

Abstract
The dnaA operon of Escherichia coli contains the genes dnaA, dnaN, and recF encoding DnaA, β clamp of DNA polymerase III holoenzyme, and RecF. When the DnaA concentration is raised, an increase in the number of DNA replication initiation events but a reduction in replication fork velocity occurs. Because DnaA is autoregulated, these results might be due to the inhibition of dnaN and recF expression. To test this, we examined the effects of increasing the intracellular concentrations of DnaA, β clamp, and RecF, together and separately, on initiation, the rate of fork movement, and cell viability. The increased expression of one or more of the dnaA operon proteins had detrimental effects on the cell, except in the case of RecF expression. A shorter C period was not observed with increased expression of the β clamp; in fact, many chromosomes did not complete replication in runout experiments. Increased expression of DnaA alone resulted in stalled replication forks, filamentation, and a decrease in viability. When the three proteins of the dnaA operon were simultaneously overexpressed, highly filamentous cells were observed (>50 μm) with extremely low viability and, in runout experiments, most chromosomes had not completed replication. The possibility that recombinational repair was responsible for the survival of cells overexpressing DnaA was tested by using mutants in different recombinational repair pathways. The absence of RecA, RecB, RecC, or the proteins in the RuvABC complex caused an additional ∼100-fold drop in viability in cells with increased levels of DnaA, indicating a requirement for recombinational repair in these cells.
The dnaA operon of Escherichia coli (Fig. 1A) contains the genes dnaA, dnaN, and recF, which encode the DNA replication initiator protein, DnaA; the β subunit of DNA polymerase III holoenzyme (Pol III); and RecF, respectively. The concentration of DnaA protein is a critical factor in determining the timing of initiation of DNA replication from oriC within the cell cycle, and it is likely that additional DnaA protein must be synthesized between rounds of replication (55). Although DnaA levels control the timing of initiation during growth and new DnaA synthesis appears to be necessary prior to new initiation events, the cell does not tolerate well increases in the amount of DnaA protein. When the concentration of DnaA is raised 1.5- to 3-fold, the number of replication forks increases, but the rate of replication decreases, and many of these additional forks appear to terminate replication prematurely (3). Katayama (24) called this an attenuation type of response to excessive initiation events, where forks stall before reaching the terminus.
FIG. 1.

Map of the dnaA operon. (A) The dnaA (p1, p2), dnaN (p3-p7, pβ*), and recF (p9, p10) gene promoters are shown. (B) E. coli chromosomal DNA fragments inserted into pLex5BA.
Such stalled replication forks in cells with increased levels of DnaA could lead to double-strand breaks (DSBs) which, if not repaired, would cause cell death. There are now many examples in which cells with arrested replication forks depend on some form of recombinational repair for survival: e.g., rep mutants (40); dnaBts, dnaEts, and dnaNts mutants at the nonpermissive temperature (18, 45, 46); and UV irradiation (11, 12). Stalled forks are often restarted by “replication fork reversal,” wherein pairing of the two newly synthesized strands creates a double-stranded end, a substrate for RecBCD and, together with pairing of the template strands, results in a Holliday junction to which the resolvase, RuvABC, binds. The nature of the stalled fork determines which proteins are involved in this process of fork reversal and replication restart. For example, RecA is required for RuvABC action when replication is arrested by inactivation of DnaB helicase (46), but RecA is not required when fork arrest is caused by the lack of Rep helicase (46) or a defect in the HolD subunit of the Pol III clamp loader (15) or in a dnaEts mutant (18).
If a newly initiated replication fork were to reach a stalled fork and copy nascent DNA at the stalled fork, this would create double-stranded ends, leading to a collapsed replication fork (6, 32, 47). Strains carrying DNA replication termination sites (Ter) at new positions in the chromosome depend on RecA, RecBC, and RuvABC (all proteins involved in recombinational repair) for viability (6, 21, 0.47) Fork breakage was not evident in these cells, although linear DNA was created when a new fork reached a fork blocked at Ter. These collapsed forks were repaired by homologous recombination rather than by replication fork reversal (6).
We predicted that if stalled or collapsed forks were being repaired by recombination proteins in cells with increased levels of DnaA, then a loss of viability would occur in cells with mutations in recombinational repair genes when the expression of DnaA was increased. There are two major recombination repair pathways in E. coli: RecFOR and RecBCD (13, 31). The RecF pathway repairs daughter-strand gaps, and the RecBC pathway repairs DSBs and disintegrating replication forks.
The UV hypersensitivity of recF mutant strains is due to the failure to resume DNA replication at replication forks disrupted by irradiation. The RecF protein, as well as UvrA, UvrC, RecA, and RecR, are required for the resumption of replication at disrupted replication forks in UV-irradiated cells (11, 12). Courcelle and Hanawalt (10) have suggested that these Rec proteins protect and maintain replication forks arrested at DNA lesions until the lesions are removed by excision repair and that recombination might not occur during this process.
RecBCD binds double-stranded DNA ends and, in conjunction with its helicase activity, the ExoV nuclease of RecBCD degrades in the 3′-5′ direction until a Chi site is reached on the chromosome where ExoV is attenuated and a weaker 5′-3′ nuclease is activated (1, 14). RecA filaments form on the resulting 3′-terminal single-stranded DNA tail with the ability to invade homologous double-stranded DNA (2), resulting in a D loop where PriA can facilitate the assembly of replication forks after first assembling a primosome (36, 37, 39).
Increased expression of Bacillus subtilis DnaA in B. subtilis cells (42) and of Vibrio harveyi dnaA in E. coli cells (5) also resulted in increased initiation events and, as in the case of E. coli, many of the additional forks appeared not to have completed replication in runout replication experiments. Autorepression of the DnaA promoters was considered as a possible explanation for the stalling of replication forks in the case of B. subtilis and, when the expression of both the dnaA and dnaN genes was increased, the growth defects observed with increased DnaA levels alone were alleviated. In exponentially growing cells, all three genes are transcribed mainly from the promoters upstream of the dnaA gene, dnaAp1 and dnaAp2, even though the dnaN and recF genes have their own promoters (Fig. 1A) (38, 43). Autoregulation of dnaA occurs through a dnaA box present between the two promoters where repression of transcription from both dnaAp1 and dnaAp2 occurs upon DnaA binding (4, 8, 29, 44).
The β subunit of Pol III acts as a clamp tethering Pol III to the DNA template (22, 50), allowing high processivity during replication of the chromosome. Leading strand replication requires that the β subunit become loaded onto DNA by the γ-complex of the Pol III holoenzyme at oriC (23). For lagging strand synthesis, the β subunit is loaded for the synthesis of each Okazaki fragment (20). Because the β subunit acts distributively during lagging strand synthesis (53), its intracellular concentration may affect the rate at which it is loaded onto the chromosome. This rate may also contribute to determining the rate of fork movement and, therefore, the length of the C period, which is the time during which DNA is replicated in the cell cycle. The concentration of DnaA at least in part establishes the rate of initiation events and, thereby, the number of replication forks. Coordinating the expression of the β subunit with that of DnaA may ensure that the cell has the correct proportion of β clamps to replication forks to maintain the observed constant C period in the cell cycle. Additionally, the β subunit is a negative regulator of the initiator protein, DnaA inactivating the non-DNA-bound form of DnaA (25). This inactivation occurs predominantly with the help of an unidentified IdaB protein when the ATP-bound active form of DnaA interacts with the β subunit of Pol III when associated with DNA (25).
The initiation of DNA replication is precisely coordinated, occurring at a specific time, once per cell cycle, during steady-state growth. When two or more origins are present in the same cell, they initiate simultaneously. The precision with which all origins in a cell initiate DNA replication can be established by measuring the number of chromosomes in each cell after allowing replication to come to completion (runout replication). This is achieved by using rifampin, which inhibits further initiation events but allows ongoing rounds of replication to complete. In a wild-type culture of E. coli, cells with coordinated and synchronous initiation of all chromosomes contain 2n (n = 0, 1, 2, 3, 4) origins. Cells with defects in the timing of initiation contain “irregular” numbers of completed chromosomes (3, 5, 6, 7, etc.), indicating asynchronous initiation of replication. These abnormal chromosomal numbers are a result of cells containing two or more origins where initiation at these origins occurs at different times or initiation does not occur at one of the origins (49). Cells that contain chromosomes that have not completed replication due to a DNA lesion or blockage in replication after rifampin treatment show broad peaks of DNA where the amount of DNA content per cell does not correspond to the amount of DNA in a single completed chromome or in multiple numbers of completed chromosomes.
Our initial results were similar to previous results (3) and indicated that increased DnaA expression leads to loss of viability and cells with chromosomes that have not completed replication in runout experiments. Although an increased amount of β clamp or RecF corrects to some extent the aberrant replication observed at higher concentrations of DnaA, with increased expression of all three proteins the cells became extremely filamentous and nonviable. Because stalled or collapsed replication forks lead to DSBs in DNA, we examined the effects of mutations in recombinational repair genes on the viability of cells with increased levels of DnaA and found that the absence of RecA, RecBC, or RuvABC proteins was extremely detrimental to these cells, indicating a requirement for recombinational repair in cells overexpressing DnaA.
MATERIALS AND METHODS
Bacterial strains.
