D. van Batenburg, and C. W. A. Pleij. 1995. The computer simulation of RNA folding pathways using a genetic algorithm. J. Mol. Biol. 250:37-51. [DOI] [PubMed] [Google Scholar]
- 11.Hacker, J., G. Blum-Oehler, I. Mühldorfer, and H. Tschäpe. 1997. Pathogenicity islands of virulent bacteria: structure, function and impact on microbial evolution. Mol. Microbiol. 23:1089-1097. [DOI] [PubMed] [Google Scholar]
- 12.Hacker, J., G. Schmidt, C. Hughes, S. Knapp, S. Marget, and W. Goebel. 1985. Cloning and characterization of genes involved in production of mannose-resistant, neuraminidase-susceptible (X) fimbriae from a uropathogenic O6:K15:H31 Escherichia coli strain. Infect. Immun. 47:434-440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hacker, J., L. Bender, M. Ott, J. Wingender, B. Lund, R. Marre, and W. Goebel. 1990. Deletions of chromosomal regions coding for fimbriae and hemolysins occur in vitro and in vivo in various extraintestinal Escherichia coli isolates. Microb. Pathog. 8:213-225. [DOI] [PubMed] [Google Scholar]
- 14.Hansen, M. J., L.-H. Chen, M. L. S. Fejzo, and J. G. Belasco. 1994. The ompA untranslated region impedes a major pathway for mRNA degradation in Escherichia coli. Mol. Microbiol. 12:707-716. [DOI] [PubMed] [Google Scholar]
- 15.Hultgren, S. J., C. H. Jones, and S. Normark. 1996. Bacterial adhesins and their assembly, p. 2730-2756. In F. C. Neidhardt, R. Curtiss III, J. L. Ingraham, E. C. C. Lin, K. B. Low, B. Magasanik, W. S. Reznikoff, M. Riley, M. Schaechter, and H. E. Umbarger (ed.), Escherichia coli and Salmonella: cellular and molecular biology, 2nd ed. American Society for Microbiology, Washington, D.C.
- 16.Jones, C. H., J. S. Pinkner, R. Roth, J. Heuser, A. V. Nicholes, S. N. Abraham, and S. J. Hultgren. 1995. FimH adhesin of type 1 pili is assembled into a fibrillar tip structure in the Enterobacteriaceae. Proc. Natl. Acad. Sci. USA 92:2081-2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kokoska, R. J., and D. A. Steege. 1998. Appropriate expression of filamentous phage f1 DNA replication genes II and X requires RNase E-dependent processing and separate mRNAs. J. Bacteriol. 180:3245-3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Korhonen, T. K., M. V. Valtonen, J. Parkkinen, V. Vaisanen-Rhen, J. Finne, F. Orskov, I. Orskov, S. B. Svenson, and P. H. Makela. 1985. Serotypes, hemolysin production, and receptor recognition of Escherichia coli strains associated with neonatal sepsis and meningitis. Infect. Immun. 48:486-491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuehn, M. J., J. Heuser, S. Normark, and S. J. Hultgren. 1992. P pili in uropathogenic E. coli are composite fibres with distinct fibrillar adhesive tips. Nature 356:252-255. [DOI] [PubMed] [Google Scholar]
- 20.Lindberg, F., B. Lund, and S. Normark. 1986. Gene products specifying adhesion of uropathogenic Escherichia coli are minor components of pili. Proc. Natl. Acad. Sci. USA 83:1891-1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Loomis, W. P., and S. L. Moseley. 1998. Translational control of mRNA processing in the F1845 fimbrial operon of Escherichia coli. Mol. Microbiol. 30:843-853. [DOI] [PubMed] [Google Scholar]
- 22.McCarthy, J. E. 1990. Post-transcriptional control in the polycistronic operon environment: studies of the atp operon of Escherichia coli. Mol. Microbiol. 4:1233-1240. [DOI] [PubMed] [Google Scholar]
- 23.Morschhäuser, J., B. E. Uhlin, and J. Hacker. 1993. Transcriptional analysis and regulation of the sfa determinant coding for S fimbriae of pathogenic Escherichia coli strains. Mol. Gen. Genet. 238:97-105. [DOI] [PubMed] [Google Scholar]
- 24.Naureckiene, S., and B. E. Uhlin. 1996. In vitro analysis of mRNA processing by RNase E in the pap operon of Escherichia coli. Mol. Microbiol. 21:55-68. [DOI] [PubMed] [Google Scholar]
- 25.Nilsson, P., and B. E. Uhlin. 1991. Differential decay of a polycistronic Escherichia coli transcript is initiated by RNase E-dependent endonucleolytic processing. Mol. Microbiol. 5:1791-1799. [DOI] [PubMed] [Google Scholar]
- 26.Nilsson, P., S. Naureckiene, and B. E. Uhlin. 1996. Mutations affecting mRNA processing and fimbrial biogenesis in the Escherichia coli pap operon. J. Bacteriol. 178:683-690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schägger, H., and G. von Jagow. 1987. Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal. Biochem. 166:368-379. [DOI] [PubMed] [Google Scholar]
- 28.Schembri, M. A., D. W. Ussery, C. Workman, H. Hasman, and P. Klemm. 2002. DNA microarrays analysis of fim mutations in Escherichia coli. Mol. Genet. Genomics 267:721-729. [DOI] [PubMed] [Google Scholar]
- 29.Schmoll, T., H. Hoschutzky, J. Morschhauser, F. Lottspeich, K. Jann, and J. Hacker. 1989. Analysis of genes coding for the sialic acid-binding adhesin and two other minor fimbrial subunits of the S-fimbrial adhesin determinant of Escherichia coli. Mol. Microbiol. 3:1735-1744. [DOI] [PubMed] [Google Scholar]
- 30.Schmoll, T., J. Hacker, and W. Goebel. 1987. Nucleotide sequence of the sfaA gene coding for the S-fimbrial protein subunit of Escherichia coli. FEMS Microbiol. Lett. 41:229-235. [Google Scholar]
- 31.Schmoll, T., J. Morschhäuser, M. Ott, B. Ludwig, I. van Die, and J. Hacker. 1990. Complete genetic organization and functional aspects of the Escherichia coli S fimbrial adhesin determinant: nucleotide sequence of the genes sfaB, C, D, E, F. Microb. Pathog. 9:331-343. [DOI] [PubMed] [Google Scholar]
- 32.Schmoll, T., M. Ott, B. Ougeda, and J. Hacker. 1990. Use of a wild-type gene fusion to determine the influence of environmental conditions on expression of the S fimbrial adhesin in an Escherichia coli pathogen. J. Bacteriol. 172:5103-5111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van Batenburg, F. H. D., A. P. Gultayev, and C. W. A. Pleij. 1995. An APL-programmed genetic algorithm for the prediction of RNA secondary structure. J. Theor. Biol. 174:269-280. [DOI] [PubMed] [Google Scholar]
- 34.von Gabain, A., J. G. Belasco, J. L. Schottel, A. C. Y. Chang, and S. N. Cohen. 1983. Decay of mRNA in Escherichia coli: investigation of the fate of specific segments of transcripts. Proc. Natl. Acad. Sci. USA 80:653-657. [DOI] [PMC free article] [PubMed] [Google Scholar]