The bacterial strains used in the present study are listed in Table 1. E. coli DH5α was used for all experiments that involved plasmid construction, isolation, and amplification. The recF349 allele of strain HL919, ruvA60 of N2057, ΔruvABC of JJC754, ΔruvC of JJC783, recB268 of N2101, and recC266 of N2103 were transferred by bacteriophage P1 transduction (56) to MG1655, producing the strains AVG349, AG2057, AVG754, AVG783, AG2101, and AG2103, respectively. E. coli PK2649 contains a sulA::lacZ fusion that allows the detection of the SOS response. Increased sensitivity to UV light was used to identify transfer of recF349, ruvA60, recB268, and recC266 among the tetracycline-resistant transductants, and ΔruvABC and ΔruvC among the chloramphenicol-resistant transductants. Plasmids were introduced into strains by electroporation (56).
TABLE 1.
E. coli strains used in this study
| Strain | Genotype | Source or reference |
|---|---|---|
| DH5α | supE44 lacU169 (φ80 lacZΔM15) hsdR17 recA1 endA1 gyrA96 thil relA | Stratagene |
| MG1655 | λ− F− | Mary Berlin |
| AB1157 | λ− F−rac− thr-1 ara-14 leuB6 Δ(gpt-proA)62 lacY1 tsx-33 supE44 galK2 hisG4(Oc) rfbD1 mgl-51 rpsL31 kdgK51 xyl-5 mtl-1 argE3(Oc) thi-1 qsr | Barbara Bachmann |
| PK2649 | trpR trpA9605 his-29 ilv pro-2 arg-427 thyA deoB Δ(argF-lac)205 λ cI ind sfiA::lacZ | Peter Kuempel (30) |
| HL919 | λ−thyA36 deoC IN(rrnD-rrnE) recF349 tnaA300::Tn10 | Phil Hanawalt (11) |
| N2057 | AB1157 ruvA60::Tn10 | Bénédicte Michel (48) |
| JJC754 | AB1157 hsdR ΔruvABC::Cm | Bénédicte Michel (45) |
| JJC783 | AB1157 hsdR ΔruvC::Cm | Bénédicte Michel (45) |
| N2101 | AB1157 recB268::Tn10 | Robert Lloyd (34) |
| N2103 | AB1157 recC266::Tn10 | Robert Lloyd (34) |
| ALS972 | MG1655 recA938::cat | This lab (54) |
| DPB271 | MG1655 recD1903::mTn10 | Stan Cohen (7) |
| ALS973 | MG1655 recA938::cat recD1903::mTn10 | This lab (54) |
| AVG349 | MG1655 recF349 tnaA300::Tn10 | This study |
| AG2057 | MG1655 ruvA60::Tn10 | This study |
| AVG754 | MG1655 ΔruvABC::Cm | This study |
| AVG783 | MG1655 ΔruvC::Cm | This study |
| AG2101 | MG1655 recB268::Tn10 | This study |
| AG2103 | MG1655 recC266::Tn10 | This study |
Plasmid construction.
Plasmids pDnaN, pDnaAN, pRecF, pAF, pNF, and pANF were all constructed from PCR products derived from the plasmid pBF101 (17). The primer pairs used for PCR were designed with noncompatible restriction enzyme sites ensuring the orientation of the insert into the vector. For plasmids that contain the dnaA gene, the plasmid pDnaA116 (28) was used as the plasmid parent. For the other plasmids, pLex5BA (28) was used as the vector source. Plasmid pLex5BA contains the lacI gene that encodes the LacI repressor that binds to two lacO sites in the Bujard promoter, PA1-03/04 (28). The addition of IPTG (isopropyl-β-d-thiogalactopyranoside) allows the transcription of genes inserted downstream of this promoter. The plasmid also contains the terminator rrnBt1t2 at the end of the gene in order to prevent readthrough after the inserted gene, the bla gene encoding for ampicillin resistance, and the ColE1 origin of replication. Plasmid pDnaA116, a derivative of pLex5BA (28), was used for increasing the DnaA expression. The dnaA gene of pDnaA116 contains a GTG→ATG mutation in the start codon, changing a weak start signal to a strong one. Plasmids pDnaAN, pAF, and pANF were constructed with this GTG-to-ATG change.
Plasmid pDnaN utilized primers dnaN (EcoRI) (5′-TAT TGA ATT CAT TTA ATC AGA ACA TTG-3′) and dnaN (BamHI) (5′-CGC GGG ATC CCA AGC GGG TGA GGG ACA-3′) to amplify the dnaN gene from plasmid pBF101 (17). This PCR product and the pLex5BA vector were digested with EcoRI and BamHI, mixed, and ligated together. The plasmid insert containing bases 2263 to 3410 of the dnaA operon (19) was sequenced for verification by using the LexL (5′-TGT TTT ATC AGA CCG CTT-3′) and LexU2 (5′-ACA ATT TCA AGC CTC-3′) primers.
Plasmid pDnaAN was constructed with the primers pre-dnaA (BglII) (5′-CAG AAG ATC TCT TGC GCA GTT TAG GCT-3′) and dnaN (HindIII) (5′-GCG GAA GCT TAA GCG GGT GAG GGA CT-3′). Both the insert and the pDnaA116 were digested with BglI and HindIII before ligation and transformation into DH5α. Sequencing was again used to verify that the plasmid contained bases 887 to 3409 (19).
Plasmid pRecF utilized the primers recF start (EcoRI) (5′-GGC GAA TTC AAT GAG ACT GTA ATG TCC C-3′) and recF end (HindIII) (5′-GGC GAA GCT TAA TCC GTT ATT TTA CCC-3′) to amplify by PCR the recF gene from pBF101 (17). Both the vector pLex5BA and the PCR product were cut with both EcoRI and HindIII, ligated, and transformed into DH5α. The plasmid insert was verified through sequencing to contain bases 3481 to 4567 of the dnaA operon (19), by using the primers LexL and LexU2.
Plasmid pAF was initially constructed with the primers recF start (RsrII) (5′-GTT GTC GGT CCG ATG AGA CTG TAA TGT CCC-3′) and recF end (HindIII) for PCR amplification of the insert. Both the insert and pDnaA116 were cut with RsrII and HindIII, and the DNA fragments were ligated and transformed into DH5α. Sequencing was used to verify that the plasmid contained bases 887 to 2453 and 3483-4567 (19). These bases were chosen to put the stop codon of dnaA in frame with the start codon of recF.
Plasmid pANF utilized the primers pre-dnaA (BglII) and recF end (HindIII) for PCR amplification of the DnaA operon. The vector pDnaA116 and the PCR product were both cut with EcoRI and HindIII, and the appropriate DNA fragments were ligated and transformed into DH5α. Upon conformation of the insert, pANF was found to have the correct insert sequence of bases 887 to 4567 (19).
Flow cytometry.
An overnight culture of each of the MG1655 strains grown in M9 enriched medium (1× M9 salts, 0.2% glucose, 1% Casamino Acids, 20 μg of uracil/ml, 5 μg of thymine/ml, 0.1% MgSO4, 0.01% CaCl2, 2 mg of thiamine/ml) containing 100 μg of ampicillin/ml was diluted 1:1,000 into prewarmed media containing 100 μg of ampicillin/ml and various amounts of IPTG. The cells were grown at 37°C with constant shaking. When the optical density at 450 nm (OD450) of each culture reached 0.2, 400 μl of the sample was added to 7 ml of 74% ethanol. In addition, 40 ml of the culture was added to chloramphenicol (200 μg/ml [final concentration]) to be prepared for Western analysis (see below). Immediately after the samples were obtained, 50 ml of culture was transferred into another flask, and rifampin (150 μg/ml) and cephalexin (50 μg/ml) were added. This treated culture was grown for 4 more h, with continuous shaking, after which 400 μl of each culture was added to 7 ml of 74% ethanol. Approximately 1.5 ml of each fixed sample was centrifuged for 10 min at 1,200 rpm at 4°C. After the supernatant was discarded, the pellets were washed in 1 ml of ice-cold staining buffer (10 mM Tris and 10 mM MgCl2 [pH 7.4] in sterile distilled H2O) and resuspended in 65 μl of staining buffer and 65 μl of staining solution (40 μg of ethidium bromide/ml and 200 μM mithramycin A). The cells were incubated on ice in the dark for 30 min and analyzed in a Bryte-HS (Bio-Rad) flow cytometer at 390 to 440 nm. The cells were measured at a rate of up to 104 cells/s. Each cell gives rise to a pulse of fluorescent light, the intensity of which is proportional to the cellular DNA content, which is displayed as a histogram of fluorescence intensity (directly related to DNA content) versus cell number.
Immunoblot analysis of DnaA, β subunit, and RecF proteins.
Samples were taken as described above in flow cytometry. Chloramphenicol was added immediately to inhibit further protein synthesis. The cells were centrifuged, and the cell pellets were resuspended in 0.5 ml of ice-cold 10% trichloroacetic acid and placed on ice for 30 min and then centrifuged again. To the pellets, 100 μl of solubilization buffer (25 mM Tris-HCl [pH 6.8], 1% sodium dodecyl sulfate [SDS], 1 N NaOH, 1 mM phenol red) was added.
The total protein of each sample was determined by using a BCA protein assay kit (Pierce) according to the manufacturer's directions. The protein concentration of each sample was then normalized to 4 mg/ml by diluting the samples into an appropriate amount of solubilization buffer. The final concentration of each sample was then brought to 2 mg/ml by adding 2× loading buffer (0.5 ml of 0.5 M Tris-HCl [pH 6.8], 0.4 ml of 100% glycerol, 0.8 ml of 10% SDS, 0.2 ml of 2-β-mercaptoethanol, 0.1 ml of 0.05% bromophenol blue, 6 ml of deionized water).
The DnaA protein (16), β subunit (obtained from Michael O'Donnell), RecF (obtained from Michael Cox), and protein samples were boiled for 3 min, mixed, and centrifuged for 3 min at 14,000 rpm. The samples, along with the standards and Kaleidoscope Marker (Bio-Rad), were separated by a SDS-PAGE containing 10.5% separating gel (3.84 ml of distilled H2O, 2.5 ml of 1.5 M Tris-HCl [pH 8.8], 100 μl of 10% SDS, 3.5 ml of 30% acrylamide-bisacrylamide [29:1], 50 μl of 10% ammonium persulfate [APS], 5 μl of tetramethylethylenediamine [TEMED]) and a 4% stacking gel (5.5 ml of H2O, 937.5 μl of 1 M Tris-HCl [pH 6.8], 75 μl of 10% SDS, 975 μl of 30% acrylamide-bisacrylamide, 37.5 μl of 10% APS, 7.5 μl of TEMED) for ca. 3 h at 25 mA by using the Bio-Rad Minigel apparatus. The separated proteins were transferred to a polyscreen PVDF-Plus transfer membrane (NEN-Dupont) by using the Bio-Rad Minitransblot apparatus at 100 V (250 mA) at 4°C for 90 min in cold high-glycine transfer buffer (25 mM Tris, 700 mM glycine, 5% electronic grade methanol [pH 8.3]).
After completion of protein transfer, the membrane was rinsed with water and blocked with 50 ml of 6% nonfat milk in 1× PBST (500 mM NaCl, 80 mM Na2HPO4, 20 mM NaH2PO4, 0.1% Tween 20) for 2 h with agitation. The membrane was then washed three additional times with the 1× PBST for 15 min each. It was then incubated in 10 ml of primary antibody (anti-DnaA [16] at 1:7,500, anti-β subunit [obtained from Charles McHenry] at 1:10,000, or anti-RecF [obtained from Michael Cox] at 1:2,500) for 1 h with shaking. The membrane was again washed with 50 ml of 1× PBST five times for 15 min each time and incubated in 10 ml of secondary antibody for 1 h at room temperature (1:10,000 goat anti-rabbit immunoglobulin G-horseradish peroxidase). Finally, the membrane was washed with 50 ml of 1× PBST-500 mM NaCl four times for 15 min each time and then in 50 ml of 1× PBST-150 mM NaCl for 15 min. The membrane was developed by using NEN-Dupont chemiluminescence reagent according to the manufacturer's protocol. A Molecular Dynamics densitometer was used to scan the X-ray film, and ImageQuant software was used to quantify the intensity of the bands by volume integration. The intensies of the RecF protein bands were relatively weak compared to the background signal of the blot, and the amounts could not be accurately determined.
Pulse-labeling analysis.
An overnight culture of each of the strains grown in M9 enriched medium containing 100 μg of ampicillin/ml was diluted (1:1,000) into prewarmed M9 enriched medium containing 100 μg of ampicillin/ml and an appropriate amount of IPTG. The cells were grown at 37°C with constant shaking. When the OD450 of each culture reached 0.2, 50 ml of the sample was mixed with cephalexin (50 μg/ml) and rifampin (150 μg/ml) was added. The cells were reincubated with continuous shaking. Starting at time zero and at 5-min intervals thereafter (in duplicate), 1 ml of the treated culture was removed from the flask and added to prewarmed tubes containing [3H]thymidine with a specific activity of 814.85 Ci/mol of thymidine (total concentration of 0.6193 μM [3H]thymidine) in a total volume of 50 μl. The reactions were mixed well and allowed to incubate at 37°C for 2 min. The reactions were terminated with 1 ml of ice-cold 10% trichloroacetic acid and 50 mM sodium pyrophosphate. The precipitate was collected by filtration onto 2.5-cm Schleicher & Schuell glass fiber filters, and the level of radioactivity was determined in a liquid scintillation counter.
Fluorescent cell staining.
Overnight cultures of cells were diluted 1:1,000 into M9 enriched medium containing IPTG and grown to an OD450 of 0.2. To stain cells with DAPI (4′,6′-diamidino-2-phenylindole), 4 ml of each culture was centrifuged, washed with 1 ml of 1× M9 salts, and resuspended into 30 μl of 1× M9 salts. Then, 10 μl of each sample was then separately spread onto clean glass slides, dried at 55°C for ca. 5 min, fixed in methanol for another 5 min, and rinsed in water. After air drying, the slides were coated with 10 μl of 10 μg of poly-l-lysine/ml and allowed to dry again. The slides were spread with 10 μl of DAPI (10 μg/ml) and viewed under simultaneous phase-contrast and fluorescence microscopy (54). Live-dead staining of cells was done according to the protocol provided by the Live/Dead BacLight Viability kit (Molecular Probes). A total of 1 ml of each culture was centrifuged and washed twice with filter-sterilized water. Equal volumes of reagents A and B from the kit were mixed and added to the cells (3 μl/ml of cells). After thorough mixing and incubation at room temperature in the dark for 15 min, 200 μl of each of the stained cells was filtered onto a separate polycarbonate membrane (0.2-μm pore size, 25 mm, black; Poretics catalog no. 11021) and washed with 1 ml of filter-sterilized water. Each filter was then placed onto a drop of lens oil on a clean glass slide and covered with a drop of lens oil and a glass coverslip. Cells were viewed by using fluorescence microscopy. Cells exhibiting a loss in cell membrane permeability take up the red propidium iodide dye saturating the green SYTO 9 dye taken up by all cells. Thus, red cells indicate the occurrence of cell death, and green cells indicate live cells.
Measuring SOS induction.
β-Galactosidase assays were carried out according to the method of Miller (41) with strain PK2649 (30), which contains a sulA::lacZ fusion that produces β-galactosidase in response to SOS induction. A positive control culture containing 0.1 μg of mitomycin C/ml was also included. Each of the plasmids was transformed into this strain and analyzed for the presence of an SOS response during IPTG induction of the proteins encoded by the plasmids. Triplicate samples were assayed for each culture.
Cell viability analysis.
An overnight culture of each of the strains grown in M9 enriched medium containing 100 μg of ampicillin/ml was diluted (1:1,000) into prewarmed M9 enriched medium containing 100 μg of ampicillin/ml. The cells were grown at 37°C with constant shaking. When the OD450 of each culture reached 0.08, IPTG was added to the cultures, and further growth was allowed for another 6 h. At 6 h, samples were taken, diluted, and plated in triplicate. Colonies were counted after growth overnight at 37°C to determine the viability of each culture.
RESULTS
Increased expression of DnaA protein leads to filamentation and cell death.
To monitor the effects of increasing the intracellular concentration of DnaA protein on chromosomal replication, cells containing the DnaA-expressing plasmid, pDnaA116, were grown in the presence of 0, 50, 75, or 100 μM IPTG. At an OD450 of 0.2, rifampin (to block further initiation events) and cephalexin (to block cell division) were added to the cultures, and replication was allowed to continue for 4 h. The amount of DNA in individual cells was analyzed by flow cytometry and found to be distributed in a broad peak rather than in discrete peaks at IPTG concentrations of ≥50 μM (Fig. 2). The level of DnaA protein induced in these cells by 50 to 100 μM IPTG compared to the wild type was 2.8- to 20.9-fold higher than normal levels (Fig. 3). These results are consistent with previously published data (3), demonstrating that as the concentration of DnaA increases, the number of replication forks increase; however, the replication velocity is reduced such that many forks do not reach the terminus during the time cells are exposed to rifampin.
FIG. 2.

Increased DnaA concentration leads to incomplete chromosomal replication. Strain MG1655 containing pDnaA116 was grown to an OD450 of 0.2 in the presence of 0, 50, 75, or 100 μM IPTG; treated with rifampin and cephalexin for 4 h; and then fixed, stained, and analyzed by flow cytometry.
FIG. 3.

Levels of DnaA or β-subunit proteins in MG1655 containing pDnaA116 or pDnaN, respectively, in response to IPTG. Immunoblot gel analysis was used to determine DnaA or β-subunit protein concentrations in cell extracts obtained after growth in the presence of IPTG to an OD450 of 0.2. Fold increase compared to cells grown in the absence of IPTG at each IPTG concentration is plotted.
Cells containing pDnaA116 grown in the presence of 100 μM IPTG and visualized by fluorescence microscopy after being stained with DAPI or the Live/Dead BacLight viability stain were elongated, with most cells containing a single long nucleoid at the cell center (Fig. 4A and Table 2). A similar filamentous phenotype was observed in E. coli dnaA cos cells that overinitiate DNA replication (26, 27) and in B. subtilis cells expressing increased DnaA levels (42). The filamentous phenotype suggests that the SOS response is induced in cells overproducing DnaA; however, the lack of expression of the sulA promoter in response to increased levels of DnaA protein indicates that the SOS response was not induced in these cells (Table 3). This result is consistent with previously observed results in E. coli dnaA cos cells showing that filamentation occurs independently of the SOS response (26). In B. subtilis, the SOS response is induced with increased levels of DnaA (42).
FIG. 4.

DnaA overproduction leads to filamentation and nucleoid elongation; however, the simultaneous overproduction of DnaA and RecF leads to nucleoid condensation and segregation but not cell division. Strain MG1655 containing pDnaA116 (A and B) or pAF (C and D) was grown in the presence of 0, 60, and 100 μM IPTG to an OD450 of 0.2. Cells were stained with DAPI (A and C) or Live/Dead BacLight bacterial viability stain (B and D) and then viewed by fluorescence microscopy. Bar, 2 μm.
TABLE 2.
Increased expression of some proteins in the dnaA operon causes cell death and filamentation
| MG1655 plasmid | 0 μM IPTG
|
100 μM IPTG
|
|||
|---|---|---|---|---|---|
| % live cellsa | Mean avg live cell length (μm)a ± SD | % live cellsa | Mean avg live cell length (μm)a ± SD | Mean avg dead cell length (μm)b ± SD | |
| pDnaA116 | 93 | 2.2 ± 0.4 | 29 | 6.5 ± 3.2 | 11.3 ± 5.3 |
| pDnaN | 92 | 2.1 ± 0.3 | 85 | 2.5 ± 0.4 | 4.2 ± 2.4 |
| pDnaAN | 83 | 2.2 ± 0.3 | 13 | 2.6 ± 0.5 | 5.6 ± 5.1 |
| pRecF | >99 | 2.3 ± 0.4 | >99 | 2.2 ± 0.5 | 5.2 ± 4.4 |
| pAF | 82 | 2.5 ± 0.6 | 38 | 2.3 ± 0.5 | 13.0 ± 10.9 |
| pNF | 89 | 2.7 ± 0.5 | 64 | 2.5 ± 0.5 | 6.8 ± 4.1 |
| pANF | 98 | 3.1 ± 1.1 | <1 | 4.1 ± 2.0 | >50 |
Live cells are defined as those cells stained green with SYTO 9 from the Live/Dead BacLight bacterial viability kit.
Dead cells are defined as cells that stained red with propidium iodide from the Live/Dead BacLight bacterial viability kit.
TABLE 3.
SOS is not induced with increased expression of DnaA
| Plasmida | Expression (Miller units)c with:
|
% Change with 100 μM IPTG | |
|---|---|---|---|
| 0 μM IPTG | 100 μM IPTG | ||
| pLex5BA | 90 | 82 | 9 |
| pLex5BA + mitomycin Cb | 1,223 | ND | NA |
| pDnaA116 | 123 | 128 | 4 |
| pDnaN | 100 | 129 | 28 |
| pDnaAN | 116 | 129 | 11 |
| pRecF | 104 | 101 | 2 |
| pAF | 120 | 107 | 11 |
| pNF | 101 | 98 | 3 |
| pANF | 117 | 170 | 46 |
All plasmids are in strain PK2649 (30), which contains a sulA::lacZ fusion that produces β-galactosidase in response to SOS induction.
That is, 0.1 μg of mitomycin C/ml.
There was less than ±3% variation in the assay results of triplicate samples for each culture. NA, not applicable; ND, not determined.
Because Live/Dead BacLight viability staining demonstrated the presence of dead cells at increased DnaA levels (Fig. 4B and Table 2), we monitored the viability of cells exposed to 100 μM IPTG for 6 h. Cell death begins to occur after 2 h of exposure to IPTG, and viability decreased by >10-fold after 6 h (Fig. 5, see difference in CFU between 2 and 6 h).
FIG. 5.

DnaA overexpression leads to cell death. MG1655 cells containing pDnaA116 were grown to an OD450 of 0.08 and then 100 μM IPTG was added. CFU were determined on solid medium in the absence of IPTG.
Increased expression of β clamp causes only slight changes in cell length and viability, although many replication forks do not complete replication.
Cells with increased expression of β clamp at 60 μM IPTG (12.4-fold) contained incomplete chromosomes (Fig. 6B), and the C period increased from 41 to 46 min (Table 4), suggesting that β clamp is not the rate-limiting protein in replication.
FIG. 6.

Strains MG1655 containing plasmid pDnaA116 (A), pDnaN (B), pDnaAN (C), pRecF (D), pAF (E), pNF (F), or pANF (G) were grown to an OD450 of 0.2 in the presence of 0, 20, 40, 60, or 100 μM IPTG; treated with rifampin and cephalexin for 4 h; and then removed, fixed, stained, and analyzed by flow cytometry.
TABLE 4.
Flow cytometric and pulse-labeling analysis
| Plasmid | IPTG concn (μM) | τa (min) | DNA/cellb | % Completed chromosomesc
|
Non-four or -eightd | Relative C period (min)e | |
|---|---|---|---|---|---|---|---|
| Four | Eight | ||||||
| pLex5BA | 0 | 36.1 | 1 | 16 | 77 | 7 | 43 |
| pDnaA116 | 0 | 33.5 | 1 | 49 | 46 | 5 | 42 |
| 20 | 34.7 | 1 | 48 | 47 | 5 | 43 | |
| 40 | 31.1 | 1.04 | 36 | 57 | 7 | 49 | |
| 60 | 33.5 | 1.11 | 17 | 60 | 23 | 65 | |
| pDnaN | 0 | 35.9 | 1 | 41 | 56 | 3 | 41 |
| 20 | 33.9 | 1 | 41 | 57 | 2 | 45 | |
| 40 | 34.0 | 1.08 | 32 | 63 | 5 | 46 | |
| 60 | 35.9 | 1.16 | 17 | 51 | 32 | 46 | |
| pDnaAN | 0 | 33.9 | 1 | 46 | 51 | 3 | 46 |
| 20 | 32.5 | 1.02 | 38 | 58 | 4 | 44 | |
| 40 | 33.4 | 1.04 | 14 | 78 | 8 | 46 | |
| 60 | 44.9 | 1.08 | 3 | 62 | 35 | 58 | |
| pRecF | 0 | 35.4 | 1 | 28 | 67 | 5 | 43 |
| 20 | 33.4 | 0.97 | 24 | 69 | 7 | 41 | |
| 40 | 32.4 | 1 | 22 | 72 | 6 | 39 | |
| 60 | 32.4 | 1.02 | 23 | 69 | 8 | 38 | |
| pAF | 0 | 35.6 | 1 | 28 | 66 | 6 | 42 |
| 20 | 34.8 | 1.02 | 23 | 69 | 8 | 45 | |
| 40 | 36.5 | 1.04 | 23 | 70 | 7 | 43 | |
| 60 | 35.9 | 1.06 | 22 | 69 | 9 | 47 | |
| pNF | 0 | 38.2 | 1 | 18 | 69 | 13 | 49 |
| 20 | 38.4 | 0.98 | 15 | 73 | 12 | 45 | |
| 40 | 37.2 | 1.01 | 14 | 73 | 13 | 43 | |
| 60 | 38.6 | 1.06 | 13 | 69 | 18 | 40 | |
| pANF | 0 | 39.1 | 1 | 46 | 51 | 3 | 48 |
| 20 | 41.2 | 1 | 38 | 58 | 4 | 44 | |
| 40 | 40.0 | 1.03 | 14 | 78 | 8 | 41 | |
| 60 | 40.8 | 1.04 | 3 | 62 | 35 | 45 | |
Generation time.
DNA/cell refers to the mean value of light fluorescence in exponentially growing cells compared to MG1655.
That is, the percentage of cells with completed chromosomes (four or eight) obtained from exponentially growing cells treated for 4 h with rifampin and cephalexin.
Non-four or -eight, percentage of cells that have not completed chromosomal replication.
For an explanation of the relative C period, see the Appendix.
Lack of β clamp is not the cause of replication fork stalling in DnaA-overexpressing cells.
The arrangement of the genes of the dnaA operon and the replicative functions of their gene products suggest that increased levels of combinations of these proteins may aid in correcting the chromosomal replication and growth defects caused by overexpressing DnaA protein. The plasmids pDnaN (dnaN), pDnaAN (dnaA and dnaN), pRecF (recF), pAF (dnaA and recF), pNF (dnaN and recF), and pANF (dnaA, dnaN, and recF) were constructed with the genes under control of the Bujard promoter, PA1-03/04 (28, 33). The DNA contents of the cells, grown in the presence of IPTG to induce expression of these proteins, were examined after runout DNA replication in the presence of rifampin and cephalexin by flow cytometry (Fig. 6). At the highest IPTG concentration tested, 100 μM, which corresponds to a 20.9-fold increase in DnaA protein and a 37.7-fold increase in β-subunit protein (Fig. 3), cells containing plasmids pDnaA116, pDnaN, pDnaAN, pAF, pNF, and pANF contained chromosomes that had not completed replication. The only cells that showed a normal chromosomal distribution when the plasmid-encoded protein was overexpressed at 100 μM IPTG were those containing pRecF (Fig. 6D). The protein concentrations of RecF could not be determined because the faint protein bands were obscured by the background signal present on the immunoblot (data not shown).
The effects of lower levels of these proteins on chromosomal replication by flow cytometry was determined. We determined whether replication velocity was affected in these overexpressing strains by measuring the “C period” (see Appendix) of cells induced at 0, 20, 40, and 60 μM IPTG; treated with rifampin and cephalexin at an OD450 of 0.2; and pulse-labeled with [3H]thymidine for 2 min at 5-min intervals. When the DnaA protein concentration was increased almost fourfold with 60 μM IPTG (Fig. 2), the amount of DNA in individual cells was distributed more broadly than in untreated cells (Fig. 6A) and the percentage of cells containing other than four or eight chromosomes after runout replication increased to 23% (Table 3), indicating that some chromosomes had not completed replication. The concomitant overproduction of DnaA and RecF, however, decreases this percentage to 9% at 60 μM IPTG (Table 4), and discrete peaks were seen by flow cytometry at four- and eight-chromosome peak distributions (Fig. 6E). Additionally, the overproduction of RecF together with β clamp also resulted in discrete peaks of chromosomal DNA that had a broad distribution when β clamp alone was overexpressed 12.4-fold at the 60 μM IPTG level, as shown by flow cytometry (compare Fig. 6B and F). The number of cells containing other than four or eight chromosomes at 60 μM IPTG decreased from 32%, seen in cells only overexpressing β clamp, to 18%, in both β clamp- and RecF-overexpressing cells (Table 4). The combined overproduction of DnaA and β clamp in cells containing pDnaAN resulted in slightly more incomplete chromosomes than in cells containing either pDnaA116 or pDnaN. This finding suggests that the lack of additional β clamp in DnaA-overexpressing cells is not the cause of fork stalling. This observation differs from previously published data (42), where DNA replication and growth defects caused by DnaA overproduction in B. subtilis could be alleviated by concomitant increased expression of DnaA and β clamp.
Increased expression of RecF with DnaA results in the condensation and segregation of nucleoids.
Increased amounts of DnaA led to filamentous cells carrying uncondensed chromosomes located in the center of each cell (Fig. 4A and Table 2), and most cells were also nonviable (red cells in Fig. 4B and Table 2). Cells overexpressing both DnaA and RecF continued to show the presence of elongated cells, indicating that cell division was still inhibited (Fig. 4 and Table 2); however, the chromosomes within these cells were condensed and distributed evenly throughout the filamentous cell lengths or toward the poles of the normal-sized cells. However, surprisingly, the viability levels measured after 6 h of 0, 60, 80, 100, 120, or 140 μM IPTG treatment remained about the same in these cells compared to DnaA-overexpressing cells (Fig. 7).
FIG. 7.

The recF349 mutation decreases viability in cells compared to the wild type overproducing DnaA protein. Strain AVG349 was constructed containing the recF349 mutation, replacing the wild-type allele of MG1655. The viability of cells after 6 h of treatment with various IPTG concentrations was determined by triplicate colony counts on solid medium in the absence of IPTG. Averages of the triplicate counts have been plotted.
Cells with the recF349 mutation show a slight decrease in viability when overproducing DnaA protein.
Flow cytometry and pulse-labeling data indicate that low levels of concomitant increased expression of RecF with DnaA protein resulted in synchronous initiation and a normal C period. We performed additional viability studies on strain AVG349, which contains the recF349 mutation. Plasmids pDnaA116 and pAF were each transformed into AVG349 and viability assessed after 6 h of growth in various IPTG levels from 0 to 140 μM. Figure 7 indicates that viability decreased ∼10-fold in the mutated RecF strain compared to the wild-type strain, both overproducing DnaA. These results indicate that RecF protein affects cell viability when DnaA is overexpressed, suggesting that RecF protein could have a role in preserving fork integrity at stalled forks in these cells; RecF has been shown to be involved in preserving stalled forks and resuming replication from disrupted replication forks (11, 12).
Concomitant overexpression of RecF and β clamp results in a decrease in cell viability.
Since concomitant RecF and β clamp overexpression, up to a level of 60 μM IPTG induction, showed discrete peaks when analyzed by flow cytometry (Fig. 6F) and the C period decreased (Table 4), we examined the effects on viability. Figure 8 shows that overexpression of RecF decreased the viability of these cells by >100-fold at the highest IPTG concentration compared to cells where only β clamp was overexpressed. This finding was quite surprising because RecF nor β clamp overproduction had very little if any effect on cell viability, even at the highest IPTG concentration of 140 μM. The most extreme response to increased expression of proteins encoded by the dnaA operon occurred when all three proteins were overexpressed, causing most cells to form extremely long filaments (>50 μm) that had lost the ability to form colonies (Table 2, Table 5; see summary in Table 6; see also Fig. 8), and the SOS response was slightly induced (Table 3).
FIG. 8.

The overexpression of β clamp and RecF proteins together decreases viability of the cells, even though each individually had little or no effect on viability alone. The viability of cells after 6 h of treatment with various IPTG concentrations was determined by triplicate colony counts on solid medium in the absence of IPTG. Average of the triplicate counts have been plotted.
TABLE 5.
Viability after 6 h in the presence or absence of IPTG
| Strain | Mutant gene(s) | Mean viability (CFU/ml) ± SD at:
|
100 μM IPTG/ 0 μM IPTG | |
|---|---|---|---|---|
| 0 μM IPTG | 100 μM IPTG | |||
| MG1655/pLex5BA | Wild type | 1.51 × 109 ± 2.35 × 108 | 1.49 × 109 ± 3.51 × 107 | 9.8 × 10−1 |
| MG1655/pDnaA116 | Wild type | 1.78 × 109 ± 2.79 × 108 | 1.30 × 107 ± 6.41 × 106 | 7.3 × 10−3 |
| AVG349/pDnaA116 | recF349 | 2.15 × 109 ± 1.33 × 108 | 1.46 × 106 ± 1.12 × 106 | 6.8 × 10−4 |
| AVG349/pAF | recF349 | 1.96 × 109 ± 4.73 × 108 | 7.03 × 106 ± 1.94 × 106 | 3.6 × 10−3 |
| ALS972/pDnaA116 | recA938 | 2.14 × 109 ± 3.99 × 108 | 1.65 × 105 ± 4.12 × 104 | 7.7 × 10−5 |
| DPB271/pDnaA116 | recD1903 | 6.10 × 108 ± 6.56 × 107 | 7.73 × 106 ± 1.65 × 106 | 1.3 × 10−2 |
| ALS973/pDnaA116 | recA938, recD1903 | 3.43 × 108 ± 4.51 × 107 | 6.77 × 102 ± 1.37 × 102 | 2.0 × 10−6 |
| AG2057/pDnaA116 | ruvA60 | 1.84 × 109 ± 1.01 × 108 | 4.33 × 104 ± 3.06 × 104 | 2.4 × 10−5 |
| AVG754/pDnaA116 | ΔruvABC | 1.82 × 109 ± 1.55 × 108 | 2.67 × 104 ± 1.53 × 104 | 1.5 × 10−5 |
| AVG783/pDnaA116 | ΔruvC | 6.07 × 108 ± 8.02 × 107 | 2.80 × 105 ± 4.58 × 104 | 4.6 × 10−4 |
| AG2101/pDnaA116 | recB268 | 4.53 × 108 ± 9.02 × 107 | 1.10 × 105 ± 3.11 × 104 | 2.4 × 10−4 |
| AG2103/pDnaA116 | recC266 | 3.50 × 108 ± 6.93 × 107 | 9.00 × 102 ± 5.20 × 102 | 2.6 × 10−6 |
| MG1655/pDnaN | Wild type | 2.66 × 109 ± 8.19 × 107 | 5.70 × 108 ± 1.18 × 108 | 2.1 × 10−1 |
| MG1655/pDnaAN | Wild type | 1.98 × 109 ± 1.54 × 108 | 6.67 × 107 ± 2.06 × 107 | 3.4 × 10−2 |
| MG1655/pRecF | Wild type | 2.23 × 109 ± 1.15 × 108 | 1.70 × 109 ± 1.63 × 108 | 7.6 × 10−1 |
| MG1655/pAF | Wild type | 2.63 × 109 ± 2.30 × 108 | 1.13 × 107 ± 8.33 × 105 | 4.3 × 10−3 |
| MG1655/pNF | Wild type | 2.35 × 109 ± 4.30 × 108 | 8.57 × 107 ± 4.30 × 107 | 3.7 × 10−2 |
| MG1655/pANF | Wild type | 1.95 × 109 ± 2.57 × 108 | 1.12 × 105 ± 7.16 × 104 | 5.7 × 10−5 |
TABLE 6.
Summary of the effects of increased levels of proteins encoded by the dnaA operon
| Protein(s)a | % Completed chromosomes at 60 μM IPTGb | % Viabilityc at:
|
C period (min)d at:
|
Cell length (μm) at 100 μM IPTGe | ||
|---|---|---|---|---|---|---|
| 60 μM IPTG | 100 μM IPTG | 0 μM IPTG | 60 μM IPTG | |||
| DnaA | 77 | 15.4 | 0.7 | 42 | 65 | 11.3 |
| β subunit | 68 | 38.3 | 21.4 | 41 | 46 | 4.2 |
| DnaA and β subunit | 65 | 31.7 | 3.4 | 46 | 58 | 5.6 |
| RecF | 92 | 80.3 | 76.2 | 43 | 38 | 5.2 |
| DnaA and RecF | 91 | 10.0 | 0.4 | 42 | 47 | 13.0 |
| β subunit and RecF | 82 | 34.8 | 3.7 | 49 | 40 | 6.8 |
| DnaA, β subunit, and RecF | 65 | 1.2 | 0.0057 | 48 | 45 | >50 |
The strains were MG1655/pDnaA116, MG1655/pDnaN, MG1655/pDnaAN, MG1655/pRecF, MG1655/pAF, MG1655/pNF, and MG1655/pANF.
The flow cytometry distribution of DNA content/cell at 100 μM IPTG suggested that most, if not all, chromosomes contained stalled replication forks except for MG1655/pRecF (see Fig. 6 and Table 4).
The viability of each strain was determined after 6 h in 60 or 100 μM IPTG and compared to cells grown in the absence of IPTG.
From Table 4.
That is, the average dead cell length (see Table 2). The average dead cell length was 3.9 μm for MG1655/pDnaA116 and 9.4 μm for MG1655/pAF at 60 μM IPTG. The average live cell length of cells grown in the absence of IPTG varied between 2.1 and 3.1 μm.
DNA break repair proteins are needed for viability in cells overexpressing DnaA.
To determine whether the RecA-dependent DNA repair pathways are necessary for viability in DnaA-overexpressing cells, we determined the viability of a recA mutant strain, ALS972, with increasing levels of DnaA. The results shown in Fig. 9 and Table 5 show that ALS972 cells had almost an 80-fold decrease in viability over the wild-type strain, MG1655, at 100 μM IPTG. Additionally, we found that the recAD mutant strain, ALS973, containing pDnaA116 showed a further decrease in viability of 240-fold from the recA mutant strain and >19,000-fold from the wild-type strain at the 100 μM IPTG level even though viability was not affected with increased expression of DnaA in the recD mutant strain, DPB271.
FIG. 9.

The overexpression of DnaA protein in the wild type and various DNA repair mutants (relevant genotype indicated) decreases viability by various degrees. The viability of cells after 6 h of treatment with various IPTG concentrations was determined by triplicate colony counts on solid medium in the absence of IPTG. The averages of the triplicate counts were plotted.
Figure 9 and Table 5 show that the Ruv protein mutant strains—AG2057 (ruvA60), AVG754 (ΔruvABC), and AVG783 (ΔruvC)—resulted in 300-, 487-, and 46-fold decreases in viability, respectively, at the 100 μM IPTG induction level compared to wild-type cells. The 300-fold decrease in viability observed in the ruvA60 mutant strain is close to the loss in viability of the ruvABC mutant strain compared to all of the other strains tested. This suggests that the additional loss of RuvB and RuvC function when RuvA is inactive does not affect cell viability greatly. The loss in viability seen in the ruvC mutant strain differs from the remaining strains in that the loss in viability begins gradually when DnaA levels increase up to an IPTG induction of 80 μM and then decline more rapidly at higher DnaA concentrations. The Ruv proteins are involved in resolving Holliday junctions formed in the process of DSB repair (DSBR). Additionally, the recB mutant strain AG2101 and the recC mutant strain AG2103, each overproducing DnaA, showed decreases in viability of ca. 120- and 14,500-fold, respectively, compared to the wild-type strain under increased DnaA levels of 100 μM IPTG. The recC266 mutant strain shows a loss in viability similar to the very low viability levels observed in the recAD mutant strain. These results suggest that homologous recombination involving the RecBC pathway is important in maintaining a certain level of viability in cells with increased levels of DnaA and that RecF also contributes somewhat to the survival of these cells.
DISCUSSION
Increased levels of DnaA lead to a decrease in the rate of DNA replication and stalled forks that do not complete replication in the presence of rifampin (3). We have confirmed these observations and find, in addition, that such cells are filamentous with extended nucleoids and have lowered viability. The block in cell division observed in cells overexpressing DnaA was not SOS dependent but could be the result of the loss of transcription of essential cell division genes. When chromosome replication is blocked in a recA mutant where the SOS response cannot be induced, cells still form filaments and the transcription of cell division genes in the mra cluster is repressed (35).
To test the possibility that the additional forks initiated as a result of overexpression of DnaA had stalled or collapsed and, in the cells still viable, were being repaired by recombination proteins, the DnaA-expressing plasmid, pDnaA116 (28), was transferred into strains carrying mutations or deletions in genes encoding the recombination and repair proteins RecA, RecBCD, RuvABC, and RecF. The results of viability studies of these strains in response to increasing levels of DnaA indicate that RecA, RecBC, and RuvABC proteins and complexes are involved in the survival of cells overexpressing DnaA, suggesting a requirement for DSBR by homologous recombination.
The ExoV activity of RecBCD, which is missing in the strains carrying the recB, recC, or recD mutations, is not important for DSBR in these cells unless RecA is absent as well. Mutants defective in RecB or RecC are deficient in homologous recombination and do not have ExoV activity, and although recD mutants are recombination proficient they do not have ExoV activity as well (for a review, see reference 31). Only a small loss in viability was observed in the absence of RecD compared to the wild type when the concentration of DnaA increased (Table 5 and Fig. 9). In a recD mutant, helicase activity is still present in the RecBC complex, but the requirement for a Chi site is eliminated, and RecA is loaded, constitutively stimulating pairing and strand exchange with homologous DNA, followed by recombination (9, 51). In the recAD mutant strain, ALS973, viability is significantly decreased. This could be due to the unwinding of DNA by the RecBC complex from breaks that cannot be repaired in the absence of RecA converting duplex DNA into a substrate for single-stranded DNA nucleases, leading to cell death.
Survival requirements for the attenuation type of response caused by an increase in initiation events, wherein forks stall before reaching the terminus, are similar to the survival requirements for replication fork collapse at Ter sites placed at ectopic positions in the E. coli chromosome (6). Both require RecA, RecBC, and RuvABC but not RecD. Linear DNA is formed in the Ter strains only after a second round of replication forks reach forks previously blocked at Ter sites, thus explaining the need for recombinational repair as opposed to replication fork reversal (6). Similarly, when the additional replication forks initiated with DnaA overexpression reach a stalled fork, double-stranded ends would be formed, leading to a collapsed replication fork and the need for the RecBCD pathway of DSBR.
When RecF protein was overexpressed in combination with DnaA, nucleoids condensed and segregated normally. However, the cells were filamentous and viability was low. In our experiments, the addition of RecF to DnaA-overexpressing cells aided in DNA condensation and the completion of replication but not viability.
When β clamp expression was increased, many chromosomes did not complete replication in runout experiments, suggesting that some forks were stalling. The profile of DNA content per cell was similar to that found with DnaA-overexpressing cells (Fig. 6) and yet β clamp-overexpressing cells were more viable (Table 6). One explanation for this observation is that the stalled forks in β clamp-overexpressing cells may be repaired by replication fork reversal, whereas in DnaA-overexpressing cells the excessive number of forks could lead to fork collision, creating a collapsed replication fork that can only be repaired by RecBCD DSBR, where the replication fork reversal is less likely to lead to permanent damage than RecBCD DSBR (32).
Ogura et al. (42) observed a recovery from growth defects caused by DnaA overproduction in the presence of increased β clamp expression in B. subtilis, and we observed this to some extent in E. coli. In cells overexpressing both DnaA and β clamp, there was a slight increase in viability compared to cells overexpressing DnaA, but chromosomes did not complete replication in runout experiments.
We have no explanation for the precipitous drop in viability observed when all three proteins encoded by the dnaA operon are overexpressed. These cells are extremely filamentous and did have a slight SOS response (Table 3). The SOS response was not induced in E. coli cells overexpressing DnaA, although these cells were filamentous (Fig. 4). Ogura et al. (42) observed, in contrast to our results with E. coli, induction of the SOS response with filamentation in B. subtilis cells overproducing DnaA. When DnaA and β clamp expression were increased simultaneously, filamentation did not occur in either B. subtilis or E. coli cells.
Since β clamps act distributively during Okazaki fragment synthesis of the lagging strand, we considered the possibility that the rate of replication may be dependent on the concentration of β clamps, as well as the speed of clamp loading and removal by the γ complex.
Additionally, the overproduction of RecF protein caused a significant loss in viability of cells overexpressing β clamp. Stationary-phase-dependent mechanisms have evolved in order to coordinate expression of dnaN and recF independently of the dnaA regulatory region (52). Therefore, if DNA replication should stall during the transition from exponential growth into stationary phase, the coordinate induction of dnaN and recF would be needed to help complete current rounds of replication. These mechanisms may be part of a developmental program aimed at maintaining DNA integrity under stressful conditions. However, our results indicate that overproduction of both of these genes during the exponential growth phase is detrimental, did not result in the completion of replication, and led to a loss in viability.
In summary, DSBs in DNA that escape repair are probably the cause of the decrease in survival that occurs with increased levels of DnaA. The decreased tolerance of cells with deficiencies in RecA, RecBC, and RuvABC suggest that cells that do survive the excessive number of new replication forks initiating with increased levels of DnaA do so through a RecBC pathway of DSBR. Co-overexpressing the proteins of the dnaA operon resulted in extreme lethality and excessive filamentation rather than correcting the deficiencies observed with DnaA overexpression.
Acknowledgments
We thank Phil Hanawalt, Peter Kuempel, Robert Lloyd, Walter Messer, and Bénédicte Michel for strains and plasmids and Doug Smith for a critical reading of the manuscript. We also thank Charles McHenry, Michael O'Donnell, and Michael Cox for purified proteins and antibodies. Elio Schaechter is especially appreciated for invaluable discussions.
These studies were supported by the National Science Foundation (United States) grant MCB-9507209 to J.W.Z. and by CICYT (Spain) grant PB98-1626 to E.C.G.
APPENDIX
A mathematical model was developed specifically to estimate the C and D periods from DNA pulse-labeling data in the present study. Details are provided here on the assumptions used in the mathematical model and how the computer code fits the experimental data. For illustration, numerical values for the standard run on a strain of E. coli K-12 with the plasmid pLex5BA are used with the general formulation of the model.
The mathematical model is a deterministic formulation based on the assumption of a perfect asynchronous culture, dividing exactly with a generation time of τ. Clearly, individual cells will vary in their growth, creating a distribution of generation times and C periods. Our model tracks the means of these distributions as the best representative times. An asynchronous, exponentially growing culture of E. coli implies that the age-structure of the cells must have twice as many cells that have just undergone cell division (age 0) than cells that are about to divide (age τ). Figure A1 shows the distribution of cells having an age from 0 to τ, wherein τ is the age of the cell at division.
FIG.

A1.
Cell distribution at ages of 0 to τ. The fraction of cells with an age less than t is given by p(t), the patterned area of the graph.
If we define p(t) to be the fraction of cells with an age less than t, then (Fig. A1)
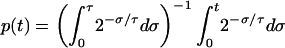 |
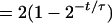 |
[Note that p(0) = 0 and p(τ) = 1, indicating that no cells have an age of <0 and that all cells are accounted for by age τ.]
The experiment works with an exponentially growing culture of cells, and when the culture reaches an OD450 of 0.2, the cells are treated with the antibiotics rifampin, which inhibits new DNA initiation events, and cephalexin, which inhibits any new cell division events (details are provided in the text). Flow cytometry analysis demonstrated that, after a 4-h treatment to permit completion of the rounds of replication, cells in all cultures had either four chromosomes, eight chromosomes, or an indeterminate amount of DNA (indicating the presence of replication forks that had not reached the terminus). Our model assumes that all of the cultures have either four or eight origins at the time when the antibiotics are added.
With this assumption, Fig. A2 provides a schematic of the key events of initiation (i), termination (t), and cell division (d) for three cell cycles of the growing cultures. The figure also indicates the number of origins present at each time in the cell cycle and shows the C and D periods leading into the division at 3τ. Note that the ordering of the termination and initiation events could be reversed in the figure, depending on the length of the C and D periods.
FIG.

A2.
Events of initiation (i), termination (t), and cell division (d) for three cell cycles.
The value of τ was measured for each of the cultures analyzed in these experiments. For pLex5BA, the experimental data gave τ = 36 min, so 3τ = 108 min. The first step in the mathematical analysis was determining the timing of initiation, i. From Fig. A2, it follows that all cells with ages 0 to i have four origins, whereas those with ages from i to τ have eight oriCs. From Fig. A1, if f is the fraction of cells with four origins, then
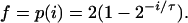 |
This is readily solved to give
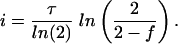 |
The model normalizes the fractions found in experiments for cells with four and eight origins and then uses these data in the formula above to compute i. Figure A2 shows that the sum of the C and D periods is given by
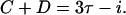 |
The experiments with pLex5BA showed that 17% of the cells had four origins and 67% of the cells had eight origins. Normalizing these values gives
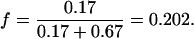 |
Thus,
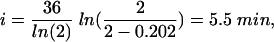 |
or an initiation event occurs between 5 and 6 min after cell division. From this information, the length of the C + D periods is calculated to be between 102 and 103 min.
The next step in our analysis is determining the C period from the pulse-labeling experiments. The mathematical model examines how an ideal asynchronously growing culture of cells that are treated with the antibiotics at t = 0 would respond to a 2-min pulse of radioactive thymidine. At t = 0, the model assumes the population has a distribution according to Fig. A1, where the value of τ is obtained from the experiment.
The program sequentially selects integer values for the C period, ranging from C = 30 to C = 70. From the calculations above, the initiation time is known, so for a given value of C, the termination time t is computed. The program divides the distribution of cells illustrated in Fig. A1 into τ distinct cohorts (1-min age groups), representing the age classes of each group of the asynchronous culture with similar characteristics. From Fig. A2, it can be determined how many DNA strands are replicating for each of the age classes. The model assumes that the thymidines are uniformly distributed and that the replication forks in each age class are advancing at the same speed. The program then computes how much radioactive labeling each age class receives and what fraction of the radioactivity is contributed by each age class.
As an example, consider the case of pLex5BA with i = 6, τ = 36, and C = 43. It follows that the D period is 59 min. At t = 0, the age classes from 0 to 6 have four oriCs present and four replication forks advancing. The age classes from 6 to 13 have eight oriCs with 12 replication forks advancing since an initiation event has occurred for cells older than 6 min (giving 8 new and the 4 old replication forks). A termination, halting four replication forks, occurs for cells age 13, so the age classes from 13 to 36 have eight replication forks.
At t = 0, the antibiotics act on all age classes of cells, preventing any new initiations. The program simulates how these age classes of cells would respond to a 2-min pulse of radioactive thymidine for each minute after t = 0 (until t = 70). As time advances, the model traces the advancing replication forks until they reach the termination point, which for a given age class occurs C minutes after its last associated initiation event. At each time t and age 0, every age class is carefully monitored for its number of replication forks. More age classes reach termination with advancing time until the last age class, which just initiated at the beginning of the simulation, no longer uptakes radioactivity at t = C. The relative strength of the theoretical radioactive signal is simulated for all times from t = 0 to 70. The result is a monotonically decreasing function as more age classes reach termination and cannot acquire the radioactive label.
Since the model gives the relative amount of radioactivity for each minute after the introduction for any given length of C period, it remains for us to fit the parameters of the C period and the amplitude of the radioactive signal to the pulse-labeling experimental data. Suppose we define the normalized mathematical model with a given C period as y(t,C) and its amplitude as α. The normalized model gives y(0,C) = 1 and y(C,C) as the first time that the function reaches 0 counts of radioactivity. The program determines the least-squares best fit between the experimental data and the mathematical model, which is given by
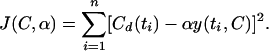 |
The experimental data are given by Cd(ti) with the units of counts per minute found at times ti when the pulse-label of radioactive thymidine is added. The parameters α and C are varied over the ranges of 500 to 15,000 and 30 to 70, respectively. The smallest value of J(C,α) gives the best fit of the theoretical model to the experimental data, i.e., the least-squares best fit.
Due to complications observed in fitting the data to the model, the first three datum points (times before 10 min) were ignored. These often gave large deviations from the model that could be the result of variations in uptake of the antibiotics or some other unknown experimental complication from the initial shock to the treated cell cultures. However, it is clear the data were behaving well after 10 min. With this information, the program finds the best fit to the data as illustrated in Fig. A3. Figure A3 shows the standard culture pLex5BA with the best-fit theoretical model overlaying the actual experimental data. The best-fit function J(C,α) found that
 |
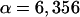 |
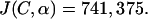 |
FIG.

A3.
Best-fit theoretical model for the C period determination of pLex5BA overlaying the actual pulse-labeled data.
These modeling simulations were run for each set of experimental data, giving the results reported in the text.
REFERENCES
- 1.Anderson, D. G., and S. C. Kowalczykowski. 1997. The recombination hot spot chi is a regulatory element that switches the polarity of DNA degradation by the RecBCD enzyme. Genes Dev. 11:571-581. [DOI] [PubMed] [Google Scholar]
- 2.Anderson, D. G., and S. C. Kowalczykowski. 1997. The translocating RecBCD enzyme stimulates recombination by directing RecA protein onto ssDNA in a chi-regulated manner. Cell 90:77-86. [DOI] [PubMed] [Google Scholar]
- 3.Atlung, T., and F. G. Hansen. 1993. Three distinct chromosome replication states are induced by increasing concentrations of DnaA protein in Escherichia coli. J. Bacteriol. 175:6537-6545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Atlung, T., E. S. Clausen, and F. G. Hansen. 1985. Autoregulation of the dnaA gene of Escherichia coli K-12. Mol. Gen. Genet. 200:442-450. [DOI] [PubMed] [Google Scholar]
- 5.Berenstein, D., K. Olesen, C. Speck, and O. Skovgaard. 2002. Genetic organization of the Vibrio harveyi dnaA gene region and analysis of the function of the V. harveyi DnaA Protein in Escherichia coli. J. Bacteriol. 184:2533-2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bidnenko, V., S. D. Ehrlich, and B. Michel. 2002. Replication fork collapse at replication terminator sequences. EMBO J. 21:3898-3907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Biek, D., and S. Cohen. 1986. Identification and characterization of recD, a gene affecting plasmid maintenance and recombination in Escherichia coli. J. Bacteriol. 167:594-603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Braun, R. E., K. O'Day, and A. Wright. 1985. Autoregulation of the DNA replication gene dnaA in Escherichia coli K-12. Cell 40:159-169. [DOI] [PubMed] [Google Scholar]
- 9.Churchill, J. J., D. G. Anderson, and S. C. Kowalczykowski. 1999. The RecBC enzyme loads RecA protein onto ssDNA asymmetrically and independently of chi, resulting in constitutive recombination activation. Genes Dev. 13:901-911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Courcelle, J., and P. C. Hanawalt. 2001. Participation of recombination proteins in rescue of arrested replication forks in UV-irradiated Escherichia coli need not involve recombination. Proc. Natl. Acad. Sci. USA 98:8196-8202. [DOI] [PMC free article] [PubMed]
- 11.Courcelle, J., C. Carswell-Crumpton, and P. C. Hanawalt. 1997. RecF and RecR are required for the resumption of replication at DNA replication forks in Escherichia coli. Proc. Natl. Acad. Sci. USA 94:3714-3719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Courcelle, J., D. J. Crowley, and P. C. Hanawalt. 1999. Recovery of DNA replication in UV-irradiated Escherichia coli requires both excision repair and RecF protein function. J. Bacteriol. 181:916-922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cox, M. M., M. F. Goodman, K. N. Kreuzer, D. J. Sherratt, S. J. Sandler, and K. J. Marians. 2000. The importance of repairing stalled replication forks. Nature 404:37-41. [DOI] [PubMed] [Google Scholar]
- 14.Dixon, D. A., and S. C. Kowalczykowski. 1993. The recombination hotspot chi is a regulatory sequence that acts by attenuating the nuclease activity of the E. coli RecBCD enzyme. Cell 73:87-96. [DOI] [PubMed] [Google Scholar]
- 15.Flores, M. J., S. D. Ehrlich, and B. Michel. 2002. Primosome assembly requirement for replication restart in the Escherichia coli holDG10 replication mutant. Mol. Microbiol. 44:783-792. [DOI] [PubMed] [Google Scholar]
- 16.Froelich, J. M., T. K. Phuong, and J. W. Zyskind. 1996. Fis binding in the dnaA operon region. J. Bacteriol. 178:6006-6012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fuller, R. S., J. M. Kaguni, and A. Kornberg. 1981. Enzymatic replication of the origin of the Escherichia coli chromosome. Proc. Natl. Acad. Sci. USA 78:7370-7374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grompone, G., M. Seigneur, S. D. Ehrlich, and B. Michel. 2002. Replication fork reversal in DNA polymerase III mutants of Escherichia coli: a role for the β clamp. Mol. Microbiol. 44:1331-1339. [DOI] [PubMed] [Google Scholar]
- 19.Hansen, F. G., E. B. Hansen, and T. Atlung. 1982. The nucleotide sequence of the dnaA gene promoter and of the adjacent rpmH gene, coding for the ribosomal protein L34, of Escherichia coli. EMBO. 1:1043-1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Herendeen, D. R., and T. J. Kelly. 1996. DNA polymerase III: running rings around the fork. Cell 84:5-8. [DOI] [PubMed] [Google Scholar]
- 21.Horiuchi, T., and Y. Fujimura. 1995. Recombinational rescue of the stalled DNA replication fork: a model based on analysis of an Escherichia coli strain with a chromosome region difficult to replicate. J. Bacteriol. 177:783-791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jeruzalmi, D., M. O'Donnell, and J. Kuriyan. 2002. Clamp loaders and sliding clamps. Curr. Opin. Struct. Biol. 12:217-224. [DOI] [PubMed] [Google Scholar]
- 23.Jeruzalmi, D., O. Yurieva, Y. Zhao, M. Young, J. Stewart, M. Hingorani, M. O'Donnell, and J. Kuriyan. 2001. Mechanism of processivity clamp opening by the delta subunit wrench of the clamp loader complex of E. coli DNA polymerase III. Cell 106:417-428. [PubMed] [Google Scholar]
- 24.Katayama, T. 2001. Feedback controls restrain the initiation of Escherichia coli chromosomal replication. Mol. Microbiol. 41:9-17. [DOI] [PubMed] [Google Scholar]
- 25.Katayama, T., T. Kubota, K. Kurokawa, E. Crooke, and K. Sekimizu. 1998. The initiator function of DnaA protein is negatively regulated by the sliding clamp of the E. coli chromosome replicase. Cell 94:61-71. [DOI] [PubMed] [Google Scholar]
- 26.Katayama, T., M. Takata, and K. Sekimizu. 1997. CedA is a novel Escherichia coli protein that activates the cell division inhibited by chromosomal DNA overreplication. Mol. Microbiol. 26:687-697. [DOI] [PubMed] [Google Scholar]
- 27.Kellenberger-Gujer, G., A. J. Podhajska, and L. Caro. 1978. A cold sensitive dnaA mutant of E. coli which overinitiates chromosome replication at low temperature. Mol. Gen. Genet. 162:9-16. [DOI] [PubMed] [Google Scholar]
- 28.Krause, M., B. Rückert, R. Lurz, W. Messer. 1997. Complexes at the replication origin of Bacillus subtilis with homologous and heterologous DnaA protein. J. Mol. Biol. 274:365-380. [DOI] [PubMed] [Google Scholar]
- 29.Kücherer, C., H. Lother, R. Kölling, M. A. Schauzu, and W. Messer. 1886. Regulation of transcription of the chromosomal dnaA gene of Escherichia coli. Mol. Gen. Genet. 205:115-121. [DOI] [PubMed]
- 30.Kuempel, P. L., J. M. Henson, L. Dircks, M. Tecklenburg, and D. F. Lim. 1991. dif, a recA-independent recombination site in the terminus region of the chromosome of Escherichia coli. New Biologist 3:799-811. [PubMed] [Google Scholar]
- 31.Kuzminov, A. 1999. Recombinational repair of DNA damage in Escherichia coli and bacteriophage λ. Microbiol. Mol. Biol. Rev. 63:751-813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuzminov, A. 2001. DNA replication meets genetic exchange: chromosomal damage and its repair by homologous recombination. Proc. Natl. Acad. Sci. USA 98:8461-8468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lanzer, M., and H. Bujard. 1988. Promoters largely determine the efficiency of repressor action. Proc. Natl. Acad. Sci. USA 85:8973-8977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lloyd, R. G., C. Buckman, and F. E. Benson. 1987. Genetic analysis of conjugational recombination in Escherichia coli K12 strains deficient in RecBCD enzyme. J. Gen. Microbiol. 133(Pt. 9):2531-2538. [DOI] [PubMed] [Google Scholar]
- 35.Liu, G., K. Begg, A. Geddes, and W. D. Donachie. 2001. Transcription of essential cell division genes is linked to chromosome replication in Escherichia coli. Mol. Microbiol. 40:909-916. [DOI] [PubMed] [Google Scholar]
- 36.Liu, J., and K. J. Marians. 1999. PriA-directed assembly of a primosome on D loop DNA. J. Biol. Chem. 274:25033-25041. [DOI] [PubMed] [Google Scholar]
- 37.Liu, J., L. Xu, S. L. Sandler, and K. J. Marians. 1999. Replication fork assembly at recombination intermediates is required for bacterial growth. Proc. Natl. Acad. Sci. USA 96:3552-3555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Macián, F., I. Pérez-Roger, and M. E. Armengod. 1994. An improved vector system for constructing transcriptional LacZ fusions: analysis of regulation of the dnaA, dnaN, recF, and gyrB genes of Escherichia coli. Gene 145:17-24. [DOI] [PubMed] [Google Scholar]
- 39.McGlynn, P., A. A. Al-Deib, J. Liu, K. J. Marians, and R. G. Lloyd. 1997. The DNA replication protein PriA and the recombination protein RecG bind d-loops. J. Mol. Biol. 270:212-221. [DOI] [PubMed] [Google Scholar]
- 40.Michel, B., S. D. Ehrlich, and M. Uzest. 1997. DNA double-strand breaks caused by replication arrest. EMBO. 16:430-438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miller, J. H. 1992. A short course in bacterial genetics. Cold Spring Harbor Laboratory Press, New York, N.Y.
- 42.Ogura, Y., Y. Imai, N. Ogasawara, and S. Moriya. 2001. Autoregulation of the dnaA-dnaN operon and effects of DnaA protein levels on replication initiation in Bacillus subtilis. J. Bacteriol. 183:3833-3841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pérez-Roger, I., M. Garcia-Sogo, J. P. Navarro-Aviñó, C. López-Acedo, F. Macián, and M. E. Armengod. 1991. Positive and negative regulatory elements in the dnaA-dnaN-recF operon of E. coli. Biochimie 73:329-334. [DOI] [PubMed] [Google Scholar]
- 44.Polaczek, P., and A. Wright. 1990. Regulation of expression of the dnaA gene in Escherichia coli: role of the two promoters and the DnaA box. New Biologist 2:574-582. [PubMed] [Google Scholar]
- 45.Seigneur, M., V. Bidnenko, S. D. Ehrlich, and B. Michel. 1998. RuvAB acts at arrested replication forks. Cell 95:419-430. [DOI] [PubMed] [Google Scholar]
- 46.Seigneur, M., S. D. Ehrlich, and B. Michel. 2000. RuvABC-dependent double-strand breaks in dnaBts mutants require RecA. Mol. Microbiol. 38:565-574. [DOI] [PubMed] [Google Scholar]
- 47.Sharma, B., and T. M. Hill. 1995. Insertion of inverted Ter sites into the terminus region of the Escherichia coli chromosome delays completion of DNA replication and disrupts the cell cycle. Mol. Microbiol. 18:45-61. [DOI] [PubMed] [Google Scholar]
- 48.Shurvinton, C. E., R. G. Lloyd, F. E. Benson, and P. V. Attfield. 1984. Genetic analysis and molecular cloning of the Escherichia coli ruv gene. Mol. Gen. Genet. 194:322-329. [DOI] [PubMed] [Google Scholar]
- 49.Skarstad, K., E. Boye, and H. B. Steen. 1986. Timing of initiation of chromosome replication in individual E. coli cells. EMBO. 5:1711-1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stukenberg, P. T., P. S. Studwell-Vaughan, and M. O'Donnell. 1991. Mechanism of the sliding β-clamp of DNA polymerase III holoenzyme. J. Biol. Chem. 266:11328-11334. [PubMed] [Google Scholar]
- 51.Thaler, D. S., E. Sampson, I. Siddiqi, S. M. Rosenberg, L. C. Thomason, F. W. Stahl, and M. M. Stahl. 1989. Recombination of bacteriophage λ in recD mutants of Escherichia coli. Genome 31:53-67. [DOI] [PubMed] [Google Scholar]
- 52.Villarroya, M., I. Pérez-Roger, F. Macián, and M. E. Armengod. 1998. Stationary phase induction of dnaN and recF, two genes of Escherichia coli involved in DNA replication and repair. EMBO 17:1829-1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu, C. A., E. L. Zechner, and K. Marians. 1992. Coordinated leading- and lagging-strand synthesis at the Escherichia coli DNA replication fork: I. Multiple effectors act to modulate Okazaki fragment size. J. Biol. Chem. 267:4030-4044. [PubMed] [Google Scholar]
- 54.Zyskind, J. W., A. L. Svitil, W. B. Stine, M. C. Biery, and D. W. Smith. 1992. RecA protein of Escherichia coli and chromosome partitioning. Mol. Microbiol. 6:2525-2537. [DOI] [PubMed] [Google Scholar]
- 55.Zyskind, J. W., and D. W. Smith. 1992. DNA replication, the bacterial cell cycle, and cell growth. Cell 69:5-8. [DOI] [PubMed] [Google Scholar]
- 56.Zyskind, J. W., and S. I. Bernstein. 1992. Recombinant laboratory manual. Academic Press, Inc., San Diego, Calif.